

Summary
CD4+ T cell differentiation into multiple T helper cell (Th) lineages is critical for optimal adaptive immune responses. This report identifies an intrinsic mechanism by which programmed death-1 receptor (PD-1) signaling imparted regulatory phenotype to Foxp3+ Th1 cells (denoted as Tbet+iTregPDL1 cells) and inducible regulatory T cells (iTregs). Tbet+iTregPDL1 cells prevented inflammation in murine models of experimental colitis and experimental graft versus host disease (GvHD). Programmed death ligand-1 (PDL-1) binding to PD-1 imparted regulatory function to Tbet+iTregPDL1 cells and iTreg cells by specifically down regulating endo-lysosomal protease asparaginyl endopeptidase (AEP). AEP regulated Foxp3 stability and blocking AEP imparted regulatory function in Tbet+iTreg cells. Also, Aep−/− iTreg cells significantly inhibited GvHD and maintained Foxp3 expression. PD-1 mediated Foxp3 maintenance in Tbet+ Th1 cells occurred both in tumor infiltrating lymphocytes (TIL) and during chronic viral infection. Collectively, this report has identified an intrinsic function for PD-1 in maintaining Foxp3 through proteolytic pathway.
Graphical abstract
Th1 cells are known for their enhanced stability, therefore, mechanisms which mediate their flexibility are poorly studied. Here, Stathopoulou et al, demonstrate that plasticity of Th1 cells to Tbet+iTreg cells is mediated by PD-1 signaling via asparaginyl endopeptidase (AEP). AEP inhibition enhances iTreg cells in GvHD and tumor models.
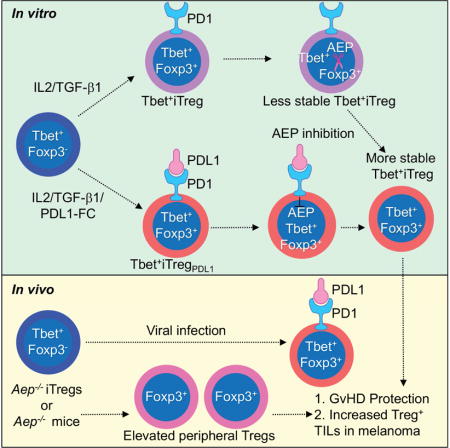
Introduction
Functional plasticity of cells belonging to the innate and adaptive immune system is necessary for the generation of robust immune responses while minimizing detrimental effects towards the host. CD4+ T cell plasticity has been extensively studied in recent years (O’Shea and Paul, 2010). A plasticity index has been proposed for the various T helper cell lineage subsets: with each subset possessing different lineage flexibility (Murphy and Stockinger, 2010). Of the numerous CD4+ T cell subsets, peripherally generated T regulatory (Tregs) cells and T helper (Th) 17 cells are regarded as plastic (Bailey-Bucktrout et al., 2013; Boniface et al., 2010; Gagliani et al., 2015; McGeachy et al., 2007; Mukasa et al., 2010; Yang et al., 2008) whereas functional stability has been attributed towards thymic derived Treg cells (tTregs) (Miyao et al., 2012), Th1 and Th2 cell lineages. Both Th2 cells (Adeeku et al., 2008; Hegazy et al., 2010; Peine et al., 2013; Taylor et al., 2006) and tTreg cells (Feng et al., 2014; Laurence et al., 2012; Zhou et al., 2009) have been demonstrated to be plastic in disease conditions. In light of these studies Th1 cells remain the lineage with the least evidence of functional flexibility. Furthermore, the molecular mechanisms that influence lineage stability in Th1 cells are poorly defined (Brown et al., 2015).
In contrast to work on cytokine signaling (O’Shea and Paul, 2010), the role of co-receptors in mediating functional plasticity has received minimal attention. One such co-inhibitory molecule that has been implicated in Th cell plasticity is programmed death ligand-1 (PDL-1 or B7-H1). In our previous work, we have found that PDL-1 can induce Foxp3 in human Th1 cells (Amarnath et al., 2011), consistent with work in murine naïve T cells (Francisco et al., 2009). In the tumor microenvironment, PDL-1 expression coincides with increased intra-tumor Foxp3+ T cells (Duraiswamy et al., 2013; Jacobs et al., 2009) suggesting that PDL-1 may play a role in maintaining Foxp3 expression in CD4+ Th subsets. PDL-1 binds to its receptor PD-1 on T cells which signals through the inhibitory phosphatase SHP1 (Chemnitz et al., 2004). SHP1 or SHP2 recruitment results in STAT dephosphorylation (Amarnath et al., 2011) (Taylor et al., 2017), potentially destabilizing the transcriptional signature of Th1 cell lineage.
In the current study, we have elucidated an intrinsic mechanism by which PD-1 signaling maintains Foxp3 in Tbet+iTreg and iTreg cells. The data presented here demonstrates that PD-1 can inhibit a functional nuclear pool of active asparaginyl endopeptidase (AEP), an endolysosomal protease previously implicated in antigen processing in dendritic cells (Dall and Brandstetter, 2016; Manoury et al., 1998; Manoury et al., 2002). We show that AEP is responsible for destabilizing Foxp3 in both iTregs and Tbet+iTreg cells. We found that PD-1 activation significantly enhanced Foxp3 expression in primed anti-viral and anti-tumor Tbet+Th1 cells, which was reversed in the presence of a blocking antibody to PDL-1. Of note, PDL-1 blockade did not reverse Tbet+Th1 cell conversion and iTreg cell induction in the absence of AEP. Therefore, this study demonstrates that downregulation of AEP is necessary for PD-1 generated Foxp3 stability.
Results
PD-1 signaling, in the absence of TGF-β1, reinforces Foxp3 expression in CD4+Tbet+Foxp3− T cells
To investigate if PD-1 maintains Foxp3 in Th1 cells, previously described Tbx21ZsGreen reporter mice were crossed with Foxp3RFP mice. Flow sorted CD4+Tbet+Foxp3−T cells (Figures 1A and 1B, S1A and S1B) were differentiated under iTreg cell conditions (Tbet+iTreg cells) or iTreg cell conditions with PDL-1 (Tbet+iTregPDL1 cells) (Figures 1A–1D). When subsets were maintained with interleukin-2 (IL-2) and transforming growth factor-β (TGF-β1), Foxp3 expression under both culture conditions was similar (Figure 1D). In culture conditions where TGF-β1 was omitted from day 4 until day 7 (Figure 1E), a significant loss in Foxp3 was noted within the Tbet+iTreg cells compared with Tbet+iTregPDL1 cells (Figures 1F and 1G). We next purified Foxp3+ cells from both groups (denoted hereafter as Tbet+iTreg cells and Tbet+ iTregPDL1 cells) and examined their function in vitro. Both populations suppressed effector T cell proliferation in vitro to a similar extent (Figures S1C–S1E). Tbet+iTregPDL1 cells secreted less IL-10 and IFN-γ, when compared to Tbet+Foxp3− cells (Figure S1F). These results indicate that PDL1 signaling maintains Foxp3 expression in Tbet+iTreg cells in the absence of TGF-β1.
Figure 1. Purified CD4+TbethiFoxp3− cells can upregulate Foxp3 under iTreg conditions.
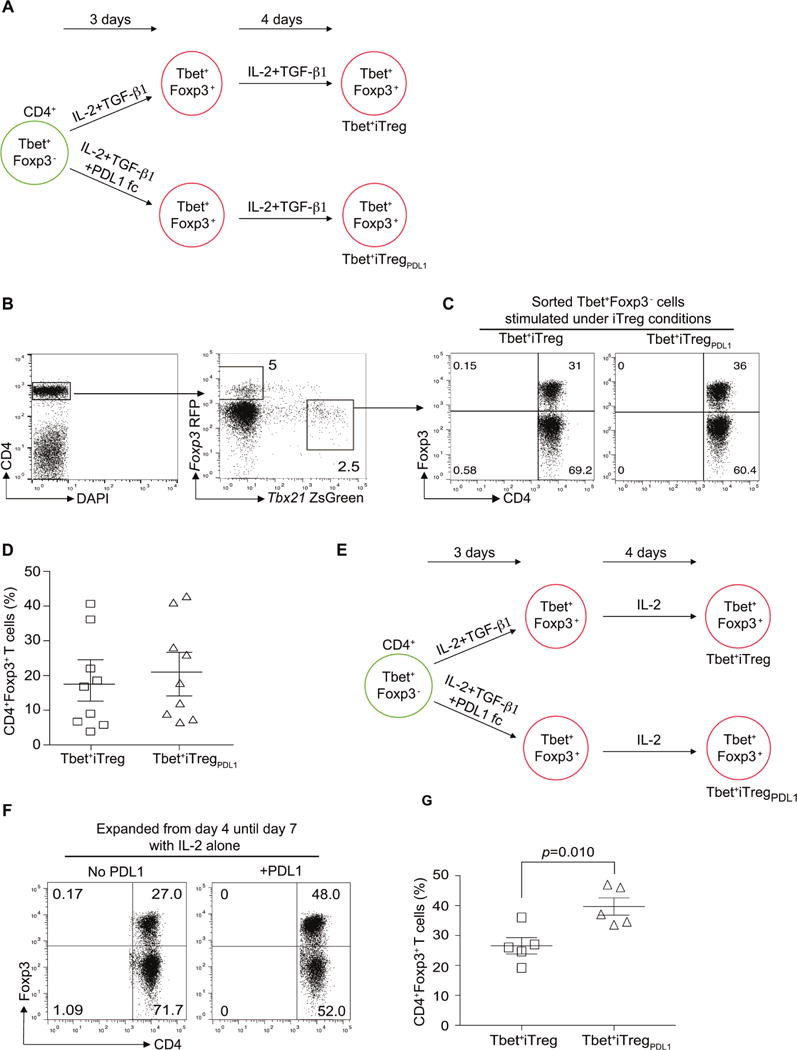
Schematic representation of cell culture conditions under which sorted CD4+TbethiFoxp3− cells were expanded (A). Flow cytometry profile of Tbx21ZsGreen Foxp3RFP expressing CD4+ T helper cells (B), Foxp3 expression of CD4+TbethiFoxp3− cells in the presence of IL-2 and TGF-β1 (Tbet+iTreg cells; C) or IL-2, TGF-β1 and PDL-1 Fc chimera (Tbet+iTregPDL1 cells; C) post differentiation and expansion. Summary of %CD4+Foxp3+ cells expression in Tbet+iTreg cells and Tbet+iTregPDL1 cells (D). Schematic representation of alternate cell culture conditions under which sorted CD4+TbethiFoxp3− cells were expanded (E). Foxp3 expression of CD4+TbethiFoxp3− cells in the presence of IL-2 (F), and summary of Foxp3 expression in different cell subsets (G). Experiments were repeated at least 5 times and each experiment was performed with n=5-9 mice. Cumulative data from all experiments are presented Mean±SEM. Please also refer to Figure S1
PDL1 induces stable regulatory phenotype in Tbet+iTregPDL1 cells during experimental autoimmune colitis and graft vs host disease (GvHD)
The in vivo regulatory potential of Tbet+iTreg cells and Tbet+iTregPDL1 cells was next evaluated. Rag2−/− mice were reconstituted with CD4+CD45RBhiCD25− naïve T effector cells alone (CD45.1+) or in conjunction with various indicated flow sorted iTreg cell populations (CD45.2+). Cohorts that received either T effector cells alone or together with Tbet+iTreg cells succumbed to colitis. By contrast, animals that received effector T cells and Tbet+iTregPDL1 cells were protected from clinical weight loss (Figure S2A). We next evaluated T effector cell function in the spleen and lamina propria lymphocytes (LPL). Adoptive transfer of Tbet+iTregPDL1 cells significantly diminished interferon -γ (IFN-γ) production in the T effectors in the spleen (Figures 2A and 2B) and in the LPL (Figure S2B). These results suggested that despite both groups of Th1 cells expressing Foxp3, only Tbet+iTregPDL1 cells had a robust regulatory phenotype in vivo that was comparable to iTreg cells generated from naïve CD4+ cells. We next investigated if the ability of Tbet+iTregPDL1 cells to prevent colitis was attributed to Foxp3 stability in these cells. Foxp3 expression was measured within the CD45.2+ cells isolated from the spleen (Figures 2C and 2D) and lamina propria (Figure S2C) at day 60 post adoptive transfer. Frequency of Foxp3 expressing Tbet+iTregPDL1 cells was increased when compared to cohorts that received Tbet+iTreg cells. Finally, we evaluated the inherent capacity of these cells to revert back to IFN-γ producers. We found that the capacity of Tbet+iTregPDL1 cells to secrete IFN-γ was significantly diminished when compared to Tbet+iTreg cells (Figure 2E).
Figure 2. In vivo function of Tbet+iTreg cells and Tbet+iTregPDL1 cells.
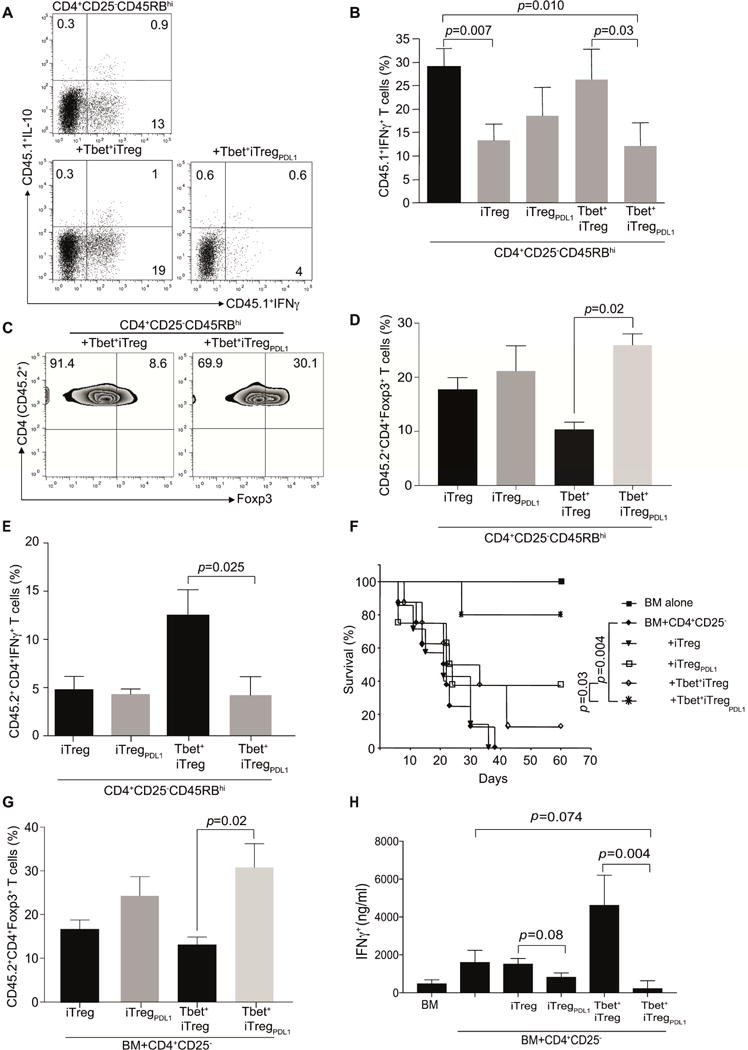
Tbet+iTreg cells and Tbet+iTregPDL1 cells were generated and then utilized for the prevention of autoimmune colitis and alloimmune GvHD. B6.Rag2−/− mice were reconstituted with CD45.1+CD4+CD45RBhiCD25− T cells (4×105 cells/mouse) either alone or along with CD45.2+iTreg cells, iTreg PDL1 cells, Tbet+iTreg cells and Tbet+iTregPDL1 cells (1×105 cells/mouse) cells. At day 60 post adoptive transfer, spleens were characterized. Representative flow plots of intracellular IFN-γ and IL-10 cytokine expression in cohorts that either received CD4+ T effector cells alone, cohorts that received Tbet+iTreg cells in addition to T effector cells or cohorts that received Tbet+iTregPDL1 cells in addition to T effector cells (A). Summary of T cell effector cytokine IFN-γ in the various different cohorts within CD45.1+ cell populations (B). Representative flow plot of Foxp3 expression in CD45.2+CD4+Tbet+iTreg cells and Tbet+iTregPDL1 cells (C), Summary of Foxp3 and IFN-γ expression in iTreg cells, iTregPDL1 cells, Tbet+iTreg cells and Tbet+iTregPDL1 cells (D–E). Function of Tbet+iTreg cells and Tbet+iTregPDL1 cells were assessed in an experimental model of GvHD. Survival curve of mice that succumbed to GvHD in the various different cohorts (F). Summary of Foxp3 expression in iTreg cells, iTregPDL1 cells, Tbet+iTreg cells and Tbet+iTregPDL1 cells on day 14 post-transplant (G). Alloreactive IFN-γ was measured using Luminex (H). Each experiment had n=3-5 mice per cohorts. Data shown is cumulative from 2 independent experiments. For survival curve, each cohort consisted of n=10 mice. Data are presented as Mean±SEM. Please also refer to Figure S2.
The potency of Tbet+iTregPDL1 cells was then tested in an allogeneic murine GvHD model. We found that BALB/c host mice that received B6 cells stimulated under iTreg cells plus PDL-1 conditions had higher survival rates, diminished weight loss and histological manifestations compared with mice that received cells stimulated under iTreg cell conditions alone, reaching significance in cohorts with Tbet+iTregPDL1 cells (Figures 2F, S2D and S2E). We next measured Foxp3 expression within the CD45.2+ iTreg, iTregPDL1, Tbet+iTreg and Tbet+iTregPDL1 cells at day 14 post-transplant and found significant Foxp3 expression was maintained within Tbet+iTregPDL1 cells (Figure 2G). We next evaluated the amount of alloreactive IFN-γ expression in the cohorts that received Tbet+ iTreg and Tbet+ iTregPDL1 cells. A significant reduction in the Tbet+iTregPDL1 cell treated cohorts (Figure 2H) was noted. These data indicate that PDL-1 induced a stable regulatory phenotype in Tbet+ iTregPDL1 cells.
PD-1 signaling downregulates asparaginyl endopeptidase in Tbet+ iTregPDL1 and iTregPDL1 cells
In order to identify PD-1 dependent molecular mechanism that was operational in Tbet+iTregPDL1 cells, we first evaluated AKT and mTOR signaling pathway as previously reported in iTreg cells (Francisco et al., 2009). No difference in the phosphorylation of AKT or mTOR pathway was observed (Figure S3A). Therefore, we next evaluated the gene expression profile of Tbet+iTreg and Tbet+iTregPDL1 cells. We found downregulation of a protease namely asparaginyl endopeptidase (AEP) in Tbet+iTregPDL1 cells using micro-array analysis (Figure S3B). Consistent with our microarray data, AEP protein expression (Figure 3A) and enzyme activity was significantly decreased in Tbet+ iTregPDL1 cells compared with Tbet+ iTreg cells (Figures S3C and S3D). The active form of AEP was evaluated by using LE28 probe [which emits a fluorescent signal when ligated to the active form of AEP (Edgington et al., 2013); Figure S3C]. We next evaluated if a functional nuclear pool of AEP was available within Tbet+iTreg cells, as previously reported (Dall and Brandstetter, 2016; Haugen et al., 2013; Kosugi et al., 2009), using LE28 by imaging flow cytometry. We found a significant increase in the presence of active nuclear AEP in Tbet+iTreg cells compared with Tbet+iTregPDL1 cells (Representative image Figures 3B and 3C; Summary of nuclear activity Figure S3E). In addition, active AEP was expressed in iTreg cells generated in the absence of PDL-1, but not in polarized Th1, Th2, CD4+CD25− or CD4+CD25+ T cells (Figures 3D and S3F). PDL-1 treated iTreg cell cultures showed relatively diminished AEP activity, which was abrogated in Pd1−/− iTreg cells (Figures 3D and S3G). Finally, we evaluated the presence of nuclear AEP by immuno blotting and confocal microscopy. In all cases, we found AEP to be expressed within the nucleus in Tbet+iTregs and iTreg cells (Figures 3E and 3F), which was limited by the addition of PDL-1 (Figure 3F).
Figure 3. PDL-1 exposure downregulates asparaginyl endopeptidase in Tbet+iTreg cells.
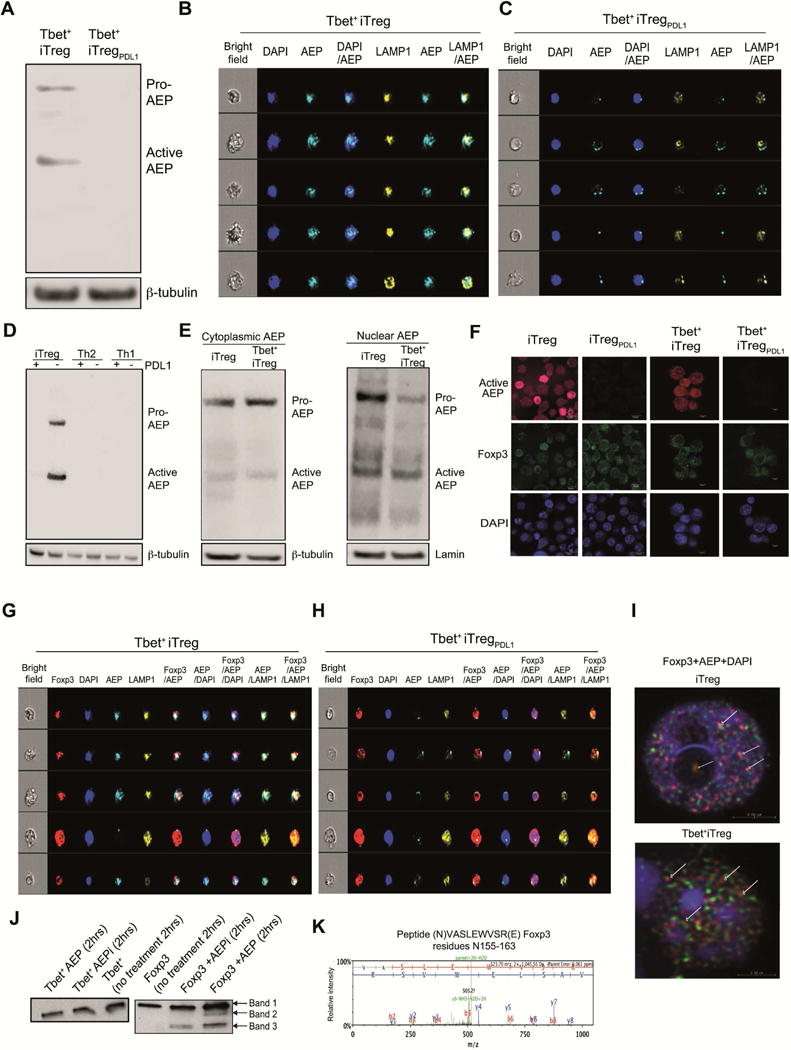
Tbet+iTreg cells and Tbet+iTregPDL1 cell lysates were generated at pH 7 and were subjected to immuno blotting. Asparaginyl endopeptidase (AEP; Legumain; LGMN) was measured in the different subsets (A). Tbet+iTreg cells and Tbet+iTregPDL1 cells were differentiated and then stained with LE28 (measuring active AEP) along with LAMP1 and DAPI and then subjected to Amnis Imaging Cytometry. Representative images of active AEP enzyme expression in the nucleus in Tbet+iTreg cells (B) and Tbet+iTregPDL1 cells (C). AEP expression in iTreg cells, iTregPDL1 cells, Th1 cells, Th1PDL1 cells, Th2 cells and Th2PDL1 cells (D). AEP expression was determined in cytoplasmic and nuclear fractions of iTreg cells and Tbet+iTreg cells (E). iTreg cells, iTregPDL1 cells, Tbet+iTreg cells and Tbet+iTregPDL1 cells were stained with DAPI, Foxp3 PE and AEP cy5. Confocal microscopy showing AEP and Foxp3 expression inside the nucleus (F; red is AEP, green is Foxp3 and blue is DAPI). Foxp3 co-localization with AEP in the nucleus in Tbet+iTreg cells (G) and Tbet+iTregPDL1 cells was shown (H). Confocal microscopy detecting co-localization of AEP and Foxp3 within the nucleus in iTreg cells and Tbet+iTreg cells (I; white arrows show AEP co-association with Foxp3 within the nucleus). Tbet cleavage (J; left panel) and Foxp3 cleavage (J; right panel) in the presence of AEP or AEP inhibitor. Spectral analysis of Foxp3 cleavage at N155 (K). Each experiment was repeated 3-5 times and representative data from one experiment is shown. Data are shown as Mean±SEM. Please also refer to Figures S3 and S4.
Using confocal microscopy and imaging flow cytometry on cells stained with fluorescent markers for Foxp3 protein and active nuclear AEP, we found that Foxp3 and AEP were co-localized in Tbet+iTreg cells and iTreg cells (Figures 3G–3I and S3H–S3O). This result prompted us to examine whether Foxp3 was a specific target of AEP or whether AEP indirectly regulated Foxp3 expression. Co-incubation of activated AEP with Foxp3 protein demonstrated that AEP directly cleaved Foxp3, but not T-bet protein (Figure 3J). In spite of possessing numerous asparaginyl sites, T-bet protein was refractory to AEP cleavage, whereas minimally cleaved Foxp3 bands (band 2 and 3) were noted by immuno blotting. However, band 3 was noted in conditions, which incorporated AEP inhibitor (Figures 3J). When we subjected band 2 and band 3 to high-throughput mass spectrometry, AEP specific peptide cleavage product was found in band 2 and not band 3. Data from this analysis identified a specific semi-tryptic cleaved peptide within band 2 which was a target for AEP [N155; AEP cleaves after Asn (N)]. No other AEP specific targets were identified within the two bands (Figure 3K and Table S1).
We next evaluated Foxp3 protein turnover within Tbet+iTreg cells in the presence of AEP inhibitor. Significant inhibition of Foxp3 protein degradation was noted in the presence of AEP inhibitor in Tbet+iTreg cells (Figure S4A; top and bottom panel). To confirm this, we evaluated Foxp3 turnover in WT and Aep−/− iTreg cells; again, Foxp3 turnover was lower in Aep−/− iTreg cells as compared to WT iTreg cells (Figure S4B; top and bottom panels).
To further investigate the direct action of AEP on Foxp3, we designed a human Foxp3 mutant 1 (all N mutated to A (alanine); 12 sites; Figures S4C and S4D). We tested if the AEP resistant mutant Foxp3 had enhanced stability in in vitro experiments. WT and mutant 1 were transduced into HEK293T cells and then rate of Foxp3 degradation in the presence of AEP and AEP inhibitor was evaluated. AEP specifically degraded WT Foxp3 protein while showing no activity on mutated Foxp3 mutant 1 protein (Figures S4E and S4F). These in vitro experiments suggest that AEP may directly act on Foxp3 protein within T cells.
Deletion of AEP specific cleavage site in Foxp3 results in prevention of alloreactive GvHD
We next evaluated the in vivo function of cells that were transduced with WT Foxp3 (mouse and human) or Foxp3 mutant 2 (human) and mouse Foxp3 mutant 1. For these experiments, we first constructed human WT and mutated Foxp3 (N154; Asn site is at 154 in human) as our mass spectrometry data were obtained from human Foxp3 protein. In addition, we also constructed WT and mutated (N153; Asn site is at 153 in mouse) mouse Foxp3. All the constructs were transduced into naïve mouse T cells and Foxp3 expression was evaluated (Figure 4A), and nuclear localization (Figure S5A) for each construct were verified at day 4 post-transduction. The transduced WT and mutant forms of Foxp3 were functional as they suppressed the capacity of the transduced cells to produce IFN-γ; IFN-γ being a specific target of Foxp3 in vitro (Figure S5B). The in vivo suppressive function of transduced T cells was tested in a murine GvHD model (Laurence et al., 2012). We found that T cells transduced with mutated versions of Foxp3 (both mouse and human) significantly prevented GvHD lethality in murine recipients as compared to WT Foxp3 (Figure 4B).
Figure 4. AEP specific Foxp3 mutants and AEP inhibition prevents GvHD.
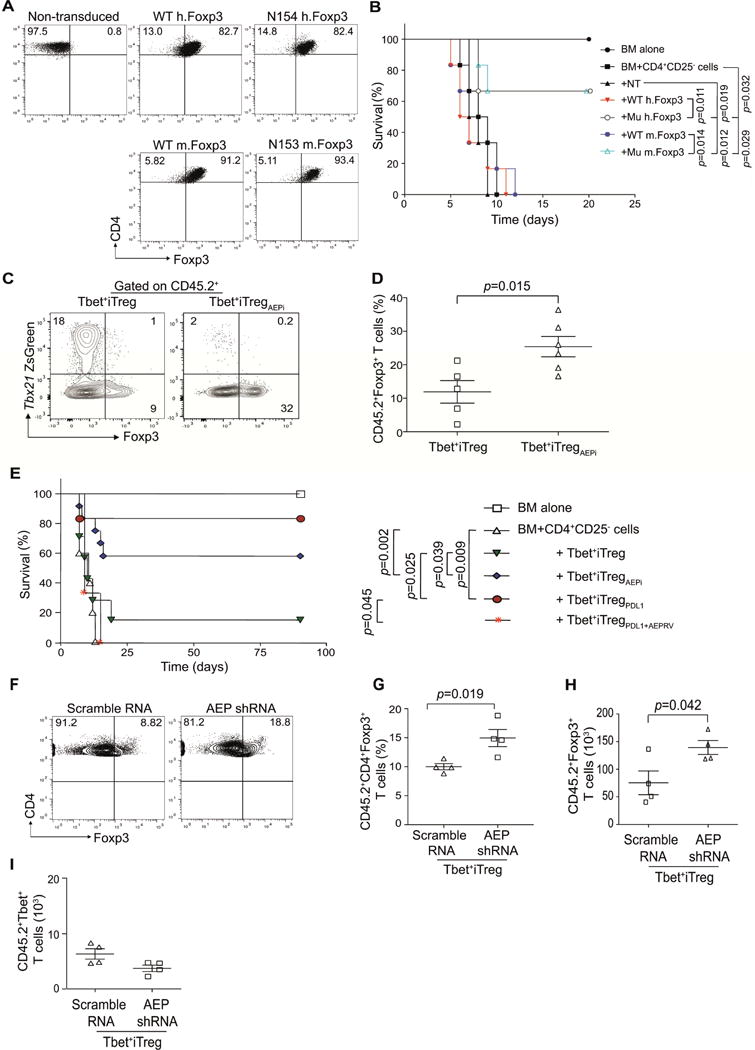
Murine CD4+CD25− T cells were transduced with WT human Foxp3, Mutant (N154) human Foxp3, WT murine Foxp3 or mutant murine Foxp3 (N153). Transduction efficiency at day 4 was measured by flow cytometry (A). Host BALB/c mice were subjected to lethal total body irradiation (TBI; 950cGy) and then reconstituted with B6 T depleted bone marrow (BM, 5×106 cells) alone, or with CD4+CD25− T cells (CD45.1 marked, 0.1×106). Certain cohorts were treated with BM plus CD4+CD25− T plus non-transduced T cells (NT, 0.1×106) or T cells transduced with WT human Foxp3 (WT hFoxp3, 0.1×106), or human mutant Foxp3 (Mu hFoxp3, N154; 0.1×106) or murine WT Foxp3 (WT mFoxp3, 0.1×106) or murine mutant Foxp3 (N153; Mu mFoxp3, 0.1×106). GvHD lethality was monitored (n=6 per cohort) (B). Host BALB/c mice were subjected to TBI and then reconstituted with bone marrow (BM, 107 cells), CD4+CD25− T effector cells (CD45.1 marked; 1×106 cells) and either Tbet+iTreg cells (1×106 cells) that were expanded with AEP inhibitor or control Tbet+iTreg cells (1×106 cells). At day 14 post-transplant, splenocytes were harvested and the Foxp3 was measured in the Tbet+iTreg cell populations (marked with CD45.2). Representative flow plots showing Foxp3 expression in the different cell populations (C); frequency of Foxp3 expression (D). A survival curve experiment was set up to test the efficacy of blocking AEP in preventing GvHD. Animals were conditioned with TBI and then reconstituted with BM alone or plus CD4+CD25− T cells. Cohorts were then treated with Tbet+iTreg cells, Tbet+iTreg cells expanded with AEP inhibitor (AEPi), Tbet+iTregPDL1 cells or Tbet+iTregPDL1 cells overexpressing AEP (E). Experiments were repeated with CD45.2+ Tbet+iTreg cells that were expanded with AEP shRNA or scramble shRNA and then tested in GvHD. Frequency and absolute numbers of CD45.2+Foxp3+ cells in the different cohorts (F–H) and absolute numbers of Tbet+ cells at day 14 post-transplant in Tbet+ iTreg cells (I). Experiments were repeated twice with n=4-6 mice for immunological studies and n=6-10 mice for survival curve. Representative data from one experiment is shown as Mean±SEM. Please also refer to Figure S5.
Inhibiting AEP activity maintains Foxp3 expression in Tbet+iTreg cells in vivo
We next tested the role of AEP in limiting Foxp3 stability in Tbet+iTreg cells using an AEP specific inhibitor (AEPi). First, the in vitro efficacy of AEPi was tested in iTreg cell cultures. Naïve CD4+CD25− T cells from WT mice were expanded under iTreg cell conditions (experimental outline Figure 1E). Certain culture conditions were supplemented with AEPi. We found that the AEPi cultures had significantly higher Foxp3 expression as compared to the control cultures, whereas no significant difference was noted in Aep−/− iTreg cell cultures, thus confirming the specificity of the AEP inhibitor (Figures S5C and S5D). Splenic Tbet+Foxp3− Th1 cells were expanded into Tbet+iTreg cells in the presence of AEP inhibitor (MV026630, 100μM) and then Foxp3 stability was tested in vivo. Murine recipients were reconstituted with CD4+CD25− naïve T cells (CD45.1+) along with Tbet+iTreg cells (CD45.2+) expanded in the presence of vehicle (DMSO) or AEP inhibitor (MV026630). At day 14 post-transplant, the frequency of Foxp3+ cells within the adoptively transferred Tbet+iTreg cells was measured. Tbet+iTreg cells generated in the presence of AEPi exhibited an enhanced frequency of Foxp3+ cells as compared to control Tbet+iTreg cells (Figures 4C and 4D). To further evaluate the role of AEP in Tbet+iTreg cell function, we over-expressed AEP in Tbet+iTregPDL1 cells using a retro-virus. AEP overexpression and nuclear localization was first confirmed (Figures S5E and S5F) in Tbet+iTregPDL1 cells. We then tested the function of overexpressed AEP by transducing Aep−/− iTreg cells with either empty vector (EV) or AEP RV. At day 7 post expansion, Aep−/− iTreg cells were capable of expressing Foxp3, which was completely abrogated when AEP was overexpressed (Figure S5G). We then evaluated the clinical outcome of over-expressing AEP (in Tbet+iTregPDL1 cells) or inhibiting AEP (in Tbet+iTreg cells) in acute GvHD. BALB/c mice that received B6 Tbet+iTregPDL1 cells with forced expression of AEP had a significantly higher mortality compared with those that received control B6 Tbet+iTregPDL1 cells. Similarly, Tbet+iTreg cells expanded with AEP inhibitor showed similar regulatory function to that of Tbet+ iTregPDL1 cells in vivo (Figure 4E). To further confirm the role of AEP in Treg cell function in vivo, a retroviral small inhibitory RNA (RV-shRNA) for AEP was utilized. AEP silencing in Tbet+iTreg cells was effective (Figures S5H–S5J). Murine recipients reconstituted with Tbet+iTreg cells transduced with AEP RV-shRNA had a significant delay in the loss of Foxp3 (Figures 4F–4H). Ablating AEP had minimal effect on the expression of Tbet in Tbet+ iTreg cells (Figure 4I). Finally, we mutated the nuclear localization sequence in AEP (AEP NLS mutant) and then evaluated Foxp3 stability. We found that overexpressing AEP NLS mutant in Aep−/− iTreg cells maintained Foxp3 expression but this was not the case in cohorts that overexpressed AEP WT protein (Figures S5K–S5M).
AEP deficiency modulates in vivo Treg cell function by maintaining Foxp3 expression
To further assess the role of AEP in Foxp3 regulation, we evaluated the efficacy of iTreg cells from Aep−/− mice in an experimental murine model of GvHD. First, we characterized the CD4+ T cell compartment within Aep−/− mice. WT and Aep−/− CD4+ T cells had a similar phenotype with respect to T cell frequency and activation, but Aep−/− mice had higher Treg cell frequency (Figures 5A–5F and S6A and S6B). By contrast, no significant difference was noted in the cytokine expression by CD4+ T cells from WT and Aep−/− mice (Figure S6C). Next, in vitro expanded iTreg cells from WT and Aep−/− naïve CD4+ T cells (Figure S6D) were used tested in GvHD. Aep−/− iTreg cells were significantly efficient at preventing GvHD compared to WT iTreg cells (Figure 5G). Of note, cohorts treated with Aep−/− iTreg cells (CD45.2+) had significantly higher numbers of Foxp3+ cells in the spleen and lymph nodes (Figures 5H and S6E and S6F) as compared to WT iTreg cell treated cohorts.
Figure 5. iTreg cells deficient in AEP inhibit GvHD and maintain Foxp3 expression in vivo.
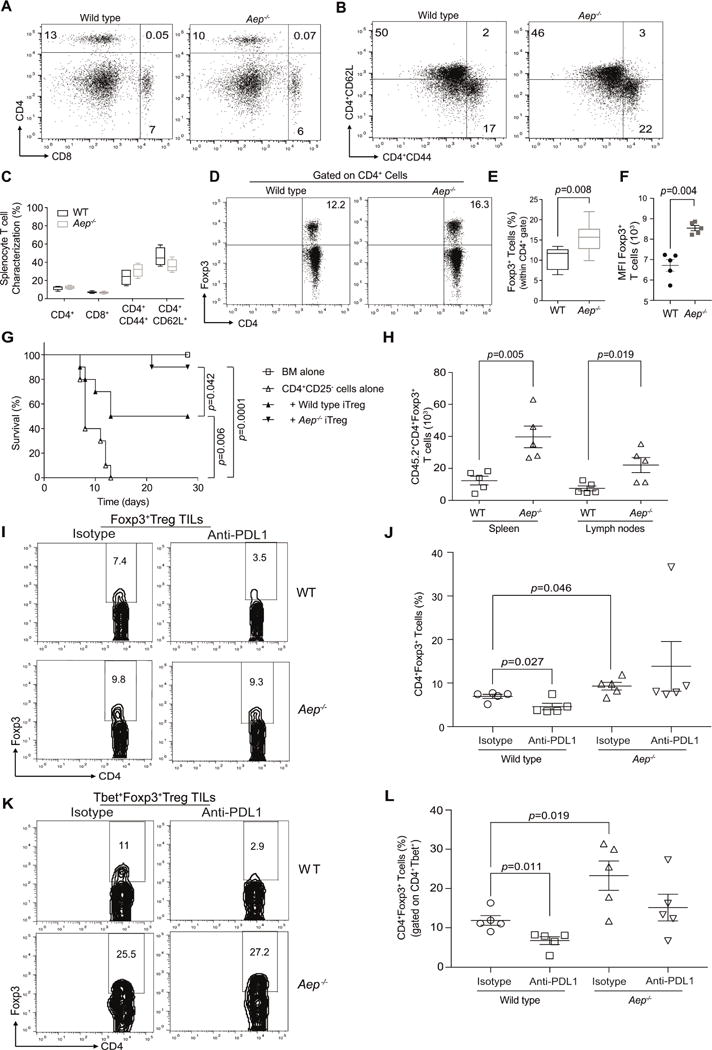
Splenocytes from WT littermate controls and Aep−/− mice were characterized for CD4+ T cells, CD8+ T cells (A), T central and effector memory phenotype of CD4+ cells were characterized using CD62L and CD44 markers (B). Cumulative data from n=4 mice on the frequency of CD4, CD8, CD44 and CD62L (C). Treg cell frequency was evaluated by intracellular Foxp3 expression (D). Cumulative data from n=5 mice of Foxp3 frequency (E), and Foxp3 mean fluorescence intensity (F). Host BALB/c mice were subjected to TBI and then reconstituted with B6 BM (107), CD4+CD25− T cells (CD45.1 marked; 0.1×106 cells) and either WT iTreg cells (0.1×106 cells) or Aep−/−iTreg cells (0.1×106 cells). Survival curve of cohorts that received the various cell populations (G). Foxp3+ cells in WT and Aep−/− iTreg cells (marked with CD45.2+) at day 14 post-transplant (H). WT and Aep−/− mice were reconstituted with 3×105 B16F10 melanoma cells subcutaneously and cohorts were treated with either isotype control or anti-PDL1 antibody at day 5, 7 and 9. Mice were euthanized on day 11 and then evaluated for Foxp3+Treg cells and Tbet+Foxp3+Treg cells within the TILs. Representative flow plots of Treg cell frequency within the tumor (I), and summary of Treg cells in the various cohorts (J). TILs were gated on CD4+Tbet+ cells and then Foxp3 expression was evaluated. Representative flow plots of Tbet+Treg cell frequency within the tumor (K), and summary of Tbet+Treg cell in the various cohorts of TILs (L). Data are shown as Mean±SEM, from one representative experiment. Each experiment was at least repeated twice and had n=5 mice for immunological studies or n=10 for survival. Please also refer to Figure S6.
In order to explore the relationship between PD-1 signaling and AEP activation within T cells, we tested the frequency of Treg cells in Aep−/− mice under disease conditions in the presence of PDL-1 blocking antibody. A syngeneic B16F10 melanoma tumor model was used whereby WT and Aep−/− mice were reconstituted with tumor cells and the cohorts were treated with either isotype or PDL-1 antibody. At day 11, tumors were resected and the tumor infiltrating lymphocytes (TILs) were tested for the frequency of Foxp3+Treg cells and Tbet+Foxp3+Treg cells. We found that Aep−/− cohorts had a small but significant increase in Foxp3+ TILs as compared to WT cohorts (Figures 5I and 5J). In the presence of anti-PDL-1, we saw a decrease in Foxp3+ TILs in the tumors of WT cohorts but no change in the Aep−/− cohorts (Figures 5I and 5J). When we gated on Tbet+ TILs, we found a similar pattern with an elevated proportion of Tbet+ TIL’s expressing Foxp3 in the Aep−/− compared with WT mouse cohorts and the addition of anti-PDL1 significantly inhibiting Foxp3 expression only in the WT cohorts (Figures 5K and 5L). Collectively, these in vivo experiments confirm our in vitro data and identified PD-1 as a regulator of Foxp3 through AEP.
Tbet+ Th1 cells primed during acute and chronic viral infections upregulate Foxp3 ex vivo
We next investigated if Th1 cells arising during viral infection can also be induced to express Foxp3. First, during acute LCMV infection, a significant percentage of Tbet+Foxp3+ cells was observed at day 14 post infection (Figures 6A and 6B and S7A) and second, Tbet+Foxp3− cells had a significant PD-1 expression (Figures S7B and S7C). These virus primed CD4+Tbet+Foxp3− cells were sorted on day 14 post-infection and cultured in vitro under iTreg cell conditions with or without PDL-1. In both cases, iTreg cell-polarizing cytokines could induce Foxp3 expression in Th1 cells and this was enhanced to a small but significant extent in the presence of PDL-1 (Figure 6C). Similarly, during chronic LCMV infection, a substantial increase was noted in Tbet+Foxp3+ cells at day 14 post infection in vivo (Figures 6D and 6E and S7D) and again, Tbet+Foxp3− cells had a significant PD-1 expression (Figures S7E and S7F). These chronic LCMV virus primed CD4+Tbet+Foxp3− cells were sorted on day 14 post-infection and cultured in vitro under iTreg cell conditions with or without PDL-1. In both cases iTreg cell-polarizing cytokines could induce Foxp3 expression in Th1 cells and this was enhanced by a small but significant extent in the presence of PDL-1 (Figures 6F and 6G). In order to confirm that PDL-1 is required for conversion of Tbet+ cells into Tbet+pTreg cells during chronic LCMV, we adoptively transferred flow sorted CD45.2+Tbet+Foxp3− cells into CD45.1+ hosts infected with chronic LCMV. Mice were treated with either isotype or anti-PDL1 antibody. At day 10, frequency of converted Tbet+Foxp3+ in the spleen was measured (Figures 6H and 6I). Anti-PDL-1 treatment significantly inhibited the conversion of Tbet+ cells into Foxp3+ cells and enhanced the proliferation of both total and GP33 specific CD45.1+ CD8+ T cells in vivo (Figures S7G–S7M).
Figure 6. Virus primed CD4+Tbet+Foxp3− cells upregulate Foxp3 under iTreg and pTreg conditions.
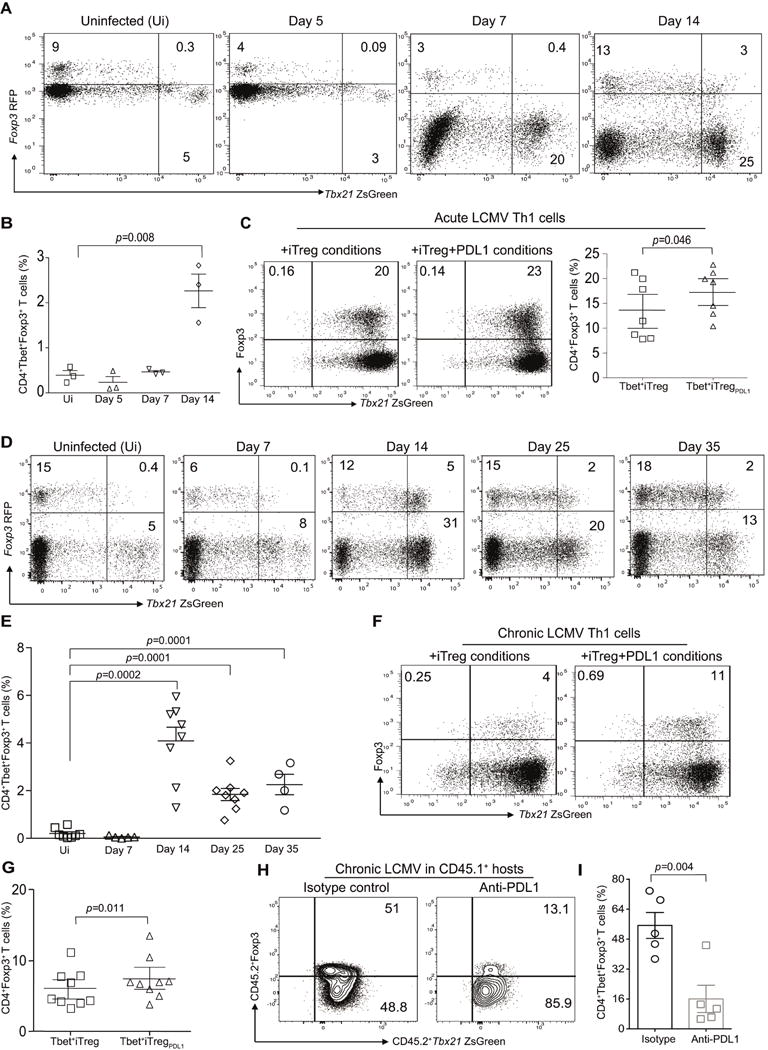
B6.Tbx21ZsgreenFoxp3RFP mice were infected with LCMV Armstrong (2×105 PFU) and then monitored for the presence of CD4+TbethiFoxp3+ cells. Representative flow plots from three individual experiments showing TbethiFoxp3+ cells in the CD4 compartment of murine recipients at various time points post infection (A). Summary of frequency of TbethiFoxp3+ T cells at various time points (B). Virus primed TbethiFoxp3− cells from acute LCMV infected mice were flow sorted at day 14 and then expanded under iTreg conditions in the absence or presence of PDL-1fc chimera. The frequency of Foxp3 expression in TbethiFoxp3− cell population in the absence or presence of PDL-1 after 7 days of ex vivo culture (C). B6.Tbx21ZsgreenFoxp3RFP mice were infected with LCMV clone 13 (2×106 PFU) and then monitored for the presence of CD4+TbethiFoxp3+ cells. Representative flow plots showing TbethiFoxp3+ cells in the CD4 compartment of murine recipients at various time points post infection (D). Summary of frequency of TbethiFoxp3+ T cells at various time points (E). The frequency of Foxp3 expression in TbethiFoxp3− cell population in the absence or presence of PDL-1 after 7 days of ex vivo culture (F). Summary of Tbet+iTreg cell differentiation (G). CD45.2+Tbet+FoxP3− cells (0.7×106 cells) were adoptively transferred into CD45.1+ murine hosts that were infected with Clone-13. Cohorts were treated with either isotype control or anti-PDL1 antibody (200μg/mouse). Splenocytes were harvested at day 10 and the frequency of CD4+Tbet+Foxp3+ pTreg cells were evaluated (H–I). Data are shown as Mean±SEM from a representative of one to three individual experiments involving n=3-9 mice per cohorts. Please also refer to Figure S7.
Tbet+pTreg cells are increased in the TILs of mice bearing B16F10 melanoma tumor
In order to identify the biological context during which Tbet+pTreg cells arise from Tbet+Th1 cells, an animal model of B16F10 melanoma, where PD-1 based therapies play an important role, was used. Adoptive transfer of Tbet+Th1 cells into murine recipients with established tumor was performed (Outline of Experimental Methodology, Figure 7A). The emergence of Tbet+pTreg cells was then evaluated in the spleen and within the TILs. A significant increase in Tbet+pTreg cells was noted within the TILs in the tumor microenvironment (Figures 7B and 7C). We next tested if PDL-1 contributed to the differentiation of Tbet+Th1 cells into Tbet+iTreg cells in the tumor microenvironment. Stimulation with PDL-1 significantly enhanced Foxp3 expression in sorted Tbet+Th1 cells from tumor bearing mice in ex vivo cultures (Figures 7D and 7E). We subsequently tested in vivo Tbet+ cell conversion in our tumor model. Rag2−/− mice were reconstituted with tumor cells followed by adoptive transfer of sorted Tbet+Foxp3− cells. Cohorts were either treated with isotype control or anti-PDL1 and then TILs were evaluated for Tbet+Foxp3+ cells. The frequency of Tbet+pTreg cells was significantly increased in the isotype treated cohorts but not in the anti-PDL1 treated murine recipients (Figures 7F–7H). The experiment was repeated with Tbet+Foxp3− cells that were transduced with either control shRNA RV or AEP shRNA RV. Consistent with our experiments performed using Aep−/− mice, AEP silencing rendered Tbet+ cells refractory to PDL1 blockade and resulted in significant conversion towards Tbet+Foxp3+ cells within the tumor microenvironment (Figures 7I and 7J). Finally, we evaluated conversion of CD45.2+Tbet+ cells in CD45.1+ hosts that were reconstituted with B16F10 melanoma tumor. Certain cohorts were treated with either isotype or anti-PDL1 antibody. In this experimental condition, we again found that Tbet+Foxp3−cells were capable of converting to Tbet+iTreg cells within the tumor microenvironment, which was efficiently blocked in the presence of PDL1 antibody (Figures 7K and 7L). In all these experiments, CCR4 expression on Tbet+Foxp3+ cells within the tumor microenvironment was minimal (data not shown). These results suggest that Tbet+ cell conversion can occur in vivo within the tumor microenvironment and PDL1 blockade reverses the conversion of Tbet+ cells into Tbet+Foxp3+ cells. However, anti-PDL-1 effect in blocking Tbet+ cell conversion to Tbet+Foxp3+ cells is abrogated in the absence of AEP.
Figure 7. Tbet+Th1 cells upregulate Foxp3 in tumor microenvironment.
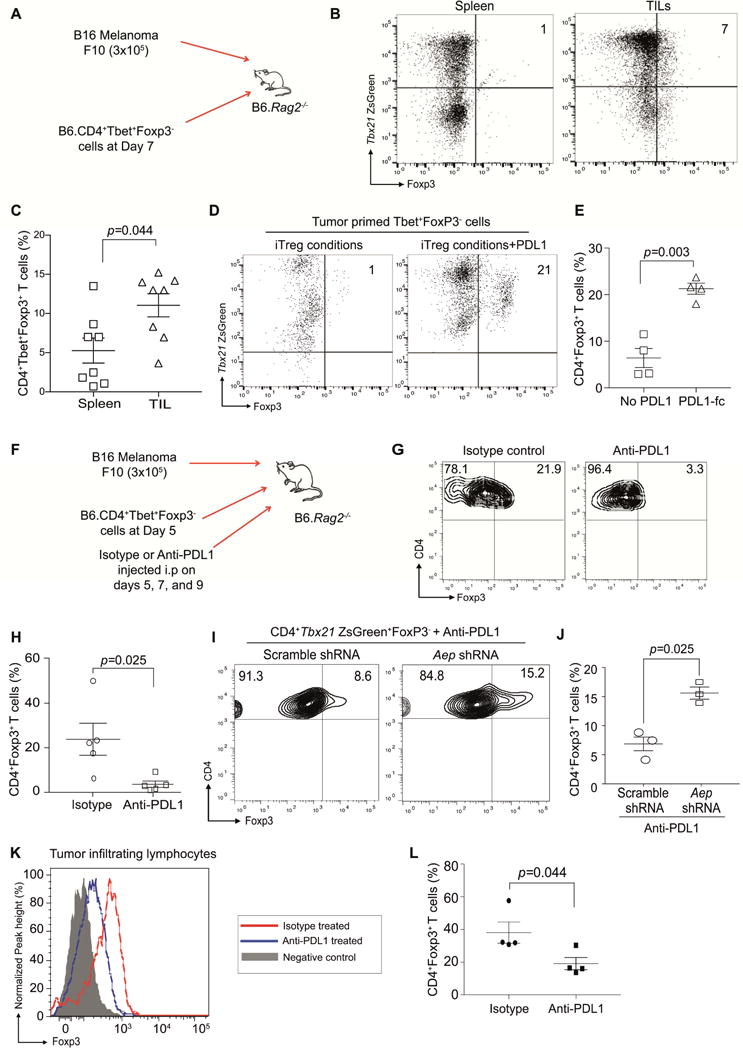
B6.Rag2−/− mice were subcutaneously injected with B16F10 melanoma cells. At day 7 post-tumor inoculation, adoptive transfer of Tbet+Th1 cells was performed (A). Mice were euthanized on day 14, splenocytes and TILs were isolated and then Tbet+Foxp3+ cell frequency was determined (B). Summary of Foxp3+Tbet+ cells from the spleen and TIL (C). Tbet+Foxp3−cells were flow sorted from the spleen and then expanded under iTreg conditions or iTreg conditions plus PDL-1fc. The upregulation of Foxp3 was then monitored by flow cytometry at day 7 post culture (D–E). B6.Rag2−/− mice were subcutaneously injected with B16F10 melanoma cells and adoptive transfer of Tbet+Th1 cells was performed at day 5. Cohorts were either treated with isotype control or anti-PDL1 antibody (250μg/mouse) at days 5, 7 and 9. Animals were euthanized at day 11. Experimental methodology outlined in (F), and the frequency of Tbet+iTreg cells was monitored within the TILs (G–H). Experiments were performed with Tbet+Foxp3− cells transduced with either scramble shRNA RV or AEP shRNA RV and then treated with anti-PDL1 antibody. Frequency of Tbet+iTreg cells within the TILs in the different cohorts (I–J). Murine CD45.1+ hosts were reconstituted with B16F10 melanoma tumors and sorted CD45.2+Tbet+Foxp3− cells. Cohorts were treated with either isotype or anti-PDL1 antibody. Frequency of Tbet+iTreg cells was measured within the TILs (K–L). Experiments were repeated at least twice and data are shown as Mean±SEM from one to two individual experiments involving n=3-8 mice per group.
Discussion
The regulation of Foxp3 in Tregs and T helper cell subsets is an active area of investigation and may help in understanding dysregulation of the immune system in disease processes. In this report, we have identified a proteolytic regulation of Foxp3 protein in iTreg cells, Tbet+iTreg cells and pTbet+Treg cells. We demonstrated that (a) PD-1 signaling maintains Foxp3 protein stability through regulating AEP, (b) AEP directly cleaves Foxp3 and results in Foxp3 instability in iTreg cells and pTreg cells and (c) inhibiting AEP resulted in enhanced Treg cell function. These data elucidate a basic mechanism that is operational in Treg cells and paves a path to the development of translational approaches for developing Treg cell based cell therapies.
The results outlined in this paper are in agreement with the existence of these Tbet+Foxp3+ T cells in vivo. However, reports on Tbet+Treg cells (Hall et al., 2012; Koch et al., 2012; Koch et al., 2009; Levine et al., 2017) propose that tTreg cells are the likely precursors of Tbet+Treg cells. The data presented here extend these observations and clearly demonstrate that an alternate pathway is involved in the upregulation of Foxp3 expression by Tbet+Foxp3− T cells.
In vivo cell tracing experiments performed in the long-term colitis model highlights a mechanism by which PDL-1 imparts regulatory function to Tbet+iTregPDL-1 cells. During colitis Tbet+iTregPDL-1 cells had sustained Foxp3 expression after 60 days in an inflammatory environment unlike their counterpart Tbet+iTreg cells. These data support a regulatory mechanism whereby differentiation of Tbet+ cells in the presence of PDL-1 can result in sustained Foxp3 expression in vivo and led us to explore the molecular mechanisms by which PD-1 signaling regulated Foxp3 stability. We found a proteolytic pathway that was operational in maintaining Foxp3 protein stability in iTregs and Tbet+iTreg cells involving direct inhibition of the activity of AEP. PD-1 inhibition of AEP was independent of CD28 signaling (Hui et al., 2017; Kamphorst et al., 2017).
The notion that a specific protease can perform an essential specific proteolytic function is controversial as cells express many proteases that exhibit considerable functional redundancy (van Kasteren and Overkleeft, 2014). However, cell type specific differences in protease function have been previously reported. For example, AEP breaks down self-antigens in DCs (Manoury et al., 2002) and Cathepsin G performs this function in B cells (Burster et al., 2004). AEP activity has been reported both in lysosomes and in the nucleus of tumor cells (Haugen et al., 2013). Functionally AEP can induce tumor cell proliferation, migration and process antigens for optimal presentation by DCs (Andrade et al., 2011; Lin et al., 2014; Manoury et al., 1998). In contrast to other lysosomal proteases, AEP is expressed in the cytosol and nucleus and the activity of AEP across both neutral and acidic pH has been previously reported (Haugen et al., 2013). These observations enable a mechanism by which Foxp3 in the nucleus can be targeted by AEP.
The imaging and biochemical data presented here demonstrated that in T cells, AEP played a specific function in cleaving Foxp3, but not Tbet. These results are in accordance with previous AEP studies where it has been shown that AEP substrates in part are not amenable to other protease activity. In addition, AEP is well known for its substrate and cleavage specificity and often AEP mediated cleavage results in a functional immunological outcome in vivo (Manoury et al., 1998; Manoury et al., 2002). In T cells, AEP adheres to this phenotype, whereby it specifically targets and cleaves Foxp3 at a single site, which results in the instability of the protein. Therefore, our study implicates the occurrence of a proteolytic mediated regulation of Foxp3 in iTregs and Tbet+iTreg cells.
The data presented here postulates a post-translational mechanism of Foxp3 protein regulation in addition to the previously described proteosomal pathway that is operational in Foxp3 regulation. However, deleting AEP was sufficient for maintaining Foxp3 protein in iTregs and Tbet+iTreg cells in vivo. In contrast to the proteosomal degradation study where an shRNA approach was used (Chen et al., 2013; van Loosdregt et al., 2013; Zhao et al., 2015), we have utilized a genetic loss of function model (Aep−/− mice) to demonstrate the stability of Foxp3 in vivo during acute inflammation. Furthermore, mutating AEP specific sites in Foxp3 protected mice from GvHD mediated lethality. Therefore, our data demonstrates that either AEP deficiency or Foxp3 mutated at AEP specific sites can enhance Treg cell function and is a primary pathway in modulating post-translational stability of Foxp3 protein.
Our results suggest that AEP inhibitor can be used to generate large-scale Treg cells that are stable and are functionally robust in vivo. The use of PDL-1 to grow Tbet+iTreg cells may be efficacious but a substantial decrease in cell numbers can occur given the role of PD-1 in inhibiting T cell proliferation. Using AEP inhibitors, this hurdle can be overcome in order to generate large numbers of antigen primed Tbet+iTreg cells that maintain regulatory function in vivo. In addition, donor derived AEP deficient Treg cells can also be generated for the treatment of GvHD.
Data from the acute and chronic LCMV infected mice further identified a role for PDL-1 in inducing Foxp3 in primed “antigen-specific” Tbet+ Th1 cells. Although insufficient cell numbers prevented us from isolating antigen specific T cells prior to ex vivo iTreg cell culture, the results presented here imply that PDL-1 can be used to generate iTreg cells from previously antigen primed Tbet+Th1 cells. Consistent with previous studies (Hall et al., 2012; Koch et al., 2012; Koch et al., 2009; Levine et al., 2017), we have also identified a unique population of Tbet+iTreg cells that expands in both acute and chronic LCMV. In summary, these experiments highlight many aspects of Tbet+iTreg cells: (a) primed CD44hiTbethiFoxp3+ cells can arise during acute and chronic LCMV infection, (b) primed CD4+CD44hiTbethiFoxp3− T cells can give rise to Tbet+Foxp3+ T cells in ex vivo cultures, and finally (c) blocking PDL-1 in vivo dampened Tbet+pTreg cell conversion, therefore confirming PDL1 as a critical mediator of Tbet+pTreg cell generation in chronic viral infection.
In melanoma, Treg cell mediated tolerance has largely been attributed to the migration of Treg cells from the periphery to the tumor site (Spranger et al., 2013), while Tbet+Th1 cell conversion is largely unexplored. Since the microenvironment in melanoma provides an abundance of PDL-1 that can result in activation of PD-1 signaling on Tbet+ TILs, we explored and found that Tbet+Th1 cell conversion indeed occurred. However, this study does not address the contribution of hematopoietic versus non-hematopoietic versus tumor tissue derived PDL-1 in inducing Treg cell conversion within the tumor microenvironment. The results presented here raise the possibility that the PDL-1 driven Treg cell generation within the TILs is dependent on AEP expression and that individuals who overexpress AEP within their Treg TIL populations may be more responsive to PD-1 and or PDL-1 based immunotherapeutics.
In conclusion, this report has identified a mechanism by which sustained PD-1 signaling induces robust regulatory function in iTreg cells through post-translational regulation of the Foxp3 protein. Therefore, this study demonstrates an insightful interaction between co-inhibitory receptor signaling and protease activity and has elucidated the importance of these two signaling pathways in maintaining T regulatory phenotype.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead contact, Dr. Shoba Amarnath.
Experimental Model and Subject Details
Mice
Female C57BL/6 (B6, H-2Kb) and Balb/c (H-2Kd) mice 8- to 10- weeks old were obtained from Frederick Cancer Research Facility, USA or Charles River, UK. Female C57BL/6 Rag2−/− and C57BL/6 Foxp3RFP mice were purchased from Jackson Laboratories, Aep−/− and WT littermate controls was kindly provided by C. Watts. B6.Tbx21Zsgreen mice were bred with B6.Foxp3RFP mice in a specific pathogen-free facility at the National Institutes of Health, USA and at Newcastle University, UK. B6.Tbx21ZsGreenFoxp3RFP mice were utilized for all T cell lineage-tracing experiments. The use of animals for this research was approved by the Animal Care and Use Committee, National Cancer Institute, and National Institute of Allergy and Infectious Diseases, NIH, and carried out in accordance with the NIH animal health and safety guidelines. Animal experiments conducted at Newcastle University were approved by the Newcastle Ethical Review committee and performed under a UK home office approved project license. Experimental methodology was in accordance to the NC3R recommendations and data are shown in accordance with ARRIVE guidelines.
Method Details
Cell sorting and Flow cytometry and tetramer staining
Unless stated otherwise, ZsGreen fluorescence was used to determine Tbet expression and RFP fluorescence was used to flow sort various cell populations. CD4+Tbet+Foxp3−Th cells were characterized by multi-parameter flow cytometry for surface markers. Cytokine phenotype of day 0 cells was measured by stimulating cells with phorbol-12-myristate 12- acetate (PMA) and ionomycin for four hours along with Golgiplug and Golgistop which was added in the last 2 hrs of incubation. Flow cytometry staining antibodies for CD4 (clone: RM4-4), CXCR3 (clone: CXCR3-173), PD-1 (clone: 29F.1A12), PDL-1 (clone: 10F.9G2), CD44 (clone: IM7), CD45.1 (clone: A20), CD45.2 (clone: 104), H 2Kb − (AF6-88.5), CD62L (clone: MEL-14), CD8 (clone: 53-6.7), CCR4 (clone: 2G12), Neuropilin-1 (clone: 3E12), Helios (clone: 22F6), CD25 (clone: PC61) and CD127 (clone: A7R34) were purchased from BioLegend. Foxp3 (clone: FJK-16s), IL-10 (clone: JES5-16E3), IFN-γ (clone: XMG1.2) and Ki67 (clone: SolA15) were from Thermo Fisher Scientific. For MHC class I tetramer staining, H-2Db GP33-41 was used at 1:100 dilutions and staining was performed at 4° C for 1 hr. Data were acquired using either an LSR II or Fortessa or FACS CANTO and analyzed using FlowJo software version 9.6.4 or 10.0.6.
In vitro cell culture
CD4+Tbet+Foxp3− T helper cells were flow sorted and stimulated in 24 well cell culture plates coated with Anti-CD3 (clone:145-2C11; 5μg/ml) for 3 days in cell culture media [RPMI, supplemented with 10% FCS, glutamine (2mM), non-essential amino acids (0.1mM), 2-mercaptoethanol (50μM), sodium pyruvate (1mM), penicillin and streptomycin (100U/ml)] along with soluble anti-CD28 (clone:37.51;2μg/ml), rmIL-2 (80ng/ml), rhTGF-β1 (2ng/ml), anti-IL-4 (BioLegend, clone:11B11;20μg/ml) and anti-IFN-γ (BioLegend, clone:XMG1.2; 20μg/ml) with or without coated PDL-1fc chimera (5μg/ml). After 3 days of culture, cells were expanded for an additional four days in the presence of IL-2 (80ng/ml) and TGF-β1 (2ng/ml) or with IL-2 alone. Cells were then characterized by intracellular flow cytometry for Foxp3 expression. Both populations were flow sorted for Foxp3+ cells and are denoted as Tbet+iTreg and Tbet+iTregPDL1 cells. Post-differentiation Tbet+iTreg and Tbet+iTregPDL1 cells were stimulated overnight with Anti-CD3 and Anti-CD28 and then supernatants were subjected to a multiplex bead array luminex assay. Control populations of iTregs (iTreg and iTregPDL1) were generated from CD4+Tbet−Foxp3− subsets, which were similarly cultured and characterized. Subsequently, expanded Foxp3+ cells from the various subsets were sorted and utilized in in vitro suppression assays and in vivo animal models of autoimmunity and alloimmunity. In certain experiments, CD4+Tbet+Foxp3− T helper cells were either cultured with AEP inhibitor MV026630 (100μM) or AEP ShRNA or Vehicle (DMSO) from day 0 of culture in addition to αCD3, αCD28, rh-IL-2 and rh-TGF-β1 prior to being used as cellular therapeutics.
In vitro Treg suppression assay
CD4+CD25− cells were isolated using the Miltenyi Biotec Treg isolation kit and utilized in a Treg suppression assay as previously described (Thornton and Shevach, 1998). Purified CD4+CD25− cells (5×104) were labeled with CellTrace Violet and then cultured in 96 well round bottom plates in 200μl complete media along with 2×105 irradiated T cell-depleted spleen cells (3000 cGy) as accessory cells. Anti-CD3 (0.5μg/ml) was added along with cultured flow sorted Foxp3+iTreg populations (iTreg, iTregPDL1, Tbet+ iTreg and Tbet+ iTregPDL1 cells) at the indicated ratios. Cells were incubated at 37° C for 72 hrs and proliferation and suppression was monitored by flow cytometry. Proliferation of responder T cells was evaluated by CellTrace Violet dilution. Percent suppression of CD4 responder T cell was calculated with values representing the ratio of total divided peaks to both divided and non-divided peaks, normalized to the anti-CD3 alone experimental group.
In vivo animal models
Experimental Autoimmune Colitis
B6.Rag2−/− female mice were reconstituted with B6. CD45.1+CD4+CD25−CD45RBhi (T effectors) populations (4×105) as previously described (Asseman et al., 1999) along with flow sorted B6.CD45.2+iTreg cells (1×105), iTregPDL1 cells (1×105), Tbet+iTreg (1×105), or Tbet+ iTregPDL1 cells (1×105). Mice were weighed weekly and loss of body weight was used as an indicator of colitis. Immune endpoints were measured in the splenocytes and in LPL. LPL were isolated as previously described (Asseman et al., 1999). Briefly, large intestine was digested using Liberase TL and DNAse I followed by percoll gradient centrifugation. LPLs were washed twice with complete media and then used for immunological assays. Both splenocytes and LPL were stimulated with PMA and ionomycin along with GolgiPlug and Golgistop for 4 hrs and then effector cytokines were measured by intracellular flow cytometry.
Experimental Allogeneic GVHD
BM was flushed from B6 donor femurs and tibias and T cell depleted (TDBM) using CD90.2 MACS beads (Miltenyi Biotec). Host allogeneic (BALB/c) female mice were conditioned with total body irradiation of 950 cGy in two divided doses three hours apart before being rescued with 107 TDBM cells together with 1×106 CD4+CD25−T cells from WT CD45.1+ B6 donors. In addition, various flow sorted iTreg populations (CD45.2+; 1×106) were adoptively transferred for prevention of GVHD. Survival was monitored as a measure of Treg cell potency. Alloreactive IFN-γ was measured as follows: Single cell suspension of splenocytes (1×106) was cultured overnight with either syngeneic or allogeneic bone marrow derived dendritic cells DCs (1× 105). Supernatant was harvested at 24 hrs and Th1 cytokines were measured by using a multiplex luminex bead array system. Allogenic IFN-γ cytokine in the supernatant was measured by subtracting the amount of IFN-γ present in the syngeneic controls.
LCMV
Six to eight weeks old B6.Tbx21ZsGreenFoxp3RFP mice were infected with Armstrong (2×105 PFU, i.p) or Clone-13 virus (2×106 PFU, i.v) as previously described (Wherry et al., 2003). Titers of virus were determined by plaque assay on Vero cells as previously described (Ahmed et al., 1984). At indicated time points, spleens were harvested and the frequency of Tbet+Foxp3+ T cells was characterized by flow cytometry. In certain experiments, primed CD4+Tbet+Foxp3− T cells were flow sorted on day 14 post-infection and then differentiated under iTreg conditions with or without PDL-1 followed by Foxp3 characterization using flow cytometry. B6.CD45.1+ murine recipients were injected with anti-CD4 (500μg/mouse; day -7) infected with Clone-13 virus along with adoptive transfer of CD45.2+Tbet+Foxp3− cells (0.7×106). Cohorts were treated with either isotype or anti-PDL1 antibody (200μg/mouse) at day 1, 5 and 9. On day 10 Tbet+Foxp3+ cells were analyzed in spleens of the infected mice.
B16F10 melanoma
B6.Rag2−/−mice, WT mice, CD45.1 and Aep−/− mice were reconstituted with 3×105 B16F10 melanoma cells (kindly provided by Dr. Pawel Muranski and Prof. Nick Restifo, NCI, NIH) and the tumor was allowed to progress for 7 days. At day 7, murine recipients were reconstituted with 2×105 Tbet+Foxp3−T effectors from B6.Tbx21ZsGreenFoxp3RFP mice. At day 14 post-tumor inoculation, mice were euthanized, spleen and TILs were isolated and analyzed for the presence of TbethiFoxp3+ cells. In certain experiments, cohorts were treated with either isotype or anti-PDL1 antibody (250μg/mouse) at days 5, 7 and 9. For shRNA experiments, Rag2−/− mice were reconstituted with tumor at day 0 along with flow sorted CD4+Tbet+Foxp3− T cells transduced with either scramble or AEP shRNA. Murine recipients were then treated with anti-PDL1 antibody at days 5, 7 and 9. Host CD45.1+ murine hosts were reconstituted with tumor at day 0 along with flow sorted CD4+Tbet+Foxp3− T cells and then treated with antibodies at days 5, 7 and 9. Splenocytes and tumors were harvested at day 11 and the frequencies of Foxp3+ cells were evaluated.
Histological Analysis
Representative samples of liver, intestine and colon were obtained from the mice that underwent GvHD and fixed in 10% phosphate buffered formalin. Samples were embedded in paraffin, sectioned and stained with hematoxylin and eosin. All slides were coded and read by an external pathologist (Dr. Michael Eckhaus) in a blinded fashion. A four-point scale of GvHD severity was used to score the samples.
Affymetrix Gene Expression Profiling
Total RNA was isolated with RNAeasy kit from Qiagen. RNA quality was checked on Agilent Bioanalyzer. All samples used for microarray analysis had high quality score (RIN >9). 100 ng of RNA was reverse transcribed and amplified using Ambion WT expression kit following manufacturer’s suggested protocol. Sense strand cDNA was fragmented and labeled using Affymetrix WT terminal labeling kit. Four replicates of each group were hybridized to Affymetrix mouse Gene ST 2.0 GeneChip in Affymetrix hybridization oven at 45° C, 60RPM for 16 hrs. Wash and stain were performed on Affymetrix Fluidics Station 450 and scanned on Affymetrix GeneChip scanner 3000. Data were collected using Affymetrix AGCC software. Statistical and clustering analysis was performed with Partek Genomics Suite software using RMA normalization algorithm. Differentially expressed genes were identified with ANOVA analysis. Genes that are up- or down-regulated more than 2 fold and with a p<0.001 were considered significant.
Immuno Blotting
Protein lysates were obtained from Tbet+ iTreg, and Tbet+ iTregPDL1 cells. Lysates were run on 10-20% SDS-PAGE gels and transferred onto nitrocellulose membrane. Membranes were blocked with 5% milk in TBST buffer (20mmol/L TrisHCl, 500 mmol/L NaCl, and 0.01% Tweeen-20) and incubated overnight at 4° C with primary antibodies (Ab) in TBST containing either 5% milk or BSA. Immune reactivity was detected by sequential incubation with HRP-conjugated secondary Ab and enzymatic chemiluminescence (Cell Signaling Technology). Primary Abs to mouse PTEN, mTOR, phospho-mTOR, Akt, phospho-AKT (Ser473 and Thr 308), Foxp3, P70S6K, phospho-p70S6K, ERK, phospho-ERK, GAPDH, β-tubulin, β-actin were procured from Cell Signaling. AEP (Legumain) was obtained from R&D systems. Images were acquired using a LiCOR FcOdyssey system or Wes Simple Protein system.
AEP enzyme activity
AEP activity in cell lysates was measured in triplicates by cleavage of the substrate z-Ala-Ala-Asn-AMC (Bachem) as previously described (Haugen et al., 2013; Johansen et al., 1999). Briefly, cell lysate (20μl) was added to black 96-well microplates. After the addition of 100μl buffer and 50μl substrate solution (final concentration is 10μM) at pH5.8, a kinetic measurement based on increase in fluorescence over 10min was performed at 30° C in a plate reader and presented as enzyme units where one unit of activity was defined as the amount of enzyme releasing 1.0μmol of product/min under the standard conditions described. Enzyme activity was then normalized to the enzyme activity of DCs. DC enzyme activity was set to 100% and then the % enzyme activity of Tbet+ iTreg and Tbet+ iTregPDL1 cells was calculated. Enzyme activity was also measured by using imaging flow cytometry using the AEP probe LE28 (Edgington et al., 2013). LE28 specifically binds to active AEP enzyme. Briefly, Tbet+ iTreg and Tbet+ iTregPDL1 cells were incubated with the AEP probe LE28 cy5 for 1 hr at 37° C, washed and then AEP activity was measured using flow cytometry.
Foxp3 co-localization Assays
The co-localization of Foxp3 in the nucleus was measured using imaging flow cytometry on the Amnis Image stream MKII. For this, cells were fixed and then stained with DAPI, Foxp3APC and LAMP-1 APC-cy7 (BioLegend, clone 1D4B). For co-localization experiments with AEP cells were stained with DAPI, Foxp3PE, LAMP-1 APC-Cy7, and LE28 cy5.
Amnis Imaging Flow cytometry
Sample Acquisition
Samples were run on an Image StreamX MKII using INSPIRE data acquisition software (Amnis EMD-Millipore) at a concentration of approximately 1×106 cells in 50μl of PBS. The system was outfitted with 2 cameras, 12 channels, 405, 488, 561, 642, and 785nm lasers, and an extended depth of field element (EDF). Brightfield was collected in channels 1 and 9, SSC was collected in channel 6 at a 785nm power of 2mW, DAPI was detected in channel 7 (430-505nm filter) at a 405nm laser power of 10mW, TbetZsgreen was detected in channel 2 (480-560nm filter) at a 488 laser power of 20mW, Foxp3 PE was detected in channel 3 (560-595 filter) at a 561nm laser power of 200mW, and LE28 Cy5 were detected in channel 11 (660-745nm filter) and LAMP1 APC-Cy7 was detected in channel 12 (745-800nm filter) respectively, at a 642nm laser power of 150mW. Acquisition gates in INSPIRE were set as follows: a single cell gate was set on a Brightfield Area versus Brightfield Aspect Ratio plot to encompass single cells and eliminate debris and aggregates, a Brightfield Gradient RMS plot was used to gate single cells which were in focus, and gates were set in Raw Max Pixel plots to eliminate events saturating the camera in each fluorescent channel used. 20,000 single, focused, non-saturating events were acquired at 60X magnification, using the EDF element.
Data Analysis
Data analyses were performed in IDEAS 6.0software (Amnis EMD-Millipore). A compensation matrix was created utilizing single color controls acquired with Brightfield and the 785 laser turned off, and all others laser powers set to the powers listed above. In IDEAS, single, focused, and nucleated (DAPI+) cells were gated and used for downstream analysis.
Determination of Foxp3 in the nucleus
A series of masks was created which enabled the determination of the amount of Foxp3 in the nucleus. First, a tight mask was created on the nuclear image by eroding the default mask in one pixel (Erode (M07, 1). Next, a mask was created which identified Foxp3 staining (Intensity (M11, 11_Foxp3, 100-4095)). Lastly, a mask was created to identify pixels which contained DAPI staining and Foxp3 staining by combining the aforementioned masks with an AND operator ((Erode (M07, 1) And Intensity(M11, 11_Foxp3, 100-4095)). The area of Foxp3 inside the nucleus was determined by creating an Area feature on the combined DAPI:Foxp3 mask and gating the Area plot on DAPI+Foxp3+ cells.
Determination of Foxp3 and AEP co-localization in the nucleus
A series of masks was created which enabled the determination of the co-localization of Foxp3 and AEP in the nucleus. First, a mask was created which identified Foxp3 staining (Intensity (M03, 3_Foxp3, 300-4095). Next, a mask was created which identified AEP staining (Intensity (M11, 11_Legumain/AEP, 362-4095). The DAPI mask used is as described in the section above. Lastly, a mask was created to identify pixels which contained DAPI, Foxp3, and AEP staining by combining the aforementioned masks with an AND operator (Intensity (M03, 3_Foxp3, 300-4095) AND (Erode (M07, 1) AND Intensity (M11, 11_Legumain). The area of the pixels containing DAPI, Foxp3, and AEP staining was determined by creating an Area feature on the combined DAPI:Foxp3:AEP mask and gating the Area plot on DAPI+Foxp3+AEP+ cells.
Confocal Microscopy
Flow sorted iTregs, iTregPDL1, Tbet+iTreg and Tbet+iTregPDL1 were first stained with AEP Cy5 followed by fixation and permeabilization. Cells were washed with permeabilization buffer and then stained with Foxp3 PE and DAPI. Images were acquired on a Leica SP8 point scanning confocal microscopy with while light super continuum lasers. Colocalization analysis were performed as previously described (Dunn et al., 2011). A comprehensive explanations of the confocal analysis is provided below:
Orthogonal projection view to enhance visualization of protein spatial location
An orthogonal projection of the image allows identification of the spatial location of a protein within the cell. It is a three-dimensional view of the cell but in a two-dimensional figure. A quadrant was drawn on a particular point in the nucleus, if all three fluorophores are present together in this 3D view, then the analysis shows this on merging the three images.
Deconvolution of the images to enhance signal to noise ratio in an orthogonal projection
Deconvolving is a recognized image processing technique to digitally reassign out of focus light to its originating focal plane. Deconvolution corrects optical aberration and provide higher resolution which enhances signal to noise ratio therefore minimizing false positive analysis. Combining deconvolution and orthogonal projection enhances understanding of the spatial location and co-localization of proteins within a cell.
Particle Analysis
Particle analysis was performed with the deconvolved images as recently shown(Kwon et al., 2017). Using this technique, we measured the number of Foxp3 and AEP particles within the nucleus. We next performed a quantitative analysis of the particles and plotted the values as follows: Each data point depicts the number of particles within the nucleus and the y axis denotes the relative volume of Foxp3 within the nucleus and similarly the relative volume of AEP within the nucleus. If cytosolic contamination occurs, it will be represented in this analysis as follows. 100% on y axis denotes that the entire particle is within the nucleus whereas 20% denotes a particle that does not explicitly localize in the nucleus.
We next quantitatively measured AEP and Foxp3 co-localization. Each data point depicts the number of particles within the nucleus that is colocalized and the y axis denotes the value which is the sum of the intensity of all voxels that colocalize with each other.
Foxp3 cleavage and Mass Spectrometry
Human Foxp3 protein and Tbet protein was purchased from Origene and then used in the cleavage assay in the presence of purified AEP protein (kindly provided by Dr. Colin Watts). Briefly, 1μg of human AEP protein was incubated in 200μl of activation buffer (0.1M NaCl, 0.1M NaOAc, pH 4.5) in 96 well round bottom plates for 30 mins at 37° C. In certain conditions, the AEP inhibitor was added (100μM). Post AEP activation, either Foxp3 protein (5μg) or Tbet protein (5μg) was added to the wells with only activation buffer or activation buffer plus AEP or activation buffer plus AEP plus inhibitor. The plate was then incubated for 2 hrs at 37°C. Samples were then reduced with LDS sample buffer and then subjected to immuno blotting. Once the proteins were run on a gel, they were transferred to a membrane and blotted for Foxp3 and Tbet. In separate experiments, gels were stained with Coomassie blue (Gel Code Blue reagent; Thermo Fisher Scientific) and then bands were cut out and subjected to mass spectrometry as previously described (Zhang et al., 2014). Briefly, bands were dehydrated using acetonitrile followed by vacuum centrifugation, reduced with 10 mM dithiothreitol and alkylated with 55mM iodoacetamide. Gel pieces were then washed alternately with 25 mM ammonium bicarbonate followed by acetonitrile. Samples were digested with trypsin overnight at 37°C. Digested samples were analyzed by LC-MS/MS using an UltiMate® 3000 Rapid Separation LC (RSLC, Dionex Corporation, Sunnyvale, CA) coupled to an Orbitrap Elite (Thermo Fisher Scientific, Waltham, MA) mass spectrometer. Peptide mixtures were separated using a gradient from 92% A (0.1% FA in water) and 8% B (0.1% FA in acetonitrile) to 33% B, in 44 min at 300 nL min−1, using a 75 mm × 250 m i.d. 1.7 mM CSH C18, analytical column (Waters). Peptides were selected for fragmentation automatically by data dependent analysis. Data produced were searched using Mascot (Matrix Science UK), against the SWISSPROT database. Data were validated using Scaffold (Proteome Software, Portland, OR). Additional Foxp3 degradation experiments were performed with WT or mutated Foxp3 expressing 293 cells. The WT Foxp3 cDNA was isolated from a BamH1 + Xho1 digest of plasmid purchased from Addgene. The Mu Foxp3 containing alanine substituted for arginine was synthesized by Integrated DNA Technologies custom gene synthesis service. The construct contained BamH1 and Xho1. These sites were used to clone both WT and mutant Foxp3 cDNAs into pCDNA3.1 and pEV Thy1.1 RV. The mutant Foxp3 construct also included a Not1 site to identify recombinant plasmids. The Foxp3 mutant cDNA was fully sequenced prior to use in experiments.
Pulse Chase Assays for Foxp3 protein turnover
Tbet+iTreg and Tbet+ iTregPDL1 cells were washed and then aliquoted into 24 well tissue culture plates at 3×106/ml. In certain experiments, WT or Aep−/− iTregs were plated into 24 well plates at 1×106/ml. Cells were treated with cycloheximide (150μg) for the indicated time points and lysates were used to measure Foxp3 and Tbet protein degradation by immuno blotting. In order to block proteosomal degradation of Foxp3 and Tbet, MG132 (0.5 μM, Sigma-Aldrich) was added to certain culture conditions. AEP inhibitor MV026630 (100 μM, kindly provided by Prof. Colin Watts and Dr. Sanders van Kasteren) was added to cultures to block specific AEP mediated degradation of Foxp3.
AEP and Foxp3 silencing and mutation assays
AEP specific shRNAs were obtained from Origene. On confirming efficient silencing of AEP in NIH3T3 cells, the most efficient shRNA was chosen for further analysis. Single-stranded oligonucleotides containing shRNA targeted to AEP or scrambled sequence, 20 bp complementary to the sequence flanking a Xho1 site within with the expression vector pEV Thy1.1 RV (gift from Dr. Vanja Lazarevic, NCI, NIH), and a Not1 site were purchased from IDT and annealed in buffer containing 1mM Tris pH 8.0, 50 mM NaCl and 1mM EDTA. The vector pEV Thy1.1 RV was digested with Xho1. The vector and the double stranded oligos were combined and a Gibson reaction (New England Biolabs) was performed. Recombinant clones were identified by Not1 restriction analysis. The integrity of the insert sequence and orientation was confirmed by DNA sequencing. Silencing of AEP was further confirmed in 3T3 cells and then was used to silencing AEP in primary murine Tbet+iTreg cells. pENTR Foxp3 was a gift from Prof. Anjana Rao [Addgene plasmid # 16363 (Wu et al., 2006)] containing WT human Foxp3 cDNA that was cloned into pEV-Thy1.1-RV using BglII and Xho1. The AEP resistant human Foxp3 was synthesized as a minigene (IDT) and cloned into the BglII and Xho1 sites of pEV-Thy1.1-RV. The N154A human Foxp3 mutant was created by inserting a 240 bp G-Block DNA fragment (IDT) between the Bcl1 and BstB1 sites using the Gibson reaction (New England Biolabs). AEP cDNA was obtained from Prof. Colin Watts and cloned into pEV-Thy1.1-RV using Xho1. For NLS experiments AEP NLS site was mutated whereby KRK was replaced to AAA at site 318-320. pMIGR-mFoxp3 was a gift from Prof. Dan Littman (Addgene plasmid # 24067, unpublished). The N153A mutant was created by inserting a 182 bp G-Block DNA fragment (IDT) between the Bcl1 and BamH1 sites using the Gibson reaction (New England Biolabs). All modified constructs were verified by sequencing before use.
Retroviral transductions of Tbet+iTreg cells were performed as follows. Briefly, cells were stimulated for 24 hrs and then washed once. Cells (0.5×106) were spin inoculated with 1ml of virus supernatant in the presence of polybrene (4μg/ml) consisting of scramble or AEP shRNA for 50 mins at 3000RPM. The cells were then placed at 37° C for 2 hrs after which an additional 2ml of complete RPMI media was added. Infection was monitored after 4 days as the percent of CD90.1 and AEP cy5 expression by flow cytometry. CD90.1+Foxp3+Tbet+ iTreg cells were flow sorted and then adoptively transferred in to a murine model of allogeneic GVHD and in vivo stability of Foxp3 in Tbet+ iTreg cells was monitored.
Statistical analysis
Statistical analysis was determined using GraphPad Prism 7 software. For experiments where two groups were compared, a two-tailed student t test was performed. For comparison of three or more groups, a one-way ANOVA was performed followed by appropriate multiple comparison tests. For survival curve analysis, Kaplan-Meier survival curve analysis followed by a log rank test was performed. Unless stated otherwise, histogram columns represent the mean values for each experiment and error bars indicate the standard error of the mean. Data presented were considered significant if p value was ≤ 0.05.
Data and Software Availability
The microarray data reported in this paper have been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession number GEO:GSE113815.
Supplementary Material
Acknowledgments
We would like to thank Dr. Tsung-Ping and Dr. Shang-Yi Tsai, National Institute on Drug Abuse, NIH for sharing protocols on pulse chase experiments and analysis. We would like to thank Dr. William G. Telford for his valuable input on Amnis flow cytometry experiments and analysis. We would like to thank Dr. Billur Akkaya, NIAID, for providing us with B6. Tbx21ZsGreenFoxp3RFP mice for chronic LCMV experiments. We would like to thank Dr. Bishop Hague, NIAID, for his assistance in flow cytometry sorting of virus infected cells. We would like to thank the core facilities at Newcastle University namely Bioimaging, Comparative Biology Centre and Flow cytometry. We would also like to that Mr. Christopher Huggins for his technical expertise. The authors would also like to thank the NIH tetramer core facility for providing the tetramers used in this study. This research was funded by the intramural research program of NCI and NIAID, NIH, USA and Newcastle University Research Fellowship, Newcastle University; MRC-Newcastle University Single Cell Unit Award; G.M is supported by MRC-DiMEN Doctoral Training Partnership program; A.L is supported by Crohn’s and Colitis Foundation of America.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
C.S, G.M, L.P.L- performed experiments, analyzed data. A.G- performed LCMV experiments, analyzed data, wrote paper. D.K-provided intellectual input, performed mass-spectrometry experiments. F.A.F-provided intellectual input, performed gene silencing assays, analyzed data, wrote paper. L.S- provided intellectual input, performed Amnis experiments, analyzed data, wrote paper. T.C, J.M-F-performed experiments, analyzed data. R.B-P-provided intellectual input, performed confocal experiments and analyzed data. L.E-M and J.Y-engineered LE28 probe. M.A.E-scored histology. M.B, C.W, S.V.K- provided intellectual input on AEP experiments, provided relevant AEP reagents, critically read and wrote manuscript. E.M.S, A.L, D.H.F, J.Z - provided intellectual input, reagents and critically read and wrote manuscript. S.A- conceptualized the project, analyzed data and wrote paper.
Declaration of Interests
The authors declare no competing interests.
References
- Adeeku E, Gudapati P, Mendez-Fernandez Y, Van Kaer L, Boothby M. Flexibility accompanies commitment of memory CD4 lymphocytes derived from IL-4 locus-activated precursors. Proc Natl Acad Sci U S A. 2008;105:9307–9312. doi: 10.1073/pnas.0704807105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed R, Byrne JA, Oldstone MB. Virus specificity of cytotoxic T lymphocytes generated during acute lymphocytic choriomeningitis virus infection: role of the H-2 region in determining cross-reactivity for different lymphocytic choriomeningitis virus strains. Journal of virology. 1984;51:34–41. doi: 10.1128/jvi.51.1.34-41.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, Foley JE, Massey PR, Felizardo TC, Riley JL, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Science translational medicine. 2011;3:111ra120. doi: 10.1126/scitranslmed.3003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade V, Guerra M, Jardim C, Melo F, Silva W, Ortega JM, Robert M, Nathanson MH, Leite F. Nucleoplasmic calcium regulates cell proliferation through legumain. J Hepatol. 2011;55:626–635. doi: 10.1016/j.jhep.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, Fehling HJ, Bluestone JA. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity. 2013;39:949–962. doi: 10.1016/j.immuni.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boniface K, Blumenschein WM, Brovont-Porth K, McGeachy MJ, Basham B, Desai B, Pierce R, McClanahan TK, Sadekova S, de Waal Malefyt R. Human Th17 cells comprise heterogeneous subsets including IFN-gamma-producing cells with distinct properties from the Th1 lineage. J Immunol. 2010;185:679–687. doi: 10.4049/jimmunol.1000366. [DOI] [PubMed] [Google Scholar]
- Brown CC, Esterhazy D, Sarde A, London M, Pullabhatla V, Osma-Garcia I, Al-Bader R, Ortiz C, Elgueta R, Arno M, et al. Retinoic Acid is essential for Th1 cell lineage stability and prevents transition to a th17 cell program. Immunity. 2015;42:499–511. doi: 10.1016/j.immuni.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burster T, Beck A, Tolosa E, Marin-Esteban V, Rotzschke O, Falk K, Lautwein A, Reich M, Brandenburg J, Schwarz G, et al. Cathepsin G, and not the asparagine-specific endoprotease, controls the processing of myelin basic protein in lysosomes from human B lymphocytes. J Immunol. 2004;172:5495–5503. doi: 10.4049/jimmunol.172.9.5495. [DOI] [PubMed] [Google Scholar]
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- Chen Z, Barbi J, Bu S, Yang HY, Li Z, Gao Y, Jinasena D, Fu J, Lin F, Chen C, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity. 2013;39:272–285. doi: 10.1016/j.immuni.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall E, Brandstetter H. Structure and function of legumain in health and disease. Biochimie. 2016;122:126–150. doi: 10.1016/j.biochi.2015.09.022. [DOI] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol. 2011;300:C723–742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013;73:6900–6912. doi: 10.1158/0008-5472.CAN-13-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington LE, Verdoes M, Ortega A, Withana NP, Lee J, Syed S, Bachmann MH, Blum G, Bogyo M. Functional imaging of legumain in cancer using a new quenched activity-based probe. J Am Chem Soc. 2013;135:174–182. doi: 10.1021/ja307083b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell. 2014;158:749–763. doi: 10.1016/j.cell.2014.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliani N, Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, de Zoete MR, Licona-Limon P, Paiva RS, Ching T, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015;523:221–225. doi: 10.1038/nature14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, Pritchard GH, Silver JS, Bouladoux N, Stumhofer JS, et al. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity. 2012;37:511–523. doi: 10.1016/j.immuni.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugen MH, Johansen HT, Pettersen SJ, Solberg R, Brix K, Flatmark K, Maelandsmo GM. Nuclear legumain activity in colorectal cancer. PLoS One. 2013;8:e52980. doi: 10.1371/journal.pone.0052980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegazy AN, Peine M, Helmstetter C, Panse I, Frohlich A, Bergthaler A, Flatz L, Pinschewer DD, Radbruch A, Lohning M. Interferons direct Th2 cell reprogramming to generate a stable GATA-3(+)T-bet(+) cell subset with combined Th2 and Th1 cell functions. Immunity. 2010;32:116–128. doi: 10.1016/j.immuni.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, Vale RD. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JF, Idema AJ, Bol KF, Nierkens S, Grauer OM, Wesseling P, Grotenhuis JA, Hoogerbrugge PM, de Vries IJ, Adema GJ. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro Oncol. 2009;11:394–402. doi: 10.1215/15228517-2008-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen HT, Knight CG, Barrett AJ. Colorimetric and fluorimetric microplate assays for legumain and a staining reaction for detection of the enzyme after electrophoresis. Analytical biochemistry. 1999;273:278–283. doi: 10.1006/abio.1999.4221. [DOI] [PubMed] [Google Scholar]
- Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–1427. doi: 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity. 2012;37:501–510. doi: 10.1016/j.immuni.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Matsumura N, Takashima H, Miyamoto-Sato E, Tomita M, Yanagawa H. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J Biol Chem. 2009;284:478–485. doi: 10.1074/jbc.M807017200. [DOI] [PubMed] [Google Scholar]
- Kwon HK, Chen HM, Mathis D, Benoist C. Different molecular complexes that mediate transcriptional induction and repression by FoxP3. Nat Immunol. 2017;18:1238–1248. doi: 10.1038/ni.3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence A, Amarnath S, Mariotti J, Kim YC, Foley J, Eckhaus M, O’Shea JJ, Fowler DH. STAT3 transcription factor promotes instability of nTreg cells and limits generation of iTreg cells during acute murine graft-versus-host disease. Immunity. 2012;37:209–222. doi: 10.1016/j.immuni.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AG, Medoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, Hoyos BE, Putintseva EV, Chaudhry A, Dikiy S, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature. 2017;546:421–425. doi: 10.1038/nature22360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Qiu Y, Xu C, Liu Q, Peng B, Kaufmann GF, Chen X, Lan B, Wei C, Lu D, et al. Functional role of asparaginyl endopeptidase ubiquitination by TRAF6 in tumor invasion and metastasis. J Natl Cancer Inst. 2014;106:dju012. doi: 10.1093/jnci/dju012. [DOI] [PubMed] [Google Scholar]
- Manoury B, Hewitt EW, Morrice N, Dando PM, Barrett AJ, Watts C. An asparaginyl endopeptidase processes a microbial antigen for class II MHC presentation. Nature. 1998;396:695–699. doi: 10.1038/25379. [DOI] [PubMed] [Google Scholar]
- Manoury B, Mazzeo D, Fugger L, Viner N, Ponsford M, Streeter H, Mazza G, Wraith DC, Watts C. Destructive processing by asparagine endopeptidase limits presentation of a dominant T cell epitope in MBP. Nat Immunol. 2002;3:169–174. doi: 10.1038/ni754. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, Huehn J, Hori S. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36:262–275. doi: 10.1016/j.immuni.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Mukasa R, Balasubramani A, Lee YK, Whitley SK, Weaver BT, Shibata Y, Crawford GE, Hatton RD, Weaver CT. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity. 2010;32:616–627. doi: 10.1016/j.immuni.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol. 2010;11:674–680. doi: 10.1038/ni.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–1102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peine M, Rausch S, Helmstetter C, Frohlich A, Hegazy AN, Kuhl AA, Grevelding CG, Hofer T, Hartmann S, Lohning M. Stable T-bet(+)GATA-3(+) Th1/Th2 hybrid cells arise in vivo, can develop directly from naive precursors, and limit immunopathologic inflammation. PLoS Biol. 2013;11:e1001633. doi: 10.1371/journal.pbio.1001633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Science translational medicine. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JJ, Mohrs M, Pearce EJ. Regulatory T cell responses develop in parallel to Th responses and control the magnitude and phenotype of the Th effector population. J Immunol. 2006;176:5839–5847. doi: 10.4049/jimmunol.176.10.5839. [DOI] [PubMed] [Google Scholar]
- Taylor S, Huang Y, Mallett G, Stathopoulou C, Felizardo TC, Sun MA, Martin EL, Zhu N, Woodward EL, Elias MS, et al. PD-1 regulates KLRG1(+) group 2 innate lymphoid cells. J Exp Med. 2017;214:1663–1678. doi: 10.1084/jem.20161653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kasteren SI, Overkleeft HS. Endo-lysosomal proteases in antigen presentation. Curr Opin Chem Biol. 2014;23:8–15. doi: 10.1016/j.cbpa.2014.08.011. [DOI] [PubMed] [Google Scholar]
- van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, Meerding J, Berkers CR, Barbi J, Grone A, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. 2013;39:259–271. doi: 10.1016/j.immuni.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. Journal of virology. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Song M, Liu X, Kang SS, Kwon IS, Duong DM, Seyfried NT, Hu WT, Liu Z, Wang JZ, et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med. 2014;20:1254–1262. doi: 10.1038/nm.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Guo H, Qiao G, Zucker M, Langdon WY, Zhang J. E3 Ubiquitin Ligase Cbl-b Regulates Thymic-Derived CD4+CD25+ Regulatory T Cell Development by Targeting Foxp3 for Ubiquitination. J Immunol. 2015;194:1639–1645. doi: 10.4049/jimmunol.1402434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.