

Abstract
Healthy skeletal muscle possesses the extraordinary ability to regenerate in response to small-scale injuries; however, this self-repair capacity becomes overwhelmed with aging, genetic myopathies, and large muscle loss. The failure of small animal models to accurately replicate human muscle disease, injury and to predict clinically-relevant drug responses has driven the development of high fidelity in vitro skeletal muscle models. Herein, the progress made and challenges ahead in engineering biomimetic human skeletal muscle tissues that can recapitulate muscle development, genetic diseases, regeneration, and drug response is discussed. Bioengineering approaches used to improve engineered muscle structure and function as well as the functionality of satellite cells to allow modeling muscle regeneration in vitro are also highlighted. Next, a historical overview on the generation of skeletal muscle cells and tissues from human pluripotent stem cells, and a discussion on the potential of these approaches to model and treat genetic diseases such as Duchenne muscular dystrophy, is provided. Finally, the need to integrate multiorgan microphysiological systems to generate improved drug discovery technologies with the potential to complement or supersede current preclinical animal models of muscle disease is described.
Keywords: disease modeling, induced pluripotent stem cells, skeletal musclemuscular dystrophy, tissue engineering
1. Introduction
Skeletal muscle comprises 35–40% of the entire human body weight and functions to generate forces that enable respiration, posture, and locomotion.[1] Healthy skeletal muscle can fully regenerate from small lacerations and tears caused by everyday activity owing to the presence of resident muscle stem cells called satellite cells (SCs).[2] Decreased ability of muscle to selfrepair, either due to aging,[3] genetic diseases,[4] or volumetric muscle loss (VML), can result in debilitating myopathies that severely impact quality of life or shorten lifespan.[5] Traditionally, small animal models and 2D cell cultures have been used to study biological processes and identify and validate pharmacological compounds to treat skeletal muscle disease.[6,7] While these models have enabled numerous discoveries and improved our knowledge of muscle biology, their translational value has been hampered by a limited ability to predict drug effects in human studies.[8] The need to study human skeletal muscle without the ethical considerations and experimental limitations of clinical research has driven new developments in the fields of human skeletal muscle tissue engineering and muscle stem cell biology. Specifically, the first engineering of functional human skeletal muscle in vitro,[9] derivation of skeletal muscle precursor cells (MPCs) from human induced pluripotent stem cells (hiPSCs),[10,11] and genetic correction of rare muscle diseases such as muscular dystrophy by CRISPR-Cas9 technology[12–14] are just some examples of recent methodological advances that open doors to enhanced understanding of human muscle pathology and treatment of disease.
In this progress report, we first review the development, structure, function, and regenerative capacity of native skeletal muscle. We then discuss the limitations of 2D cell culture models to study skeletal muscle pathophysiology in vitro and highlight the progress made in engineering functional skeletal muscle tissues from primary and pluripo-tent stem cells. Lastly, we discuss the current applications and future utility of tissue-engineered muscles in studying muscle regeneration, modeling disease, and predicting drug outcomes for improved therapy.
2. Skeletal Muscle Development, Function, and Regeneration
2.1. Muscle Development
2.1.1. Embryonic Skeletal Muscle Specification
With the exception of the craniofacial and extra-ocular mus-cles,[15] skeletal muscle progenitor cells are derived from the paraxial mesoderm, which forms from the posterior primitive streak during gastrulation.[16] Paraxial mesoderm specification and formation is regulated by FGF, Wnt and BMP signaling and results in the sequential expression of the early mesoderm marker T (Brachyury) and paraxial mesoderm markers mesogeninl, Tbx6 and Pax3 (Figure 1).[16–18] The segmentation clock, which generates oscillations of FGF, Notch and Wnt signals, regulates somite formation from paraxial mesoderm progenitors.[19] The somite then develops into two distinct compartments, the ventral sclerotome that gives rise to cartilage and bone, and the dermomyotome that gives rise to skeletal muscle, brown fat, and dermis of the back.[20] The dermomyotome consists of three distinct divisions, the central dermomyotome (which gives rise to brown fat, back dermis, and trunk muscles), the dorsomedial lip, and the ventrolateral lip.[16,19] The first muscle mass to form is the myotome, which forms from the delamination and migration of MPCs from the dorsomedial and ventrolateral lips.[21] Transcription factor Pax3, which is initially expressed in paraxial mesoderm progenitors, becomes restricted to the dermomyotome and induces expression of c-met, the functional receptor for HGF/SCF.[22] Mice that do not express Pax3, c-met, or HGF do not form musculature due to the lack of migration and delamination that precludes myotome development.[21] The myotome itself can be divided into an epaxial region, which gives rise to the muscles of back, and a hypaxial region that gives rise to other trunk muscles and the limb bud.[21]
Figure 1.
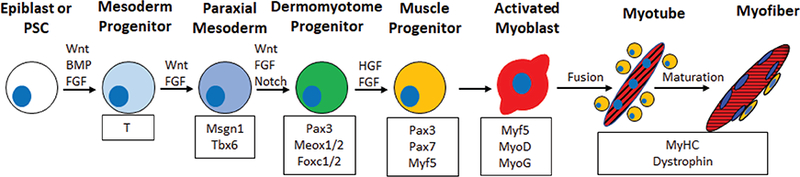
Skeletal muscle development and specification. Schematic shows the developmental stage (top), factors that regulate developmental transitions (above arrows), muscle-specific proteins expressed at different developmental stages (bottom). Wnt and FGF signals primarily regulate differentiation to the paraxial mesoderm and muscle progenitor stage. Upon differentiation, activated myoblasts fuse together while a subset of muscle progenitors (yellow cells) does not differentiate but resides on the outside of the myofiber to contribute to future muscle regeneration events.
2.1.2. Skeletal Muscle Differentiation
Skeletal muscle commitment and differentiation of is regulated by the myogenic regulatory factor (MRF) family of basic helix loop helix transcription factors Myf 5, MyoD, Mrf4, and myo-genin.[23] Myogenic commitment only occurs after somatic cells migrate into the myotome where Pax3 expression is downregu-lated and Myf5, MyoD, and Mrf4 are upregulated to generate committed embryonic myoblasts.[24–26] Muscle formation is prevented if all three but not two of these transcription factors are deleted, indicating their redundancy and overlapping func-tions.[24,25,27] Following commitment to the myogenic lineage, terminal muscle differentiation requires the expression of myo-genin that acts co-operatively with MyoD and Mrf4 to promote the fusion of myoblasts into multinucleated myotubes.[28,29] Following the formation of primary myotubes, the central dermomyotome loses its epithelial character and Pax3+/Pax7+cells migrate into the myotome.[30] These cells then either retain Pax7 expression to become adult muscle stem cells or commit to differentiation and provide fetal myoblasts to generate secondary myotubes that complete embryonic and fetal muscle development.[26,30,31]
2.1.3. Primary Myotube Formation
Primary myotubes are the first muscle fibers to form by the fusion of embryonic myoblasts that span from tendon to tendon in all animals,[32] and continue to grow from fusion of fetal and adult myoblasts in the postnatal period.[33] In small rodents this process occurs in 2 d but in humans and large animals the period of primary myotube formation can last up to 7 d to accommodate the need for larger numbers and length of primary myotubes.[34] While primary myotubes ultimately comprise only a minority of final adult myofiber numbers, they provide the structural support for the development of secondary myotubes and are critical for muscle development.
2.1.4. Secondary Myotube Formation
Secondary myotubes form by the adhesion of fetal myoblasts to primary myotubes and the subsequent fusion of additional myoblasts along the length of the primary myotube to form a new, secondary myotube.[35] In small animals, secondary myotubes grow longitudinally along the primary myotube by cell fusion to span from tendon to tendon, but in larger animals secondary myotubes may form intramuscular myo-myonal junctions and not span from tendon to tendon.[36] The number of secondary myotubes that can form along a primary myotube determines the final muscle size, with 5 to 9 formed in small animals,[37,38]and more than 20 in large animals.[34,38,39] While the final number of primary myotubes can give an indication of the final muscle fiber number, it is the number of secondary myotubes able to form around a single primary myotube that is the main determinant of final muscle size across species.
Innervation is critical for complete muscle development to occur, with axons entering the premuscle mass before myotube formation begins.[37,40] Innervation has the most profound effect on muscle development with denervation almost completely preventing and hyperinnervated muscle increasing secondary myotube formation.[40] The final critical step in adult muscle formation leading to normal innervation is motoneuron death. Skeletal muscle activity regulates motor neuron survival with increasing levels of muscle activity decreasing motor neuron survival.[41] Primary myotubes are polyneuronally innervated and massive motor neuron death occurs during late embryonic and early postnatal development until all myotubes display single motor neuron innervation.[37,40]
2.1.5. Postnatal Muscle Growth
Postnatal growth of skeletal muscle is characterized by an increase in the length and cross-sectional area (hypertrophy) of myofibres. The continuous passive tension applied to skeletal muscle by bone growth and elongation during embryogenesis and neonatal development increases muscle weight, muscle length, and myofilament organization.[42] Muscle hypertrophy occurs by the continual fusion of new myoblasts, as evidenced by the increases in myotube nuclei up to three months post-natally,[43] and by increased cytoplasmic volume per myonu-cleus.[44] At birth, satellite cells comprise ≈30% of sublaminal nuclei in newborn mice but this number drops to 2–5% in adult mouse muscle as the myonuclei number per fiber increases.[45] Currently, the postnatal muscle growth period is the least characterized stage of muscle development with the mechanisms by which final satellite cell number is achieved remaining largely unknown. In conjunction with increase in murine postnatal muscle size, motor nerve activity and locomotor ability increase throughout postnatal development, both reaching adult levels by postnatal day 21.[46–48] Neural input regulates muscle fiberphenotype, with slow fiber neural input typically displaying a chronic tonic activation pattern and fast fiber input showing infrequent phasic activity.[46–48]
2.2. Muscle Architecture and Contractile Function
2.2.1. Extracellular Matrix Composition in Development and Growth
Skeletal muscle is composed of two major ECM structures, the basal lamina and intramuscular connective tissue (iMCT).[49,50] The basal lamina interacts directly with muscle cells and provides a supportive scaffold to spatiotemporally regulate muscle progenitor migration, proliferation, differentiation, and quies-cence.[50] Throughout all stages of muscle development, the basal lamina consists of a predominantly laminin-rich matrix, collagen IV, nidogen/entactin, and perlecan.[50,51] Within the somite, der-momyotome, and myotome, the developmental laminins-111 and 511 are the predominant laminin isoforms that generate the myotomal basement membrane, which itself is surrounded by a fibronectin matrix.[52,53] Prior to MyoD expression, α6β1 integrin is the predominant integrin expressed in epaxial muscle progenitors and is required, along with laminin-111, for proliferation and maintenance of an undifferentiated state in epaxial MPCs.[52,53] Upon MyoD expression, α6β1 integrin is replaced with α7β1 integrin as myoblast fusion occurs and primary myotubes form.[54,55] In contrast, migration and proliferation of hypaxial MPCs are dependent upon fibronectin and the fibronectin-binding alpha 4 and alpha 5 integrins.[55,56] In both epaxial and hypaxial muscles, laminin-111 and fibronectin are gradually replaced by the mature muscle laminin-211 isoform upon myo-tube formation that persists throughout adulthood.[54,57]
The iMCT surrounds the basal lamina and acts to transmit forces exerted passively among muscle fibers and to transmit forces to the tendon for muscle movement.[49] The iMCT is composed of three layers, 1) the endomysium that ensheathes individual skeletal muscle fibers, the basal lamina, capillaries, and nerves and is composed of randomly aligned collagen fibers to permit movement; 2) the perimysium, a multilayered sheet of collagen fibers, that runs transversely to groups of skeletal muscle fibers that it encloses; and 3) the epimysium, which encloses the entire skeletal muscle mass and is typically formed of two wavy sheets of collagen fibrils.[49,58,59] Each iMCT layer is formed primarily of type I and III fibrillar collagens with small amounts of type II, V, VI, IX, XII, and XIV collagen expressed in a muscle and species-specific manner.[49,58,59] During bovine muscle development, the collagen fibrils bind more tightly together in the endomysium, become thicker in the perimysium, and form a more regular wavy pattern in the epimy-sium.[59] These structural changes and increases in collagen cross-linking density and number lead to progressive increases in the mechanical strength of the iMCT during development and the early postnatal period.[58–63]
2.2.2. Myofiber Structure
Adult skeletal muscle tissue exhibits highly ordered architecture, which allows the generation of force and movement for posture and locomotion. At the ultrastructural level, myofibers in skeletal muscles consist of spatially registered sarcomeres, basic contractile units containing a central myosin-rich dark anisotropic (A) band and two actin-dominated light isotropic (I) bands.[64,65] At the junction of the A and I bands are the transverse tubules (Telivery of calcium to the sarcomere from sarcoplasmic reticulum (SR).[66] Where the T-tubules and SR meet, the SR membrane enlarges, fuses, and forms expanded chambers called terminal cisternae that contain high levels of calcium necessary for muscle contraction.[67] At the micro and macroscopic scale, multinucle-ated skeletal muscle fibers range from millimeters to centimeters in length while being densely packed with sarcomeres.
2.2.3. Regulation of Force Generation
Upon release of calcium from SR into myofibrils, calcium binds to troponin C causing a conformational change that removes tropomyosin from the myosin allowing actin to bind to myosin. This is followed by adenoside triphosphate (ATP) hydrolysis resulting in a conformational change, called the power stroke, that pulls actin along the myosin toward the center of the sarcomere, shortening the sarcomere and generating muscle con-traction.[68] For muscle relaxation, calcium is actively pumped back into the SR by the sarcoplasmic-endoplasmic reticulum Ca2+ ATPase pumps. Removal of calcium from cytoplasm and dissociation from troponin and tropomyosin results in reblocking of the myosin binding sites.
Skeletal muscle force output is primarily regulated by the neural input into the muscle via summation and motor unit recruitment. Summation relates to the increase in tension generated upon action potentials that arrive before the muscle can relax, resulting in increased force generation until maximum force is generated with a fused tetanus. Slow fibers achieve maximum force at lower nerve firing frequencies than fast fibers due to predominance of slow isoform sarcomeric and calcium handling proteins.[69] By a process of motor unit recruitment, groups of muscle fibers innervated by individual motor neurons are recruited in greater numbers and ascending order of size, enabling a single muscle to produce both delicate and explosive movements.[70] Finally, as explained by the sliding filament theory,[71] contractile force is also regulated by the muscle length where at optimal lengths the overlap of myosin and actin filaments within each sarcomere is maximal and permits greatest cross-bridge cycling.
2.3. Skeletal Muscle Regeneration
First identified by Mauro in 1961,[72] the ability of adult skeletal muscle to regenerate is dependent upon Pax7-expressing SCs that reside in niches beneath the basal lamina of myofibers.[73,74] In healthy adult muscle, SCs are typically quiescent but become activated in response to injury and disturbance of the niche.[75] Satellite cell activation is characterized by a shift to a G1 proliferative state,[76] expression of Myf5,[77] phosphorylation of p38α/β mitogen-activated protein kinase, and consequent expression of MyoD.[78] Activated SCs divide asymmetrically and can adopt two fates: 1) retain Pax7 expression and return to a quiescent state to contribute to future muscle regeneration; or 2) commit to terminal muscle differentiation by losing Pax7 expression, upregulating myogenin, and fusing into damaged fibers or forming de novo fibers.[79] Importantly, the ability to retain regenerative ability is dependent upon asymmetric division and successful replenishment of the SC pool.[80,81]
In the last decade, the importance of additional cell types that support, enable, and regulate muscle regeneration has become more appreciated. Skeletal muscle regeneration is intricately regulated by the innate immune response, with deletion of monocytes completely preventing muscle regeneration.[82] The first stage of muscle regeneration is characterized by neutrophil infiltration that results in degradation of damaged muscle fibers. Concurrently, macrophages are recruited to damaged muscle and adopt an Ml phenotype that is characterized by proinflammatory cytokine secretion, which in turn promotes SC activation and proliferation.[83,84] Macrophage polarization then shifts to an M2 macrophage phenotype, which is critical for the repair process as it shifts SCs toward differentiation.[85] Additional cell types such as fibroadipogenic progenitors also contribute to muscle regeneration by promoting myofiber formation through controlled ECM deposition and paracrine factors.[86,87]
The ability of skeletal muscle to regenerate is not infinite and can be overwhelmed by volumetric muscle loss following trauma or large volume tissue resection. In these cases, muscle is incapable of regenerating the affected region and is irreversibly replaced by a fibrotic scar.[88] Muscle regeneration is also impaired in aged or dystrophic muscle due, in part, to satellite cell dysfunction induced by telomere shortening and changes in the systemic milieu.[89–92] Additionally, muscle fiber atrophy, loss of muscle fibers, decreased number and size of motor units, and increased fibrosis and fat accumulation all contribute to loss of function in aged and dystrophic muscle.[93–96]
3. Current Skeletal Muscle Tissue Engineering Methods
3.1. Need for Tissue-Engineered Skeletal Muscle
Traditionally, skeletal muscle biology has been studied using muscle biopsies from healthy or diseased individuals, animal models, or 2D cell culture experiments. While the use of human subjects and tissues is ideal, the type of studies that can be performed are limited by tissue availability and ethical considerations. To overcome this limitation, most studies have been performed in rodent models of muscle disease and increasingly in zebrafish and drosophila models. Use of these models has massively contributed to our understanding of muscle biology and pathology; however, the ability to accurately replicate severity of human disease and predict drug responses has been poor.[8,97,98] As an alternative to animal experiments, cell culture studies utilizing primary cells or immortalized cell lines, such as the murine C2C12 or rat L6 myoblasts, have been used for decades to study muscle biology. Traditionally, these cultures involve expanding myoblasts in a high-serum media and then inducing differentiation into myotubes by decreasing serum content.[99] The benefits of 2D cell culture are the ability to completely control the cell’s microenvironment and perform rapid drug screening studies. However, traditional muscle cell cultures give rise to developmentally immature myotubes with limited physiological relevance. This functional immaturity is partly a result of using stiff substrates (glass, plastic) for cell growth and differentiation. In their seminal work, Engler et al. demonstrated that all cells are exquisitely sensitive to their physical environment (Figure 2).[100] and that culture of myotubes on soft substrates with muscle-like (12–18 kPa) stiffness can improve myotube structure.[101] Furthermore, stiff substrates not only decrease muscle-specific gene expression but also result in detachment of spontaneously contractile myotubes, to greatly shorten culture duration. Using ECM proteins (e.g., entactin, matrigel) to enhance cell adhesion can alleviate this problem,[102] however, myotube contractions are still hindered by rigid 2D substrates that fail to reproduce the complex 3D environment of native muscle. Thus, the ability to engineer a biomimetic 3D muscle environment in vitro could overcome several limitations of traditional 2D cell culture systems.[103] As discussed below, 3D muscle cultures permit far longer culture times,[9,104,105] increased myotube size,[106] increased protein content,[106] and improved maturation of myosin heavy chain (MHC) gene expression compared to 2D cultures.[104] Importantly, engineered muscles enable the measurements of contractile function (i.e., muscle strength and fatigue resistance), which are more representative of in vivo muscle physiology and pathology than the sole changes in protein or gene expression, as typically assessed in 2D cell cultures. For a utility in preclinical drug development, 3D tissue-engineered muscle should: 1) be prepared from human cells, 2) replicate the structure, function, and regenerative capacity of native muscle, 3) accurately model phenotype of various congenital and acquired diseases, and 4) be amenable to iniaturization and online functional and metabolic monitoring for enhanced experimental throughput.
Figure 2.
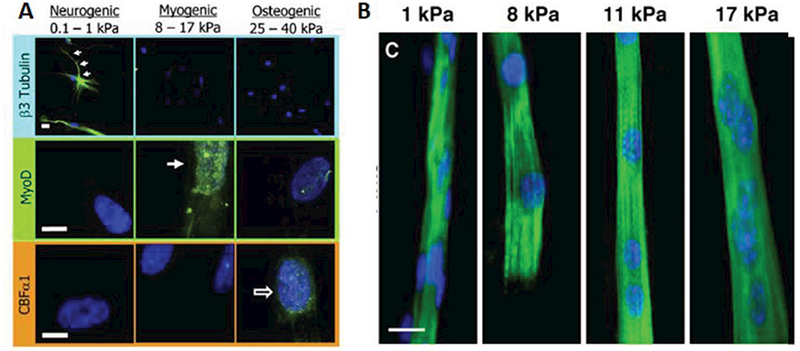
Stiffness regulates myogenic differentiation and maturation. A) Tissue-specific substrate stiffness regulates mesenchymal stem cells expression of neuronal cytoskeletal marker β3 tubulin (top), muscle transcription factor MyoD1 (middle), or osteoblast transcription factor CBFαl (bottom). Reproduced with permission.[100] Copyright 2006, Cell Press. B) C2C12 myoblasts stained for sarcomeric MHC (green) and DAPI (blue) display improve structural maturation (cross-striations) when cultured on micropatterned collagen substrates with muscle-specific stiffness (8–12 kPa). Reproduced with permission.[101] Copyright 2004, Rockefeller University Press.
3.2. Biomaterials for Tissue Engineering of Skeletal Muscle
To engineer a functional muscle in vitro, the classical tissue engineering paradigm is to create a biomimetic microenvironment that provides cells with a tissue-specific ECM combined with the biochemical and mechanical signals that promote rapid tissue maturation and function. Specifically, optimal ECM should be not only biocompatible, but also contain a high surface area for cell adhesion, support mechanical integrity of the tissue, minimize diffusion distances, and degrade once there is no need for structural support.[107,108] As described below, the most commonly used biomaterials for engineering skeletal muscle are native ECM proteins such as collagen, laminin, and fibrin.[99,109] Alternatively, synthetic polymers such as poly-L-lactic, polylactic-glycolic, and polyurethane or decellularized muscles can be used as scaffolds for muscle tissue genera-tion.[99,110,113] Traditionally, during tissue fabrication, undifferentiated myoblasts are embedded at high density in 3D scaffolds (Figure 3A), in which cellular forces exerted on the scaffold or scaffold topography provide alignment cues yielding accelerated myotube fusion and differentiation.[114,115] Alternatively, in a scaffold-free approach (Figure 3B), myoblasts are differentiated into myotubes as 2D monolayers followed by their self-assembly into cylindrical tissues or free-floating tissue sheets.[116,117]
Figure 3.
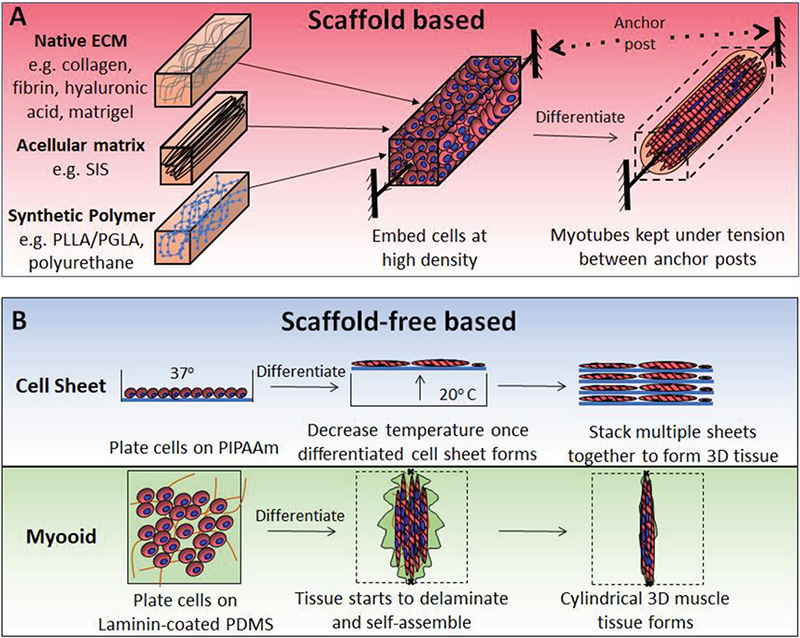
Methods for the in vitro engineering of 3D muscle tissues. Schematics showing A) scaffold and B) scaffold-free methods for engineering skeletal muscle. Abbreviations: SIS, small intestine submucosa; PIPAAm, poly(N-isopropylacrylamide); and PDMS, polydimethylsiloxane.
3.2.1. Collagen
The first 3D tissue-engineered muscle was produced by embedding predifferentiated avian myotubes in collagen I gel,[106] the most abundant ECM protein in native muscle.[49,118] Okano and Vandenburgh groups further showed that mouse C2C12 myoblasts could also proliferate and differentiate within collagen hydrogels.[119–121] In each of these studies, the cell and hydrogel mixture were maintained under constant bulk tension between two anchor points that functioned to both increase myotube alignment and provide a growth stimulus. Compared to traditional 2D cell culture techniques, these studies demonstrated that the 3D environment permitted longer culture times, increased myotube size and protein content suggestive of a more mature muscle. Further studies have shown that embedding primary human or rodent myoblasts within collagen hydrogels also permits the formation of engineered muscle tissue.[122–124]
While collagen I is the most abundant ECM component of native skeletal muscle, myofibers in healthy muscle directly interact with basal lamina proteins, which prompted approaches that supplement Matrigel (a commercially available basal lamina extract isolated from Engelbreth-Holm-Swarm tumors) at the time of tissue formation. Adding Matrigel to collagen hydrogels improved engineered muscle structure;[125] however, resulting low contractile forces indicated that high collagen content has adverse effects on muscle’s functional maturation.[126] This is consistent with findings that fibrosis, in which collagen levels are highly elevated, is associated with poor regeneration and function of native muscle.[127] Furthermore, collagen I is not easily remodeled, does not stimulate endogenous ECM secretion, and may not provide the optimal stiffness for muscle differentiation.[128] Consequently, the collagen hydrogel is being used less frequently for engineering of functional skeletal muscle as superior ECM choices have been identified.
3.2.2. Fibrin
Fibrin is the major structural component of a blood clot that plays a vital role in the wound healing response where it is completely replaced over time by cell-synthesized ECM.[129] Unlike collagen, fibrin is a preferred matrix for tissue engineering of skeletal muscle due to its ability to be extensively remodeled, degraded, and replaced by endogenously derived ECM.[130–132] Additionally, fibrin fibrils have a stiffness comparable to that of native muscle and several orders of magnitude lower than collagen fibrils.[133,134] Fibrin also promotes angiogenesis and neurite extension, which is critical for the formation of a fully functional engineered muscle for in vivo applications.[135–139] The disadvantages of fibrin as a scaffold are lot-to-lot variability and variable degradation rate, which can be readily overcome by lot testing and control of fibrinolysis using cross-linkers or anti-fibrinolytic compounds such as aminocaproic acid and aprotinin.[140]
The Dennis laboratory reported the first fibrin-based engineered skeletal muscles using a method where adult rat myogenic cells were plated on top of the fibrin hydrogel rather than embedded within the matrix.[141] The cells remodeled the fibrin and formed spontaneously contractile myotubes that generated specific forces greater than those reported for collagen gel-based muscle but still six to eight times lower than those of native adult muscle. The addition of Matrigel to the fibrin gel further increases force generation capability of engineered muscle.[126] Under similar conditions, high passive stiffness of collagen but not fibrin hydrogel induced premature rupturing of strongly contracting engineered muscle tissues providing further evidence for superiority of fibrin over collagen for muscle tissue engineering.[126]
3.2.3. Scaffold-Free Self-Assembled Muscle Tissues
An alternative approach to use of hydrogels/scaffolds is to allow cells to secrete their own ECM and self-organize into a 3D tissue. The first self-organized engineered muscle used saran wrap as a cell culture substrate in combination with exogenous fibroblasts to secrete sufficient ECM to enable the tissue self-assembly.[142] This method was further improved by replacing saran wrap with laminin to generate functional engineered tissues termed myooids.[116] The longer time needed for aligned muscle formation (≈35 d), small tissue size, and challenges with scale-up have limited the utilization of this method compared to hydrogel-based methods. More rapid and directed myotube alignment and fusion have been reported by using micropatterned surfaces, resulting in improved muscle dif-ferentiation.[143–146] Differentiating primary rat myoblasts on an aligned micropatterned surface prior to transfer to a thin fibrin gel increased force production compared to nonaligned controls.[147] More recently, unaligned cell sheets have been produced by co-culturing muscle cells and fibroblasts on the ther-moresponsive polymer poly(N-isopropylacrylamide).[117] When sufficient ECM has been generated, cell sheets can be detached from culture plates by lowering temperature and layered with other muscle, vascular, or neuronal cell sheets to generate relatively thick tissue sheets.[117,148,149] To date, contractile forces of these tissues have not been reported.
4. Methods to Improve Engineered Muscle Function
4.1. Contractile Function of Engineered Muscle
Replicating the native adult muscle structure and function would require obtaining specific forces above 200 mN mm−2 and myofiber diameters of 50–100 μm.[150] All reported functional engineered muscle tissues demonstrate basic contractile physiological properties such as positive force-frequency and length-tension relationships; however, specific forces typically range from 0.4 to 20 mN mm−2 with myofiber diameters being less than 25 μm.[9,116,126,141,151] Notably, even when similar tissue-engineering methodologies are used, specific forces vary widely depending on the source of myogenic cells, increasing from C2C12, to human, to adult rat, to neonatal rat cells.[152] Engineered muscles also retain the contractile and metabolic phenotype specific to the muscle from which cells are isolated demonstrating that epigenetic differences in vivo are retained in vitro.[153,154] The ability to engineer muscle with a particular fiber type is important for studying certain diseases such as Pompe disease, Duchenne muscular dystrophy (DMD), and sarcopenia that preferentially target fast muscle fibers.[155–157] Further improvements in contractile output of engineered muscle could be achieved by electrical and mechanical stimulation and improved nutrient delivery as outlined below.
4.2. Electrical Stimulation to Increase Engineered Muscle Function
Complete muscle development and maintenance of adult muscle mass and function requires functional innervation and electrochemical signals from motor neurons. In vitro, electrical field stimulation is usually applied to engineered muscle tissues as a surrogate for neuronal activity, whereby optimized pulse width and energy to minimize muscle damage can allow chronic stimulation over multiple weeks of culture.[154,158,159] Two-week electrical stimulation to mimic the neuronal inputs into slow or fast muscles shifted the kinetic phenotype of engineered muscle to slow or fast type, but shifts in MHC isoforms were not achieved.[154] On the other hand, replicating the adult muscle soleus innervation by inducing tetanic contractions for longer than 60 s yielded a shift from a fast to a slow muscle phenotype at MHC level.[47,159] Overall, while electrical or optogenetic stimulation of engineered muscle yields both a two to threefold increase in force generation and measurable increases in cellular protein content,[160] it does not appear to significantly advance the maturation of myofibers beyond embryonic/neonatal phenotype. This prompted studies that explored potential maturation benefits of motoneurons and neuromuscular junction formation beyond electrical input. Specifically, treatment of engineered muscle with the synapto-genic molecule agrin promoted acetylcholine receptor formation and clustering and doubled contractile force generation.[161] Co-culture with motor neurons or spinal cord explants resulted in the formation of immature neuromuscular junctions and improved both the structure and force generation of engineered muscle.[162–164] Nevertheless, the observed enhancement in contractile strength does not appear to coincide with a significant shift to adult-like MHC profile or increased myofiber size.
While the focus on engineered muscle innervation in vitro has primarily surrounded motor input, skeletal muscle also possesses intrafusal spindle fibers and sensory motoneurons that regulate proprioception and reflex initiation, forming a feedback system that is essential for regulating motor input. Using primary human myoblasts, intrafusal fibers can be generated in a defined serum-free media that yields classical intrafusal fiber morphology and protein expression.[165,166] When co-cultured with proprioceptive sensory neurons derived from human neuroprogenitors, intrafusal fibers can support the formation of mechanosensory-like nerve terminal structures. Importantly, electrophysiological studies in this system demonstrated repetitive firing patterns characteristic of functional sensory innervation in vivo.[165] These models are promising new tools for studying human proprioception but true validation of their functionality in response to changes in fiber length and tension have yet to be reported.
4.3. Mechanical Stimulation to Increase Engineered Muscle Function
Skeletal muscle is highly adaptive to changes in mechanical tension and loading during both embryonic development and adulthood. During embryonic growth, skeletal muscle undergoes continuous passive stretch that is essential for muscle development. In vitro modeling of this process using continuous ramp stretch induces the secretion of the IGF-1 splice variant, mechano-growth factor, that may promote tissue maturation.[167,168] In adult muscle, the absence or reduction in passive tension via hindlimb unloading or spaceflight induces significant muscle atrophy.[169] Similarly, decreasing engineered muscle tension by shortening its length yields significant myofiber atrophy and loss of contractile strength.[170] Vandenburgh and co-workers were first to demonstrate that cyclic loading induces muscle hypertrophy and increase in protein and DNA content.[122,171–173] Several studies since then have investigated the effect of cyclic stretch on engineered muscle, showing equivocal effects on tissue function and differentia-tion.[174–176] The wide range of protocols performed in different culture systems and different cell differentiation stages as well as the lack of direct comparisons across different methods has made interpretation of these studies difficult. Regardless, similar to use of electrical stimulation alone, no breakthroughs in myofiber size or force generation have been reported with use of mechanical stimulation alone or its combination with electrical stimulation.[113]
4.4. Increasing Nutrient Delivery to Engineered Muscle
A long-pursued goal of tissue engineering has been to provide tissues with sufficient oxygen and nutrients to maintain their function and support increasing energy demands during growth and maturation. This is of critical importance for engineering metabolically active skeletal muscle tissues where oxygen diffusion in vitro is typically limited to 100–150 μm,[177] beyond which necrotic core is formed at the tissue center.[120] Alterations in tissue architecture by generating smaller tissues or incorporating pores to minimize diffusion distances significantly increases tissue function (Figure 4).[115,178] Similarly, supplementing culture media with the synthetic oxygen carrier, perfluorocarbon, increases oxygen tension and differentiation of engineered muscle.[179] Alternatively, oxygen and nutrient delivery to tissues can be improved by dynamic culture in which media is displaced or agitated to improve mass transfer around the tissue.[180] In engineered skeletal and cardiac muscle tissues, dynamic culture on a rocking platform significantly increased force generation and improves overall tissue organization,[181–183] yielding engineered neonatal rat muscles with the highest specific forces reported to date and comparable to those of native neonatal muscle.[151] While dynamic culture conditions are a cheap, simple, and effective method to increase nutrient delivery in media volumes greater than 1 mL, the miniaturized organ-on-a-chip models with small media volume will require the use of microfluidic perfusion systems to both enhance nutrient delivery to engineered muscle and enable paracrine communication among multiple microtissues/organs.[184,185]
Figure 4.

Methods to increase nutrient delivery to engineered muscle. A) Representative image of a larger-scale engineered muscle bundle that leads to the formation of necrotic core. Reproduced with permission.[126] Copyright 2011, Elsevier. B) Miniaturization of muscle bundle from (A) to eliminate necrotic core formation. Reproduced with permission.[9] Copyright 2015, eLife Sciences Publications. C) Network patch configuration diminishes diffusion distances and increases nutrient delivery to engineered muscle. Reproduced with permission.[115] Copyright 2009, Elsevier. D) Schematic of dynamic culture whereby media displacement increases nutrient supply to engineered muscle. E) Dynamic culture (Dyn, bottom panel) of engineered muscle bundles increases cross-sectional muscle mass (F-actin, green) compared to static (Stat) culture. Reproduced with permission.[181] Copyright 2014, Elsevier. F) Dynamically cultured tissues of size show in (B) display cross-sectional area uniformly filled with myotubes (F-actin, green).
In vivo, nutrient delivery to skeletal muscle is achieved via the dense vascular system. Attempts to generate vascularized engineered muscle in vitro have been hampered by the different culture media required to maintain endothelial cells (ECs) and muscle cells in culture and interference of ECs with myoblast fusion.[186] Using C2C12 and human umbilical vein endothelial cells (HUVECs), it was shown that addition of a supporting fibroblast cell population enhanced formation of vascular lumen structures in vitro and that increasing culture time prior to implantation increased vessel density and accelerated implant perfusion and anastomosis with host vasculature in vivo.[112,187] A recent study showed that co-culture of C2C12 muscle bundles and HUVEC vascular bundles increased both muscle force production and vascular sprouting and lumen for-mation· suggesting a beneficial paracrine cross-talk between the two cell types,[188] which remains to be validated with human muscle cells. Importantly, the ability of the formed vascular networks to increase nutrient and oxygen delivery and enable survival of larger muscle constructs in vitro has yet to be shown.
5. Engineering Regenerative Muscle Tissues
Studies of skeletal muscle in vitro usually involve the isolation and expansion of primary myogenic progenitors followed by their differentiation into multinucleated myotubes. Myogenic progenitor isolation is performed either by enzymatic digestion or cell outgrowth from muscle fibers.[99] Regardless of the isolation method, obtained satellite cells cultured in vitro rapidly activate, express the myogenic transcription factor MyoD, and enter the cell cycle.[189] While the proliferation and expansion of satellite cells is desired, the rapid loss of SC identity and selfrenewal with time of culture prevents the ability to accurately study their function in vitro over prolonged periods.[190–192] A very small fraction of the in vitro expanded SCs that retain native-like characteristics are slow-dividing and when injected in vivo show good engraftment and re-population of the satellite cell niche. However, the rare occurrence of these cells precludes their use as a cell source for tissue engineering or cellular therapy necessitating alternative isolation methods for maintenance of SC identity and function.
A fundamental question in skeletal muscle cell culture is why satellite cells become rapidly activated and lose engraft-ment ability following in vitro culture.[193] One of the first factors identified is the role of substrate stiffness, with SCs cultured on gels with muscle-like stiffness retaining significantly higher engraftment ability after short-term expansion.[194] Adequate stiffness alone cannot help retain full engraftment potential of SCs, suggesting that other factors contribute to the loss of myogenic potential. A second factor regulating SC activation is metabolism, with glycolysis favoring SC activation and MyoD expression in a SIRT1 dependent manner,[195] with use of galactose media delaying and minimizing the expression of MyoD by decreasing glycolysis and SIRT1 activation. A third factor regulating SC activation is the activation of p38 MAPK, which is responsible for MyoD induction and is elevated in aged muscle.[196,197] Small molecule inhibition of p38 MAPK improves the engraftment ability of cultured SCs derived from aged mice or human muscle biopsies.[198,199] While these approaches have minimized activation of SCs, the SCs still proliferate and are not deactivated and returned to a quiescent state in vitro. Recently, human SC quiescence was achieved for 3 d on collagen I fibers coated with laminin and integrin α4β1 in a five-factor serum-free media.[200] The cells showed superior engraftment in vivo compared to traditional expansion methods, but the media could still not induce quiescence of activated SCs, which would be required for cell therapy and engineered tissue approaches to study muscle regeneration.
Furthermore, while the above studies have shown that a select population of expanded muscle progenitors can retain engraftment and myogenic potential in vivo, there has been little evidence for the regenerative ability of these cells in vitro. Through a combination of short-term expansion on matrigel-coated surface and dynamic 3D culture, significant numbers of neonatal rat satellite cells were retained within an SC-like niche in engineered muscle tissues (Figure 5).[151] These SCs displayed two native functional properties: 1) continual fusion into myotubes over a four week culture period as seen during postnatal development, and 2) the ability to contribute to muscle regeneration following cardiotoxin (CTX) injury in vitro. In response to CTX, a snake toxin typically used for in vivo muscle regeneration studies, significant myotube injury and death resulted in a loss of force generation (Figure 5). Remarkably, over a period of 10 d, both the contractile function and muscle mass returned to preinjury levels, while MRFs exhibited spati-otemporal expression profiles typical of muscle regeneration in vivo. Importantly, both pre and postinjury, a large population of the Ki67− SCs were found to reside within native-like niches, suggesting attainment of a quiescent state. While this study represents a key advance in demonstrating the ability of satellite cells to maintain functionality in vitro, it utilized cells that were derived from neonatal rat muscles and only expanded for 2 d before tissue fabrication. Thus, this approach remains to be extended to adult rodent and human cells and, ideally, include significant cell expansion in vitro.
Figure 5.
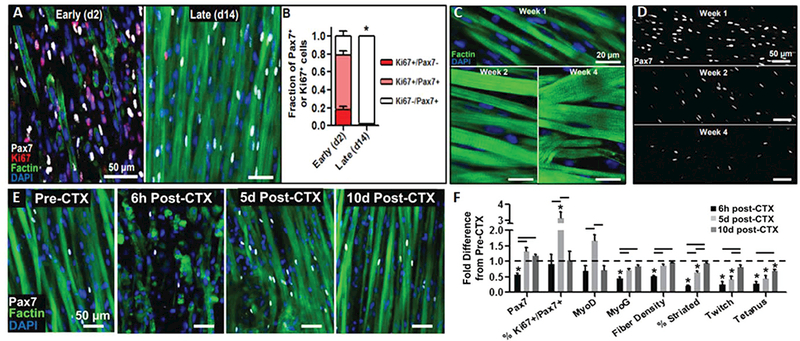
Presence of functional satellite cells in neonatal rat-derived tissue-engineered muscle. A) Representative images and B) corresponding quantification showing that highly proliferative Pax7+ cells present upon tissue formation (day 2) enter quiescence within tissue-engineered muscle dynamically cultured for 14 d. C,D) Representative images C) showing progressive myotube hypertrophy and D) decrease in the number of Pax7+ cells with time of culture (1–4 weeks). E) Representative images of engineered muscle structure and Pax7+ cell dynamics and F) corresponding quantification of myogenic transcription factors, muscle differentiation markers, and muscle function demonstrating robust regenerative response after cardiotoxin injury in vitro (6 h to 10 d post-CTX). Reproduced with permission.[151] Copyright 2014, Elsevier.
6. Disease Modeling with Engineered Human Muscle
Congenital skeletal muscle diseases (myopathies) can develop due to a wide range of genetic mutations that often effect the contractile or metabolic apparatus of muscle cells. Traditionally, myopathies have been modeled in vivo using genetically modified animals that have significantly contributed to the mechanistic understanding of disease, but often fail to fully replicate human pathology. For example, animal models are often generated by complete knockout of the gene of interest, while corresponding human diseases do not completely lack the underlying protein but have a truncated or nonfunctional form. Second, a broad range of mutations can cause the same disease in humans that cannot be rapidly generated in mice. Third, the severity of disease can be modulated in a patient-specific fashion by disease modifiers located in other genes, while studies in mice are usually performed in a single genetic background.[201–203] Fourth, animal models typically only replicate certain aspects of human disease, with the severity, tissues affected, and onset of disease potentially differing from that in humans.[97] Fifth, the majority of drugs identified to work in animals fail to translate to the clinic due to differential toxicity between animal species.[8] Engineering functional muscle from a patient’s cells has the potential to overcome some of these limitations and lead to a new era of personalized medicine. Ideally, this could be achieved by: 1) having access to an abundant patient-specific cell source from which to generate engineered muscle, 2) being able to replicate most or all clinically relevant aspects of disease in vitro, and 3) identifying new drugs that will translate to clinic more efficiently than use of current animal models.
6.1. Induced Pluripotent Stem Cells as a Source for Muscle Tissue Engineering
A recent report on the first engineering of functional human skeletal muscle tissues using primary myogenic cells isolated from healthy donor muscle biopsies[9] represents an important step toward our ability to study pharmacological and physiological responses of diseased human muscle in vitro. Still, the use of primary engineered muscle tissues for disease modeling may encounter the ethical concerns and difficulties with obtaining muscle biopsies from patients with debilitating muscle diseases. The hiPSC technology, on the other hand, offers the possibility for generating skeletal muscle tissues from an unlimited source of cells derived from patients’ blood or skin samples. Combined with the rapid progress in genome engineering techniques,[204–206] hiPSC lines can be derived to permit direct comparison among various disease mutations in an isogenic background. Thus, generation of functional skeletal muscle cells and tissues from pluripotent stem cells has been actively pursued by researchers in the field for both in vitro and potential in vivo applications, as outlined below.
6.1.1. MyoD Overexpression
Seminal studies by Constantinides et al. more than 40 years ago have shown that fibroblasts can be converted to muscle cells by treatment with demethylating agent 5-azacytidine.[207] A decade later, it was shown that this conversion was due to the activation of the transcription factor MyoD, with overexpression of MyoD alone also converting fibroblasts to muscle cells.[208,209] When tested in different tissue-specific cells, myogenic differentiation only occurred in some cell types demonstrating that a specific genetic landscape is required for transdifferentiation (Table 1). Overexpression of MyoD in mouse embryonic stem cells also generated myoblasts and myotubes but only after embryoid body (EB) formation,[210,211] a step in which pluripotent stem cells spontaneously differentiate into the three germ layers,[212] with a subset of cells further differentiating into the paraxial mesoderm.[213] Similar findings have been reported for human ESCs, where inducible MyoD over expression generated skeletal muscle cells only following EB formation.[214–216] Recently, the use of a PiggyBac transposon system to overexpress MyoD directly reprogramed undifferentiated iPSCs into myoblasts without the need for EB formation or paraxial mesoderm dif-ferentiation.[217] Underlying mechanisms are unclear, but the high and stable expression of MyoD transgene may be required. In support, daily transfections of high levels of synthetic MyoD mRNA also enabled direct myogenic differentiation of iPSCs,[218] albeit at lower efficiency (≈40%) than the PiggyBac transposon system (70–90%). MyoD overexpression in hiPSCs has since been used to generate muscle cells from patients with Duchenne muscular dystrophy,[219,220] Miyoshi myopathy,[217] carnitine palmitoyltransferase-II deficiency,[221] and Pompe disease.[222–225] Encouragingly, generated muscle cells from iPSCs exhibited some of typical disease features including impaired calcium handling, impaired membrane resealing, and greater glycogen accumulation (Table 2). However, the MyoD overexpression system does not yield expandable progenitors or satellite cells, which precludes its use for accurate in vitro modeling of muscle regeneration. Additionally, MyoD-overexpressed iPSCs can engraft in myofibers in vivo,[226] but they do not repopulate SC niche, thus having a limited therapeutic prospect.
Table 1.
Summary of transgenic methods to reprogram pluripotent stem cells into skeletal muscle. Compilation of selected published works using transgenic methods to generate skeletal muscle from pluripotent stem cells (PSCs) listed in chronological order. For each study, key parameters including PSC species (H: human; M: mouse), PSC type, culture format (EB: embryoid body), the transcription factor(s) overexpressed, cell surface markers used for cell sorting, diseases modeled (DMD: Duchenne muscular dystrophy), and if in vivo transplantations were performed. Key findings and methodology such as transfection method and disease phenotypes seen are listed in the comments column.
| Study | Species | Cell type | Culture method | Transgene | Cell sorting | Disease model | In vivo | Comments |
|---|---|---|---|---|---|---|---|---|
| Dekel et al. (1992)[211] |
M | ESC | EB | MyoD | N/A | N/A | N/A | Electroporation, mixed populations of myoblasts and myotubes |
| Shani et al. (1992)[210] | M | ESC | EB | MyoD | N/A | N/A | N/A | Electroporation, mixed populations of myoblasts and myotubes |
| Darabi et al. (2008)[227] | M | ESC | EB | Pax3 | PDGFRa+ Flk1− | N/A | Yes | Tet-on system, increased muscle function and dys expression in mdx mice |
| Warren et al. (2010)[218] | H | iPSC | EB | MyoD | N/A | N/A | N/A | Synthetic MyoD mRNA transfection |
| Darabi et al. (2011)[228] | M | ESC | EB | Pax3, Pax7 | PDGFRa+ Flk1− | N/A | Yes | Tet-on system, increased muscle function and dys expression in mdx mice |
| Darabi et al. (2012)[229] | H | ESC, iPSC | EB | Pax7 | GFP | N/A | Yes | Tet-on system, Pax7+ cells in SC-like niche in vivo |
| Tedesco et al. (2011)[277] | H, M | iPSC | 2D | MyoD | N/A | N/A | Yes | TMX-ERT system, mesoangioblast-like cells, i/a delivery improved mdx muscle function |
| Goudenege et al. (2012)[226] | H | ESC | EB | MyoD | N/A | N/A | Yes | Adenoviral vector, dys+ fibers |
| Tanaka et al. (2013)[217] | H | iPSC | 2D | MyoD | N/A | Miyoshi Myopathy | N/A | PiggyBac transposon tet-on system, defective membrane resealing in diseased myotubes |
| Albini et al. (2013)[215] | H | ESC | EB | MyoD, Baf60c | N/A | N/A | N/A | Lentiviral vector, Baf60c improved muscle generation efficiency |
| Li et al. (2015)[249] |
H | iPSC | 2D | MyoD | N/A | DMD | N/A | PiggyBac transposon Tet-on system, DMD correction via genome editing |
| Shoji et al. (2015)[219] |
H | iPSC | 2D | MyoD | N/A | DMD | N/A | PiggyBac transposon Tet-on system, DMD correction via exon skipping |
| Sato et al. (2016)[222] | H | iPSC | 2D | MyoD | N/A | Pompe | N/A | PiggyBac transposon Tet-on system, increased glycogen content in Pompe cells |
| Yoshida et al. (2017)[223] | H | iPSC | 2D | MyoD | N/A | Pompe | N/A | PiggyBac transposon Tet-on system, decreased glycogen clearance in Pompe cells |
Table 2.
Summary of disease models using human muscle cells. Compilation of published works using human cells to study muscle diseases listed in chronological order. For each study, key parameters including cell type (iPSC-MyoD indicates MyoD overexpression; iPSC-SMD indicates small molecule differentiation; and primary cells), culture method, disease modeled (LGMD2B, Limb girdle muscular dystrophy 2B; CPT-II, carnitine palmitoyltransferase II; DMD, Duchenne muscular dystrophy; BMD, Becker muscular dystrophy), reported phenotype (CK, creatine kinase; GAA, acid alpha-glucosidase), treatment or correction tested (AON, antisense oligonucleotides; HAC, human artificial chromosome; TFEB, transcription factor EB), and observed therapeutic effects have been shown.
| Study | Cell type | Culture method | Disease model | Reported phenotype | Treatment or correction | Therapeutic effect |
|---|---|---|---|---|---|---|
| Tanaka et al. (2013)[217] | iPSC -MyoD | 2D | LGMD2B | Defective membrane resealing, decreased dysferlin protein level | Full length dysferlin expression via PiggyBac Transposon | Restored membrane resealing similar to healthy control |
| Yasuno et al. (2014)[221] | iPSC-MyoD | 2D | CPT-II deficiency | Increased palmitoylcarnitine accumulation | Bezafibrate | Decreased palmitoylcarnitine accumulation in both CTL and CPT-II myotubes |
| Abujarour et al. (2014)[220] | iPSC-MyoD | 2D | DMD BMD | Not reported | Wnt7a and IGF-1 | Hypertrophy of DMD and BMD myotubes |
| Li et al. (2015)[249] |
iPSC-MyoD | 2D | DMD | No dystrophin protein | TALEN and CRISPR exon skipping or exon knock-in | Restoration of full-length dystrophin with exon knock-in, minimal off-target mutagenesis |
| Shoji et al. (2015)[219] | iPSC-MyoD | 2D | DMD | No dystrophin protein, increased CK release and calcium influx | AON exon 45 skipping | Induction of dystrophin protein, decreased CK and Ca2+ influx |
| Choi et al. (2016)[233] |
iPSC-SMD | 2D | DMD | Increased TGF-β signaling, impaired myoblast fusion | HAC with full length dystrophin, combined BMP4 and TGF-β inhibition | Increased dystrophin expression and myoblast fusion by HAC, improved fusion in patient-specific manner by drug inhibition |
| Nesmith et al. (2016)[247] | Primary | 3D | DMD | Delayed and impaired fusion, decreased contractile function | N/A | N/A |
| Sato et al. (2016)[222] | iPSC-MyoD | 2D | Pompe | Decreased GAA enzyme activity and increased glycogen accumulation | Lentiviral GAA and TFEB |
Synergistically increased glycogen clearance |
| Yoshida et al. (2017)[223] | iPSC-MyoD | 2D | Pompe | Decreased GAA activity, no increase in glycogen accumulation, decreased glycogen clearance | Recombinant human GAA |
Increased glycogen clearance in serum-starved cells |
| Van der Wal et al. (2017)[224] | Ipsc -SMD | 2D | Pompe | Decreased GAA transcript and enzyme activity | AONs for exon 2 inclusion | Increased GAA activity up to 3.3-fold, no change in lysosome size or accumulation |
6.1.2. Pax3/Pax7 Overexpression
During embryonic muscle development and adult muscle regeneration, expressions of Pax3 and Pax7, respectively, precede that of MyoD. Thus, to better mimic muscle development, the Perlingeiro lab was the first to generate MPCs by doxycycline-inducible expression of Pax3 in ESC-derived EBs.[227] Induction of Pax3 permitted generation of early myogenic progenitors with high Myf5 but low MyoD and Myogenin expression that could be differentiated into multinucleated myotubes. In vivo implantation of MPCs required selection of a PDGR+/FLK− cell fraction to avoid teratoma formation and yielded improved muscle function in a mouse model of Duchenne muscular dystrophy (mdx mice). Further studies demonstrated that muscle progenitors could be derived via inducible Pax3 or Pax7 overexpression from mouse or human ES and iPS cells with no difference in ability to generate MPCs, engraft in vivo, improve force generation in mdx mice, and contribute to regeneration after multiple muscle injuries.[228,229] Unlike MyoD-reprogramed iPSCs, Pax3/7 induced MPCs can repopulate SC niches in vivo; however, the need to generate EBs, use of serum, low differentiation efficiency, and requirement for lentiviral transduction may limit the potential clinical translation of this system in the future.
6.1.3. Small Molecule Differentiation/Transgene-Free Methods
While the transgenic reprogramming methods to generate skeletal muscle cells from hiPSCs show promise for studying and reating muscle diseases, they involve random genomic integration that can induce uncontrolled epigenetic modifications and heterogeneity in isogenic cultures. Thus, developmentally based, transgene-free, small molecule differentiation (SMD) protocols for generation of skeletal muscle cells have been pursued by various researchers in the field. Initial reports using mouse ESCs demonstrated spontaneous muscle development with correct temporal MRF expression and formation of multinucleated myotubes within EBs.[230] More recently, PDGFRα positive cells were isolated from human EBs and shown to be able to expand and generate myotubes in vitro and survive in vivo.[214] Since lineage specification in EBs is difficult to control and the differentiation efficiency is serum and cell line dependent, the EB-based methods are increasingly being replaced by monolayer-based differentiation protocols.
The first stage of SMD in hiPSC monolayers is the stepwise induction of late primitive streak, mesoderm, and paraxial mesoderm lineages (Figure 1).[16] For induction of mesoderm progenitors, the roles of FGF, insulin, Wnt, BMP, and activin signaling have been extensively characterized for their ability to induce the pan-mesodermal marker T and generate various mesoderm subpopulations.[231] Late primitive streak progenitors are initially induced with activin, FGF, and a PI3K inhibitor, followed by Wnt activation through GSK3 inhibition to generate paraxial mesoderm.[232] Small molecule drug screens in Mesogenin1 and Tbx6 reporter lines of paraxial mesoderm[11,233] have further revealed that the GSK3 inhibition or Wnt activation can generate paraxial mesoderm without late primitive streak induction, although BMP4 may also need to be inhibited to prevent spontaneous transition to the lateral plate mesoderm.[11] Despite the use of similar small molecules to generate paraxial mesoderm, the concentrations and exposure times differ between laboratories and likely need to be tailored to specific cell lines.[10,11,233,234] Interestingly, relative to other GSK3 inhibitors, CHIR99021 has been the most effective inducer of paraxial mesoderm.[233] However, the precise molecular mechanisms that underlie its effectiveness are still not fully understood.
Despite robust methods for induction of paraxial mesoderm, transition of these cells into muscle progenitors still represents a challenge as spontaneous transition to Pax3+ cells occurs with variable and relatively low efficiency.[10,11,234,235] Growth factors such as bFGF, HGF, and EGF have been thus added to expansion media to maximize myogenic proliferation and minimize premature differentiation in an attempt to generate high numbers of Pax3+ and Pax7+ cells. Recently, a small molecule screen identified GSK3-β inhibition, bFGF, and forskolin as a cocktail to efficiently induce hiPSCs to skeletal muscle cells.[236]The most efficient muscle differentiation from pluripotent stem cells was reported when hESCs were sequentially treated with: 1) muscle induction media to generate 80% Pax3+ cells and 20% Pax7+ cells in 10 d, 2) a myoblast expansion media (containing Alk4, 5 and 7 inhibitor, PDGF, bFGF, oncostatin, and IGF-1) that increased myogenic commitment to 50–60% MyoD positive cells, and 3) a final media supplemented with insulin, necrosulfonamide (to decrease cell death), oncostatin, and ascorbic acid to induce terminal muscle differentiation.[235] Alternatively, timed inhibition of Notch signaling known to promote myogenic commitment of presomitic mesoderm progenitors was used to increase the proportion of MyoD cells; however, the maturity of the resulting myotubes appeared to be inferior to other SMD methods.[233]
One disadvantage of the transgene-free protocols is that they generate a heterogeneous population of cells consisting of Pax7+/Myf5+ muscle progenitors, committed MyoD+ myogenic cells, and terminally differentiated myogenin+/MyHC+ cells. The ideal protocol would however generate a pure population of progenitor cells resembling native satellite cells. To address this issue, several studies have identified cell surface markers to purify muscle progenitors capable of further expansion in vitro, including CD73+,[237] PDGFR+/FLK−,[227] and cMET+/CXCR4+/ACHR+ selection.[234] More recently, a screen of over 300 cell surface markers identified CD82 and CD318 as novel cell surface markers of SCs in human skeletal muscle.[238] By RNA-Seq comparison of hPSCs with and without Pax7 overexpression, CD54, integrin α9β1, and syndecan 2 (SDC2) were identified and validated as cell surface markers for Pax7+ progenitors.[239] These cells were capable of contributing to muscle regeneration in vivo but their ability to be expanded in vitro was not demonstrated. Overall, the above studies indicate that iPSC-derived MPCs may have specific surface markers different from well-known primary myogenic cell markers, such as CD56,[239] although dual CD56+/CD57− selection was able to robustly enrich for hiPSC-derived myogenic progenitors.[233]
6.2. Modeling DMD In Vitro
To date, the only reported genetic disease model studied using 3D engineered muscle tissues has been DMD, an X-linked recessive disease that affects ≈1 in 5000 live births.[240] DMD is characterized by mutations in dystrophin gene leading to severe muscle weakness, loss of ambulation by age 11 to 12, and premature death in the late 20s to early 30s.[241] More than 4000 mutations have been identified in the DMD gene, with the majority comprising of exon deletions (≈68%) or duplications (≈11%) that result in fame-shift mutations and premature translation termination.[242,243] The severity of the DMD phenotype and efficacy of pharmacological treatments is significantly impacted by gene modifiers,[202] with effectors of TGF-beta signaling strongly implicated with disease pro-gression.[201,203,244,245] Together, the wide range of mutations and strong effects of gene modifiers on disease severity and response to treatment would greatly benefit from the use of patient-specific engineered muscle tissues to effectively model the disease and develop personalized therapeutic approaches.
The first tissue-engineered muscle model of DMD was generated by embedding immortalized cells derived from the mdx mouse in a collagen gel.[246] The 31 drugs reported to improve function in the mdx mouse model or DMD patients were then tested for their ability to enhance force generation in engineered tissues, of which 11 drugs showed positive effects including clinically used prednisolone and deflazacort. Though promising, the treatments were only performed for 3–4 d and the 11 identified drugs also increased force generation in healthy control tissues, suggesting that the force increase may stem from increased differentiation and maturation of the myotubes and not a disease-specific effect that may be clinically relevant. Recently, primary human DMD myoblasts were shown to exhibit decreased alignment on microtopographic cues, which likely contributed to their reduced fusion, smaller myotube size, and weaker contractions compared healthy myoblasts.[247] Still, histopathological and functional features of a more advanced DMD phenotype such as myofiber branching and hypercontractions, fibrosis and lipid accumulation, and increased membrane permeability and susceptibility to injury have not been demonstrated. An important consideration for studying DMD in vitro is that significant membrane-bound expression of dystrophin occurs only in mature myotubes, and is yet to be reported in human cells in vitro.[248] Therefore, achieving advanced structural and functional maturation in engineered human muscle tissues will be critical for the ability to perform in vitro predictive screening of candidate therapies for DMD.
While the above studies have utilized primary myogenic cells from mice or humans, most future studies will utilize hiPSCs due to the ethical concerns associated with obtaining DMD muscle biopsies. Recently, myotubes differentiated from DMD hiPSCs have been shown to exhibit several disease-related features including impaired calcium handling, increased creatine kinase release, and branched myotube morphology.[11,219,233] By applying DNA microarray analysis to hiPSC-derived myotubes generated from five DMD patients, Choi et al.[233] have demonstrated increased TGFβ signaling in all five lines, as well as patient-dependent variation in the expression of osteopontin, a potent genetic modified or disease severity. Moreover, myotube formation was significantly impaired in all DMD lines and could be partially restored in certain donors by dual Smad inhibition suggesting that patient-specific therapeutic responses could be identified using this system. While this platform holds promise for future personalized medicine applications, the efficiency of myotube formation was extremely low, requiring caution when extrapolating the results to clinical setting.
6.2.1. Using Genome Editing and Tissue Engineering to Study and Treat DMD
TALEN and CRISPR-Cas9 are genome editing tools that can induce permanent modifications to the dystrophin gene to maintain the open reading frame and thus theoretically do not need lifelong repeated treatments. In DMD hiPSCs, it was demonstrated that both TALEN or CRISPR mediated exon skipping could restore dystrophin protein levels with minimal off-site effects and that corrected clones with minimal mutational load could be selected for potential future in vivo applications.[249] Tissue engineering offers benefits in testing the efficacy of different genome correction methods as shown using a functionally deficient, engineered DMD cardiac tissue model made of hiPSCs in which exons 8–9 were deleted by CRISPR-Cas9.[250] Three different exon-deletion strategies were then tested for their ability to restore the open reading frame of dystrophin and rescue deficit in contractile function. It was found that different approaches had significantly different effects, with exon 3–7 deletion being most efficient in restoring cardiomyocyte functionality close to that of control unedited tissues. Similar approaches could be applied to functional hiPSC-derived engineered muscle tissues, once methods for their fabrication are developed.
6.3. Using Engineered Muscle Tissues to Improve Drug Screening Fidelity
For several decades cell culture models have been used in drug screening assays to identify novel therapeutic candidates, which were then tested in small and large animal models. However, only 10% of drugs that pass preclinical animal tests are eventually used clinically due to unforeseen drug toxicity or insufficient therapeutic benefits, resulting in an average drug development cost of≈1 billion dollars[8,251,252] Culturing human cancer cells in a 3D microenvironment has been shown to yield more predictive drug screening assays than use of 2D cultures or animal models, suggesting that 3D human tissue models may be able to better model clinical drug responses[253,254] For skeletal muscle, desired drug response would likely include improved contractile function and/or tissue regeneration. In 2D cultures, these outcomes can only be measured indirectly via expression of differentiation marker levels, fusion index, or cell viability. The potential to improve cell maturation within 3D culture environment,[9] measure contractile function, and study SC-based muscle regeneration[151] renders 3D engineered human muscle tissues particularly attractive for in vitro screening of drug toxicity and efficacy.
Of specific importance for potential drug development applications is the fact that human engineered muscle tissues appear to behave like native tissues in response to known myotoxic or mitochondrial toxic drugs.[9,255] Unforeseen mitochondrial toxicity is the most common cause for failure of candidate drugs identified from in vitro drug screens in preclinical animal models.[256] This can be attributed to the Crabtree effect,[257] where in vitro cultured cells typically utilize glycolysis to generate ATP despite the presence of abundant oxygen and functional mitochondria.[258] The Crabtree effect can be circumvented by replacing glucose in the cell culture media with galactose, which decreases energy obtained from glycolysis and forces cells to rely on oxidative phosphorylation to meet their energy demands.[259] A hallmark of Type II diabetes is impaired oxidative metabolism, which in 2D cultures was only seen when myotubes were grown in galactose media.[260] While there have been no reports on growing engineered muscles tissues in galactose, use of 25 × 10−3 m glucose increases force generation of engineered muscle compared to 5 × 10−3 m glucose.[261] The conventional paradigm for improving engineered muscles is to increase their force generation, but this clearly should be achieved while best maintaining physiological conditions. Future cell culture media will likely undergo modifications to better mimic human plasma composition and to allow serum-free culture, both of which should generate more predictive drug responses in vitro and improve reproducibility among different laboratories[262–266]
In addition to improved sensitivity to mitochondrial toxicity, optimal drug screening assays should also model complex organ-organ interactions that ultimately govern drug metabolism and toxicity in the body. Recent advances in organ-on-a-chip technologies can allow multiple engineered tissue types to be multiplexed by: 1) direct coupling via microfluidic flow or 2) functional coupling via sequential culturing and transferring media from one tissue type to another. Direct coupling better replicates the in vivo milieu but considerations such as organ-specific flow rates, rapid accumulation of metabolic waste, and the requirement of a common media for all organs pose significant technical challenges[267,268] Direct coupling has been achieved with three to four organs,[269,270] including one system that incorporated skeletal muscle, cardiac muscle, liver, and neuronal tissues and demonstrated expected responses to known toxic drugs.[271] The benefit of these microphysiological platforms for identifying unexpected drug toxicity was shown in a system integrating the heart, lung, and liver, whereby adding bleomycin to cardiomyocytes alone showed no toxic effect, but coupling with the lung resulted in severe cardiomyocyte toxicity.[270] Additionally, functional coupling of hepatocytes and skeletal muscle demonstrated that liver tissues could prevent the potent myotoxic effects of terfenadine, an anti-histamine known to have cardiac arrhythmogenic effects.[272] Improved predictive capacity in future studies will require addition of other tissue-engineered organs, including the gut and intestine to model first-pass metabolism that regulates the concentration and chemical form of any drug that enters the bloodstream.[273]
7. Current and Future Applications of Engineered Muscle Tissues
Compared to the traditional monolayers, 3D engineered muscle tissues offer the possibility for longer-term culture, improved cell maturation, and ability to measure functional outcomes. For these reasons, engineered muscle is the current gold standard for in vitro studies of human skeletal muscle function, development, and disease. Importantly, the ability to generate large numbers of miniaturized functional skeletal muscle tissues from patient-specific cells that respond to pharmacological interventions in a comparable manner to native muscle has the potential to start a new era of personalized medicine. For example, a patient requiring pharmacological treatment or gene therapy could have skeletal muscle microtissues engineered from a primary biopsy or iPSCs and utilized in a high-throughput functional screen to identify best therapeutic options (Figure 6). Nevertheless, the future use of engineered muscle tissues for high-fidelity preclinical drug screening studies awaits the development of improved methods to: 1) expand primary human satellite cells in vitro without loss of self-renewing or myogenic capacity and 2) increase efficacy of generating hiPSC-derived Pax7+ progenitors via small molecule differentiation and surface marker purification. Additionally, human engineered muscles must be significantly matured in vitro to achieve an adult-like phenotype, while their regenerative capacity must be verified in response to single and repeated muscle injuries. While significant advances toward overcoming these challenges have been already made through the combination of unique engineering approaches and improved understanding of underlying biology, it is important to recognize that the native muscle development in humans takes many years. Therefore, the ability to generate engineered human muscle tissues with stable adult-like structure, function, and metabolism will likely pose many unforeseen challenges and may require unconventional and potentially nonbiomimetic approaches to ultimately achieve the success.
Figure 6.
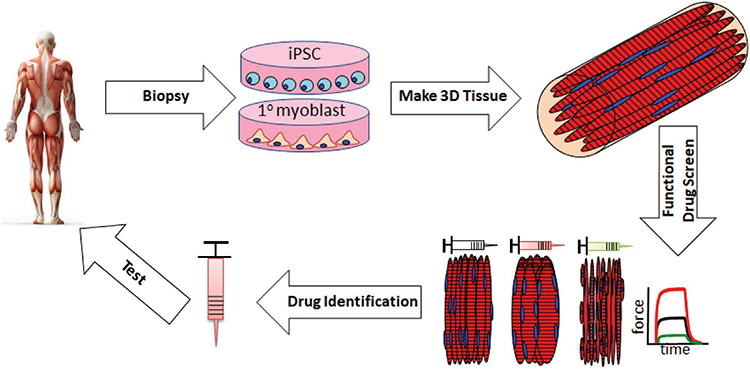
Potential use of engineered muscle tissues for personalized medicine applications. Tissue biopsies taken from a patient can be made into engineered muscle tissues via primary myoblast or hiPSC-based technologies and different pharmaceutical or gene therapy strategies can be rapidly tested for safety and efficacy in vitro. Upon identification and further testing in animal models in vivo and/or human multiorgan body-on-a-chip models in vitro, the candidate therapy can undergo ultimate validation in clinical studies.
Furthermore, methods to integrate multiple human microtissues on the same platform will be essential to better replicate the intricate organ-organ cross-talk and will require the development of a “universal” culture media that simultaneously supports maturation and function of multiple tissue types. Ideally, this media should also be serum-free and promote oxidative metabolism to negate the Crabtree effect. In addition to standardization of cell and tissue preparation and culture conditions, pharmacological studies in hiPSC-based microphysiological systems must undergo frequent quality control. Recent studies have demonstrated that hiPSCs accumulate large and varied numbers of mitochondrial mutations within clones of the same isogenic lineage that can significantly impact mitochondrial function.[274,275] Resulting changes in cellular respiration would adversely affect engineered muscle function and outcomes of drug studies, and thus require frequent screening of mitochondrial DNA and metabolic function to prevent such issues. Given the heterogeneous nature of mutations, multiple clones from each hiPSC line should be pretested for resultant muscle function and robust guidelines established to ensure reproducibility and efficacy of the screening procedures.
The above advances in human muscle cell culture are also expected to promote the development of regenerative therapies for muscle disease and injury. Recently, freshly isolated satellite and endothelial cells were embedded within a hydrogel and after bioreactor culture for 24 h implanted into a murine model of VML injury.[276] When combined with chronic exercise, this treatment yielded partial innervation of grafted muscle and improved host function. Similarly, hiPSC-derived muscle cells and mesoangioblasts improved muscle function when implanted in dystrophic mice, thus suggesting that muscle progenitor cell delivery can induce therapeutic benefits in small animal models.[227,229,277] Significant scale-up of myogenic cell numbers by robust expansion of primary SCs or generation of hiPSC-derived progenitors may pave the way for testing these approaches in larger animals and clinics. On the other hand, therapies utilizing mature functional engineered muscle tissues face a significantly larger number of challenges. Fundamental issues with efficient restoration of SC niche, tissue prevascularization without functional loss, rapid anastomosis with host vasculature, and functional innervation upon implantation need to be all addressed before considering potential for translation[109,278]
8. Conclusion
In this report, we have highlighted the considerable progress that has been made in the fields of skeletal muscle tissue engineering and stem cell biology. These advances have provided researchers with novel tools and models with which to study and treat human muscle disease. Future progress in pluripo-tent stem cell and genome editing technologies and development of high-fidelity human multiorgan microphysiological systems are expected to further accelerate basic discoveries and translational efforts, ultimately benefiting millions of muscle disease patients worldwide.
Acknowledgements
This work was supported by the NIH Grants AR070543 and AR065873 from the National Institute of Arthritis and Musculoskeletal and Skin Disease (NIAMS) and grants UH3TR000505 and UG3TR002142 from the NIH Common Fund for the Microphysiological Systems Initiative and NIAMS. The content of the manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Biographies
Alastair Khodabukus is a postdoctoral scholar in the department of Biomedical Engineering at Duke University. He earned his Ph.D. at the university of Dundee in 2011 from the department of Molecular Physiology. His research interests involve optimizing the function and maturation of engineered human muscle tissues made of primary and iPSC-derived progenitors to study muscle physiology and disease.

Neel Prabhu is a third-year undergraduate student studying Biomedical Engineering at Duke University. He joined the laboratory of Prof. N. Bursac in 2015, where he now develops tissue engineered models of congenital muscle disease as an Edmund T. Pratt Jr. Research Fellow. With dual interests in medicine and translational science, Neel is currently applying to medical schools to pursue a career as a physician-scientist.

Nenad Bursac is a Professor of Biomedical Engineering and Cell Biology at Duke University. During his Ph.D. and postdoctoral work at MIT and JHU, he developed first engineered mammalian heart tissues and methods to study cardiac arrhythmias in a dish. Currently, his research involves design of high-fidelity human organ-on-a-chip systems to study pathophysiology of striated muscles in vitro and development of novel cell and gene-based therapies for cardiac and skeletal muscle regeneration in vivo.

Footnotes
Conflict of Interest
The authors declare no conflict of interest.
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adhm.201701498.
References
- [1].Janssen I, Heymsfield SB, Wang ZM, Ross R, J. Appl. Physiol 2000, 89, 81. [DOI] [PubMed] [Google Scholar]
- [2].Dumont NA, Wang YX, Rudnicki MA, Development 2015, 142, 1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Alway SE, Myers MJ, Mohamed JS, Front. Aging Neurosci. 2014,6, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wallace GQ, McNally EM, >Annu. Rev. Physiol 2009, 71, 37. [DOI] [PubMed] [Google Scholar]
- [5].Grogan BF, Hsu JR,J. Am. Acad. Orthop. Surg 2011, 19 (Suppl. 1), S35. [DOI] [PubMed] [Google Scholar]
- [6].McGreevy JW, Hakim CH, McIntosh MA, Duan D, Dis. Models Mech 2015, 8, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cabrera PV, Pang M, Marshall JL, Kung R, Nelson S SF.Stalnaker H, Wells L, Crosbie-Watson RH, Baum LG,J. Biol. Chem 2012, 287,22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J, Nat. Biotechnol 2014, 32, 40. [DOI] [PubMed] [Google Scholar]
- [9].Madden L, Juhas M, Kraus WE, Truskey GA, Bursac N, eLife 20144, e04885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shelton M, Metz J, Liu J, Carpenedo RL, Demers SP, Stanford WL, Skerjanc IS, Stem Cell Rep. 2014, 3, 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chal J, Oginuma M, Al Tanoury Z, Gobert B, Sumara O, Hick A, Bousson F, Zidouni Y, Mursch C, Moncuquet P, Tassy O, Vincent S, Miyanari A, Bera A, Garnier JM,Guevara G, Hestin M, Kennedy L, Hayashi S, Drayton B,Cherrier T, Gayraud-Morel B, Gussoni E, Relaix F, Tajbakhsh S, Pourquie O, Nat. Biotechnol 2015, 33, 962. [DOI] [PubMed] [Google Scholar]
- [12].Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L,Zhang F, Vandenberghe LH, Church GM, Wagers AJ, Science 2016, 351, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nelson CE, Hakim CH, Ousterout DG, Thakore PI,Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X,Ran FA, Yan WX, Asokan A, Zhang F, Duan D,Gersbach CA, Science 2016, 351, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Long C, Amoasii L, Mireault AA, McAnally JR, Li H,Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R,Olson EN, Science 2016, 351, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sambasivan R, Kuratani S, Tajbakhsh S, Development 2011, 138, 2401. [DOI] [PubMed] [Google Scholar]
- [16].Chal J, Pourquie O, Development 2017, 144, 2104. [DOI] [PubMed] [Google Scholar]
- [17].Yamaguchi TP, Takada S, Yoshikawa Y, Wu N, McMahon AP, Genes Dev. 1999, 13, 3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ciruna B, Rossant J, Dev. Cell 2001, 1, 37. [DOI] [PubMed] [Google Scholar]
- [19].Hubaud A, Pourquie O, Nat. Rev. Mol. Cell Biol 2014, 15, 709. [DOI] [PubMed] [Google Scholar]
- [20].Musumeci G, Castrogiovanni P, Coleman R, Szychlinska MA,Salvatorelli L, Parenti R, Magro G, Imbesi R, Acta Histochem. 2015, 117, 313. [DOI] [PubMed] [Google Scholar]
- [21].Endo T, Bone 2015, 80, 2. [DOI] [PubMed] [Google Scholar]
- [22].Epstein JA, Shapiro DN, Cheng J, Lam PY, Maas RL, Proc. Natl. Acad. Sci. USA 1996, 93, 4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zammit PS, Semin. Cell Dev. Biol 2017, 72, 19. [DOI] [PubMed] [Google Scholar]
- [24].Tajbakhsh S, Rocancourt D, Buckingham M, Nature 1996, 384, 266. [DOI] [PubMed] [Google Scholar]
- [25].Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R, Cell 1993, 75, 1351. [DOI] [PubMed] [Google Scholar]
- [26].Buckingham M, Curr. Opin. Genet. Dev 2006, 16, 525. [DOI] [PubMed] [Google Scholar]
- [27].Kassar-Duchossoy L, Gayraud-Morel B, Gomes D, Rocancourt D,Buckingham M, Shinin V, Tajbakhsh S, Nature 2004, 431, 466. [DOI] [PubMed] [Google Scholar]
- [28].Rawls A, Valdez MR, Zhang W, Richardson J, Klein WH,Olson EN, Development 1998, 125, 2349. [DOI] [PubMed] [Google Scholar]
- [29].Nabeshima Y, Hanaoka K, Hayasaka M, Esumi E, Li S,Nonaka I, Nabeshima Y, Nature 1993, 364, 532. [DOI] [PubMed] [Google Scholar]
- [30].Buckingham M, Bajard L, Daubas P, Esner M, Lagha M,Relaix F, Rocancourt D, Anat. Embryol 2006, 211 (Suppl. 1), 51. [DOI] [PubMed] [Google Scholar]
- [31].Yoshida N, Yoshida S, Koishi K, Masuda K, Nabeshima Y,J. Cell Sci 1998, 111 (Pt 6), 769. [DOI] [PubMed] [Google Scholar]
- [32].Dennis MJ, Ziskind-Conhaim L, Harris AJ, Dev. Biol 1981, 81, 266. [DOI] [PubMed] [Google Scholar]
- [33].Harris AJ, Duxson MJ, Fitzsimons RB, Rieger F, Development 1989, 107, 771. [DOI] [PubMed] [Google Scholar]
- [34].Wilson SJ, McEwan JC, Sheard PW, Harris AJ,J. Muscle Res. Cell Motil 1992, 13, 534. [DOI] [PubMed] [Google Scholar]
- [35].Duxson MJ, Usson Y, Development 1989, 107, 243. [DOI] [PubMed] [Google Scholar]
- [36].Richmond FJ, MacGillis DR, Scott DA,J. Neurophysiol 1985, 53, 868. [DOI] [PubMed] [Google Scholar]
- [37].Ross JJ, Duxson MJ, Harris AJ, Development 1987, 100, 383. [DOI] [PubMed] [Google Scholar]
- [38].Ontell M, Hughes D, Bourke D, Am. J. Anat 1988, 181, 279. [DOI] [PubMed] [Google Scholar]
- [39].Wigmore PM, Stickland NC, J. Anat 1983, 137 (Pt 2), 235. [PMC free article] [PubMed] [Google Scholar]
- [40].Ross JJ, Duxson MJ, Harris AJ, Development 1987, 100, 395. [DOI] [PubMed] [Google Scholar]
- [41].Pittman RH, Oppenheim RW, Nature 1978, 271, 364. [DOI] [PubMed] [Google Scholar]
- [42].Olwin BB, Arthur K, Hannon K, Hein P, McFall A, Riley B, Szebenyi G, Zhou Z, Zuber ME, Rapraeger AC, Riley B, Szebenyi G, Fallon JF, Mol. Reprod. Dev 1994, 39, 90. [DOI] [PubMed] [Google Scholar]
- [43].Wigmore PM, Baillie HS, Khan M, Morrison EH, Mayhew TM, Muscle Nerve 1992, 15, 1301. [PubMed] [Google Scholar]
- [44].Debras E, Prod’homme M, Rieu I, Balage M, Dardevet D, Grizard J, Nutrition 2007, 23, 267. [DOI] [PubMed] [Google Scholar]
- [45].Schultz E, Anat. Rec 1974, 180, 589. [DOI] [PubMed] [Google Scholar]
- [46].Westerga J, Gramsbergen A, Brain Res. Dev. Brain Res 1994, 80, 233. [DOI] [PubMed] [Google Scholar]
- [47].Eken T, Elder GC, Lomo T, J. Neurophysiol 2008, 99, 1899. [DOI] [PubMed] [Google Scholar]
- [48].Navarrete RM, Vrbova G, Dev. Brain Res 1983, 8, 11. [Google Scholar]
- [49].Kjaer M, Physiol. Rev. 2004, 84, 649. [DOI] [PubMed] [Google Scholar]
- [50].Thorsteinsdottir S, Deries M, Cachaco AS, Bajanca F, Dev. Biol 2011, 354, 191. [DOI] [PubMed] [Google Scholar]
- [51].Frantz C, Stewart KM, Weaver VM, J. Cell Sci. 2010, 123, 4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bajanca F, Luz M, Raymond K, Martins GG, Sonnenberg A, Tajbakhsh S, Buckingham M, Thorsteinsdottir S, Development 2006, 133, 1635. [DOI] [PubMed] [Google Scholar]
- [53].Anderson C, Thorsteinsdottir S, Borycki AG, Development 2009, 136, 3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cachaco AS, Pereira CS, Pardal RG, Bajanca F, Thorsteinsdottir S, Dev. Dyn 2005, 232, 1069. [DOI] [PubMed] [Google Scholar]
- [55].Bajanca F, Luz M, Duxson MJ, Thorsteinsdottir S, Dev. Dyn 2004, 231, 402. [DOI] [PubMed] [Google Scholar]
- [56].Brand-Saberi B, Krenn V, Grim M, Christ B, Anat. Embryol 1993, 187, 17. [DOI] [PubMed] [Google Scholar]
- [57].Gullberg D, Velling T, Lohikangas L, Tiger CF, Front. Biosci 1998, 3, D1039. [DOI] [PubMed] [Google Scholar]
- [58].Nishimura T, Meat Sci. 2015, 109, 48. [DOI] [PubMed] [Google Scholar]
- [59].Nishimura T, Ojima K, Liu A, Hattori A, Takahashi K, Tissue Cell 1996, 28, 527. [DOI] [PubMed] [Google Scholar]
- [60].McCormick RJ, Meat Sci. 1994, 36, 79. [DOI] [PubMed] [Google Scholar]
- [61].Lepetit J, Meat Sci. 2007, 76, 147. [DOI] [PubMed] [Google Scholar]
- [62].Tanzer ML, Science 1973, 180, 561. [DOI] [PubMed] [Google Scholar]
- [63].Rowe RW, Tissue Cell 1981, 13, 681. [DOI] [PubMed] [Google Scholar]
- [64].Hanson J, Huxley HE, Nature 1953, 172, 530. [DOI] [PubMed] [Google Scholar]
- [65].Huxley HE, Biochim. Biophys. Acta 1953, 12, 387. [DOI] [PubMed] [Google Scholar]
- [66].Flucher BE, Dev. Biol 1992, 154, 245. [DOI] [PubMed] [Google Scholar]
- [67].Rossi D, Barone V, Giacomello E, Cusimano V, Sorrentino V, Traffic 2008, 9, 1044. [DOI] [PubMed] [Google Scholar]
- [68].Sellers JR,J. Muscle Res. Cell Motil 2004, 25, 475. [DOI] [PubMed] [Google Scholar]
- [69].Schiaffino S, Reggiani C, Physiol. Rev 2011, 91, 1447. [DOI] [PubMed] [Google Scholar]
- [70].Henneman E, Somjen G, Carpenter DO, J. Neurophysiol 1965, 28, 599. [DOI] [PubMed] [Google Scholar]
- [71].Huxley AF, Prog. Biophys. Biophys. Chem 1957, 7, 255. [PubMed] [Google Scholar]
- [72].Mauro A,J. Biophys. Biochem. Cytol 1961, 9, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-Morel B, Guenou H, Malissen B, Tajbakhsh S, Galy A, Development 2011, 138, 3647. [DOI] [PubMed] [Google Scholar]
- [74].Lepper C, Partridge TA, Fan CM, Development 2011, 138, 3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Juhas M, Bursac N, Curr. Opin. Biotechnol 2013, 24, 880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mourikis P, Sambasivan R, Castel D, Rocheteau P, Bizzarro V, Tajbakhsh S, Stem Cells 2012, 30, 243. [DOI] [PubMed] [Google Scholar]
- [77].Yan Z, Choi S, Liu X, Zhang M, Schageman JJ, Lee SY, Hart R, Lin L, Thurmond FA, Williams RS,J. Biol. Chem 2003, 278, 8826. [DOI] [PubMed] [Google Scholar]
- [78].Jones NC, Tyner KJ, Nibarger L, Stanley HM, Cornelison DD, Fedorov YV, Olwin BB,J. Cell Biol. 2005, 169, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Yin H, Price F, Rudnicki MA, Physiol. Rev 2013, 93, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Jiang C, Wen Y, Kuroda K, Hannon K, Rudnicki MA, Kuang S, Dis. Models Mech 2014, 7, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Du H, Shih CH, Wosczyna MN, Mueller AA, Cho J, Aggarwal A, Rando TA, Feldman BJ, Nat. Commun 2017, 8, 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B, J. Exp. Med 2007, 204, 1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Madaro L, Bouche M, BioMed Res. Int 2014, 2014, 438675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Aurora AB, Olson EN, Cell Stem Cell 2014, 15, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Saclier M, Cuvellier S, Magnan M, Mounier R, Chazaud B, FEBS J 2013, 280, 4118. [DOI] [PubMed] [Google Scholar]
- [86].Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM, Nat. Cell Biol. 2010, 12, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, Chawla A, Cell 2013, 153, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Turner NJ, Badylak SF, Cell Tissue Res. 2012, 347, 759. [DOI] [PubMed] [Google Scholar]
- [89].Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA, Nature 2005, 433, 760. [DOI] [PubMed] [Google Scholar]
- [90].Decary S, Mouly V, Hamida CB, Sautet A, Barbet JP, Butler-Browne GS, Hum. Gene Ther. 1997, 8, 1429. [DOI] [PubMed] [Google Scholar]
- [91].Tichy ED, Sidibe DK, Tierney MT, Stec MJ, Sharifi-Sanjani M, Hosalkar H, Mubarak S, Johnson FB, Sacco A, Mourkioti F, Stem Cell Rep. 2017, 9, 1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Sacco A, Mourkioti F, Tran R, Choi J, Llewellyn M, Kraft P, Shkreli M, Delp S, Pomerantz JH, Artandi SE, Blau HM, Cell 2010, 143, 1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ryall JG, Schertzer JD, Lynch GS, Biogerontology 2008, 9, 213. [DOI] [PubMed] [Google Scholar]
- [94].Morse CI, Thom JM, Reeves ND, Birch KM, Narici MV, J. Appl. Physiol 2005, 99, 1050. [DOI] [PubMed] [Google Scholar]
- [95].Hughes VA, Frontera WR, Wood M, Evans WJ, Dallal GE, Roubenoff R, Fiatarone Singh MA, J. Gerontol. A: Biol. Sci. Med. Sci 2001, 56, B209. [DOI] [PubMed] [Google Scholar]
- [96].Porter MM, Vandervoort AA, Lexell J, Scand. J. Med. Sci. Sports 1995, 5, 129. [DOI] [PubMed] [Google Scholar]
- [97].Partridge TA, FEBS J 2013, 280, 4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Rodrigues M, Echigoya Y, Fukada SI, Yokota T,J. Neuromuscul. Dis 2016, 3, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Juhas M, Ye J, Bursac N, Methods 2016, 99, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677. [DOI] [PubMed] [Google Scholar]
- [101].Engler AJ, Griffin MA, Sen S, Bonnemann CG, Sweeney HL, Discher DE, J. Cell Biol. 2004, 166, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Funanage VL, Smith SM, Minnich MA, J. Cell. Physiol 1992, 150, 251. [DOI] [PubMed] [Google Scholar]
- [103].Cheng CS, Davis BN, Madden L, Bursac N, Truskey GA, Exp. Biol. Med 2014, 239, 1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Rao L, Qian Y, Khodabukus A, Ribar T, Bursac N, Nat. Commun 2018, 9, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Khodabukus A, Baar K, Tissue Eng., Part C 2009, 15, 501. [DOI] [PubMed] [Google Scholar]
- [106].Vandenburgh HH, Karlisch P, Farr L, In Vitro Cell. Dev. Biol 1988, 24, 166. [DOI] [PubMed] [Google Scholar]
- [107].Bach AD, Beier JP, Stern-Staeter J, Horch RE, J. Cell. Mol. Med 2004, 8, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Rosso F, Giordano A, Barbarisi M, Barbarisi A, J. Cell. Physiol 2004, 199, 174. [DOI] [PubMed] [Google Scholar]
- [109].Y Shadrin I, Khodabukus A, Bursac N, Cell. Mol. Life Sci 2016, 73, 4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Conconi MT, De Coppi P, Bellini S, Zara G, Sabatti M, Marzaro M, Zanon GF, Gamba PG, Parnigotto PP, Nussdorfer GG, Biomaterials 2005, 26, 2567. [DOI] [PubMed] [Google Scholar]
- [111].De Coppi P, Bellini S, Conconi MT, Sabatti M, Simonato E, Gamba PG, Nussdorfer GG, Parnigotto PP, Tissue Eng. 2006, 12, 1929. [DOI] [PubMed] [Google Scholar]
- [112].Levenberg S, Rouwkema J, Macdonald M, Garfein ES, Kohane DS, Darland DC, Marini R, van Blitterswijk CA, Mulligan RC, D’Amore PA, Langer R, Nat. Biotechnol 2005, 23, 879. [DOI] [PubMed] [Google Scholar]
- [113].Liao IC, Liu JB, Bursac N, Leong KW, Cell. Mol. Bioeng 2008, 1, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Bian W, Juhas M, Pfeiler TW, Bursac N, Tissue Eng., Part A 2012, 18, 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Bian W, Bursac N, Biomaterials 2009, 30, 1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Dennis RG, Kosnik PE, In Vitro Cell. Dev. Biol.: Animal 2000, 36, 327. [DOI] [PubMed] [Google Scholar]
- [117].Takahashi H, Okano T, Adv. Healthcare Mater. 2015, 4, 2388. [DOI] [PubMed] [Google Scholar]
- [118].Light N, Champion AE, Biochem.J 1984, 219, 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Okano T, Satoh S, Oka T, Matsuda T, ASAIO.J 1997, 43, M749. [PubMed] [Google Scholar]
- [120].Okano T, Matsuda T, Cell Transplant. 1997, 6, 109. [DOI] [PubMed] [Google Scholar]
- [121].Shansky J, Del Tatto M, Chromiak J, Vandenburgh H, In Vitro Cell. Dev. Biol.: Animal 1997, 33, 659. [DOI] [PubMed] [Google Scholar]
- [122].Powell CA, Smiley BL, Mills J, Vandenburgh HH, Am. J. Physiol.: Cell Physiol 2002, 283, C1557. [DOI] [PubMed] [Google Scholar]
- [123].Brady MA, Lewis MP, Mudera V, J. Tissue Eng. Regener. Med 2008, 2, 408. [DOI] [PubMed] [Google Scholar]
- [124].Vandenburgh H, Shansky J, Benesch-Lee F, Barbata V, Reid J, Thorrez L, Valentini R, Crawford G, Muscle Nerve 2008, 37,438. [DOI] [PubMed] [Google Scholar]
- [125].Sato M, Ito A, Kawabe Y, Nagamori E, Kamihira M, J. Biosci. Bioeng 2011, 112, 273. [DOI] [PubMed] [Google Scholar]
- [126].Hinds S, Bian W, Dennis RG, Bursac N, Biomaterials 2011, 32, 3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Stearns-Reider KM, D’Amore A, Beezhold K, Rothrauff B, Cavalli L, Wagner WR, Vorp DA, Tsamis A, Shinde S, Zhang C, Barchowsky A, Rando TA, Tuan RS, Ambrosio F, Aging Cell 2017, 16, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Grefte S, Vullinghs S, Kuijpers-Jagtman AM, Torensma R, Von den Hoff JW, Biomed. Mater 2012, 7, 055004. [DOI] [PubMed] [Google Scholar]
- [129].Mosesson MW, Thromb J. Haemostasis 2005, 3, 1894. [DOI] [PubMed] [Google Scholar]
- [130].Grassl ED, Oegema TR, Tranquillo RT,J. Biomed. Mater. Res 2002, 60, 607. [DOI] [PubMed] [Google Scholar]
- [131].Knapp DM, Helou EF, Tranquillo RT, Exp. Cell Res. 1999, 247, 543. [DOI] [PubMed] [Google Scholar]
- [132].Ross JJ, Tranquillo RT, Matrix Biol. 2003, 22, 477. [DOI] [PubMed] [Google Scholar]
- [133].Yang L, van der Werf KO, Koopman BF, Subramaniam V, Bennink ML, Dijkstra PJ, Feijen J, J. Biomed. Mater. Res. A 2007, 82, 160. [DOI] [PubMed] [Google Scholar]
- [134].Collet JP, Shuman H, Ledger RE, Lee S, Weisel JW, Proc. Natl. Acad. Sci. USA 2005, 102, 9133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Urech L, Bittermann AG, Hubbell JA, Hall H, Biomaterials 2005, 26, 1369. [DOI] [PubMed] [Google Scholar]
- [136].Dietrich F, Lelkes PI, Angiogenesis 2006, 9, 111. [DOI] [PubMed] [Google Scholar]
- [137].Liu JY, Swartz DD, Peng HF, Gugino SF, Russell JA, Andreadis ST, Cardiovasc. Res 2007, 75, 618. [DOI] [PubMed] [Google Scholar]
- [138].Sakiyama SE, Schense JC, Hubbell JA, FASEB J 1999, 13, 2214. [DOI] [PubMed] [Google Scholar]
- [139].Sakiyama-Elbert SE, Hubbell JA, J. Controlled Release 2000, 65, 389. [DOI] [PubMed] [Google Scholar]
- [140].Khodabukus A, Baar K, Tissue Eng., Part C 2009, 15, 501. [DOI] [PubMed] [Google Scholar]
- [141].Huang YC, Dennis RG, Larkin L, Baar K, J. Appl. Physiol 2005, 98, 706. [DOI] [PubMed] [Google Scholar]
- [142].Strohman RC, Bayne E, Spector D, Obinata T, MicouEastwood J, Maniotis A, In Vitro Cell. Dev Biol. 1990, 26, 201. [DOI] [PubMed] [Google Scholar]
- [143].Li B, Lin M, Tang Y, Wang B, Wang JH, Biomech J. 2008, 41, 3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Clark P, Dunn GA, Knibbs A, Peckham M, Int. J. Biochem. Cell Biol. 2002, 34, 816. [DOI] [PubMed] [Google Scholar]
- [145].Evans DJ, Britland S, Wigmore PM, Dev Genes Evol. 1999, 209, 438. [DOI] [PubMed] [Google Scholar]
- [146].Huang NF, Patel S, Thakar RG, Wu J, Hsiao BS, Chu B, Lee RJ, Li S, Nano Lett. 2006, 6, 537. [DOI] [PubMed] [Google Scholar]
- [147].Lam MT, Huang YC, Birla RK, Takayama S, Biomaterials 2009, 30, 1150. [DOI] [PubMed] [Google Scholar]
- [148].Takahashi H, Shimizu T, Nakayama M, Yamato M, Okano T, Biomaterials 2013, 34, 7372. [DOI] [PubMed] [Google Scholar]
- [149].Nagamori E, Ngo TX, Takezawa Y, Saito A, Sawa Y, Shimizu T, Okano T, Taya M, Kino-oka M, Biomaterials 2013, 34, 662. [DOI] [PubMed] [Google Scholar]
- [150].Close RI, Physiol. Rev 1972, 52, 129. [DOI] [PubMed] [Google Scholar]
- [151].Juhas M, Engelmayr GC Jr., Fontanella AN, Palmer GM, Bursac n., Proc. Natl. Acad. Sci. USA 2014, 111, 5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Khodabukus A, Baar K, Cells Tissues Organs 2016, 202, 159. [DOI] [PubMed] [Google Scholar]
- [153].Khodabukus A, Baar K,J. Cell. Physiol 2015, 230, 1750. [DOI] [PubMed] [Google Scholar]
- [154].Huang YC, Dennis RG, Baar K, Am. J. Physiol.: Cell Physiol 2006, 291, C11. [DOI] [PubMed] [Google Scholar]
- [155].Raben N, Fukuda T, Gilbert AL, de Jong D, Thurberg BL, Mattaliano RJ, Meikle P, Hopwood JJ, Nagashima K, Nagaraju K, Plotz PH, Mol. Ther 2005, 11, 48. [DOI] [PubMed] [Google Scholar]
- [156].Webster C, Silberstein L, Hays AP, Blau HM, Cell 1988, 52, 503. [DOI] [PubMed] [Google Scholar]
- [157].Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ, Am. J. Physiol.—Endocrinol. Metab 2007, 292, E151. [DOI] [PubMed] [Google Scholar]
- [158].Khodabukus A, Baar K, Tissue Eng., Part C 2012, 18, 349. [DOI] [PubMed] [Google Scholar]
- [159].Khodabukus A, Baehr LM, Bodine SC, Baar K, J. Cell. Physiol 2015, 230, 2489. [DOI] [PubMed] [Google Scholar]
- [160].Raman R, Cvetkovic C, Uzel SG, Platt RJ, Sengupta P, Kamm RD, Bashir R, Proc. Natl. Acad. Sci. USA 2016, 113, 3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Bian W, Bursac N, FASEB J. 2012, 26, 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Larkin LM, Van der Meulen JH, Dennis RG, Kennedy JB, In Vitro Cell. Dev Biol.: Animal 2006, 42, 75. [DOI] [PubMed] [Google Scholar]
- [163].Morimoto Y, Kato-Negishi M, Onoe H, Takeuchi S, Biomaterials 2013, 34, 9413. [DOI] [PubMed] [Google Scholar]
- [164].Martin NR, Passey SL, Player DJ, Mudera V, Baar K, Greensmith L, Lewis MP, Tissue Eng., Part A 2015, 21, 2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Guo X, Colon A, Akanda N, Spradling S, Stancescu M, Martin C, Hickman JJ, Biomaterials 2017, 122, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Colon A, Guo X, Akanda N, Cai Y, Hickman JJ, Sci. Rep. 2017, 7, 17202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Cheema U, Yang SY, Mudera V, Goldspink GG, Brown RA, Cell Motil. Cytoskeleton 2003, 54, 226. [DOI] [PubMed] [Google Scholar]
- [168].Cheema U, Brown R, Mudera V, Yang SY, McGrouther G, Goldspink G, J. Cell. Physiol 2005, 202, 67. [DOI] [PubMed] [Google Scholar]
- [169].Baldwin KM, Haddad F, Pandorf CE, Roy RR, Edgerton VR, Front. Physiol 2013, 4, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Lee PH, Vandenburgh HH, Tissue Eng, Part A 2013, 19, 2147. [DOI] [PubMed] [Google Scholar]
- [171].Vandenburgh H, Kaufman S, Science 1979, 203, 265. [DOI] [PubMed] [Google Scholar]
- [172].Vandenburgh HH, Kaufman S,J. Cell. Physiol 1981, 109, 205. [DOI] [PubMed] [Google Scholar]
- [173].Vandenburgh HH, Dev. Biol 1982, 93, 438. [DOI] [PubMed] [Google Scholar]
- [174].Handschin C, Mortezavi A, Plock J, Eberli D, Adv Drug Delivery Rev. 2015, 82–83, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Dugan JM, Cartmell SH, Gough JE, Tissue Eng J. 2014, 5, 2041731414530138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Boonen KJ, Langelaan ML, Polak RB, van DW Schaft der Baaijens FP, Post MJ,J. Biomech 2010, 43, 1514. [DOI] [PubMed] [Google Scholar]
- [177].Karande TS, Ong JL, Agrawal CM, Ann. Biomed. Eng 2004, 32, 1728. [DOI] [PubMed] [Google Scholar]
- [178].Bian W, Liau B, Badie N, Bursac N, Nat. Protoc 2009, 4, 1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Fujita H, Shimizu K, Morioka Y, Nagamori E, J. Biosci. Bioeng 2010, 110, 359. [DOI] [PubMed] [Google Scholar]
- [180].Rangarajan S, Madden L, Bursac N, Ann. Biomed. Eng. 2014, 42, 1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Juhas M, Bursac N, Biomaterials 2014, 35, 9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Jackman CP, Carlson AL, Bursac N, Biomaterials 2016, 111, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Shadrin IY, Allen BW, Qian Y, Jackman CP, Carlson AL, Juhas ME, Bursac N, Nat. Commun 2017, 8, 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Chromiak JA, Shansky J, Perrone C, Vandenburgh HH, In Vitro Cell. Dev. Biol.: Animal 1998, 34, 694. [DOI] [PubMed] [Google Scholar]
- [185].Truskey GA, Achneck HE, Bursac N, Chan H, Cheng CS, Fernandez C, Hong S, Jung Y, Koves T, Kraus WE, Leong K, Madden L, Reichert WM, Zhao X, Stem Cell Res. Ther 2013, 4 (Suppl. 1), S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Gholobova D, Decroix L, Van Muylder V, Desender L, Gerard M, Carpentier G, Vandenburgh H, Thorrez L, Tissue Eng., Part A 2015, 21, 2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Koffler J, Kaufman-Francis K, Shandalov Y, Egozi D, Pavlov DA, Landesberg A, Levenberg S, Proc. Natl. Acad. Sci. USA 2011, 108, 14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Osaki T, Sivathanu V, Kamm RD, Biomaterials 2018, 156, 65. [DOI] [PubMed] [Google Scholar]
- [189].van Velthoven CTJ, de Morree A, Egner IM, Brett JO, Rando TA, Cell Rep. 2017, 21, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Machida S, Spangenburg EE, Booth FW, Cell Proliferation 2004, 37, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Ono Y, Masuda S, Nam HS, Benezra R, Miyagoe-Suzuki Y, Takeda S, J. Cell Sci 2012, 125, 1309. [DOI] [PubMed] [Google Scholar]
- [192].Beauchamp JR, Morgan JE, Pagel CN, Partridge TA, J. Cell Biol 1999, 144, 1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, Buckingham M, Science 2005, 309, 2064. [DOI] [PubMed] [Google Scholar]
- [194].Gilbert PM, Havenstrite KL, Magnusson KE, Sacco A, Leonardi NA, Kraft P, Nguyen NK, Thrun S, Lutolf MP, Blau HM, Science 2010, 329, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Ryall JG, Dell’Orso S, Derfoul A, Juan A, Zare H, Feng X, Clermont D, Koulnis M, Gutierrez-Cruz G, Fulco M, Sartorelli V, Cell Stem Cell 2015, 16, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, Karin M, Wang JY, Puri PL, Mol. Cell. Biol 2000, 20, 3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, Olwin BB, Nat. Med 2014, 20, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Charville GW, Cheung TH, Yoo B, Santos PJ, Lee GK, Shrager JB, Rando TA, Stem Cell Rep. 2015, 5, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM, Nat. Med 2014, 20, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Quarta M, Brett JO, DiMarco R, De Morree A, Boutet SC, Chacon R, Gibbons MC, Garcia VA, Su J, Shrager JB, Heilshorn S, Rando TA, Nat. Biotechnol 2016, 34, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Lamar KM, Bogdanovich S, Gardner BB, Gao QQ, Miller T, Earley JU, Hadhazy M, Vo AH, Wren L, Molkentin JD, McNally EM, PLoS Genet. 2016, 12, e1006019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Lamar KM, McNally EM, J. Neuromuscul. Dis 2014, 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Flanigan KM, Ceco E, Lamar KM, Kaminoh Y, Dunn DM, Mendell JR, King WM, Pestronk A, Florence JM, Mathews KD, Finkel RS, Swoboda KJ, Gappmaier E, Howard MT, Day JW, McDonald C, McNally EM, Weiss RB, United Dystrophinopathy P, Ann. Neurol 2013, 73, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Wang Y, Liang P, Lan F, Wu H, Lisowski L, Gu M, Hu S, Kay MA, Urnov FD, Shinnawi R, Gold JD, Gepstein L, Wu JC,J. Am. Coll. Cardiol 2014, 64, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Kime C, Mandegar MA, Srivastava D, Yamanaka S, Conklin BR, Rand TA, Curr. Protoc. Hum. Genet 2016, 88, (Unit 21), 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Arias-Fuenzalida J, Jarazo J, Qing X, Walter J, Gomez-Giro G, Nickels SL, Zaehres H, Scholer HR, Schwamborn JC, Stem Cell Rep. 2017, 9, 1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Constantinides PG, Jones PA, Gevers W, Nature 1977, 267, 364. [DOI] [PubMed] [Google Scholar]
- [208].Weintraub H, Tapscott SJ, Davis RL, Thayer MJ, Adam MA, Lassar AB, Miller AD, Proc. Natl. Acad. Sci. USA 1989, 86, 5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Davis RL, Weintraub H, Lassar AB, Cell 1987, 51, 987. [DOI] [PubMed] [Google Scholar]
- [210].Shani M, Faerman A, Emerson CP, Pearson-White S, Dekel I, Magal Y, Symp. Soc. Exp. Biol 1992, 46, 19. [PubMed] [Google Scholar]
- [211].Dekel I, Magal Y, Pearson-White S, Emerson CP, Shani M, New Biol. 1992, 4, 217. [PubMed] [Google Scholar]
- [212].Kurosawa H, J. Biosci. Bioeng 2007, 103, 389. [DOI] [PubMed] [Google Scholar]
- [213].Fehling HJ, Lacaud G, Kubo A, Kennedy M, Robertson S, Keller G, Kouskoff V, Development 2003, 130, 4217. [DOI] [PubMed] [Google Scholar]
- [214].Hwang Y, Suk S, Lin S, Tierney M, Du B, Seo T, Mitchell A, Sacco A, Varghese S, PLoS One 2013, 8, e72023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Albini S, Coutinho P, Malecova B, Giordani L, Savchenko A, Forcales SV, Puri PL, Cell Rep. 2013, 3, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].de la Serna IL, Carlson KA, Imbalzano AN, Nat. Genet 2001, 27, 187. [DOI] [PubMed] [Google Scholar]
- [217].Tanaka A, Woltjen K, Miyake K, Hotta A, Ikeya M, Yamamoto T, Nishino T, Shoji E, Sehara-Fujisawa A, Manabe Y, Fujii N, Hanaoka K, Era T, Yamashita S, Isobe K, Kimura E, Sakurai H, PLoS One 2013, 8, e61540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, Meissner A, Daley GQ, Brack AS, Collins JJ, Cowan C, Schlaeger TM, Rossi DJ, Cell Stem Cell 2010, 7, 618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Shoji E, Sakurai H, Nishino T, Nakahata T, Heike T, Awaya T, Fujii N, Manabe Y, Matsuo M, Sehara-Fujisawa A, Sci. Rep 2015, 5, 12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Abujarour R, Bennett M, Valamehr B, Lee TT, Robinson M, Robbins D, Le T, Lai K, Flynn P, Stem Cells Transl. Med 2014, 3, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Yasuno T, Osafune K, Sakurai H, Asaka I, Tanaka A, Yamaguchi S, Yamada K, Hitomi H, Arai S, Kurose Y, Higaki Y, Sudo M, Ando S, Nakashima H, Saito T, Kaneoka H, Biochem. Biophys. Res. Commun 2014, 448, 175. [DOI] [PubMed] [Google Scholar]
- [222].Sato Y, Kobayashi H, Higuchi T, Shimada Y, Ida H, Ohashi T, Mol. Ther.—Methods Clin. Dev 2016, 3, 16054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Yoshida T, Awaya T, Jonouchi T, Kimura R, Kimura S, Era T, Heike T, Sakurai H, Sci. Rep 2017, 7, 13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].van der Wal E, Bergsma AJ, van Gestel TJM, In ‘t Groen SLM, Zaehres H, Arauzo-Bravo MJ, Scholer HR, van der Ploeg AT, Pijnappel W, Mol. Ther. Nucleic Acids 2017, 7, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Sato Y, Kobayashi H, Higuchi T, Shimada Y, Ida H, Ohashi T, Stem Cells Transl. Med 2017, 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Goudenege S, Lebel C, Huot NB, Dufour C, Fujii I, Gekas J, Rousseau J, Tremblay JP, Mol. Ther 2012, 20, 2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Darabi R, Gehlbach K, Bachoo RM, Kamath S, Osawa M, Kamm KE, Kyba M, Perlingeiro RC, Nat. Med 2008, 14, 134. [DOI] [PubMed] [Google Scholar]
- [228].Darabi R, Santos FN, Filareto A, Pan W, Koene R, Rudnicki MA, Kyba M, Perlingeiro RC, Stem Cells 2011, 29, 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Darabi R, Arpke RW, Irion S, Dimos JT, Grskovic M, Kyba M, Perlingeiro RC, Cell Stem Cell 2012, 10, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Rohwedel J, Maltsev V, Bober E, Arnold HH, Hescheler J, Wobus AM, Dev. Biol 1994, 164, 87. [DOI] [PubMed] [Google Scholar]
- [231].Murry CE, Keller G, Cell 2008, 132, 661. [DOI] [PubMed] [Google Scholar]
- [232].Mendjan S, Mascetti VL, Ortmann D, Ortiz M, Karjosukarso DW, Ng Y, Moreau T, Pedersen RA, Cell Stem Cell 2014, 15, 310. [DOI] [PubMed] [Google Scholar]
- [233].Choi IY, Lim H, Estrellas K, Mula J, Cohen TV, Zhang Y, Donnelly CJ, Richard JP, Kim YJ, Kim H, Kazuki Y, Oshimura M, Li HL, Hotta A, Rothstein J, Maragakis N, Wagner KR, Lee G, Cell Rep. 2016, 15, 2301. [DOI] [PubMed] [Google Scholar]
- [234].Borchin B, Chen J, Barberi T, Stem Cell Rep. 2013, 1, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Caron L, Kher D, Lee KL, McKernan R, Dumevska B, Hidalgo A, Li J,Yang H, Main H, Ferri G, Petek LM, Poellinger L, Miller DG, Gabellini D, Schmidt U, Stem Cells Transl. Med 2016, 5, 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Xu C, Tabebordbar M, Iovino S, Ciarlo C, Liu J, Castiglioni A, Price E, Liu M, Barton ER, Kahn CR, Wagers AJ, Zon LI, Cell 2013, 155, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Barberi T, Bradbury M, Dincer Z, Panagiotakos G, Socci ND, Studer L, Nat. Med 2007, 13, 642. [DOI] [PubMed] [Google Scholar]
- [238].Uezumi A, Nakatani M, Ikemoto-Uezumi M, Yamamoto N, Morita M, Yamaguchi A, Yamada H, Kasai T, Masuda S, Narita A, Miyagoe-Suzuki Y, Takeda S, Fukada S, Nishino I, Tsuchida K, Stem Cell Rep. 2016, 7, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Magli A, Incitti T, Kiley J, Swanson SA, Darabi R, Rinaldi F, Selvaraj S, Yamamoto A, Tolar J, Yuan C, Stewart R, Thomson JA, Perlingeiro RCR, Cell Rep. 2017, 19, 2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Mendell JR, Lloyd-Puryear M, Muscle Nerve 2013, 48, 21. [DOI] [PubMed] [Google Scholar]
- [241].Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C, D. M. D. C. C. W. Group, Lancet Neurol. 2010, 9, 77. [DOI] [PubMed] [Google Scholar]
- [242].Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K,Dawkins H, Lamont L, Roy AJ, Chamova T, Guergueltcheva V, Chan S, Korngut L, Campbell C, Dai Y, Wang J, Barisic N, Brabec P, Lahdetie J, Walter MC, Schreiber-Katz O, Karcagi V, Garami M, Viswanathan V, Bayat F, Buccella F, Kimura E, Koeks Z, van den Bergen JC, Rodrigues M, Roxburgh R, Lusakowska A, Kostera-Pruszczyk A, Zimowski J, Santos R, Neagu E, Artemieva S, Rasic VM, Vojinovic D, Posada M, Bloetzer C, Jeannet PY, Joncourt F, Diaz-Manera J, Gallardo E, Karaduman AA, Topaloglu H, El Sherif R Stringer A, Shatillo AV, Martin AS, Peay HL, Bellgard MI, Kirschner J, Flanigan KM, Straub V, Bushby K, Verschuuren J, Aartsma-Rus A, Beroud C, Lochmuller H, Hum. Mutat 2015, 36, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, T Den Dunnen J, Muscle Nerve 2006, 34, 135. [DOI] [PubMed] [Google Scholar]
- [244].Bello L, Kesari A, Gordish-Dressman H, Cnaan A, Morgenroth LP, Punetha J, Duong T, Henricson EK, Pegoraro E, McDonald CM, Hoffman EP, Cooperative I International Neuromuscular Research Group, Ann. Neurol 2015, 77, 684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Pegoraro E, Hoffman EP, Piva L, Gavassini BF, Cagnin S, Ermani M, Bello L, Soraru G, Pacchioni B, Bonifati MD, Lanfranchi G, Angelini C, Kesari A, Lee I, Gordish-Dressman H, Devaney JM, McDonald CM, Cooperative G International Neuromuscular Research, Neurology 2011, 76, 219.21178099 [Google Scholar]
- [246].Vandenburgh H, Shansky J, Benesch-Lee F, Skelly K, Spinazzola JM, Saponjian Y, Tseng BS, FASEB J. 2009, 23, 3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Nesmith AP, Wagner MA, Pasqualini FS, O’Connor BB, Pincus MJ, August PR, Parker KK, J. Cell Biol. 2016, 215, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Serena E, Zatti S, Zoso A, Lo Verso F, Tedesco FS, Cossu G, Elvassore N, Stem Cells Transl. Med 2016, 5, 1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, Tanaka M, Amano N, Watanabe A, Sakurai H, Yamamoto T, Yamanaka S, Hotta A, Stem Cell Rep. 2015, 4, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Kyrychenko V, Kyrychenko S, Tiburcy M, Shelton JM, Long C, Schneider JW, Zimmermann WH, Bassel-Duby R, Olson EN, JCI Insight 2017, 2, 95918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Park J, Wetzel I, Dreau D, Cho H, Adv. Healthcare Mater. 2017, 7, 1700551. [DOI] [PubMed] [Google Scholar]
- [252].Perel P, Roberts I, Sena E, Wheble P, Briscoe C, Sandercock P, Macleod M, Mignini LE, Jayaram P, Khan KS, BMJ 2007, 334, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Riedl A, Schlederer M, Pudelko K, Stadler M, Walter S, Unterleuthner D, Unger C, Kramer N, Hengstschlager M, Kenner L, Pfeiffer D, Krupitza G, Dolznig H, J. Cell Sci 2017, 130, 203. [DOI] [PubMed] [Google Scholar]
- [254].Zanoni M, Piccinini F, Arienti C, Zamagni A, Santi S, Polico R, Bevilacqua A, Tesei A, Sci. Rep 2016, 6, 19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Davis BN, Santoso JW, Walker MJ, Cheng CS, Koves TR, Kraus WE, Truskey GA, Tissue Eng, Part C 2017, 23, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Dykens JA, Will Y, Drug Discovery Today 2007, 12, 777. [DOI] [PubMed] [Google Scholar]
- [257].Crabtree HG, Biochem. J 1929, 23, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Rodriguez-Enriquez S, Juarez O, Rodriguez-Zavala JS, Moreno-Sanchez R, Eur. J. Biochem 2001, 268, 2512. [DOI] [PubMed] [Google Scholar]
- [259].Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y, Toxicol. Sci 2007, 97, 539. [DOI] [PubMed] [Google Scholar]
- [260].Aguer C, Gambarotta D, Mailloux RJ, Moffat C, Dent R, McPherson R, Harper ME, PLoS One 2011, 6, e28536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Khodabukus A, Baar K,J. Cell. Physiol 2015, 230, 1226. [DOI] [PubMed] [Google Scholar]
- [262].Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A Jr., Lewis CA, Sabatini DM, Cell 2017, 169, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Fujita H, Endo A, Shimizu K, Nagamori E, Biotechnol. Bioeng 2010, 107, 894. [DOI] [PubMed] [Google Scholar]
- [264].Gawlitta D, Boonen KJ, Oomens CW, Baaijens FP, Bouten CV, Tissue Eng. 2008, 14, 161. [DOI] [PubMed] [Google Scholar]
- [265].Khodabukus A, Baar K, J. Cell. Biochem 2014, 115, 2198. [DOI] [PubMed] [Google Scholar]
- [266].McKim JM Jr., Comb. Chem. High Throughput Screening 2010, 13, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Wikswo JP, Curtis EL, Eagleton ZE, Evans BC, Kole A, Hofmeister LH, Matloff WJ, Lab Chip 2013, 13, 3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Wikswo JP, Block3rd FE, Cliffel DE, Goodwin CR, Marasco CC, Markov DA, McLean DL, McLean JA, McKenzie JR,Reisere RS, Samson PC, Schaffer DK, Seale KT, Sherrod SD, IEEE Trans. Biomed. Eng 2013, 60, 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Maschmeyer I, Lorenz AK, Schimek K, Hasenberg T, Ramme AP, Hubner J, Lindner M, Drewell C, Bauer S, Thomas A, Sambo NS, Sonntag F, Lauster R, Marx U, Lab Chip 2015, 15, 2688. [DOI] [PubMed] [Google Scholar]
- [270].Skardal A, Murphy SV, Devarasetty M, Mead I, Kang HW, Seol YJ, Shrike Zhang Y, Shin SR, Zhao L, Aleman J, Hall AR, Shupe TD, Kleensang A, Dokmeci MR, Jin Lee S, Jackson JD, Yoo JJ, Hartung T, Khademhosseini A, Soker S, Bishop CE, Atala A, Sci. Rep 2017, 7, 8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Oleaga C, Bernabini C, Smith AS, Srinivasan B, Jackson M, McLamb W, Platt V, Bridges R, Cai Y, Santhanam N, Berry B, Najjar S, Akanda N, Guo X, Martin C, Ekman G, Esch MB, Langer J, Ouedraogo G, Cotovio J, Breton L, Shuler ML, Hickman JJ, Sci. Rep 2016, 6, 20030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Vernetti L, Gough A, Baetz N, Blutt S, Broughman JR, Brown JA, Foulke-Abel J, Hasan N, In J, Kelly E, Kovbasnjuk O, Repper J, Senutovitch N, Stabb J, Yeung C, Zachos NC, Donowitz M, Estes M, Himmelfarb J, Truskey G, Wikswo JP, Taylor DL, Sci. Rep 2017, 7, 42296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Lee SH, Sung JH, Adv. Healthcare Mater 2017, 7, 1700419. [Google Scholar]
- [274].Kang E, Wang X, Tippner-Hedges R, Ma H, Folmes CD, Gutierrez NM, Lee Y, Van Dyken C, Ahmed R, Li Y, Koski A, Hayama T, Luo S, Harding CO, Amato P, Jensen J, Battaglia D, Lee D, Wu D, Terzic A, Wolf DP, Huang T, Mitalipov S, Cell Stem Cell 2016, 18, 625. [DOI] [PubMed] [Google Scholar]
- [275].Perales-Clemente E, Cook AN, Evans JM, Roellinger S, Secreto F, Emmanuele V, Oglesbee D, Mootha VK, Hirano M, Schon EA, Terzic A, Nelson TJ, EMBO J. 2016, 35, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Quarta M, Cromie M, Chacon R, Blonigan J, Garcia V, Akimenko I, Hamer M, Paine P, Stok M, Shrager JB, Rando TA, Nat. Commun 2017, 8, 15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Tedesco FS, Hoshiya H, D’Antona G, Gerli MF, Messina G, Antonini S, Tonlorenzi R, Benedetti S, Berghella L, Torrente Y, Kazuki Y, Bottinelli R, Oshimura M, Cossu G, Sci. Transl. Med 2011, 3, 96–78. [DOI] [PubMed] [Google Scholar]
- [278].Bursac N, Juhas M, Rando TA, Annu. Rev. Biomed. Eng 2015, 17, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]