

Abstract
Tacrolimus exhibits inter-patient pharmacokinetic variability attributed to CYP3A5 isoenzymes and the efflux transporter, P-glycoprotein. Most African American renal transplant recipients require higher tacrolimus doses compared to whites to achieve similar troughs when race-adjusted recommendations are used. An established guideline provides tacrolimus genotype dosing recommendations based on CYP3A5*1(W/T) and loss of protein function variants: CYP3A5*3 (rs776746), CYP3A5*6 (rs10264272), CYP3A5*7 (rs41303343) and may provide more comprehensive race-adjusted dosing recommendations. Our objective was to develop a tacrolimus population pharmacokinetic model evaluating demographic, clinical and genomic factors in stable African American and white renal transplant recipients. A secondary objective investigated race-based tacrolimus regimens and genotype-specific dosing. Sixty-seven recipients receiving oral tacrolimus and mycophenolic acid ≥ 6 months completed a 12-hour pharmacokinetic study. CYP3A5*3,*6,*7 and ABCB1 1236T>C, 2677T>GA, 3435T>C polymorphisms were characterized. Patients were classified as extensive, intermediate and poor metabolizers using a novel CYP3A5*3*6*7 metabolic composite. Modeling and simulation was performed with NONMEM 7.3. A two-compartment model with first-order elimination and absorption with lag time best described the data. The CYP3A5*3*6*7 metabolic composite was significantly associated with tacrolimus clearance (P-value<0.05), which was faster in extensive (mean: 44.3 L∙hr−1) and intermediate (28.6 L∙hr−1) metabolizers than poor metabolizers (19.7 L∙hr−1). Simulations support CYP3A5*3*6*7 genotype-based tacrolimus dosing to enhance general race-adjusted regimens, with dose increases of 1.5-fold and 2-fold, respectively, in intermediate and extensive metabolizers for comparable exposures to poor metabolizers. This model offers a novel approach to determine tacrolimus dosing adjustments that maintain comparable therapeutic exposure between African American and white recipients with different CYP3A5 genotypes.
Keywords: Tacrolimus, Population pharmacokinetics, Race, CYP3A5 Genotypes
INTRODUCTION
Tacrolimus is a calcineurin inhibitor immunosuppressant, which is widely prescribed with mycophenolic acid (MPA),corticosteroids, or alternate immunosuppressant agents (e.g., azathioprine) to prevent allograft rejection in solid organ transplant recipients.1, 2 Tacrolimus exhibits a narrow therapeutic index with troughs ranging from 4 to 15 ng/ml for renal transplant recipients based upon prescribing full or calcineurin inhibitor minimization protocols.3-8 Considerable inter- and intra-patient variability in tacrolimus pharmacokinetics exists that can be attributed to race, sex, clinical factors and drug-drug interactions that impact allograft survival.3-6 Compared to whites, most African American transplant recipients require larger tacrolimus doses to achieve therapeutic trough concentrations.9-11 Therapeutic drug monitoring of trough concentrations is the standard of care to ensure patient treatment safety and allograft survival.8 However, there exists a poor correlation between tacrolimus dosage and troughs, requiring additional research into factors influencing patient drug exposure.3, 4, 68, 12
After oral administration, tacrolimus is rapidly absorbed and extensively bound to erythrocytes in the blood stream.13 Tacrolimus is highly metabolized in the liver and small intestine primarily by cytochrome P450 (CYP) 3A5 enzymes.6, 14, 15 It is also a substrate for the efflux transporter P-glycoprotein (P-gp).3, 6, 9 Fifteen metabolites of tacrolimus can be formed and excreted through the biliary route, with less than 1% of the parent drug remaining unchanged.16
Single nucleotide polymorphisms (SNPs) identified in the CYP3A5 and ABCB1 genes are associated with modulation of CYP3A5 enzyme and P-gp function respectively, which contribute to the inter-patient variability in tacrolimus pharmacokinetics.9, 17-19 Genetic variations in the CYP3A5 gene have explained 40 to 50% of the variability in tacrolimus clearance.9 The variant CYP3A5*3 (rs776746) has been commonly associated with reduction in the functional CYP3A5 enzyme or loss of protein function and is detected primarily in whites.20 In African Americans, CYP3A5 variants including CYP3A5*6 (rs10264272) and CYP3A5*7 (rs41303343), are also associated with loss of protein function, and may contribute to interracial variability in tacrolimus pharmacokinetics. 20, 21 The Clinical Pharmacogenetics Implementation Consortium (CPIC) has assigned CYP3A5 phenotypes based on *allele diplotypes.22 Patients are identified as extensive metabolizers (*1/*1), intermediate metabolizers (*1/*3, *1/*6, *1/*7), or poor metabolizers (*3/*3, *6/*6, *7/*7, *3/*6, *3/*7, *6/*7). Based on these phenotypes, CPIC has provided CYP3A5 genotype based tacrolimus dosing guidelines to help achieve target trough concentrations for solid organ transplants in adults and children. For CYP3A5 poor metabolizers, therapy should be initiated with the standard recommended dose, whereas for CYP3A5*1 expressers which include intermediate and extensive metabolizers, a suggested regimen should be increased to 1.5-2 times the standard dose without exceeding 0.3 mg/kg/day.22
The FDA initial dosing recommendations for oral tacrolimus are based on the type of organ transplant, race and time post-transplant, which is 0.1 mg/kg/day, administered as 0.05 mg/kg every 12 hours, for adult kidney transplant recipients in combination with mycophenolic acid.23 However, higher tacrolimus dosing regimens are recommended for black patients compared to whites based on the time post-transplant. These recommendations are supported by several reports comparing clinical troughs and bioavailability between African American and white healthy subjects and renal transplant recipients.24, 25
Over the last two decades, more than 50 population analyses have been conducted to investigate tacrolimus pharmacokinetics and identify significant covariates explaining inter-patient variability to better guide clinicians in defining an optimal dosing regimen.26 Factors commonly reported to influence tacrolimus pharmacokinetics include: total body weight (TBW), hematocrit, time post-transplant, hepatic function, and CYP3A5*3 polymorphisms.26 In addition, approximately 50% of population-based pharmacokinetic analyses were performed using tacrolimus trough concentrations with limited inclusion of critical patient covariates.26 To our knowledge, no population analysis has been conducted comparing whites and African Americans recipients.
The primary objectives of this study are to (i) develop a population pharmacokinetic model using intensively sampled tacrolimus profiles in stable African Americans and white renal transplant patients receiving tacrolimus minimization dosing with and MPA, and (ii) explore the effects of major demographic, clinical, and targeted genomic covariates on tacrolimus pharmacokinetic parameters. Using the final pharmacokinetic model, the secondary objective evaluates the CPIC and FDA tacrolimus dosing recommendations incorporating CYP3A5*3*6*7 genotypes and race.
METHODS
Ethical approval and study design
The study protocol was approved by the University at Buffalo Health Sciences Institutional Review Board before enrollment (IRB# PHP0599703-4). The clinical trial was conducted in accordance with the Declaration of Helsinki, and is consistent with the Principles of the Declaration of Istanbul as outlined in the “Declaration of Istanbul on Organ Trafficking and Transplant Tourism”. Written informed consent was obtained from all individual patients included in the study.
Sixty-seven stable renal transplant recipients (35 African Americans and 32 whites) receiving maintenance oral tacrolimus (Prograf®) and MPA as enteric-coated mycophenolate sodium (ECMPS; Myfortic®) for ≥ 6 months were included in a cross-sectional, open-label, 12-hour pharmacokinetic study. The study was conducted at the University at Buffalo Research Center at the Erie County Medical Center from February 2010 until June 2011. Clinical stability was based on physical exam, comprehensive metabolic panel, fasting lipid panel and complete blood count at enrollment and on study morning. Tacrolimus doses were administered twice daily and adjusted to 4 to 9ng/mL trough concentrations using a minimization dosing protocol, time post-transplant and clinical response. ECMPS dose was adjusted based upon clinical response. Medication adherence was verified at enrollment and as well as one week and 48 hours prior to the study by research personnel with medication diaries provided by patients. Ethnicity for two previous generations was verified. The inclusion criteria were: 1) ≥ 6 months post-renal transplant; 2) age 25-70 years; 3) first or second time deceased-donor or living allograft recipient; 4) stabilized on same dose of immunosuppressive drugs for ≥ 7 days prior to study; 5) serum creatinine ≤ 3.25 mg/dL with no change in serum creatinine > 0.25 mg/dL during prior 2 visits; and 6) leukocyte count ≥ 3000/mm3 and hemoglobin ≥ 8.0 g/dL. Exclusion criteria were: 1) infection within 2 weeks; 2) acute rejection within 2 weeks; 3) concomitant drugs interfering with tacrolimus or MPA absorption; 4) concomitant cytochrome P450 3A4/3A5 or P-gp inhibitors or inducers within 4 weeks; and 5) significant cardiovascular, gastrointestinal, hematologic, psychiatric, neurological or oncological diseases that would limit participation.
Patients were studied at steady-state concentrations of tacrolimus and ECMPS, with the receipt on the same dose for ≥ 7 days prior to study. Patients took immunosuppressives between 5:30 to 6:30 PM followed by a 12-hour fast prior to study. Patients were admitted at 6:00 AM, vital signs were documented and an intravenous angiocatheter inserted. A 0 hour blood sample (~15 mL) was collected prior to immunosuppressive administration for drug troughs and fasting laboratory tests. Study medications were administered orally from a single lot of tacrolimus and ECMPS by 7:00 AM. Patients remained upright throughout the study. Standardized low fat meals were provided after 4 hours. Anti-hypertensive drugs were administered after 1.5 hours, with insulin, anti-lipidemic, and other medications were administered 4 hours after the immunosuppressives. Blood samples (~7 mL) were collected at times 0 (pre-dose), 1, 2, 3, 4, 6, 8, 10, and 12 hours post-dose. Whole blood samples were aliquoted immediately and stored at -70°C until analysis.
An additional 20 ml of blood was collected in Cell Preparation Tubes (CPT®- BD Vacutainer) pre-dose with separation of peripheral blood mononuclear cells (PBMCs), which were harvested; immediately frozen in liquid nitrogen and stored at -70°C until genotype analysis.
Bioanalytics
Blood tacrolimus concentrations were measured using the ARCHITECT chemiluminescent microparticle immunoassay system (Abbott, Abbott Park, IL). Assay validation was completed with a calibration standard curve ranging from 1 to 30 ng/mL with 0.5 ng/mL as the lower limit of detection. Three quality controls (QC) were prepared at 3.0, 12.0, and 25.0 ng/mL (Bio-Rad, Hercules, CA, USA) and were co-analyzed with all samples and standards. The inter-day coefficient of variation (CV) for each QC was < 4%, and intra-day CV was < 5%. Selected trough and peak concentrations (N=40 samples) were analyzed using a validated LC/MS/MS assay by an external analytical laboratory and compared to the results generated from the ARCHITECT tacrolimus assay with excellent agreement (R2=0.98). For tacrolimus LC/MS/MS assay, the inter-day and intra-day CV were < 5% at the low and high concentration QC.
Genotyping assays
All patient samples were viable and analyzed in a genomics laboratory (University of New England Genomics, Analytics, and Proteomics Core). Laboratory personnel performing the genotyping analysis were blinded to patient treatment. Genomic DNA was isolated from PBMCs using the Wizard® Genomic DNA Purification system (Promega, Madison, WI) following manufacturer instructions. A total of 10 ng of genomic DNA was used in genotyping reactions for the following SNPs ABCB1: 1236C>T (rs1128503), 2677G>T/A (rs2032582), 3435C>T (rs1045642) and CYP3A5*3 (rs776746), CYP3A5*6 (rs10264272), and CYP3A5*7 (rs41303343) using TaqMan allelic discrimination assays (Applied Biosystems, Foster City, CA) with a CFX96 Real-Time Polymerase Chain reaction Detection System (Bio-Rad). Each SNP was analyzed in duplicate experiments. Allele frequencies were confirmed in Hardy-Weinberg equilibrium when adjusted for race.
CYP3A5*3*6*7 metabolic composite
CYP3A5 exhibits significant allelic heterogeneity. Three SNPs CYP3A5*3, CYP3A5*6 and CYP3A5*7 result in loss of protein gene expression. Loss of function by any of these SNPs can occur independent of allelic status at the other two loci. A CYP3A5*3*6*7 metabolic composite based upon the combined allelic status for each loss of function SNP was used to characterize each patient as extensive (cEMCYP3A5), intermediate (cIMCYP3A5) or poor metabolizers (cPMCYP3A5) as depicted in Figure 1. This composite followed the tacrolimus dosing guidelines provided by CPIC with the exception of the double heterozygous condition, which is not addressed by CPIC.22 Metabolic status using poor, intermediate and extensive metabolizer categories was also included for CYP3A5*1 and *3 genotypes only and are summarized in Supplemental Data.
Figure 1.
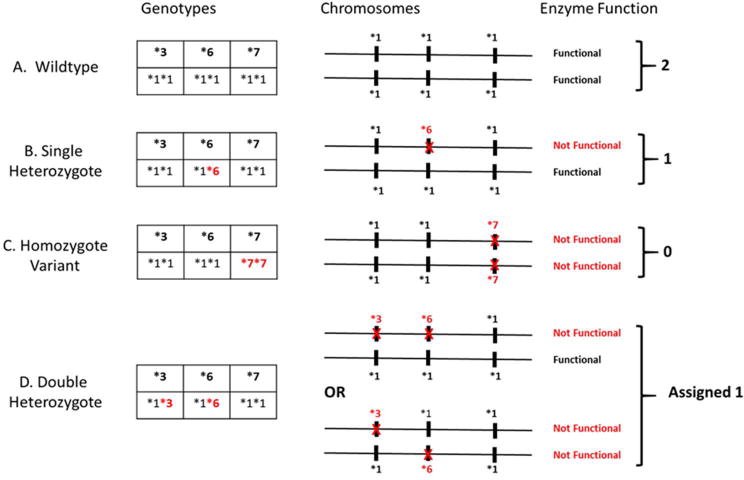
CYP3A5*3*6*7 Metabolic Composite scoring algorithm. Any of the three SNPs, CYP3A5*3, CYP3A5*6, and CYP3A5*7 independently result in loss of protein gene expression from the carrying chromosome. Metabolic composite status for each patient based upon the combined allelic status from each chromosome is summarized in fig. A–D above. A depicts an Extensive Metabolizer (CEMCYP3A5) with two completely functional genes. C depicts one possible example of a Poor Metabolizer (CPMCYP3A5): individuals who carry a loss of function allele on both chromosomes. Similarly B and D represents examples of possible Intermediate Metabolizers (CIMCYP3A5): individuals who are heterozygous at one or more loci. Note for the double heterozygote case (D) the genotyping assay used could not differentiate between cis and trans SNPs; therefore, the trans condition was conservatively assigned the intermediate metabolizer though such individuals would have no functional enzyme.
Pharmacokinetic modeling
Population pharmacokinetic modeling and simulation were performed using nonlinear mixed effects modeling with NONMEM 7.3.0 (ICON Development Solutions, Ellicott City, MD, USA). The first-order conditional estimation with interaction method was used to estimate the parameters. One- and two-compartment models with first-order absorption were compared, defined by apparent pharmacokinetic parameters in the absence of intravenous data. The use of transit compartments and a lag-time for describing drug absorption were explored. Between-subject variability (BSV) was assumed to be log-normally distributed and tested by an exponential model on pharmacokinetics parameters. Additive, proportional, and mixed residual variability models were tested. Model selection was based upon the goodness-of-fit plots, precision of parameters estimates (relative standard error; RSE), and changes in the minimum objective function value (OFV). The R-based tool Xpose 4.5.3 and GraphPad Prism 6.04 (GraphPad Software Inc, La Jolla, CA) were used for graphical presentations and statistical analysis.
Covariate analysis
Covariate analysis was performed to explore the potential effects of patient characteristics on model parameters associated with BSV. All patient characteristics listed in Table 1 were investigated as potential covariates with the exception of study dose and patient number. No data was missing among patients. Individual SNPs for ABCB1 and CYP3A5*3 polymorphisms as well as the CYP3A5*3*6*7 metabolic composite were examined. Significant covariates were selected using a classical stepwise approach.27 The influence of continuous covariates on the pharmacokinetic parameters was modeled according to a power model scaled to the population median covariate value. The influence of categorical covariates was modeled using a fractional change due to the covariate value. Model refinements (addition of BSV and correlations between BSV) were tested before and after the covariate analysis.
Table 1.
| Parameters‖ |
CYP3A5*3/*6/*7 metabolic composite
|
P-values† | |||
|---|---|---|---|---|---|
| Extensive | Intermediate | Poor | |||
| No. of Patients, n (%) | 8.0 (12) | 24 (36) | 35 (52) | – | |
| Study dose (mg) | 5.7 (1.5) | 3.9 (1.7) | 2.5 (1.1) | (Ext vs Poor) *** (Int vs Poor) ** |
|
| Race | African-American, n (%) | 8.0 (100) | 21 (88) | 6.0 (17) | *** |
| White, n (%) | 0.0 (0.0) | 3.0 (12) | 29 (83) | ||
| Sex | Male, n (%) | 4.0 (50) | 15 (63) | 19 (54) | ns |
| Female, n (%) | 4.0 (50) | 9.0 (37) | 16 (46) | ||
| Age (years) | 46 (8.3) | 49 (11) | 50 (13) | ns | |
| Total body weight (kg) | 89 (22) | 90 (21) | 85 (19) | ns | |
| Body mass index (kg/m2) | 31 (6.2) | 30 (6.8) | 30 (5.5) | ns | |
| Albumin (g/dL) | 4.1 (0.3) | 4.1 (0.2) | 4.1 (0.3) | ns | |
| Glucose (mg/dL) | 92 (19) | 122 (77) | 115 (69) | ns | |
| Triglycerides (mg/dL) | 134 (39) | 125 (67) | 154 (157) | ns | |
| Total Cholesterol (mg/dL) | 158 (24) | 150 (26) | 161 (51) | ns | |
| HDL (mg/dL) | 46 (14) | 52 (15) | 50 (19) | ns | |
| LDL (mg/dL) | 86 (21) | 74 (22) | 81 (32) | ns | |
| eGFR (mL/min/1.73m2) | 46 (14) | 54 (17) | 58 (14) | ns | |
| Serum creatinine (mg/dL) | 1.9 (0.6) | 1.6 (0.4) | 1.3 (0.3) | (Ext vs Poor)** (Int vs Poor) ** |
|
| Hemoglobin (g/dL) | 12 (1.2) | 12 (1.4) | 12 (1.4) | ns | |
| Hematocrit (%) | 37 (3.8) | 38 (4.7) | 38 (3.6) | ns | |
| Platelet (cells/mm3) | 231 (67) | 195 (59) | 196 (46) | ns | |
| Leukocytes (cells/mm3) | 5.7 (1.9) | 4.9 (1.6) | 5.4 (2.1) | ns | |
| ABCB1 1236T>C | CC, n (%) | 4.0 (50) | 11 (46) | 14 (40) | ns |
| CT, n (%) | 4.0 (50) | 7.0 (29) | 14 (40) | ||
| TT, n (%) | 0.0 (0.0) | 6.0 (25) | 7.0 (20) | ||
| ABCB1 2677T>G,A | GG, n (%) | 7.0 (88) | 17 (71) | 13 (37) | * |
| GT, n (%) | 1.0 (12) | 5.0 (21) | 15 (43) | ||
| TT, n (%) | 0.0 (0.0) | 2.0 (8.2) | 7.0 (20) | ||
| ABCB1 3435T>C CC, n (%) | 4.0 (50) | 13 (54) | 13 (37) | ns | |
| CT, n (%) | 4.0 (50) | 9.0 (38) | 15 (43) | ||
| TT, n (%) | 0.0 (0.0) | 2.0 (8.0) | 7.0 (20) | ||
| Time post-transplant (years) | 2.3 (1.8) | 3.4 (1.9) | 2.9 (3.0) | ns | |
| Diabete presence, n (%) | 2.0 (25) | 11 (46) | 12 (34) | ns | |
| ACEI use, n (%) | 1.0 (13) | 10 (42) | 9.0 (26) | ns | |
| ARB use, n (%) | 1.0 (13) | 3.0 (13) | 4.0 (11) | ns | |
| BB use, n (%) | 7.0 (88) | 21 (88) | 22 (63) | ns | |
| CCB use, n (%) | 6.0 (75) | 18 (79) | 20 (49) | * | |
| Prednisone use, n (%) | 2.0 (25) | 7.0 (29) | 5.0 (14) | ns | |
| Statin use, n (%) | 1.0 (13) | 10 (42) | 12 (34) | ns | |
Data represented as mean (standard deviation) or frequency (percentage)
HDL High-density lipoprotein, LDL Low-density lipoprotein, eGFR Glomerular filtration rate 47, ACEI Angiotensin converting enzyme inhibitor, ARB Angiotensin receptor blocker, BB Beta-blocker, CCB Calcium-channel blocker
P-value is based on χ2 test for categorical variables, and on Kruskal-Wallis test (Dunn’s multiple comparison test) for continuous variables. ns: non-significant,
p-value < 0.05,
p-value < 0.01,
p-value < 0.001.
Model evaluation
In addition to the standard goodness-of-fit plots, other methods were used to evaluate model performance. The precision of the final estimated pharmacokinetic parameters was evaluated using a nonparametric bootstrap resampling method.28 1000 replicates of the analysis dataset were generated by bootstrap and run with the final model to obtain the median and 95% percentile of all parameter estimates, which were compared to those of the final model. To evaluate the model predictive performance while accounting for different administered doses among patients and identified covariates, internal validation was performed using a prediction-corrected visual predictive check (PC-VPC).29 1000 replicates of the dataset were simulated conditional on the final pharmacokinetic parameters. The observed data and their 5th, 50th, and 95th percentiles were overlaid on the 90th confidence interval of the 5th, 50th, and 95th percentiles of the simulations to visually assess model performance. All data were corrected using the median of the population prediction within the associated time bin.
Dosing adjustment simulations based upon published guidelines
Using the final model, simulations (N = 1000) were conducted to explore the effect of significant covariates on tacrolimus exposure after oral administration of a standard dose of 0.075 mg/kg/day, given as 0.0375 mg/kg every 12 hours, which was selected as the most frequently administered standard tacrolimus dose based upon literature review.26 Individual exposures were assessed by the area under the concentration curve between 0 and 12 hours at steady state (AUCss,0-12hr):
with Dose and CL/F as the individual study dose and model-estimated apparent clearance.
To evaluate the CPIC tacrolimus dosing guidelines, simulations (N=1000) were performed for cIMCYP3A5 and cEMCYP3A5 with new dose levels consisting of 1.5 and 2-fold change compared to cPMCYP3A5. Tacrolimus exposure using simulated AUCss,0-12hr and 12-hour trough concentrations were compared across all CYP3A5*3*6*7 metabolic composite and dose level groups. They were also compared to the suggested therapeutic ranges for tacrolimus AUC of 120 to 200 hr·ng/mL and trough concentrations of 4-12 ng/mL targeted for kidney transplant recipients greater than 6 months post-transplant using a tacrolimus minimization protocol. These ranges were selected based upon the tacrolimus minimization protocol used by the UB Transplant Center, literature review.4, 7, 30-32 This analysis was also completed for CYP3A5*1 and *3 phenotypic groups.
Model simulations were also conducted based on the results reported in the FDA-approved monograph for African Americans and Caucasians for different time post-transplant.23 Tacrolimus exposure (AUCss,0-12hr) and trough concentrations were generated using individual pharmacokinetic parameters estimated from the model after administration of 0.13 mg/kg/day for whites and 0.19 mg/kg/day for African Americans (i.e., 0.065 and 0.095 mg/kg every 12 hours). Both were compared to the targeted therapeutic ranges.(20) This analysis was also completed for CYP3A5*1 and *3 phenotypic groups.
RESULTS
Data Summary and Patient Characteristics
A total of 594 tacrolimus steady-state concentrations from 67 patients were available for population pharmacokinetic analysis. The study cohort included 38 males and 29 females, 48% whites and 52% African Americans, with a median age of 46 years, and a median TBW of 85.9 kg. All concentrations were above the limit of quantification for the assay. Dose-normalized concentration-time profiles exhibited a large degree of variability within the CYP3A5*3*6*7 metabolic composite groups (Figure 2). The distribution of the CYP3A5*3*6*7 metabolic composite groups was highly correlated with race with 91% of whites identified as cPMCYP3A5 and 83% of African Americans identified as cIMCYP3A5 or cEMCYP3A5. Patient characteristics were grouped according to CYP3A5*3*6*7 metabolic composites and summarized in Table 1. The Supplemental Data (Section II) summarizes patient demographics and clinical characteristics in Table S2 stratified by CYP3A5*1 and *3 genotype groups.
Figure 2.
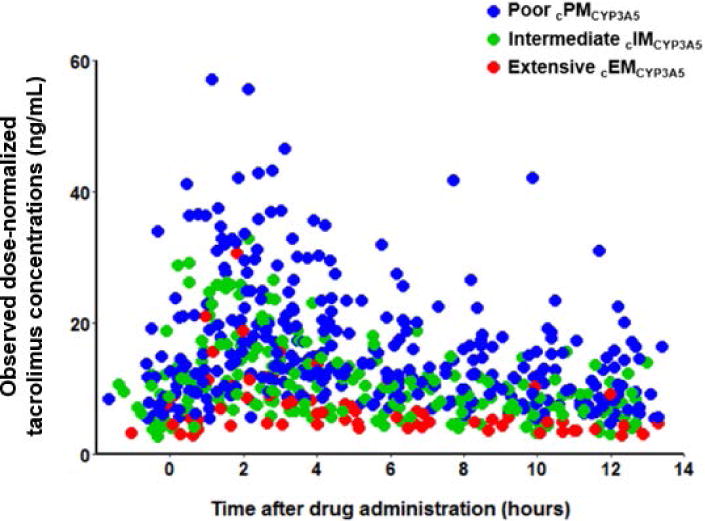
Observed tacrolimus dose-normalized concentrations (ng/mL) compared to time after drug administration stratified by CYP3A5*3*6*7 metabolic composite. For each patient, tacrolimus concentrations were divided by the recipient’s dose and multiplied by the mean study dose for the population which was 3.4 mg. The red, green, and blue dots represent individual tacrolimus concentrations for CYP3A5 extensive (cEMCYP3A5), intermediate (cIMCYP3A5) and poor metabolizers (cPMCYP3A5).
Population pharmacokinetic modeling
A two-compartment model with first-order absorption and elimination, and an absorption lag-time, best-described tacrolimus pharmacokinetics. BSV was included on the absorption rate constant (ka), central clearance and volume of distribution (CL/F, V/F), and intercompartmental clearance (CLp/F). The associated shrinkages were small (< 20%) and supported an unbiased covariate inclusion. A proportional error model best described residual variability. Visual inspection of individual pharmacokinetic parameter estimates versus covariates showed potential trends for CYP3A5*3*6*7 metabolic composite, race, African Americans females, and ABCB1-2677 polymorphisms on CL/F, serum creatinine on CLp/F, TBW and ABCB1-3435 polymorphisms on V/F, and presence of diabetes on ka. CYP3A5*3*6*7 metabolic composite exhibited the strongest effect on CL/F (Figure 3). In the forward building step, inclusion of CYP3A5*3*6*7 metabolic composite on CL/F, TBW on V/F, and diabetes on ka produced a significant decrease in OFV (> 3.84, P-value < 0.05). After backward elimination, only CYP3A5*3*6*7 metabolic composite and TBW were retained as significant covariates (P-value < 0.01), resulting in a total decrease in the OFV by 33.6 points (Supplemental Table S1). Individual CL/Fi and V/Fi values were predicted from the final model by the following equations:
in which H1 = 1 for cIMCYP3A5 and H2 = 1 for cEMCYP3A5 (0 otherwise), and ηi terms are the estimated BSV on CL/F and V/F.
Figure 3.
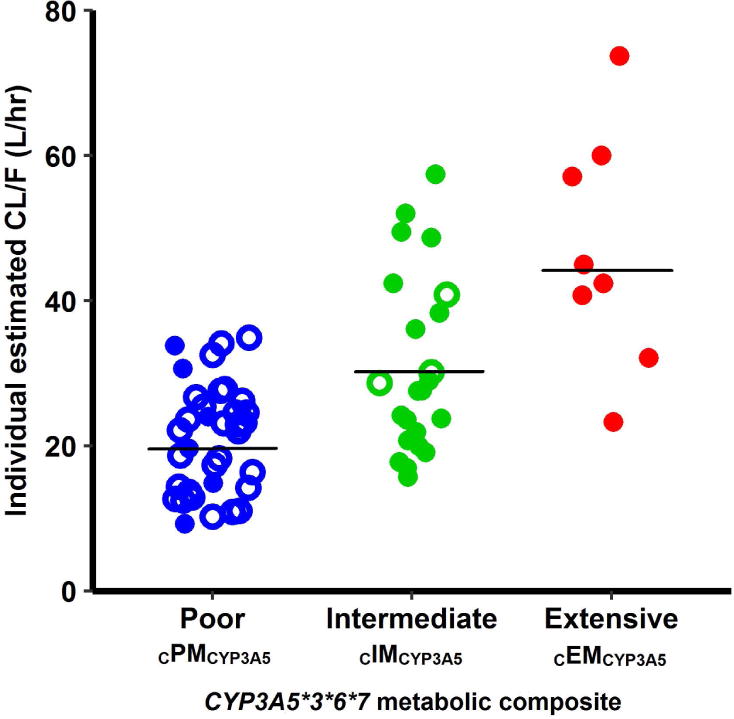
Individual base model estimated tacrolimus apparent clearance values (CL/F, symbols) and mean values (black lines) stratified by CYP3A5*3*6*7 metabolic composite. Poor metabolizers (CPMCYP3A5) exhibit lower CL/F (mean 19.8 L.hr−1), compared to intermediate CIMCYP3A5 (mean 29.5 L.hr−1) and extensive CEMCYP3A5 metabolizers (mean 45.0 L.hr−1). Open-circles represent white patients and closed-circles represent African-American patients.
Typical values of CL/F for cPMCYP3A5, cIMCYP3A5 and cEMCYP3A5 were 19.7, 28.6, and 44.3 L.hr1. Inclusion of covariates on CL/F and V/F reduced the magnitude of BSV from 46.6 to 37% (CL/F) and from 83.2 to 76.7% (V/F). No correlation between random effects was significant. All final pharmacokinetic parameters were well estimated (RSE < 30%) and are summarized in Table 2. The supplemental data summarized in Tables S3 and S4 reflect the model using CYP3A5 *1 and *3 genotype groups only. Although there is a slight bias for the population-level fit (Fig. S1a), the goodness-of-fit plots (i.e., observed concentrations versus population and individual predictions, conditional weighted residuals versus population predictions and time) did not show significant misspecification or bias in the final model (Supplemental Figure S1). Results of the nonparametric bootstrap are listed in Table 2. Mean values for all parameters were within 5% of those obtained from the final model. The 95% confidence interval was relatively narrow and did not include the value zero suggesting reliable model-parameter estimates. The PC-VPCs for all patients and stratified by CYP3A5*3*6*7 metabolic composite showed that the central tendency and variability of tacrolimus concentrations in the study population were well predicted by the final model (Figure 4).
Table 2.
Estimated population pharmacokinetic parameters of tacrolimus and bootstrap validation
| Population modeling | Bootstrap (n=1000) | ||||
|---|---|---|---|---|---|
| Parameter - Symbol | Estimates | RSE (%) | Median | 95% CI | Biasa (%) |
| CL/F (L.hr−1) − | 19.7 | 6.30 | 19.6 | 17.5 – 22.1 | −0.10 |
| CYP3A5*3*6*7 on CL/F | |||||
| CIMCYP3A5 − | 1.45 | 9.90 | 1.46 | 1.20 – 1.78 | 0.01 |
| CEMCYP3A5 − | 2.25 | 13.6 | 2.28 | 1.64 – 3.01 | 0.03 |
| V/F (L) − | 234 | 10.2 | 231 | 190 – 276 | −3.00 |
| CLp/F (L.hr−1) − | 52.6 | 9.50 | 53.2 | 44.1 – 63.6 | 0.60 |
| Vp/F (L) − | 403 | 20.1 | 404 | 275 – 572 | 1.00 |
| ka (hr−1) − | 4.21 | 20.3 | 4.14 | 2.77 – 5.74 | −0.07 |
| Lag time (hr) − | 0.828 | 3.80 | 0.830 | 0.740 – 0.873 | 0.002 |
| Between-subject variability | |||||
| BSV CL/F (CV%) − | 37.0 | 6.20 | 36.1 | 30.9 – 40.7 | −0.9 |
| BSV V/F (CV%) − | 76.7 | 12.1 | 76.0 | 56.0 – 93.5 | −0.7 |
| BSV CLp/F (CV%) − | 48.6 | 16.5 | 47.8 | 29.4 – 64.9 | −0.8 |
| BSV ka (CV%) − | 69.4 | 16.5 | 68.6 | 48.3 – 93.9 | −0.8 |
| Residual variability | |||||
| Proportional error − | 9.00 | 5.60 | 8.90 | 8.04 – 9.83 | −0.1 |
RSE (%) Percent relative standard error of the estimate; 95% CI 95th Confidence interval: 2.5 and 97.5th percentiles, BSV Between-subject variability expressed as percent coefficient of variation (CV%); CL/F =Apparent elimination clearance; CLp/F Apparent distribution clearance; V/F Apparent central volume of distribution; Vp/F Apparent peripheral volume of distribution; ka Absorption rate.
The bias of each parameter was calculated by computing the difference between the median value derived from the bootstrap and the final parameter estimate.
99% of the bootstraps runs minimized successfully (990/1000).
Final pharmacokinetic model (typical individual values):
and
where H1 = 1 for CYP3A5 intermediate metabolizers (cIMCYP3A5) and H2 = 1 for CYP3A5 extensive metabolizers (cEMCYP3A5). TBW Total body weight (kg)
Figure 4.
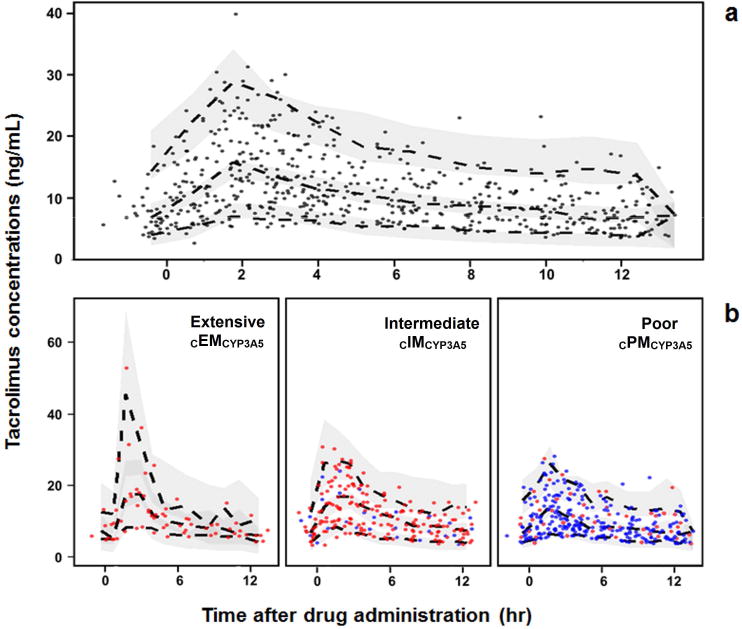
Prediction-corrected visual predictive check (PC-VPC) for the final tacrolimus pharmacokinetic model. PC-VPC for all patients a), and PC-VPC stratified by CYP3A5*3*6*7 metabolic composite b). Solid circles represent the observed data. The dashed lines represent the 5th, 50th, and 95th percentiles of the observed data, and the shaded areas represent the 90–percent confidence intervals around the 5th, 50th, and 95th percentiles of the predicted data. Symbol colors in panel b represent white (blue) and African-American (red) patients.
Dosing adjustment simulations using published guidelines
After administration of a common clinical dose of 0.075 mg/kg/day of tacrolimus, administered as 0.0375 mg/kg every 12 hours, model simulations showed differences in steady-state concentration-time profiles across CYP3A5*3*6*7 metabolic groups (Figure 5) suggesting a need for dosing adjustment. cPMCYP3A5 exhibited higher tacrolimus exposure (mean AUCss,0-12hr 170 hr.ng/mL) and trough concentrations (mean 10.9 ng/mL) than cIMCYP3A5 (AUCss,0-12hr 116 hr.ng/mL and trough 6.70 ng/mL) and EMCYP3A5 (AUCss,0-12hr 80.8 hr.ng/mL and trough 4.05 ng/mL).
Figure 5.
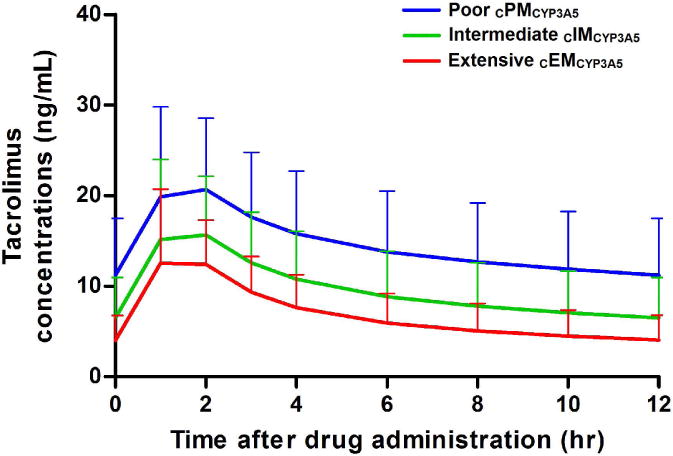
Simulations (n = 1000) of oral tacrolimus concentration-time profiles at steady-state after administration of 0.075 mg/kg/day (0.0375 mg/kg every 12 hours), represented by the mean and standard error bars according to CYP3A5*3*6*7 metabolic composite. Poor metabolizers (CPMCYP3A5) exhibit higher exposures (mean AUCss,0–12hr 170 hr.ng/mL) and 12 hr trough concentrations (mean 10.9 ng/mL) compared to intermediate(CIMCYP3A5) (AUCss,0–12hr 116 hr.ng/mL and trough 6.70 ng/mL) and extensive (CEMCYP3A5) metabolizers (AUCss,0–12hr 80.8 hr.ng/mL and trough 4.05 ng/mL).
Comparison with model-simulated tacrolimus exposures and troughs following 1.5- and 2-fold increase in doses for cIMCYP3A5 and cEMCYP3A5 are shown in Figures 6a and b. With the 0.075 mg/kg/day dosing regimen (0.0375 mg/kg every 12 hours), 47% of cPMCYP3A5 exhibited AUCss,0-12hr within the therapeutic range, whereas the majority of cIMCYP3A5 and cEMCYP3A5 showed underexposure of 62% and 88%, respectively. In 40% of the cIMCYP3A5 recipients using a 1.5-fold increased dose, similar tacrolimus exposures to cPMCYP3A5 were predicted, but a higher risk of toxicity or overexposure was determined in 52% of this group with a 2-fold higher dose. In 44% of cEMCYP3A5 recipients, a 2-fold higher dose predicted similar exposure to cPMCYP3A5.
Figure 6.
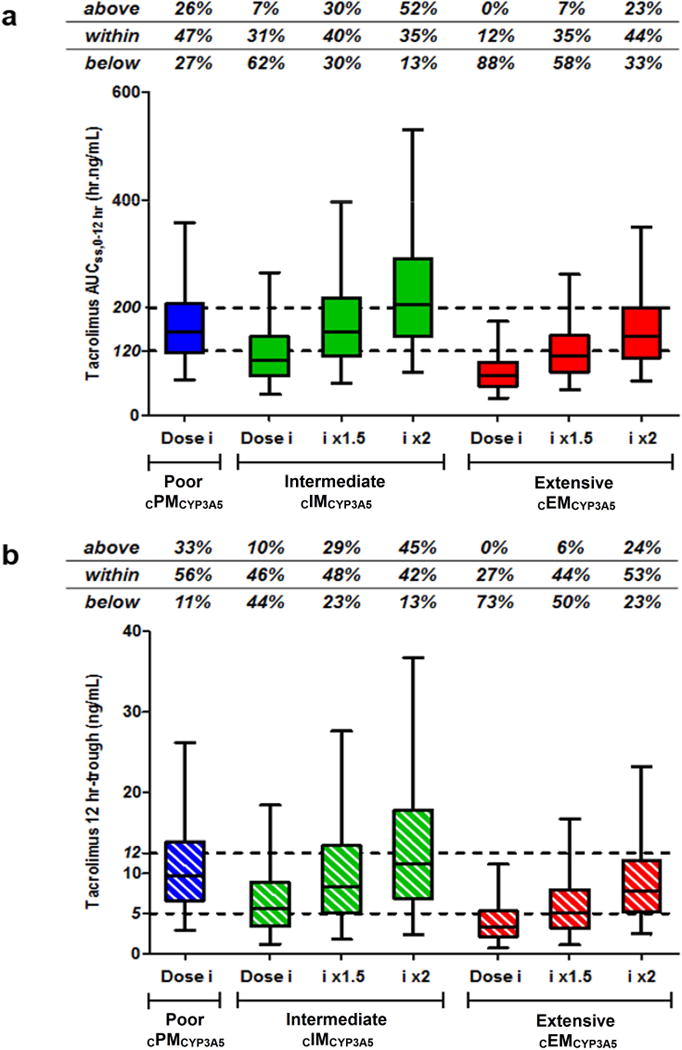
Boxplots of model-simulated tacrolimus AUCss,0–12hr a) and 12-hour trough concentrations b) stratified by CYP3A5*3*6*7 metabolic composite, obtained with different dosing regimens (i.e., Dose i = 0.075 mg/kg/day given as 0.0375 mg/kg every 12 hours, dose i x1.5, and x2).20 The dashed lines represent the targeted therapeutic ranges: between 120 and 200 hr.ng/mL for AUCss,0–12hr, and between 5 and 12 ng/mL for 12-hour trough concentrations.28 Percentages of patients exhibited AUCss,0–12hr and trough concentrations above, within and below the therapeutic range are indicated in each graph.
Model-simulated exposures and trough concentrations for African Americans in this study receiving a tacrolimus dose of 0.19 mg/kg/day (0.095 mg/kg every 12 hours) as recommended in FDA monograph are shown in Figures 7a and b. Most of African Americans cEMCYP3A5 had tacrolimus exposures within the therapeutic range. 81% and 100% of African Americans classified as cIMCYP3A5 and cPMCYP3A5 exhibited overexposure. Simulations for whites receiving a tacrolimus dose of 0.13 mg/kg/day resulted in overexposure for most patients (data not shown). For all simulations, similar conclusions were observed considering troughs.
Figure 7.
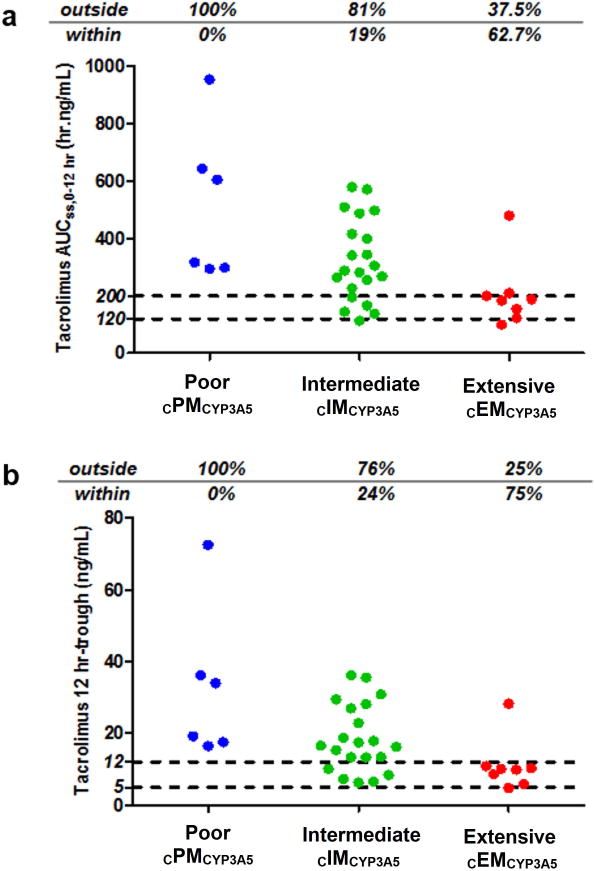
Individual model-simulated tacrolimus AUCss,0–12hr a) and 12-hour trough concentrations b) stratified by CYP3A5*3*6*7 metabolic composite for the study population African American patients receiving a dosing regimen of 0.19 mg/kg/day (0.095 mg/kg every 12 hours).21 The dashed lines represent the targeted therapeutic ranges: between 120 and 200 hr.ng/mL for AUCss,0–12hr, and between 5 and 12 ng/mL for 12-hour trough concentrations.28 Percentages of patients exhibited AUCss,0–12hr and trough concentrations above, within and below the therapeutic range are indicated in each graph.
The Supplemental Data summarizes the dosing adjustment simulations performed for CYP3A5*1 and *3 genotype groups in Figures. S6, S7, and S8.
DISCUSSION
This population-based pharmacokinetics model identifies a significant impact of multiple CYP3A5 genotypes on tacrolimus clearance using the novel CYP3A5*3*6*7 metabolic composite. Notable differences were found in tacrolimus pharmacokinetics in patients identified as cPMCYP3A5, cIMCYP3A5 and cEMCYP3A5, and reinforces the need for well-defined dosing adjustments to achieve targeted troughs and AUCss,0-12hr. The pharmacokinetic analysis resulted in a two-compartment model with delayed absorption and first-order elimination to describe the full concentration-time profiles. This model is consistent to other pharmacokinetic models using intensive sampling.26 Our final pharmacokinetic parameters and associated BSV are in agreement with previous estimates for tacrolimus.26 Most tacrolimus population pharmacokinetic models using only trough concentrations resulted in one-compartment models and results in predictive limitations.33-36 The effect of TBW was included on V/F with an allometric coefficient of 1 as previously reported.35, 37-39 Time post-transplant and hematocrit had no impact in our tacrolimus pharmacokinetic model in spite of significance found in previous models.26 This finding is likely due to the narrow inclusion criteria range for hemoglobin and hematocrit and the clinical stability of these patients.26
The distribution of CYP3A5 genotypes differs by race, which may explain differences in tacrolimus pharmacokinetics between whites and African Americans who require higher tacrolimus doses to achieve comparable concentrations.9, 10, 40 In our covariate analysis, the impact of race on tacrolimus clearance was significant until the inclusion of the CYP3A5*3*6*7 metabolic composite and a high correlation was found between these two covariates. White patients were primarily poor metabolizers (cPMCYP3A5) with none categorized as cEMCYP3A5. African Americans were mainly intermediate (cIMCYP3A5) and extensive (cEMCYP3A5) metabolizers. These findings are consistent with the incidence for the loss of function CYP3A5*3 variant of greater than 90% that is reported in whites and an approximate frequency of 30% found in African Americans.41 However, it is important to consider the other variant alleles CYP3A5*6 and CYP3A5*7 that also result in a loss of function of the respective enzymes. The CYP3A5*6 and CYP3A5*7 polymorphisms are rare in whites, but exhibit higher frequencies in African Americans of approximately 10%. Therefore, these variants contribute to the increased probability of non-functional enzymes resulting in poor metabolic phenotypes in African Americans and have important clinical implications.41 The different racial distributions of the CYP3A5 variants support the use of the CYP3A5*3*6*7 metabolic composite for more comprehensive investigation of tacrolimus dosing adjustments.
The CYP3A5*3*6*7 metabolic composite was the most significant covariate explaining tacrolimus pharmacokinetic variability in our model. Significant differences in CL/F were found relative to the three phenotypes with an approximate 1.5-fold and 2-fold increase for cIMCYP3A5 and cEMCYP3A5 compared to cPMCYP3A5. The impact of the loss of function variants CYP3A5*3, *6 and *7 on tacrolimus pharmacokinetics has been reported in two studies investigating only African American kidney transplant recipients using only trough concentrations.10, 21 Numerous tacrolimus population-based pharmacokinetic analyses were performed to explore the impact of the CYP3A5*3 genotypic variant alone, without consideration of CYP3A5*6 and *7 genotypes with limited inter-racial comparisons.26 Therefore, to assess the superiority of the CYP3A5*3*6*7 metabolic composite compared to CYP3A5*3 variant only, as source of tacrolimus pharmacokinetic inter-individual variability, we performed the same pharmacokinetic analysis considering CYP3A5*1 and *3 genotypes (See supplemental Data, section II). Significant differences on tacrolimus clearance were found between the three CYP3A5*3 genotypic groups: poor metabolizers (*3/*3), intermediate metabolizers (*1/*3) and extensive metabolizers (*1/*1) (Supplemental Figure S3). Tacrolimus clearance estimates for the CYP3A5*3 variant status were similar to those of the CYP3A5*3*6*7 metabolic composite (Supplemental Table S4). However, the inclusion of the metabolic composite explained more inter-individual variability for tacrolimus clearance. In addition, using the metabolic composite provided a comprehensive identification of patients with the loss of function variants since the presence of CYP3A5*6 and *7 are often missed in African Americans who may be categorized as intermediate or extensive metabolizers when the CYP3A5*3 variant is only analyzed.42 Unlike our analysis, the majority of the studies exploring the CYP3A5*3 variant, show differences in tacrolimus clearance between CYP3A5 non-expressers (*3/*3) or poor metabolizers in comparison to CYP3A5*1 expressers, which combine both intermediate (*1/*3) and extensive (*1/*1) metabolizers. These findings may be explained by the limited incidence of *1/*1 variant in other study populations that primarily enrolled white patients.37, 43-46 In these analyses, CYP3A5*1 expressers exhibited a 1.2- to 2.3-fold increase in tacrolimus clearance compared to CYP3A5 non-expressers.26 Significant differences between the three CYP3A5*3 genotype groups were found in two studies involving Korean adult35 and Mexican pediatric renal transplant patients.47 When combining these results with the African American recipients10, these analyses reported a 1.5 to 2 fold faster tacrolimus clearance in intermediate and extensive metabolizers using tacrolimus trough concentrations only. 10, 35, 47 Our results had a similar trend from the model developed using intensive sampling to generate a more accurate tacrolimus clearance.
Simulations were conducted with the final pharmacokinetic model to explore the impact of the CYP3A5*3*6*7 metabolic composite on tacrolimus steady-state exposure and trough concentrations (Figures 5 and 6). A standard dose of 0.075 mg/kg/day (0.0375 mg/kg every 12 hours) was selected for our simulations based on the studies reviewed by Brook et al.26 For comparison, the mean tacrolimus dose used in our study was 0.082 mg/kg. The primary goal of these simulations was to evaluate the dose adjustments required to achieve similar exposures between cPMCYP3A5, cIMCYP3A5 and cEMCYP3A5. Using the final model, simulations revealed that using an initial dose of 0.075 mg/kg/day (0.0375 mg/kg every 12 hours), about 50% of PMCYP3A5 would achieve steady-state exposures and troughs within the targeted therapeutic range (Figure 6).30 However, based on this dosing regimen, 62% of cIMCYP3A5 and 88% of cEMCYP3A5 were at a risk for sub-therapeutic exposures which may increase the risk of allograft rejection. The CPIC guidelines recommend a 1.5- to 2-fold increase in the standard tacrolimus dose for intermediate and extensive metabolizers to achieve target troughs. To further individualize the CPIC guidelines, our simulations suggest that well-defined dosing increases should be considered to include 1.5-fold for cIMCYP3A5 and 2-fold for cEMCYP3A5 using the standard dose (Figure 6). For instance, a 2-fold increased dose for cIMCYP3A5 could result in a high proportion of patients exceeding the therapeutic range with an increased risk for toxicities or over immunosuppression as depicted in Figure 6. Similar findings were obtained when performing the same simulations with the tacrolimus pharmacokinetic model based on the CYP3A5*1 and *3 variant (see supplemental Figures S6 and S7).
The updated FDA prescribing information suggests race adjusted tacrolimus dosing for black kidney recipients based on time post-transplant.23 Using our final pharmacokinetic model, simulations in the African Americans patients demonstrated that the FDA recommended tacrolimus dose regimen of 0.19 mg/kg twice daily at 12 months post-transplant could result in overexposure for all cPMCYP3A5 and 81% of cIMCYP3A5. See Figure 7. Similar results were also found with the same simulations performed with the pharmacokinetics model based on CYP3A5*3 variant alone (Supplemental Figure S8). Therefore, consideration of the pertinent CYP3A5 variants concurrent to race may optimize dosing strategy and subsequent regimen adjustments in the African American population. Other genotype factors may explain tacrolimus inter-patient variability, such as CYP3A4 variants and P-gp, which modulate tacrolimus metabolism and cellular distribution.48 The genotype, CYP3A4*22 (rs35599367) associated with reduced enzymatic activity has been combined with the CYP3A5*3 variant to define poor, intermediate and extensive metabolizers to explain tacrolimus pharmacokinetic variability.49, 50 These studies reported that well-defined doses for each metabolic group was required to achieve comparable tacrolimus exposure. In our model, P-gp may not have been adequately represented by using individual ABCB1 polymorphisms in the covariate analysis. Use of ABCB1 haplotypes may provide a more comprehensive investigation of this efflux transporter and the associated inter-patient pharmacokinetic variability of tacrolimus.51
Although this study included a small number of patients with no validation group included, the model developed has several advantages. This study was designed using intensive serial sampling strategy in clinically stable patients which provides more accurate estimates of tacrolimus pharmacokinetic parameters. All patients were enrolled based upon well-defined inclusion and exclusion criteria. In addition, medication adherence and steady state dosing conditions were carefully confirmed prior to study periods. All patients provided documentation of race for two previous generations.
CONCLUSION
The population pharmacokinetics model developed for tacrolimus identified the significant contribution of the CYP3A5*3*6*7 metabolic composite to predict inter-patient variability between white and African American renal transplant recipients. Differences between these groups can be explained by the racial differences in the distribution of CYP3A5*3*6*7 polymorphisms and metabolic enzyme activity. Well-defined dosing adjustments between the three metabolic phenotype groups comparing early to late post-transplant periods should be further investigated since extensive and intermediate metabolizers require higher doses than poor metabolizers. This model predicted tacrolimus exposures when applied to race specific FDA dosing guidelines and CPIC recommendations. This model predicted troughs and AUCss,0-12hr using a genotype-based dosing approach between African American and white transplant recipients to incorporate precision medicine into immunosuppression. Development of a Bayesian estimator based on this population pharmacokinetics model with targeted CYP3A5 genotypes could provide an efficient and feasible predictive tool for clinicians to facilitate personalized dosing of tacrolimus.
Supplementary Material
Acknowledgments
The assistance of the following individuals is greatly appreciated: Louise Cooper RPh, Vanessa Gray RN, Jean Meyers and Joseph Kabacinski, MBA from Erie County Medical Center and renal providers at the UB-MD Renal Division.
This study was supported by grants from NIDDK ARRA R21: DK077325-01A1 (KMT-PI) and an Investigator Initiated Research Grant (KMT-PI) from Astellas Pharma Global Development, Inc.
Footnotes
Disclosure:
The authors of this manuscript have no conflicts of interest or financial relationships to disclose during the time this study was ongoing.
References
- 1.Matas AJ, Smith JM, Skeans MA, et al. OPTN/SRTR 2013 Annual Data Report: Kidney. Am J Transplant. 2015;15(Suppl 2):1–34. doi: 10.1111/ajt.13195. [DOI] [PubMed] [Google Scholar]
- 2.Bowman LJ, Brennan DC. The role of tacrolimus in renal transplantation. Expert Opin Pharmacother. 2008;9(4):635–643. doi: 10.1517/14656566.9.4.635. [DOI] [PubMed] [Google Scholar]
- 3.Vanhove T, Annaert P, Kuypers DR. Clinical determinants of calcineurin inhibitor disposition: a mechanistic review. Drug Metab Rev. 2016;48(1):88–112. doi: 10.3109/03602532.2016.1151037. [DOI] [PubMed] [Google Scholar]
- 4.de Jonge H, Naesens M, Kuypers DR. New insights into the pharmacokinetics and pharmacodynamics of the calcineurin inhibitors and mycophenolic acid: possible consequences for therapeutic drug monitoring in solid organ transplantation. Ther Drug Monit. 2009;31(4):416–435. doi: 10.1097/FTD.0b013e3181aa36cd. [DOI] [PubMed] [Google Scholar]
- 5.Shuker N, Shuker L, van Rosmalen J, et al. A high intrapatient variability in tacrolimus exposure is associated with poor long-term outcome of kidney transplantation. Transpl Int. 2016;29(11):1158–1167. doi: 10.1111/tri.12798. [DOI] [PubMed] [Google Scholar]
- 6.Knops N, Levtchenko E, van den Heuvel B, Kuypers D. From gut to kidney: transporting and metabolizing calcineurin-inhibitors in solid organ transplantation. Int J Pharm. 2013;452(1–2):14–35. doi: 10.1016/j.ijpharm.2013.05.033. [DOI] [PubMed] [Google Scholar]
- 7.Ekberg H, Tedesco-Silva H, Demirbas A, et al. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med. 2007;357(25):2562–2575. doi: 10.1056/NEJMoa067411. [DOI] [PubMed] [Google Scholar]
- 8.Schiff J, Cole E, Cantarovich M. Therapeutic monitoring of calcineurin inhibitors for the nephrologist. Clin J Am Soc Nephrol. 2007;2(2):374–384. doi: 10.2215/CJN.03791106. [DOI] [PubMed] [Google Scholar]
- 9.Tang JT, Andrews LM, van Gelder T, et al. Pharmacogenetic aspects of the use of tacrolimus in renal transplantation: recent developments and ethnic considerations. Expert Opin Drug Metab Toxicol. 2016;12(5):555–565. doi: 10.1517/17425255.2016.1170808. [DOI] [PubMed] [Google Scholar]
- 10.Sanghavi K, Brundage RC, Miller MB, et al. Genotype-guided tacrolimus dosing in African-American kidney transplant recipients. Pharmacogenomics J. 2017;17(1):61–68. doi: 10.1038/tpj.2015.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Felipe CR, Silva HT, Machado PGP, Garcia R, da Silva Moreira SR, Pestana JOM. The impact of ethnic miscegenation on tacrolimus clinical pharmacokinetics and therapeutic drug monitoring. Clin Transplant. 2002;16(4):262–272. doi: 10.1034/j.1399-0012.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 12.Ekberg H, Mamelok RD, C PT, F V, H T-S, P D. The challenge of achieving target drug concentrations in clinical trials: experience from the Symphony study. Transplantation. 2009;87(9):1360–1366. doi: 10.1097/TP.0b013e3181a23cb2. [DOI] [PubMed] [Google Scholar]
- 13.Nagase K, Iwasaki K, Nozaki K, Noda K. Distribution and protein binding of FK506, a potent immunosuppressive macrolide lactone, in human blood and its uptake by erythrocytes. J Pharm Pharmacol. 1994;46(2):113–117. doi: 10.1111/j.2042-7158.1994.tb03752.x. [DOI] [PubMed] [Google Scholar]
- 14.Iwasaki K. Metabolism of tacrolimus (FK506) and recent topics in clinical pharmacokinetics. Drug Metab Pharmacokinet. 2007;22(5):328–335. doi: 10.2133/dmpk.22.328. [DOI] [PubMed] [Google Scholar]
- 15.Vanhove T, Annaert P, Kuypers D. Clinical determinants of calcineurin inhibitor disposition: a mechanistic review. Drug Metab Rev. 2016;48(1):88–112. doi: 10.3109/03602532.2016.1151037. [DOI] [PubMed] [Google Scholar]
- 16.Möller A, Iwasaki K, Kawamura A, et al. The disposition of 14C-labeled tacrolimus after intravenous and oral administration in healthy human subjects. Drug Metab Dispos. 1999;27(6):633–636. [PubMed] [Google Scholar]
- 17.Koch I, Weil R, Wolbold R, et al. Interindividual variability and tissue-specificity in the expression of cytochrome P450 3A mRNA. Drug Metab Dispos. 2002;30(10):1108–1114. doi: 10.1124/dmd.30.10.1108. [DOI] [PubMed] [Google Scholar]
- 18.Hesselink DA, van Schaik RHN, van der Heiden IP, et al. Genetic polymorphisms of the CYP3A4, CYP3A5, and MDR-1 genes and pharmacokinetics of the calcineurin inhibitors cyclosporine and tacrolimus. Clin Pharmacol Ther. 2003;74(3):245–254. doi: 10.1016/S0009-9236(03)00168-1. [DOI] [PubMed] [Google Scholar]
- 19.van Gelder T, van Schaik R, Hesselink D. Pharmacogenetics and immunosuppressive drugs in solid organ transplantation. Nat Rev Nephrol. 2014;10(12):725–731. doi: 10.1038/nrneph.2014.172. [DOI] [PubMed] [Google Scholar]
- 20.Kuehl P, Zhang J, Lin Y, et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001;27(4):383–391. doi: 10.1038/86882. [DOI] [PubMed] [Google Scholar]
- 21.Oetting WS, Schladt DP, Guan W, et al. Genomewide Association Study of Tacrolimus Concentrations in African American Kidney Transplant Recipients Identifies Multiple CYP3A5 Alleles. Am J Transplant. 2016;16(2):574–582. doi: 10.1111/ajt.13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birdwell KA, Decker B, Barbarino JM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin Pharmacol Ther. 2015;98(1):19–24. doi: 10.1002/cpt.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tacrolimus - FDA prescribing information, side effects and uses. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/050709s031lbl.pdf.
- 24.Neylan J. Racial differences in renal transplantation after immunosuppression with tacrolimus versus cyclosporine. FK506 Kidney Transplant Study Group. Transplantation. 1998;65(4):515–523. doi: 10.1097/00007890-199802270-00011. [DOI] [PubMed] [Google Scholar]
- 25.Mancinelli L, Frassetto L, Floren L, et al. The pharmacokinetics and metabolic disposition of tacrolimus: a comparison across ethnic groups. Clin Pharmacol Ther. 2001;69(1):24–31. doi: 10.1067/mcp.2001.113183. [DOI] [PubMed] [Google Scholar]
- 26.Brooks E, Tett SE, Isbel NM, Staatz CE. Population Pharmacokinetic Modelling and Bayesian Estimation of Tacrolimus Exposure: Is this Clinically Useful for Dosage Prediction Yet? Clin Pharmacokinet. 2016;55(11):1295–1335. doi: 10.1007/s40262-016-0396-1. [DOI] [PubMed] [Google Scholar]
- 27.Boeckmann AJ, Sheiner LB, Beal SL. NONMEM users guide, Part V, Introductory guide. San Fransisco (CA): University of California: NONMEM Project Group; 1994. [Google Scholar]
- 28.Parke J, Holford NH, Charles BG. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects population models. Comput Methods Programs Biomed. 1999;59(1):19–29. doi: 10.1016/s0169-2607(98)00098-4. [DOI] [PubMed] [Google Scholar]
- 29.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–151. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallemacq P, Armstrong VW, Brunet M, et al. Opportunities to optimize tacrolimus therapy in solid organ transplantation: report of the European consensus conference. Ther Drug Monit. 2009;31(2):139–152. doi: 10.1097/FTD.0b013e318198d092. [DOI] [PubMed] [Google Scholar]
- 31.Scholten E, Cremers S, Schoemaker R, et al. AUC-guided dosing of tacrolimus prevents progressive systemic overexposure in renal transplant recipients. Kidney Int. 2005;67(6):2440–2447. doi: 10.1111/j.1523-1755.2005.00352.x. [DOI] [PubMed] [Google Scholar]
- 32.Tornatore KM, Meaney CJ, Consiglio JD, Wilding G, Chang SS, Venuto RC. Sex And Race Influences on Tacrolimus Pharmacokinetics in Renal Transplant Recipients: American Society of Nephrology annual Meeting 2015. JASN. 2015;26(Suppl) [Google Scholar]
- 33.Antignac M, Barrou B, Farinotti R, Lechat P, Urien S. Population pharmacokinetics and bioavailability of tacrolimus in kidney transplant patients. Br J Clin Pharmacol. 2007;64(6):750–757. doi: 10.1111/j.1365-2125.2007.02895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Golubovic B, Vucicevic K, Radivojevic D, Kovacevic SV, Prostran M, Miljkovic B. Total plasma protein effect on tacrolimus elimination in kidney transplant patients–population pharmacokinetic approach. Eur J Pharm Sci. 2014;52:34–40. doi: 10.1016/j.ejps.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 35.Han N, Yun HY, Hong JY, et al. Prediction of the tacrolimus population pharmacokinetic parameters according to CYP3A5 genotype and clinical factors using NONMEM in adult kidney transplant recipients. Eur J Clin Pharmacol. 2013;69(1):53–63. doi: 10.1007/s00228-012-1296-4. [DOI] [PubMed] [Google Scholar]
- 36.Staatz CE, Willis C, Taylor PJ, Tett SE. Population pharmacokinetics of tacrolimus in adult kidney transplant recipients. Clin Pharmacol Ther. 2002;72(6):660–669. doi: 10.1067/mcp.2002.129304. [DOI] [PubMed] [Google Scholar]
- 37.Guy-Viterbo V, Baudet H, Elens L, et al. Influence of donor-recipient CYP3A4/5 genotypes, age and fluconazole on tacrolimus pharmacokinetics in pediatric liver transplantation: a population approach. Pharmacogenomics. 2014;15(9):1207–1221. doi: 10.2217/pgs.14.75. [DOI] [PubMed] [Google Scholar]
- 38.Staatz CE, Willis C, Taylor PJ, Lynch SV, Tett SE. Toward better outcomes with tacrolimus therapy: population pharmacokinetics and individualized dosage prediction in adult liver transplantation. Liver Transpl. 2003;9(2):130–137. doi: 10.1053/jlts.2003.50023. [DOI] [PubMed] [Google Scholar]
- 39.Sam WJ, Aw M, Quak SH, et al. Population pharmacokinetics of tacrolimus in Asian paediatric liver transplant patients. Br J Clin Pharmacol. 2000;50(6):531–541. doi: 10.1046/j.1365-2125.2000.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobson PA, Oetting WS, Brearley AM, et al. Novel polymorphisms associated with tacrolimus trough concentrations: results from a multicenter kidney transplant consortium. Transplantation. 2011;91(3):300–308. doi: 10.1097/TP.0b013e318200e991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bains RK, Kovacevic M, Plaster CA, et al. Molecular diversity and population structure at the Cytochrome P450 3A5 gene in Africa. BMC Genet. 2013;14:34. doi: 10.1186/1471-2156-14-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oetting W, Schladt D, Guan W, et al. Genomewide Association Study of Tacrolimus Concentrations in African American Kidney Transplant Recipients Identifies Multiple CYP3A5 Alleles. Am J Transplant. 2016;16(2):574–582. doi: 10.1111/ajt.13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Press RR, Ploeger BA, den Hartigh J, et al. Explaining variability in tacrolimus pharmacokinetics to optimize early exposure in adult kidney transplant recipients. Ther Drug Monit. 2009;31(2):187–197. doi: 10.1097/FTD.0b013e31819c3d6d. [DOI] [PubMed] [Google Scholar]
- 44.Benkali K, Rostaing L, Premaud A, et al. Population Pharmacokinetics and Bayesian Estimation of Tacrolimus Exposure in Renal Transplant Recipients on a New Once-Daily Formulation. Clin Pharmacokinet. 2010;49(10):683–692. doi: 10.2165/11535950-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 45.Moes DJAR, van der Bent SAS, Swen JJ, et al. Population pharmacokinetics and pharmacogenetics of once daily tacrolimus formulation in stable liver transplant recipients. Eur J Clin Pharmacol. 2016;72(2):163–174. doi: 10.1007/s00228-015-1963-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asberg A, Midtvedt K, van Guilder M, et al. Inclusion of CYP3A5 genotyping in a nonparametric population model improves dosing of tacrolimus early after transplantation. Transpl Int. 2013;26(12):1198–1207. doi: 10.1111/tri.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jacobo-Cabral C, Garcia-Roca P, Romero-Tejeda E, et al. Population pharmacokinetic analysis of tacrolimus in Mexican paediatric renal transplant patients: role of CYP3A5 genotype and formulation. Br J Clin Pharmacol. 2015;80(4):630–641. doi: 10.1111/bcp.12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Capron A, Mourad M, De Meyer M, et al. CYP3A5 and ABCB1 polymorphisms influence tacrolimus concentrations in peripheral blood mononuclear cells after renal transplantation. Pharmacogenomics. 2010;11(5):703–714. doi: 10.2217/pgs.10.43. [DOI] [PubMed] [Google Scholar]
- 49.Woillard JB, Mourad M, Neely M, et al. Tacrolimus Updated Guidelines through popPK Modeling: How to Benefit More from CYP3A Pre-emptive Genotyping Prior to Kidney Transplantation. Front Pharmacol. 2017;8:358. doi: 10.3389/fphar.2017.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andreu F, Colom H, Elens L, et al. A New CYP3A5*3 and CYP3A4*22 Cluster Influencing Tacrolimus Target Concentrations: A Population Approach. Clin Pharmacokinet. 2017;56(8):963–975. doi: 10.1007/s40262-016-0491-3. [DOI] [PubMed] [Google Scholar]
- 51.Venuto RC, Meaney CJ, Chang S, et al. Association of Extrarenal Adverse Effects of Posttransplant Immunosuppression With Sex and ABCB1 Haplotypes. Medicine (Baltimore) 2015;94(37):e1315. doi: 10.1097/MD.0000000000001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130(6):461–470. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.