

Abstract
Cell migration is strongly influenced by the organization of the surrounding 3D extracellular matrix. In particular, within fibrous solid tumors, carcinoma cell invasion may be directed by patterns of aligned collagen in the extra-epithelial space. Thus, studying the interactions of heterogeneous populations of cancer cells, that includes the stem/progenitor-like cancer stem cell subpopulation, and aligned collagen networks, is critical to our understanding of carcinoma dissemination. Here, we describe a robust method to generate aligned collagen matrices in vitro that mimic in vivo fiber organization. Subsequently, a protocol is presented for seeding aligned matrices with distinct carcinoma cell subpopulations and performing live cell imaging and quantitative analysis of cell migration. Together, the engineered constructs and the imaging techniques laid out here provide a platform to study cancer stem cell migration in 3D anisotropic collagen with real time visualization of cellular interactions with the fibrous matrix.
Keywords: Cancer Stem Cell, Cell Migration, 3D Collagen Matrices, Collagen Alignment
Introduction
Contact guidance is a prominent method of directed cell migration, observed in normal as well as diseased states, where cell movement is guided by the architecture and organization of the extracellular environment (Clark et al. 1991, Dunn and Ebendal 1978, Kim et al. 2012). In connective tissues and tumor stroma, extracellular matrix (ECM) structural anisotropy often appears in the form of parallel collagen fibers that generate aligned tracks for directional cell migration (Grinnell et al. 2006, Dickinson, Guido and Tranquillo 1994, Provenzano et al. 2006, Ray et al. 2017a). This is particularly important in breast cancer where aligned collagen architectures provide physical conduits for carcinoma invasion leading to metastasis and poor patient outcomes (Provenzano et al. 2006, Conklin et al. 2011). In addition, heterogeneity within cancer cell populations strongly influence invasion and metastatic capacity. In this context, cancer stem cells (CSCs) are a subpopulation of transformed cells that are thought to possess stem/progenitor-like properties, leading to increased tumor-initiating and metastatic capacity (Nguyen et al. 2012, Al-Hajj et al. 2003, Charafe-Jauffret et al. 2009). Thus, biophysical interactions between CSCs and their microenvironment, particularly during cell invasion, are a largely unexplored field that likely has profound implications in disease progression.
In our recently published work (Ray et al. 2017b), we report real-time biophysical interactions between breast carcinoma cells along with their CSC subpopulation and engineered 3D pro-invasive collagen patterns in vitro using two-photon microscopy for simultaneous live cell and collagen imaging. This unit describes the major techniques we employed or developed in (Ray et al. 2017b) including a novel method for generating biomimetic, aligned collagen tissue constructs, characterization of collagen matrix architecture, and subsequent live cell imaging and analysis of 3D cell migration.
NOTE: The protocols presented in this unit assume basic cell culture knowledge on the part of the end user such as sterile technique, culturing, detaching and counting adherent cells as well as access to related laboratory equipment such as biosafety cabinets, incubators, pipets, etc.
Basic Protocol 1: Fabrication of aligned and isotropic collagen matrices
The protocol for aligning collagen matrices by constrained fibroblast-mediated compaction (Ray et al. 2017b) is adapted from a previously reported method by Tranquillo and co-workers (Morin et al. 2013, Riemenschneider et al. 2016) to generate aligned microvessels in fibrin gels. Aligned matrices are generated by constrained compaction, while corresponding control isotropic matrices with randomly oriented fibers are formed by unconstrained compaction. Our findings show that this method is robustly applicable across multiple fibroblast cell types including commercially available cell lines (Ray et al. 2017b).
Materials
6-well tissue culture plate (e.g. Corning, cat. no. 353046)
24-well tissue culture plate (e.g. Corning, cat. no. 353047)
Stainless steel spoon spatula and microspatula
High-vacuum grease (UV sterilized) (Dow Corning)
Hydrophobic polyethylene sheet (Interstate specialty products, cat. no. POR-4896)
Benchtop glass bead sterilizer (e.g. Inotech Steri 250 Sterilizer)
Sub-confluent fibroblast cells on a standard tissue culture dish/flask (primary human adjacent normal breast fibroblasts (Asterand Bioscience) or primary mouse fibroblasts from mammary carcinoma or WI-38 lung fibroblasts (ATCC))
Culture medium for the chosen cell type (fibroblast lines used by authors were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), Penicillin/streptomycin and Plasmocin)
0.5% Trypsin/0.53 mM EDTA (e.g. Corning, cat. no. MT25052Cl)
1X Phosphate-buffered saline (PBS) (Calcium and magnesium-free) (e.g. ThermoFisher Scientific, cat. no. 10010-023)
High-density rat-tail collagen (Corning, cat. no. CB354249)
100 mM HEPES buffer in 2X PBS (e.g. ThermoFisher Scientific, cat. no. 15630080)
35 mm tissue culture plate (e.g. Corning, cat. no. 430165)
2 pairs of blunt, straight forceps
Prepare aligned gel templates
Cut 1.0 × 0.5 cm rectangular pieces (spacers) from the hydrophobic polyethylene sheet. Seal spacers in sterilization pouch and autoclave.
-
Trace 2.5 × 1.0 cm rectangular regions on the bottom surface of three wells of a 6-well plate.
The current protocol is designed for 3 aligned gel constructs. To make more, simply scale up.
-
Heat the flat end of a stainless steel spatula for approximately 20-30 seconds using a glass bead sterilizer at 300°C.
Hold spatula well away from heated end or wrap handle with insulating material to avoid burns.
-
Use the heated spatula to partially melt the well surface around the entire outlined region. Reheat spatula as necessary (Fig. 1A).
The well surface should be melted approximately a quarter to half-way to the bottom of the well. This melted periphery serves as a barrier to retain the cell-laden collagen mixture within the demarcated region rather than spilling out into the well but should not be so deep as to perforate the well.
-
The polyethylene spacers have a smooth and a porous surface. To each of a pair of autoclaved polyethylene spacers, apply vacuum grease to its smooth surface using a microspatula. Secure the two spacers porous-side up to the far ends of the etched rectangle (Fig. 1B).
Nonporous surface appears more reflective than porous surface and feels smooth to the touch.
Fig. 1. Engineered construct for collagen alignment.

(A) Modify wells in 6-well plates by etching out rectangular sections 2.5 × 1.0 cm in dimension on the bottom with the heated flat end of a spatula; (B) Attach hydrophobic, porous polyethylene pieces (spacers) at the two ends of the rectangular region with vacuum grease; (C and D) Plate the gel mixture onto the spacers before drawing the mixture out onto the rectangular region, allowing the two ends to meet in the middle; (E) Allow the gel to start setting at room temperature for 20 mins and then carefully transfer to the 37°C incubator.
Prepare fibroblast-seeded collagen gels
-
From a sub-confluent culture of fibroblasts, detach cells, bring them to a single cell suspension, and calculate the cell density.
We have successfully used primary human adjacent normal breast fibroblasts (Asterand Bioscience), primary mouse fibroblasts from mammary carcinoma and WI-38 lung fibroblasts from ATCC), however any contractile fibroblast line should be sufficient to produce aligned matrices with appropriate optimization of cell density and contraction time (Ray et al. 2017b).
-
Calculate the volume of high-density collagen needed to produce 1.2 mL of 3 mg/mL collagen. An equal volume of 100 mM HEPES in 2X PBS will be used to neutralize the collagen, and cell culture medium with cells will make up the remainder of the volume.
For example, for a high-density collagen stock of 10 mg/mL, mix 360 μL of collagen, 360 μL of 100 mM HEPES in 2X PBS, and 480 μL of cells in culture medium to achieve a final volume of 1.2 mL.
-
Calculate the volume of the counted single cell suspension that contains 240,000-360,000 cells. Transfer this volume to a 15 mL conical tube and centrifuge for 3 minutes at 240 RCF. Remove supernatant and resuspend cell pellet in culture medium to the previously calculated volume necessary to make up the total volume of collagen mixture
For example, resuspend pellet in 480 μL culture medium for the previous example
For human or mouse fibroblasts seeding densities of 200,000 cell/mL or 300,000 cell/mL respectively have been generally used by the authors.
-
On ice, mix the calculated volumes of collagen and 100 mM HEPES in 2X PBS in a 1.5 mL microcentrifuge tube. Add the cell/media solution from step 3 and mix thoroughly without introducing bubbles. Allow cell-seeded collagen mixture to begin polymerizing for 5 minutes at room temperature.
To account for collagen viscosity, pipet slowly. When dispensing suspension, allow collagen to pool at the base of the pipet tip for several seconds before clearing the pipet.
-
Collect 350 μL of cell-seeded collagen mixture into a pipet tip. Apply approximately 150 uL to each spacer to completely cover each of the spacers of one demarcated rectangular region. Allow mixture to seep into the porous material for 5-10 seconds. Using the pipet tip, draw collagen out of each of the spacers and into the rectangular region such that the collagen meets in the middle and fills the demarcated region (Fig. 1C, D). Repeat in the two remaining rectangular regions of the 6-well plate.
To avoid excessive collagen polymerization within the tube, this step should be completed within approximately 5 minutes.
For isotropic controls (i.e., collagen gels prepared similarly but without anisotropic or biased distribution of fibers), pipet 350 μL of cell-seeded collagen mixture into a 24-well plate well. Note that the total volume of the gel mixture is ~15% more than the total volume pipetted to make the constructs. This additional volume accounts for pipetting errors and the gel mixture that remains stuck to the walls of the microcentrifuge tube and the pipet tip.
-
Allow gels to partially polymerize at room temperature for 20 minutes (Fig. 1E), and then transfer plate to a 37°C incubator for 3 hours.
Be very careful that the gelling collagen does not spill out of the rectangular region during the transfer.
-
Overlay gels with 3 mL of growth medium (enough to submerge the construct entirely) and return to 37°C incubator for 16-24 hours to allow for vertical compaction.
For isotropic controls, overlay with 1 mL of growth medium.
Detach and transfer gels
Trace a 1.5 × 1.0 cm region on the bottom of a 35 mm tissue culture plate. Apply vacuum grease beyond the far ends of the rectangle (Fig. 2D inset).
Aspirate the media from one of the wells containing a polymerized collagen construct.
-
Using two straight tweezers, grip each of the spacers and gently detach from the bottom of the plate (Fig. 2A). Slide the spacers towards the center of the well, such that the collagen is resting on top of the spacers (Fig. 2B). Gently slide the gel back and forth to ensure detachment from the bottom of the well.
The collagen gel is highly fragile, and should be handled gently. Avoid pinching collagen between the spacers as this will damage the gel.
With the gel resting on top of the spacers, carefully lift the collagen construct from the 6-well plate and transfer it to the center of the 35 mm plate (Fig. 2C, D).
-
Slowly pull one of the spacers from the center of the plate towards the pre-applied vacuum grease and secure the spacer to the bottom of the plate (Fig. 2E). Repeat with the other spacer (Fig. 2F).
The aligned gel must be detached from the surface of the well and the spacers must be re-secured in order for the gel to undergo lateral compaction necessary for collagen reorganization. Since the original well will be slightly wet after aspiration of the media, the gel must be transferred to a fresh, dry surface in order to re-secure the spacers with vacuum grease.
Overlay gel with 3 mL of growth medium.
Repeat for remaining gels.
For isotropic controls, run a 10 μL pipet tip around the edge of the well. Gently tap the bottom of the plate to detach gel.
-
Transfer the detached gels to a 37°C incubator for 24-48 hours to allow for lateral compaction.
Limit liquid movement during transfer to avoid detaching gels from the spacers. By and large, the appearance of a typical taut, dog-bone shape is a good indicator that the matrix has been optimally aligned. Leaving the gels to contract further may lead to detachment from the spacer(s) wherein the fiber organization will be lost quickly thereafter. However, note that the macroscopic shape change is neither a necessary nor a sufficient condition for fiber alignment. The optimal amount of incubation time must be determined on a case-to-case basis and fiber reorganization may be monitored by imaging collagen fibers.
Fig. 2. Detachment and transfer of aligned matrix constructs.

(A) Hold the vertically compacted gel at the spacers by two pairs of blunt forceps; (B) Detach the spacers from the bottom, slide the gel along with the spacers to detach the gel completely from the bottom; (C) With the gel resting on top of the held spacers, transfer the gel to a new 35 mm plate; (D inset): A fresh 35 mm plate prepared beforehand by applying vacuum grease at 2 locations 1.5 cm apart; (D) The transferred gel resting on the 35 mm plate; (E) Transferred gel with one end pulled out and secured with the previously applied vacuum grease; (F) Transferred gel with both ends pulled out and secured.
Basic Protocol 2: Decellularization and characterization of engineered matrices
The aligned constructs are generated using fibroblast-mediated contractility. In order to seed cells of interest and study their behavior in these matrices, one needs to first remove the matrix-reorganizing cells from the constructs. This is achieved by a two-step decellularization protocol involving cell lysis in a hypotonic, detergent-containing solution and subsequent degradation and removal of nucleic acids (Hoshiba et al. 2012, Ray et al. 2017b).
Materials
1X PBS (Calcium and magnesium-free) (e.g. ThermoFisher Scientific, cat. no. 10010-023)
ddH2O (Tissue-culture grade)
Cell lysis solution (See Reagents and Solutions)
RNase/DNase solution (See Reagents and Solutions)
Slice anchors (optional; e.g. Warner Instruments, cat. no. 64-0256)
Rhodamine Phalloidin (or other variants; e.g. ThermoFisher Scientific, cat. no. A22287)
Hoechst dye (or other variants; e.g. ThermoFisher Scientific, cat. no. H3750)
Proteinase K (e.g. Millipore Sigma, cat. no. 3115844001)
Reagents for extraction of DNA
Reagents and solutions
- Cell lysis solution
- 1.0% v/v Triton X-100 (e.g. Millipore Sigma, cat. no. 10789704001)
- 5 mM EDTA (e.g. Corning, cat. no. 46-034-Cl)
- 10 mM Tris-HCl (pH 7.6) in ddH2O (e.g. Promega, cat. no. PR-H5131)
- Nuclease solution
- 300 μg/mL DNase I (e.g. Applichem, cat. no. NC0208431)
- 300 μg/mL RNase A in 1X PBS (e.g. ThermoFisher Scientific, cat. no. 12091021)
Cell Lysis
-
Remove medium from each contracted aligned or isotropic collagen matrix
Be careful not to damage the matrix while aspirating the media; we recommend using a pipet for this step instead of a vacuum aspirator.
-
(Optional) Apply a tissue slice anchor over the gel to limit detachment of gel from spacers during rinse steps (Fig. 3A, B).
Keeping the aligned matrix attached to the spacers is one way of immobilizing the gel during subsequent reseeding and imaging steps. Alternately, one could simply immobilize it with a slice anchor in subsequent reseeding and imaging steps.
Rinse serum-containing media from the gel by applying 1X PBS for 1 minute. Remove PBS and rinse in fresh PBS for 3 minutes, then repeat for 5 minutes.
-
Completely submerge gel in ice-cold cell lysis solution and incubate at 4°C for 24 hours.
The sample may be shaken gently in an orbital shaker during this and subsequent steps; however, it usually results in detachment of the aligned constructs from the spacers.
Fig. 3. Handling and imaging of decellularized matrices.
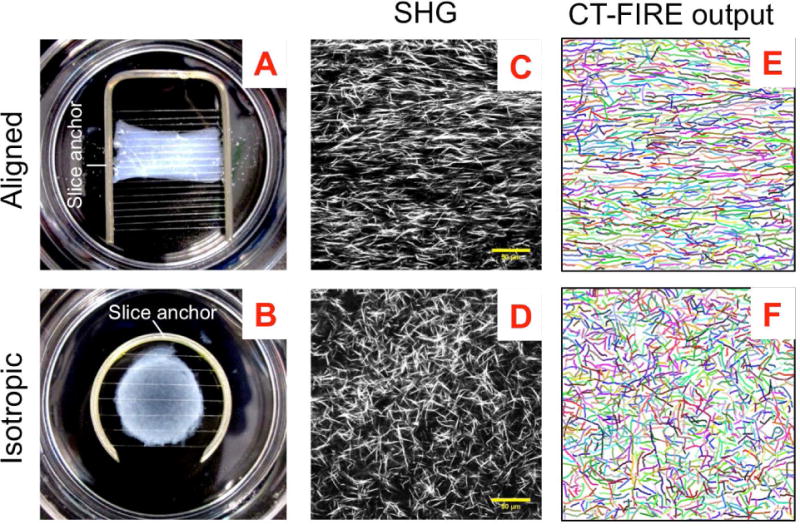
(A and B) Aligned and isotropic collagen constructs on tissue culture dishes immobilized with slice anchors for handling and imaging. (C and D) SHG images showing typical collagen fiber organization in the aligned and isotropic constructs and (E and F) corresponding CT-FIRE software output showing recognized fibers whose length, width, straightness and angular distribution can be obtained. (Scale bar, 50 um)
Nucleic acid removal
Remove cell lysis solution and rinse gel in ddH2O for 3 minutes, 5 minutes x3, then 10 minutes, replacing ddH2O after each wash to remove any residual cell lysis solution.
Completely submerge gel in nuclease solution and incubate at 37°C for 12–16 hours. It is possible that the RNase may be dispensable in this step since RNA naturally is quite unstable in the extracellular environment.
Remove nuclease solution and rinse gel in 1X PBS for 3 minutes, 5 minutes x3, then 10 minutes, replacing PBS after each wash.
Store gels in 1X PBS, sealed and at 4°C until imaging can be performed.
Assessment of decellularization efficiency
-
1
To assess the efficiency of decellularization, cut a sample aligned matrix into two halves. Perform the abovementioned decellularization steps on one half while leaving the other half non-decellularized by adding ddH2O and 1X PBS only instead of the cell lysis and nuclease solution, respectively.
-
2a
Fix and stain both fragments for F-actin (phalloidin) and DNA (Hoechst dye) by standard methods (Ray et al. 2017b, Provenzano et al. 2008)
or,
-
2b
Extract DNA from both fragments by proteinase K digestion and subsequent phenol-chloroform extraction and isopropanol precipitation using standard methods.
-
3a
Image and quantify amount of F-actin retained from fluorescence intensities.
or,
-
3b
Quantify the amount of DNA retained by measuring extracted DNA using a spectrophotometer.
Characterization of collagen fiber morphology and organization
Image the collagen fibers in the decellularized aligned and isotropic constructs by second harmonic generation (SHG) at 880 nm two-photon excitation with acquisition in the blue channel (Fig. 3C, D)
-
Process the SHG images using CT-FIRE (http://loci.wisc.edu/software/ctfire), a freely available fiber quantification software with enhanced edge detection capabilities to obtain length, width straightness and angle of each individual fiber (Fig. 3E, F).
Compare the original and the fiber-extracted images to ensure accuracy of the software’s output.
In general, default settings were used, with the exception that ‘fiber line width’ was changed to 1.5. This parameter would have to be adjusted on a case to case basis depending on the signal-to-noise ratio of the images acquired. For more information regarding the use of this software, please consult the user manual on the website listed above.
Plot and analyze fiber angle distributions to quantify the degree of alignment in several fields of view for each construct.
Basic Protocol 3: Timelapse imaging and analysis of cell migration
Timelapse imaging allows visualization of cell dynamics, specifically, in this case, cellular movement through isotropic or anisotropic 3D collagen networks and the interaction of collagen fibers with individual cells. The protocol below is specifically designed for timelapse imaging on a multiphoton microscope with an upright water immersion lens, and overcoming specific challenges this setup poses in maintaining the temperature, pH and fluid interface during the experiment. Alternate methods are also mentioned wherever appropriate. Likewise, the post-processing and 3D cell tracking is done entirely in the freely available Fiji software. Alternately, commercially available software such as Imaris may be used to perform 3D rendering and cell tracking.
Materials
Ultra-low adhesion 6-well plates (Corning, cat. no 07-200-601)
Breast cancer stem cell (BCSC) media (See reagents and solutions below)
0.25% Trypsin/2.21 mM EDTA (e.g. Corning, cat. no. MT25053Cl)
1X PBS (Calcium and magnesium-free) (e.g. ThermoFisher Scientific, cat. no. 10010-023)
Slice anchors (optional; Warner Instruments) or porous polyethylene spacers (see Basic Protocol 1)
Leibovitz’s L-15 medium with 10% FBS (e.g. ThermoFisher Scientific, cat. no. 11415064)
Heating element with 35mm clearance and corresponding O-rings (Bioscience Tools)
Temperature controller (Bioscience Tools)
Miniature temperature probe (Bioscience Tools)
Parafilm
Razor blade
Adhesive paper tape
Bubble wrap or objective heater
Matlab or equivalent mathematical computing software
Reagents and Soutions
BCSC media
DMEM/F12 (1:1) (e.g. ThermoFisher Scientific, cat. no. 10565018)
2% B27 supplement (e.g. ThermoFisher Scientific, cat. no. 17504044)
20 ng/mL Epidermal Growth Factor (e.g. Corning, cat. no. 354001)
5 μg/mL insulin (e.g. Millipore Sigma, cat. no. I1882)
10 ng/mL human basic fibroblast growth factor (e.g. Peprotech, cat. no. 100-18C)
Seeding cells into the decellularized matrix
-
1.a.i
Grow stable GFP-expressing MDA-MB-231 cells on a standard tissue culture dish (whole population) until 80-90% confluent.
Refer to (Ray et al. 2017a, Ray et al. 2017b) for methods to stably transfect MDA-MB-231 cells with GFP. Alternately, cell permeable dyes such as CellTracker (Thermo Fisher) may be added prior to the experiment.
Or,
-
1.b.i
Plate GFP-expressing MDA-MB-231 cells on ultra-low adhesion plates in 2 mL special BCSC media (see recipe above) at a density of 20,000 cells per 6-well and leave undisturbed for 4 days to allow cells to form CSC-enriched spheroids.
Adherent cultures will contain a heterogeneous population of carcinoma cells, while the low-adhesion cultures will be enriched for cells with the CSC phenotype. Cells from adherent culture can be used as a control to assess shifts in CSC behavior compared to the whole cell population.
Plate out at least 6 such wells to get sufficient cell numbers after 4 days. Alternately, one could use a plating density of 10,000 per well and use 12 such wells. When using a cell line that typically has a very low percentage of CSCs, serial passaging of the spheroids can be done to improve CSC enrichment in the spheroids.
-
2.a.i
Trypsinize adherent MDA-MB-231 cells from the tissue culture dish, count and bring to a single cell suspension in 1 mL complete medium.
Or,
-
2.b.i
Carefully aspirate MDA-MB-231 spheroids out of the low adhesion wells, pool all the fluid together and centrifuge.
To collect spheroids only beyond a certain size, one can filter the spheroid suspension through the appropriate sized cell strainer and the use the retained spheroids.
-
2.b.ii
Aspirate the supernatant and resuspend the pellet in 0.5 mL 1X PBS (without calcium and magnesium) and centrifuge.
-
2.b.iii
Aspirate the supernatant and resuspend the pellet in 0.5 mL 0.25% trypsin/2.21 mM EDTA and incubate at 37°C for at least 5 and up to 10 mins, with intermittent agitation by pipetting up and down every 1-2 mins until the spheroids are broken into a single cell suspension.
The successful dissociation of the spheroids into a single cell suspension may be monitored by intermittently pulling out 2-3 μL of the suspension onto a coverslip/dish and examining the cells under the microscope.
Enzymatic dissociation of spheroids is known to degrade cell membrane proteins, however only a handful of studies have reported non-enzymatic mechanical dissociation methods for spheroids and have not been explored by the authors.Enzyme-free dissociation solutions have been explored but have not been used successfully by the authors.
-
2.b.iv
Count the number of cells, neutralize the trypsin with 1.5 mL complete serum-containing medium, centrifuge and resuspend the pellet in 0.1-0.5 mL complete medium.
-
3
Immobilize decellularized isotropic control or aligned matrices onto a 35 mm tissue culture plate before cell seeding.
The aligned constructs could be left attached to the polyethylene posts for this experiment. In that case, the corresponding control matrices would also need to be tacked down with the same polyethylene spacers using vacuum grease. Alternately, both the aligned and control matrices may be immobilized by placing slice anchors on top of the gels (Fig. 3A, B)
-
4
Plate 100,000 cells of either kind onto the 35 mm dish containing the immobilized aligned or control matrix, and allow cells to seed and migrate into the matrix for 48h before imaging.
Timelapse imaging of cells and surrounding collagen matrix
-
Immediately prior to imaging, aspirate DMEM-based growth medium and overlay cells in L-15 medium supplemented with 10% FBS.
L-15 is a CO2-independent medium buffered in air. This is ideal for use in an imaging setup that is not equipped with a 5% CO2 environmental chamber. L-15 media in our hands worked very well for various cancer cell lines (ATCC recommends this medium for culturing MDA-MB-231 cells). One can check other cell types for their compatibility to L-15 by growing them side by side in L-15 and the standard growth medium. Note that cells in L-15 must be incubated in an air-buffered incubator for proper maintenance of pH. Switching to L-15 medium during the experiment is dispensable if the microscope is setup with a CO2-controlled environmental chamber.
-
Affix the designated O-ring around the 35 mm dish containing the sample to be imaged, and then place the ensemble onto the heating element on the microscope stage (Fig. 4A, B)
The heating element provides temperature control and is not necessary when a temperature controllable environmental chamber is built-in to the setup.
Cut out a square piece of parafilm about 2″ in length. In the center of the square, cut out a small approximately circular hole slightly smaller than the diameter of the objective lens using a razor blade. Attach paper tapes to the corners of the piece of parafilm. (Fig. 4C inset)
-
Carefully affix the parafilm to the objective like a collar so that the lens sticks out through the hole in the parafilm (Fig. 4C). In this configuration, bring down the objective into the medium and focus on the cells. (Fig. 4D)
The parafilm is meant to provide a flexible lid that allows the objective to pass through it. This helps to limit evaporation and fluid loss when performing long duration timelapse imaging on an upright microscope. Alternately, one could design a perfusion system which prevents excess fluid loss from the system.
Find positions of interest within the sample by visualizing GFP-positive cells by multiphoton fluorescence (emission: green) and collagen by SHG (emission: blue) at 880 nm two-photon excitation. Typically, regions in which cells have migrated into the collagen matrix, i.e. are completely encased in 3D, are chosen and images are acquired at 5 μm step size in an 80-100 μm deep Z-section.
Once stage positions have been stored, spread the parafilm piece around the 35 mm dish and attach loosely to the stage with the paper tapes. (Fig. 4E)
-
(Optional) Make a tiny hole in the parafilm away from the lens with a pair of sharp-edged tweezers and insert the miniature temperature probe through it until the sensor is just submerged in the media. (Fig. 4E)
The temperature probe provides a reading of the instantaneous temperature in the media. Using this, one can adjust the input power of the heating element to obtain a stable 37°C temperature at the sample. Typically, it requires a few runs to optimize the settings required for temperature stability and once that is obtained, one need not perform this step for every run.
-
Wrap the objective lens with a layer of bubble wrap to provide insulation. (Fig. 4F)
Bubble wrap, being made of plastic and air is an excellent, cheap insulator that prevents the entire outer surface of the objective lens from becoming a heat sink. Alternately, an objective heater may be used with a separate temperature controller while taking care not to overheat the objective lens as it may cause damage to the lens.
-
Bring the lens back to the stored stage positions, adjust the input power to the heating element, and let the system equilibrate for 2-3 hours before imaging. For cancer cell migration in 3D, images may be taken at 15-20 min intervals with total imaging duration of at least 8, preferably 12 hours, to capture meaningful behavior since 3D migration is much slower as compared to 2D migration.
The equilibration time is to allow the temperature of the setup to stabilize. Starting the imaging after allowing this time considerably reduces stage drift. Note that you may have to readjust the stage positions before starting the time series imaging cycle as there will be some stage drift during this equilibration period. Alternately, one could begin imaging right away (for time sensitive experiments) and manually readjust for the stage drift intermittently in the initial few hours.
Fig. 4. Setup for long-term timelapse multiphoton laser scanning microscopy.

(A inset) O-ring for uniform heat dissipation; (A) Sample to be imaged with the O-ring secured around it; (B) Place the ensemble on a heating element for temperature control; (C inset) Square parafilm piece with an objective-sized circular hole in the middle and paper tapes attached to its four corners and (C) the same piece secured to the objective ensuring that the lens sticks out through the bottom; (D) Bring the lens down and find stage positions of interest in this configuration; (E inset) Miniature temperature probe for monitoring the temperature of the sample; (E) Secure the parafilm like a lid and make a tiny puncture with a pair of sharp tweezers and gently slide the temperature probe through it; (F) Final setup with the temperature probe and a layer of bubble wrap around the objective to provide insulation.
Post-processing raw data, cell tracking and analysis of migration
-
Segregate the whole image set into hyperstacks with the correct channel, z and t dimensions.
For example, for an experiment where 3 stage positions were imaged for 40 time points each with 21 Z positions in 2 separate channels, the whole data set would be segregated into 3 5D hyperstacks each containing x, y, z (21 slices) and t (40 slices) information for 2 separate channels
The easiest way to obtain the proper hyperstacks from the raw data will depend on the particular image acquisition software and the sequence in which the images are stored and the metadata associated with it. In our case, it involved loading the whole image set into a stack and then converting the stack into a hyperstack in proper order.
-
For each stage position, correct the hyperstack for any stage drift by using the “Correct 3D Drift Plugin” in Fiji (Parslow, Cardona and Bryson-Richardson 2014) using the SHG channel for registration.
More information on this plugin may be found here: https://imagej.net/Correct_3D_Drift
-
Extract the GFP channel only from the registered hyperstack to make a sub-hyperstack and use this to track cells using TrackMate.
Tutorials on cell tracking using TrackMate may be found here: http://imagej.net/Getting_started_with_TrackMate For our purposes, we used non-branching tracks and only counted tracks which at least existed for 12 out of the 16h of image acquisition. This method of selecting tracks based on their duration is also an efficient way to prune out erroneous tracks that may be generated by artifacts of imaging or automated tracking.
-
The TrackMate output is a matrix of X, Y, Z, t data for each individual cell. Import this data into column vectors or an array in Matlab or other mathematical computing software for analysis of the tracks. The analysis will be specific to the application and the questions asked in a given experiment.
For most cell migration analysis, it is useful to obtain the speed and motility of the cells. This can be done by fitting a random walk model or a persistent random walk model to the cell trajectories (Othmer, Dunbar and Alt 1988, Harms et al. 2005). Also refer to (Dickinson and Tranquillo 1993) and (Ray et al. 2017b) for detailed descriptions on the curve fitting procedure. Matlab code used by authors will be shared on request.
- Normalize cell tracks in aligned matrices to the local direction of alignment by rotating the tracks to be centered along the direction of median collagen fiber alignment. To do this:
- Calculate the average fiber angle at each stage position by using CT-FIRE (as described in Basic Protocol 2) analysis on the XY maximum intensity projected SHG image.
- Convert the trajectory points into polar coordinates
- Rotate the angle of each position by the amount needed to align them with the local fiber alignment
- Convert the modified trajectory points back to Cartesian coordinates for downstream analysis.
There exists a small, inherent variation in the direction of alignment of the fibers from one stage position to the next. When tracks are obtained from several distinct positions and pooled together as a group, these variations may contribute to widening the angular distribution of the tracks i.e., the cell trajectories may appear less oriented to the fiber alignment than they actually are. Hence it is important to orient the tracks to the local fiber alignment direction and then pool them together if required. Moreover it also provides some flexibility while imaging in that the direction of fiber alignment need not be perfectly along the x or y axis and can be corrected in the post-processing and analysis stage.
Commentary
Background information
Contact guidance, especially from organized collagen fibers in solid tumors is a vital regulator of cancer cell movement and invasion through the local stroma, and therefore strongly influences metastatic dissemination (Provenzano et al. 2006, Lee et al. 2015, Wang et al. 2002). Thus, next generation in vitro systems are required to study cancer cell migration in these physiologically relevant aligned ECM structures. Historically, to organize collagen into aligned fiber networks in vitro a handful of methods have been employed including alignment by flow in a narrow channel (Lee et al. 2006, Riching et al. 2014), magnetic alignment either by flow of magnetic beads (Guo and Kaufman 2007, Provenzano et al. 2008) or by using high power magnets (Ceballos et al. 1999) and by mechanical stretching of collagen matrices (Riching et al. 2014). These methods have drawbacks such as introducing the confounding factor of confinement (alignment in narrow channels), being unamenable to laser scanning microscopy (magnetic beads), causing damage to the fiber structure (mechanical stretching) or being inaccessible to benchtop preparation (high power magnets). It is important to note that stromal collagen in mature, desmoplastic tumors is rarely disorganized or randomly oriented, and stromal cells such as carcinoma-associated fibroblasts are thought to be the chief architects of matrix organization. Thus, our technique, apart from avoiding the pitfalls of previous methods, also provides a biomimetic path to aligning collagen in vitro by using the contractility of primary carcinoma-associated fibroblasts isolated from mammary tumors. Indeed, the pore size distribution and effective density of these reorganized final constructs are much closer to in vivo stromal collagen than is possible to achieve by simple gelling of collagen in vitro (Ray et al. 2017b). This method, however, introduces some inherent variation in degree and direction of fiber alignment and pore size distribution within a single construct. It may be thought of as a useful compromise between the highly variable in vivo ECM and precisely controllable reductionist models.
For timelapse imaging of cell movement through the 3D collagen matrices, we used two-photon microscopy with simultaneous multiphoton fluorescence excitation and SHG to image carcinoma cells, including the CSC subpopulations, and collagen respectively. Using a two-photon setup offers several advantages in this application over conventional confocal microscopy: it enables scatter-free imaging deeper into the engineered tissues which is essential for 3D tracking of cells. In addition, we can perform label-free detection of the collagen network as the cells migrate along and interact with the fibers, which opens the door for further analysis of fiber deformation, etc. in relation to adjacent cellular dynamics. However, most two-photon setups involve an upright microscope with water immersion objective lenses. This poses some unique challenges to maintain the temperature, buffering and fluid interface during the long-term imaging experiments that we have performed here. We report here a cheap and easy method (total setup cost ~$1000) to keep the sample under physiologic conditions while performing long duration timelapse imaging with the described microscope setup. Certainly, alternative ways to engineer solutions exist, such as environmental chambers with or without CO2 control, perfusion systems and objective heaters. However, we believe our inexpensive but effective protocol will make such experiments more accessible to the broader research community. Similarly, we also present a 3D migration analysis platform using the freely available and widely used Fiji software package.
Fibroblasts are mechanosensitive cells which respond readily to the force distributions and structural anisotropy in the ECM (Calvo et al. 2013, Dickinson et al. 1994). The exact dynamics of the reorganization process that leads to the formation of aligned matrices in the described method remains to be characterized. However, based on the knowledge in the field of mechanotransduction, it can be postulated that the collagen is able to withstand more contractile force along the axis joining the two spacers due to the constraints. The fibroblasts responding to this mechanical cue reorient along this axis, thereby generating maximum forces and aligning the collagen along that direction. Although the experiments were done without activation of fibroblasts to produce fibrous collagen, we did not explicitly characterize whether any collagen in the final construct is produced by the seeded fibroblasts. This is a possibility since it is known that anisotropically organized fibroblasts tend to produce collagen fibers in the direction of cell orientation (Wang et al. 2003).
Critical parameters and troubleshooting
Fabrication and handling of engineered matrices
Collagen reorganization and the concomitant degree of fiber alignment is dependent on the ability of embedded cells to produce contractile force. Although, in principle, any contractile cell type can be used for this protocol, cell seeding density and compaction times may need to be individually optimized for each cell type.
When preparing fibroblast-seeded gels, it is critical to first mix the collagen and 2X PBS/HEPES prior to introducing cells. Mixing reagents in the wrong order may result in cell death due to improper pH and/or osmolarity. Further, introduction of bubbles in the collagen mixture will result in suboptimal fiber structure. The formation of bubbles can be limited by pipetting slowly to account for collagen viscosity. If bubbles occur during pipetting, try to remove them by gentle suction or push them to the side of the construct using a pipet tip. In addition, special care should be taken during preparation to ensure the gel is well connected to the corners of the spacers, as this is where detachment most readily occurs. To do this, pipet the cell-seeded collagen mixture onto the spacers such that the entire spacer is covered. Allow mixture to seep in for several seconds before drawing the mixture out into the demarcated region. If necessary, pipet the cell-seeded collagen mixture back over the spacers several times to encourage a robust association of the mixture to the spacers.
To avoid damaging the isotropic control gels during detachment from the bottom of the plate, use a small pipet tip to apply pressure towards the outside of the well with one steady motion around the well. Then, gently tap the plate and swirl the media to detach the gel from the well bottom. If the gel folds in on itself, it is typically still partially attached to the well bottom. A pipet tip can be used as a last resort for detachment, but is much more likely to result in damage to the gel. The collagen matrices, including the final decellularized tissues are quite fragile in general and need to be handled with care. The slice anchors described previously are an excellent tool to immobilize the matrix in the dish without inducing compression or damage while assessing the quality of alignment or timelapse imaging of cell migration.
Seeding matrices with CSCs
When growing CSC-enriched spheroids, the initial seeding density into low adhesion plates must be low to allow clonal growth of individual spheroids and minimize aggregation. The extent of CSC enrichment by spheroid formation may be monitored by serial passaging of the spheroids and assessment of molecular marker expression. Starting from a cell line that has a low percentage of CSCs, serial passaging of spheroids should enrich for the CSC population. One should see progressively less debris and cell death and enhanced spheroid formation within a few passages. ALDH1, Nanog, CD44 and CD24 are among the well-known markers of the breast CSC phenotype; probing of the cells in spheroids may be performed to determine the molecular signature of the spheroid-forming CSCs for a given cell line.
It is critical to ensure that cells from CSC-enriched spheroids are in a single-cell suspension before counting and reseeding onto the aligned matrices. On the other hand, prolonged exposure to trypsin may be detrimental to the viability of the cells. Hence, we recommend a short exposure to trypsin with intermittent agitation. Ensure that no residual BCSC media is retained and that cells have been washed with PBS without calcium and magnesium before adding the trypsin to ensure easier disintegration of the spheroids.
Timelapse imaging and analysis of 3D cell migration
For timelapse imaging of cell migration, maintaining the temperature and pH throughout the experiment is critical. We recommend the use of the temperature probe to monitor temperature and measurement of pH before and after the experiment in a pilot study to optimize the settings for particular ambient conditions. The imaging setup we describe here uses the sample placed in a 35 mm dish; by changing the heating element and using similar adaptable sealing techniques one could extend this protocol to be applicable for larger tissue culture plates as well. However, an important aspect to keep in mind is that one is fundamentally limited to stage positions within a few square millimeters of each other due to the small window in which the objective can move. One could attempt to stretch the parafilm slightly to allow for more room, although that opens up the possibility of greater evaporation and fluid loss during a long experiment. Phototoxicity is another common issue with long term timelapse imaging using a laser scanning microscope. The laser power, resolution and pixel dwell should be adjusted to minimize toxicity to the cells. An easy way to assess the health of the cells during the experiment is to analyze their instantaneous velocities versus time. On an average, across many cells, it should remain a constant over the course of the experiment in the absence of external agents that affect cell migration.
Some amount of stage drift is unavoidable in the current setup and in fact, in most timelapse microscopy setups. Before tracking of cells, it is imperative to register the hyperstacks in 3D using plugins as mentioned before. However, in some cases, it may not be possible to accurately register tracks in an automated fashion, usually due to some complex, irregular pattern of stage drift. An alternate manual correction may be employed in such cases: try to find a fiduciary marker (usually a junction of fibers around which there are no cells and hence is stationary), manually track that junction over time in the unregistered stack/hyperstack and generate a reference track for that marker. Then track all the cells normally and while analyzing simply subtract the reference track from each one to correct for the stage drift.
Anticipated results
Basic Protocol 1 and 2
Following protocol 1 and 2 one would expect to produce highly aligned or randomly organized decellularized collagen matrices. These matrices would be several times more collagen dense than the original 3 mg/ml gels due to compaction. Roughly speaking, one could expect up to 3-fold increase in collagen density for the final matrix, estimated from the reduction in total volume due to compaction. It is difficult to achieve a 100% success rate with the fabrication of aligned constructs owing to the nature of the method. For beginners, one could expect 25-50% success, which should improve to 75% or more with experience. It is also important to note that not the entire aligned construct will show the same high degree of alignment. Usually, the middle portion in the top half of the gel is the most aligned and the angle of alignment changes as one moves towards any of the spacers from the center. Decellularization by our protocol is usually quite effective, leaving only minor debris that stains faintly with Hoechst and a few small phalloidin-positive fragments (Ray et al. 2017b). Depending upon the application one could agitate the constructs more during the decellularization process, which should help clear out majority of the debris.
Basic Protocol 3
The results to be expected from the cell migration experiments would depend on the specific cell types and conditions used. In general, one could expect to see more directional migration in aligned matrices following the contact guidance cues as compared to random migration for cells in the isotropic matrices. Note that for collagen gels prepared as thin discs or rectangles as described here, the z motility is likely to be only a fraction of the motility in x and(or) y, perhaps owing to the anisotropic pore size distributions, especially in the aligned constructs. For MDA-MB-231 breast cancer cells, we found that the organization of the collagen does not alter the total motility of the cells, rather it only redistributes the motility to be primarily along one axis (Ray et al. 2017b). Our results also show that the CSC subpopulation in these cells are highly motile and migrate with much higher speed than the control population (Ray et al. 2017b). However, further studies are required to determine if these findings are also valid for other cell types.
Time considerations
Basic Protocol 1
Spacer Preparation: 60 minutes (one time for many experiments)
Aligned Template Preparation: 30 minutes.
Gel Preparation: 45 minutes.
Gel Polymerization/Vertical Compaction: 16–24 hours.
Gel Transfer: 30 minutes.
Lateral Compaction: 1–2 days.
Generally, spacers and aligned templates are prepared at least one day prior and the completed template is sterilized under UV light immediately before gel preparation. Gels are adequately polymerized for transfer after 16 hours, however, 24 hours of polymerization results in sturdier gels that are less liable to damage. For maximal alignment, gels should be allowed to undergo lateral compaction for up to 2 days, but should be decellularized well before detachment of the gel from either of the spacers occurs.
Basic Protocol 2
Cell Lysis: 15 minutes + 24 hour incubation.
Nucleic Acid Digestion: 35 minutes + 12–16 hour incubation.
Debris Removal: 35 minutes.
Imaging collagen fiber morphology: 10–15 minutes per sample.
Basic Protocol 3
Growing CSC-enriched spheroids: 4–7 days.
Seeding cells onto matrices: up to 1 hour.
Incubation: 48 hours.
Live cell Imaging setup: 1 hour.
Equilibration: 2–3 hours.
Timelapse Imaging: 12–16 hours.
CSC-enriched spheroids should be allowed to form at least for 4 days. More time needs to be allowed if serially passaged spheroids are used. For our cells, 48 hours of incubation was ideal to allow cells to infiltrate into the matrix but still maintain sparse enough distribution to allow single cell tracking in 3D. The imaging setup, equilibration and total imaging time could change based on ambient conditions and the specific application.
Significance Statement.
Cells must navigate complex three-dimensional environments during invasion. Indeed, within breast and pancreatic tumors aligned collagen architectures provide paths for carcinoma invasion that promote metastasis and poor patient outcomes. Here, we describe a robust method to generate aligned collagen matrices in vitro that mimic in vivo fiber organization, and quantify 3D migration of cancer stem cells, which are suggested to have tumor initiating capacity and capacity to successfully form metastases. Thus, this platform facilitates our ability to elucidate fundamental physical and molecule mechanisms governing cancer stem cell behavior and their role in cancer progression. Further, these assays are readily adaptable to a variety of cells and biological questions related to stem cell biology and three-dimensional cell migration.
Acknowledgments
PPP and this work was supported by a Research Scholar Grant, RSG-14-171-01-CSM from the American Cancer Society. This work was also supported by the NIH (R01CA181385 to PPP and U54CA210190 University of Minnesota Physical Sciences in Oncology Center Project 2 to PPP), UMN College of Science and Engineering (PPP) and Masonic Cancer Center (PPP), and grants from the UMN Institute for Engineering in Medicine (PPP), and the Randy Shaver Research and Community Fund (PPP). AR was supported by a UMN Doctoral Dissertation Fellowship. RKM is supported by a NSF graduate research fellowship 00039202. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or other funding agencies. We thank members of the Provenzano laboratory for insightful comments regarding this work, the Tranquillo lab for lending equipment and expertise and the Odde lab for sharing knowledge on timelapse imaging and cell migration analysis.
Footnotes
Conflict of Interest
There are no conflicts of interest to declare.
References
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637–46. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos D, Navarro X, Dubey N, Wendelschafer-Crabb G, Kennedy WR, Tranquillo RT. Magnetically aligned collagen gel filling a collagen nerve guide improves peripheral nerve regeneration. Experimental neurology. 1999;158:290–300. doi: 10.1006/exnr.1999.7111. [DOI] [PubMed] [Google Scholar]
- Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur MH, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, Birnbaum D, Wicha MS. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69:1302–13. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark P, Connolly P, Curtis AS, Dow JA, Wilkinson CD. Cell guidance by ultrafine topography in vitro. J Cell Sci. 1991;99(Pt 1):73–7. doi: 10.1242/jcs.99.1.73. [DOI] [PubMed] [Google Scholar]
- Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–32. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson RB, Guido S, Tranquillo RT. Biased cell migration of fibroblasts exhibiting contact guidance in oriented collagen gels. Ann Biomed Eng. 1994;22:342–56. doi: 10.1007/BF02368241. [DOI] [PubMed] [Google Scholar]
- Dickinson RB, Tranquillo RT. Optimal estimation of cell movement indices from the statistical analysis of cell tracking data. AIChE Journal Volume 39, Issue 12. AIChE Journal. 1993;39:1995–2010. [Google Scholar]
- Dunn GA, Ebendal T. Contact guidance on oriented collagen gels. Exp Cell Res. 1978;111:475–9. doi: 10.1016/0014-4827(78)90196-9. [DOI] [PubMed] [Google Scholar]
- Grinnell F, Rocha LB, Iucu C, Rhee S, Jiang H. Nested collagen matrices: a new model to study migration of human fibroblast populations in three dimensions. Exp Cell Res. 2006;312:86–94. doi: 10.1016/j.yexcr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Guo C, Kaufman LJ. Flow and magnetic field induced collagen alignment. Biomaterials. 2007;28:1105–14. doi: 10.1016/j.biomaterials.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Harms BD, Bassi GM, Horwitz AR, Lauffenburger DA. Directional persistence of EGF-induced cell migration is associated with stabilization of lamellipodial protrusions. Biophys J. 2005;88:1479–88. doi: 10.1529/biophysj.104.047365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiba T, Yamada T, Lu H, Kawazoe N, Chen G. Maintenance of cartilaginous gene expression on extracellular matrix derived from serially passaged chondrocytes during in vitro chondrocyte expansion. J Biomed Mater Res A. 2012;100:694–702. doi: 10.1002/jbm.a.34003. [DOI] [PubMed] [Google Scholar]
- Kim DH, Provenzano PP, Smith CL, Levchenko A. Matrix nanotopography as a regulator of cell function. J Cell Biol. 2012;197:351–60. doi: 10.1083/jcb.201108062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Downes A, Chau YY, Serrels B, Hastie N, Elfick A, Brunton V, Frame M, Serrels A. In vivo imaging of the tumor and its associated microenvironment using combined CARS / 2-photon microscopy. Intravital. 2015 doi: 10.1080/21659087.2015.1055430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Lin R, Moon J, Lee LP. Microfluidic alignment of collagen fibers for in vitro cell culture. Biomed Microdevices. 2006;8:35–41. doi: 10.1007/s10544-006-6380-z. [DOI] [PubMed] [Google Scholar]
- Morin KT, Smith AO, Davis GE, Tranquillo RT. Aligned human microvessels formed in 3D fibrin gel by constraint of gel contraction. Microvasc Res. 2013;90:12–22. doi: 10.1016/j.mvr.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133–43. doi: 10.1038/nrc3184. [DOI] [PubMed] [Google Scholar]
- Othmer HG, Dunbar SR, Alt W. Models of dispersal in biological systems. J Math Biol. 1988;26:263–98. doi: 10.1007/BF00277392. [DOI] [PubMed] [Google Scholar]
- Parslow A, Cardona A, Bryson-Richardson RJ. Sample drift correction following 4D confocal time-lapse imaging. J Vis Exp. 2014 doi: 10.3791/51086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys J. 2008;95:5374–84. doi: 10.1529/biophysj.108.133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Lee O, Win Z, Edwards RM, Alford PW, Kim DH, Provenzano PP. Anisotropic forces from spatially constrained focal adhesions mediate contact guidance directed cell migration. Nat Commun. 2017a;8:14923. doi: 10.1038/ncomms14923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Slama ZM, Morford RK, Madden SA, Provenzano PP. Enhanced Directional Migration of Cancer Stem Cells in 3D Aligned Collagen Matrices. Biophys J. 2017b;112:1023–1036. doi: 10.1016/j.bpj.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching KM, Cox BL, Salick MR, Pehlke C, Riching AS, Ponik SM, Bass BR, Crone WC, Jiang Y, Weaver AM, Eliceiri KW, Keely PJ. 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys J. 2014;107:2546–58. doi: 10.1016/j.bpj.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemenschneider SB, Mattia DJ, Wendel JS, Schaefer JA, Ye L, Guzman PA, Tranquillo RT. Inosculation and perfusion of pre-vascularized tissue patches containing aligned human microvessels after myocardial infarction. Biomaterials. 2016;97:51–61. doi: 10.1016/j.biomaterials.2016.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Jia F, Gilbert TW, Woo SL. Cell orientation determines the alignment of cell-produced collagenous matrix. J Biomech. 2003;36:97–102. doi: 10.1016/s0021-9290(02)00233-6. [DOI] [PubMed] [Google Scholar]
- Wang W, Wyckoff JB, Frohlich VC, Oleynikov Y, Huttelmaier S, Zavadil J, Cermak L, Bottinger EP, Singer RH, White JG, Segall JE, Condeelis JS. Single cell behavior in metastatic primary mammary tumors correlated with gene expression patterns revealed by molecular profiling. Cancer Res. 2002;62:6278–88. [PubMed] [Google Scholar]