

Abstract
Given that dysregulation of mechanics contributes to diseases ranging from cancer metastasis to lung disease, it is important to develop methods for screening the efficacy of drugs that target cellular forces. Here, we use nanoparticle-based tension sensors to quantify the mechanical response of individual cells upon drug treatment. As a proof-of-concept, the activity of bronchodilators is tested on human airway smooth muscle cells derived from seven donors, four of which are asthmatic. It is revealed that airway smooth muscle cells isolated from asthmatic donors exhibit greater traction forces compared to the control donors. Additionally, the mechanical signal is abolished using myosin inhibitors or further enhanced in the presence of inflammatory inducers, such as nicotine. Using the signal generated by the probes, single-cell dose-response measurements are performed to determine the “mechano” effective concentration (mechano-EC50) of albuterol, a bronchodilator, which reduces integrin forces by 50%. Mechano-EC50 values for each donor presented discrete readings that were differentially enhanced as a function of nicotine treatment. Importantly, donor mechano-EC50 values varied by orders of magnitude, suggesting significant variability in their sensitivity to nicotine and albuterol treatment. To the best of our knowledge, this is the first study harnessing a piconewton tension sensor platform for mechanopharmacology.
Keywords: mechanopharmacology, molecular tension sensors, integrins, airway smooth muscle cells
TOC entry
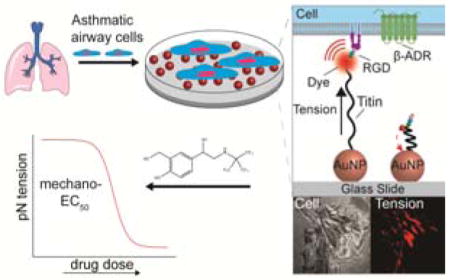
A Nanoparticle based piconewton tension sensor is applied to study the mechanopharmacology in airway smooth muscle (ASM) cells. Cells derived from asthmatic donors displayed a more contractile phenotype compared to normal ASM cells. Single cell dose-response mechanical measurements provided the mechano-EC50 of bronchodilators for cells derived from normal and asthmatic donors.
1. Introduction
Cellular forces play a central role in many biological processes ranging from immune recognition to cell differentiation and wound healing.[1] Accordingly, mechanical dysregulation contributes to a range of human diseases, especially in processes that involve mechanically active cells such as smooth muscle cells, cardiomyocytes, and cancer cells.[2] The development of drugs that directly target the underlying mechanically-mediated pathology represents a nascent and growing focus of drug development. Underscoring this growing interest, the term mechanopharmacology was recently introduced to describe the study of drugs that target cell mechanics or the study of how a cell’s mechanical state influences its sensitivity to a particular drug molecule.[3]
One of the challenges in mechanopharmacology pertains to the lack of molecular tools that can directly quantify forces in living cells; thus moving away from traditional tissue-scale muscle strip contractility assays[4] or muscle thin film measurements.[5] As a result, many drugs that target mechanical processes are assayed using secondary chemical reporters, such as intracellular calcium, cyclic nucleotides, and protein expression levels, all of which are indirectly associated with mechanics.[6]
There are two categories of single cell techniques that have been applied to study mechanopharmacology. The first category includes methods that measure changes in intrinsic mechanical properties (i.e. young modulus) of the cells upon drug treatment, for examples atomic force microscopy (AFM),[7] optical/magnetic tweezers[8] and magnetic twisting cytometry (MTC).[9] Here, the “experimenter” applies forces (e.g. tensile, bending and twisting forces) to deform the cells and thus probe their mechanical properties. Recently introduced microfluidic-based cell deformability cytometry based on inertial forces are promising for mechano-phenotyping and mechanopharmacology,[10] but they are more appropriate for screening cells that are anchorage independent. The second category of techniques can be described as passive methods, reporting the cell-generated traction forces on an artificial substrate. Thus far, traction force microscopy (TFM) and micro-post array detectors (mPADs) are widely used for passive measurements and are particularly important for studying cell-extracellular matrix (ECM) interactions[11] and juxtacrine signaling pathways.[12] Importantly, TFM screening of small molecule-treated cells has shown promise in discovering new drugs that modulate cell contractility.[13] However, the main challenges in TFM are the inherent nN force sensitivity, the need to create homogenous polymer films, and the downstream analysis is computationally intensive.
To circumvent some of the limitations of TFM, we introduced molecular tension force microscopy (MTFM) to map pN traction forces exerted by individual cells.[14] In general, MTFM probes are comprised of a molecular spring, such as DNA,[15] polyethylene glycol (PEG)[16] or protein,[17] flanked by a fluorophore and a quencher. The head of the spring is conjugated to a ligand that targets a receptor of interest, while the tail of the spring is anchored to the substrate. Forces applied to the ligand separate the fluorophore and quencher, resulting in restoration of fluorescence signal.
In this manuscript, we demonstrate that MTFM can serve as a viable mechanopharmacology platform to study the impact of drugs on cell mechanics. Given that the majority of lung diseases include modulation of lung mechanics, we chose to focus on human airway smooth muscle (ASM) cells. Within the lung tissue, contractile forces are transmitted through the cellular cytoskeleton to the extracellular matrix (ECM) through focal adhesion (FA) complexes that are comprised of hundreds of signaling and structural proteins nucleating around integrin receptors.[18] Integrins play an important role in lung physiology by regulating various downstream signaling pathways that mediate ASM cell phenotype, proliferation, hypertrophy, as well as cell adhesion and migration.[19] ASM cell contraction significantly contributes to the development and progression of asthma, a chronic lung disease characterized by airflow obstruction and bronchoconstriction.[20] β-agonists, such as albuterol, are the first-line therapy to relieve asthma symptoms by acutely relaxing ASM cells in the event of a bronchospasm.[21] Since the mechanical contractility mediates the symptoms of asthma, we hypothesized that adhesion receptor tension measurements would provide a direct approach to determine bronchodilator efficacy. In addition, an ongoing debate in asthma exists between whether enhanced mechanical contractility of individual ASM cells or increased numbers of ASM cells (airway thickening) plays a greater role in the asthma phenotype.[22] Thus, mechanical contractility measurements at the single cell level are required to answer this question, and in part, this particular need motivates the present work.
2. Results and Discussion
We first characterized the integrin traction forces of human ASM cells using a gold nanoparticle (AuNP) titin-based MTFM probe.[17] The titin-based sensor provides the most chemically stable unfolding probe developed thus far.[17] The core of the titin sensor is comprised of an I27 immunoglobulin domain, a β-sandwich protein with 89 residues, derived from the sarcomeric protein of striated muscle that exhibits spring-like properties.[23] We engineered this I27 domain with an N-terminal RGD (Arg-Gly-Asp) peptide, which binds to integrin receptors on the cell surface, and two tandem cysteines at the C-terminus.[2b,17] The cysteine residues allow for immobilization onto 9 nm AuNPs on a PEGylated glass coverslip [0.5% (w/v) lipoic acid-NHS for gold binding and 5% (w/v) mPEG-NHS for passivation] (Figure 1A and B). Additionally, we site-specifically incorporated an organic fluorophore (Cy3) to the I27 protein. The surface immobilized AuNP can effectively quench the fluorescence of Cy3 through nanometal surface energy transfer (NSET) when I27 is folded into its native β-sandwich structure.[24] The RGD-Cy3-I27 probe (30 nM) was incubated on AuNPs in the presence of passivating mPEG-SH at 1:5 ([RGD-Cy3-I27]:[mPEGSH]) molar stoichiometry for 1 h at room temperature to “backfill” the surface of AuNP. Each particle presents an average of ~5 protein sensors, which correspond to a density of ~1500 protein sensors/μm2 as previously reported.[17] These doubly PEGylated surfaces are non-fouling and thus greatly minimize non-specific protein adsorption.
Figure 1.

Measuring airway smooth muscle (ASM) cell integrin forces using titin-based molecular tension probes. A) Schematic illustration of the RGD-Cy3-I27 MTFM sensor and its mechanism of reporting integrin forces. B) Representative AFM image of 9 nm AuNPs immobilized on a PEGylated [5% (w/v) mPEG-NHS and 0.5% (w/v) lipoic acid-PEG-NHS] glass substrate. Scale bar, 200 nm. C–D) Representative RICM and fluorescence images of human ASM cells on the tension sensing substrate before and after treatment with ROCK kinase inhibitor Y-27632 (40 μM) or ML-7 (40 μM) for 30 min. Scale bar, 10 μm. E) Plot shows the normalized tension signal, defined as Idrug/Iuntreated, of the same cells (same ROI) before and after ROCK inhibitor and ML-7 treatment. Masks were created to isolate tension signal from background. Lines represent mean±SEM from n = 9 cells for Y-27632, and n = 5 cells for ML-7 treatment from three independent surface preparations, **P <0.01 (Y-27632) and ***P < 0.001 (ML-7) by Wilcoxon signed-rank test. F) Representative RICM and fluorescence images of human ASM cells transfected with GFP-vinculin incubated on the tension sensor before and after treatment with ROCK kinase inhibitor Y-27632 (40 μM) for 3 min. Scale bar, 10 μm.
When human ASM cells, derived from the lungs of healthy human donors (Table S1, Supporting Information) were cultured on the RGD-Cy3-I27 probe surface for 1 h, we observed significant fluorescence enhancement at the cell perimeter as indicated by reflection interference contrast microscopy (RICM) (Figure 1C). This enhancement in fluorescence signal demonstrates that integrin-mediated forces mechanically unfolded the I27 domain and separated the Cy3 from the AuNP quencher. To verify whether sensor unfolding is due to direct integrin engagement, we generated a variant protein sensor, in which the RGD was mutated to RGE. This single point mutation abrogated cell adhesion, showing that the RGD motif is required for integrin binding and I27 unfolding (Figure S1A, Supporting Information). Treatment with Rho-associated protein kinase inhibitor (Y-27632, Figure 1C) and myosin light chain kinase inhibitor (ML-7, Figure 1D) greatly suppresses the tension signal by ~80 and ~65%, respectively (Figure 1E). Representative tension images collected over a 6 h duration showed that the cell morphology and tension distribution were dynamic as cells spread on the substrate (Figure S2, Supporting information). Together, these results confirm that the I27 tension probe reports real time integrin traction forces in ASM cells.
To confirm that the unfolding of the I27 tension probe coincides with FA proteins, we cultured ASM cells expressing GFP-tagged FA proteins such as vinculin and paxillin for 1 h and compared the distribution of these molecules with the tension signal. We observed the Cy3 tension signal highly mirrors the distribution of FA proteins vinculin (Figure 1F) and paxillin (Figure S1B, Supporting Information), and also with the tips of actin filaments (Figure S1C, Supporting Information), indicating the unfolding of the tension probe is driven by traction forces generated and transmitted by the cytoskeleton and FA proteins.
Asthma is characterized by thickening of airway walls and enhancement in contractility, but it is unknown whether individual ASM cells exhibit altered contractility at the single cell level. Intrinsic differences between healthy and asthmatic ASM cells exist, such as dysregulation in Ca2+ levels[20,22,25] and an increase in mass;[25b,26] therefore, we wanted to investigate the mechanical differences among these types of samples at the single cell level. Accordingly, we measured integrin-mediated I27 unfolding in ASM cells isolated from healthy individuals and asthmatic patients (Table S1, Supporting Information). ASM cells were plated on the tension sensing substrates for 1 to 2 h. Tension imaging revealed that the signal was similarly localized to distal edges of both normal and asthmatic cells and this pattern mirrored that of the location of FAs (Figure 2A). However, when we integrated the fluorescence intensity generated by single cells derived from seven donors, we observed that ASM cells from asthmatic patients generated greater levels of tension signal compared to the normal donors (Figure 2C and 2D).
Figure 2.

Integrin mediated forces are enhanced in the asthmatic human ASM cells and in the presence of nicotine. A) Representative RICM and integrin-mediated tension images of normal and asthmatic human ASM cells incubated on the RGD-Cy3-I27 tension probe for 2 h. Scale bar, 10 μm. B) Representative RICM and tension images of normal and asthmatic ASM cells treated for 72 h with nicotine (50 μg/ml), for 24 h with doxycycline (4 μg/ml) and incubated on the titin-based tension probe for 2 h. Scale bar, 10 μm. C) Scatter plot quantifying the integrated fluorescence intensity of FAs from normal and asthmatic donors with and without addition of nicotine for 72 h. Each circle represents data collected from a single cell. The red line indicates the mean, while the box shows the SEM, *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001 using Student’s t-test. D) Scatter plot showing the integrated fluorescence intensity of FAs pooled from 3 normal and 4 asthmatic donors with and without addition of nicotine for 72 h. ****P<0.0001 using Mann-Whitney test.
To directly compare the contractility of normal and asthmatic ASM cells, we employed a clamped version of I27 tension probes that is mechanically locked by a disulfide bridge (RGD-Cy3-I27G32C-A75C). The cells were cultured on the clamped MTFM probe surfaces for 2 h. At this stage, the probes were partially unfolded by integrin forces but unable to reach the fully extended state, thereby only leading to partial fluorescence enhancement. To infer the relative cellular forces applied by integrins, the kinetics of RGD-Cy3-I27G32C-A75C disulfide reduction (fluorescence increase) were recorded as a function of the concentration of dithiothreitol (DTT). Single exponential fitting yielded a rate constant of protein unclamping (kobs) of 18.9 M−1s−1 for ASM cells, in contrast to 25.8 M−1s−1 for asthmatic ASM cells (Figure S3A, Supporting Information). The difference in the unclamping rate indicates that asthmatic ASM cells exhibit enhanced contractility. To investigate the cellular mechanism of the enhanced contractility in asthmatic cells, we assayed the total myosin light chain expression (tMLC) in both normal (n = 3) and asthmatic ASM cells (n = 4) by western blot. We found that asthmatic cells have higher expression level of the tMLC than the normal cells (Figure S3B, Supporting Information). This further supports our tension measurement showing that asthmatic ASM cells are more contractile than normal ASM cells. Nonetheless, our data is consistent with bronchial smooth muscle cell (BSMC) shortening experiments demonstrating that BSMC from asthmatic subjects exhibit enhanced contractility.[27]
Given that certain inhaled substances, such as a cigarette smoke, promote a contractile phenotype of human ASM cells by increasing Ca2+ concentrations and enhancing the expression levels of contractile proteins,[28] we investigated how chronic delivery of nicotine changes the cell adhesion in normal and asthmatic ASM cells. We first treated both normal and asthmatic ASM cells with a daily dose of nicotine at 50 μg/ml (total nicotine = 0.5 mg in 10 mL culture media) for three consecutive days at a 24 h interval and measured the changes in the cell morphology using optical microscopy (Figure S4A, Supporting Information). In this experiment, we observed that both cell types became more elongated and aligned when treated with nicotine compared to untreated cells. This phenotypic transition suggests changes in cell mechanics. To validate these observations at the protein level, we measured the amounts of phosphorylated myosin light chain (pMLC) by western blot (Figure S4B, Supporting Information) and found a significant increase of pMLC in nicotine treated normal ASM cells. These results confirmed that nicotine treatment of ASM cells promoted a phenotypic change accompanied by enhanced expression of contractile proteins. To further elucidate how nicotine modulates integrin traction forces, we plated nicotine treated cells on the tension sensing substrate. Unexpectedly, we observed loss in Cy3 intensity below the background level (Figure S5A, Supporting Information). This was exclusively observed after chronic nicotine treatment. As previous studies on nictotine treated vascular smooth muscle cells showed enhanced degradation of ECM by upregulating matrix metalloproteinases (MMPs),[29] we speculated that the negative signal was due to probe degradation by local release of MMPs. To test this hypothesis, we prepared binary surfaces comprised of RGD-Cy3-I27 and RGE-A647-I27 tension probes (Figure S5B, Supporting Information). Here, the mutant RGE probes do not engage integrins, and therefore do not experience tension from the integrin receptors. However, both the RGD and the RGE probes are sensitive to local release of MMPs as they only differ by a single point mutation. When ASM cells spread on the binary sensor surface after chronic nicotine treatment (t = 72 h, 1 dose per 24 h interval), we observed the negative fluorescence signal under the distal edges of the cells in both RGD (Cy3) and RGE (A647) fluorescence channels, suggesting that a subset of these sensors were degraded by proteases. By using integrin blocking antibodies, we further determined that degradation of I27-tension probes was dependent on α5β1 integrin signaling (Figure S5C, Supporting Information).[30] Further evidence of protease release is revealed by the pre-treatment of ASM cells with doxycycline (4 μg/ml, 24 h), a known inhibitor of MMP expression[31] (Figure S5D, Supporting Information). Inhibiting MMP expression led to a marked increase in Cy3 tension signal and the absence of dark signal at the cell perimeter. Note that doxycycline treatment did not significantly change the integrated tension signal (Figure S5D, Supporting Information). Because it was shown that nicotine upregulates MMP-2 and MMP-9 through the α7-nAChR,[29c,32] we set out to examine the role of α7-nAChR in nicotine-driven protease expression. When ASM cells were treated with nicotine and α-bungarotoxin, an irreversible antagonist of α7- nAChR, for 72 h and plated on the tension sensing surface, there was only positive fluorescence signal under the cell perimeter and no detectable dark signal (Figure S5E, Supporting Information). Taken together, these findings verify that nicotine drives the release of MMPs in direct proximity to mechanically active α5β1 integrins in an α7-nAChR-dependent process.
We next compared the traction forces of normal and asthmatic cells after chronic treatment (t = 72 h) with nicotine. Nicotine treated ASM cells were incubated with the MMP inhibitor doxycycline 24 h before the measurement. On the next day, the cells were harvested and plated on tension sensing surfaces for measurement. Representative RICM and tension images are shown in Figure 2B and their respective fluorescence signals are plotted in Figure 2C and 2D. The data show that stimulation with nicotine caused the cells to become more polarized and this further enhanced integrin tension in both normal and asthmatic ASM cells. There was approximately a 60–90% increase in tension signal in both cell types, suggesting that the number of receptors applying tension above the I27 unfolding threshold increases following nicotine treatment. Note that because the tension probe only responds to a threshold of tension that leads to I27 unfolding, the tension signal is not linearly proportional to the integrated traction forces.
Our next goal was to apply the protein tension probes as a readout to study the mechanopharmacology of asthma. We tested the effects of known bronchodilator drugs on integrin tension by plating asthmatic ASM cells on the tension sensing substrates for 2 h and then adding albuterol (100 μM) and isoproterenol (100 μM), short acting bronchodilators that target the β2 adrenergic receptor, for 15 min. Treatment with these bronchodilators rapidly extinguished the tension signal (Figure 3A–C). The results show that addition of the ASM relaxants led to a significant decrease in cell contractility and thus caused the refolding of protein tension probes. In all subsequent experiments, we used albuterol to investigate the mechanopharmacology of ASM cells.
Figure 3.

Bronchodilators dose-dependently inhibit tension signal in both normal and asthmatic ASM cells. A–B) Representative RICM and tension images of asthmatic ASM cells seeded on the RGD-Cy3-I27 tension sensor and treated with 100 μM of albuterol and 100 μM of isoproterenol for 15 min. Scale bar, 10 μm. C) Plots show the normalized tension signal, generated by individual cells before and after bronchodilator treatment. Line represents mean±SEM. from n = 17 cells for albuterol treatment, and n = 13 cells for isoproterenol treatment from four independent surface preparations ***P<0.001 and ****P<0.0001 by Wilcoxon signed-rank test. D) Representative RICM, integrin tension, and overlay of RICM and tension images for a single, asthmatic ASM cell adhered on the RGD-Cy3-I27 tension surface and treated with a stepwise addition of albuterol (0.01 nM–1 mM). Scale bar, 10 μm. E–F) Representative dose-dependent curves for (E) normal donors and (F) asthmatic donors with and without the presence of nicotine (50 μg/ml). R2 values associated with both fits were >0.98. EC50 was determined by sigmoidal fitting. Error bars represent SEM of EC50 values obtained from n = 10 cells for each donor collected from ten surfaces. G) Scatter plot quantifying EC50 values from dose-dependent curves of normal (n = 3 donors) and asthmatic ASM cells (n = 4 donors) stimulated with nicotine (50 μg/ml) for 72 h and doxycycline (4 μg/ml) for 24 h, cultured on the tension sensor and treated with the stepwise addition of the albuterol. Each circle represents data collected from a single cell. The red line indicates the mean, while the box shows the SEM, n.s. = not signifcant, *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001 using Student’s t-test. H) EC50 values were pooled from all normal and asthmatic donors with and without the addition of nicotine for 72 h, ****P<0.0001 by Mann Whitney test.
To quantify the dose-response function for albuterol, we titrated the drug and measured the corresponding tension signal. Briefly, we cultured donor ASM cells on the tension sensing substrate for 2 h and then added graded doses of albuterol that ranged from 0.01 nM to 1 mM. The albuterol was added to the cell media at 5 min intervals (Figure 3D), which was sufficient to allow the tension signal to approach equilibrium. Tension maps in the Cy3 channel showed a gradual decrease of the fluorescence intensity under the cell perimeter with increasing drug concentration (Figure 3D and Figure S6A, Supporting Information for the images of cells treated with nicotine). We then plotted the integrated tension signal as a function of albuterol concentration for individual cells. Sigmoidal fitting of the data produced a functional EC50 (concentration of a drug that gives half-maximal response), or mechano-EC50, for each cell incubated in the presence and absence of nicotine. These single cell mechano-EC50 measurements were performed for the ASM cells derived from normal (Figure 3E) and asthmatic (Figure 3F) donors before and after nicotine treatment. Note that each data point is collected from a single cell on a different substrate. Figure 3G summarizes mechano-EC50 values of albuterol pooled from the dose-dependent curve of individual ASM cell from normal or asthmatic donors. All together, these findings reveal that 1) asthmatic-donor derived cells had higher average EC50 than normal cells, and 2) nicotine increased mechano-EC50 values for both normal and asthmatic cells. Moreover, the mechano-EC50 for normal donor cells (EC50 = 305.8±27.8 nM) was four-fold greater than that for untreated cells (EC50 = 70.5±9.7 nM) (Figure 3H). The nicotine-induced enhancement of tension is likely under-estimated because we found that addition of doxycycline to the cells decreases EC50 values by around 30% (Figure S7, Supporting Information). Strikingly, some donors displayed up to one order of magnitude enhancement in the EC50 of albuterol following nicotine treatment, while other donors showed minor changes in EC50. Our single-cell mechanopharmacology measurement indeed highlights the heterogeneity within the same population (and across different populations) of cells in response to drugs. Nonetheless, these results show that albuterol dose-dependently inhibited traction forces in both asthmatic cells and asthmatic cells treated with nicotine; however, a higher concentration of albuterol was required to inhibit the tension for nicotine-treated cells, suggesting nicotine drives a more contractile phenotype in both normal and asthmatic ASM cells. Importantly, the mechano-EC50 value is a more direct, functional readout of albuterol potency than conventional assays measuring secondary chemical reporters. For example, Hirst and co-workers reported that the albuterol concentrations producing half-maximum attenuation of eosinophil-activating cytokines, including Granulocyte-macrophage colony-stimulating factor (GM-CSF), Chemokine (C-C motif) ligand 5 (CCL5), and eotaxin, from human ASM cells were in the range of 10–20 nM using ELISA.[33] Hall and colleagues showed that the concentration of albuterol necessary for driving half-maximum cyclic AMP formation in human ASM cells was 0.6 μM.[34]
3. Conclusion
In summary, we used our recently developed nanoparticle tension probes to quantify integrin-mediated tension in ASM cells isolated from normal and asthmatic donors. We show that individuals with asthma develop ASM cells that are more contractile than their non-asthmatic counterparts on our 2D tension sensing substrates. Kinetic unclamping experiments also confirmed this finding. Traction forces in both types of donors were further promoted in the presence of nicotine. Interestingly, we also discovered that activation of α7-nAChR using nicotine induces local release of MMPs, where the α5β1 integrin pulling events occur. Most importantly, we present a MTFM-based assay that quantifies the mechano-EC50 of drugs at the single cell level. We showed that albuterol has a four-fold higher mechano-EC50 value for the asthmatic ASM cells than the normal ones, indicating that asthmatic ASM cells are less sensitive to albuterol treatment. The highest mechano-EC50 values were associated with cells that were incubated in the presence of nicotine, suggesting that some individuals are highly sensitive to nicotine, and this exposure promotes a more contractile phenotype of ASM cells in vivo.
There are three limitations to the present work. First, the assay is performed using glass substrates, which lead to enhanced cell contractility and may bias the measured mechano-EC50 values compared to physiological settings. Second, we performed these measurements using low densities of cells to facilitate single cell quantification. However, cell-cell contact may modulate traction forces and is not investigated in the current studies. Third, the tension probes used here are less suitable for long term tension tracking (> 6 h). This is beause the probe is comprised of a genetically engineered protein immoblized through thiol-gold interaction, and the activity of biological thiols in the media along with proteases can lead to probe dissociation and/or degradation.
Nonetheless, this approach is a platform technology and, in principle, could be broadly used for screening different drugs that modulate cancer cell invasion and cardiac myopathies. As a proof-of-concept demonstration, we determined the mechano-EC50 values for Rho-associated protein kinase and myosin light chain kinase inhibitors in ASM cells (Figure S8, Supporting Information). Several other possible applications of this assay include diagnosing diseases characterized by abnormal cellular mechanics, and discovering drugs that modulate cell traction forces. Lastly, we envision that this approach may be used to complement quantitative polymerase chain reaction (qPCR) and immunohistochemistry of endobronchial biopsies, where patients’ samples are cultured to determine the mechanical drug efficacy. Obtaining ASM cells for ex vivo analysis is feasible in humans[35] as well as large animals.[36] Although implementing bronchoscopy to obtain endobronchial biopsies to characterize an individual’s ASM does require specific clinical infrastructure, this infrastructure already exists in many hospitals and academic centers.
4. Experimental methods
Reagents
Methyllycaconitine (MLA), ROCK inhibitor (Y-27632, ≥98.0%), dimethyl sulfoxide (99%, DMSO), MLCK inhibitor (ML-7), dithiothreitol (99%, DTT), (3-aminopropyl) trimethoxysilane (97%, APTMS), the fluorescent dye dibenzocyclooctyne-Cy3 (DBCO-Cy3), potassium phosphate monobasic (≥99.0%), isoproterenol hydrochloride, α-bungarotoxin-tetramethylrhodamine, unlabeled α-bungarotoxin (BGT), doxycycline hyclate (≥98%), salbutamol (albuterol) and nicotine (≥99%) were purchased from Sigma-Aldrich (St. Louis, MO). The anti-nicotinic acetylcholine receptor α7 antibody (ab10096) was obtained from Abcam (Cambridge, United Kingdom). The Slide-A-Lyzer™ Dialysis Cassettes and the fluorescent dyes: Alexa647 N-hydroxysuccinimidyl ester and Alexa647 DIBO alkyne were purchased from Life Technologies (Grand Island, NY). 4-Azido-L-phenylalanine was purchased from Chem-Impex International (Wood Dale, IL). Ni-NTA Agarose (#30210) was purchased from Qiagen (Valencia, CA). Lipofectamine® LTX with Plus™ Reagent for cell transfection was purchased from Life Technologies (Carlsbad, CA). Number two glass coverslips were purchased VWR (Radnor, PA). Lipoic Acid-PEG-NHS (MW 3400) and mPEG-NHS (MW 2000) were purchased from Nanocs (New York, NY). AuNPs of approximately 9 nm in diameter were purchased from Nanocomposix (San Diego, CA). Sodium bicarbonate was purchased from EMD chemicals (Philadelphia, PA). P2 gel size exclusion beads were purchased from BioRad (Hercules, CA).
Cell culture and transfection
Diseased and healthy human airway smooth muscle (ASM) cells isolated from asthmatic and healthy donors using endobronchial biopsies were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Mediatech) supplemented with 10% Fetal bovine serum (FBS, Mediatech), penicillin G (100 IU/ml, Mediatech) and streptomycin (100 μg/ml, Mediatech) at 37 °C in the presence of 5% CO2. Cells were passaged at ~90 % confluency and plated at a density of 50%. ASM cells were transiently transfected with either GFP-actin, GFP-vinculin or GFP-paxillin using Lipofectamine® LTX with Plus™ reagent by mixing 1 μg of DNA with Lipofectamine® LTX and Plus™ Reagent for each well in a 24-well plate and incubated for 24–48 h.
Western blot
After overnight serum starvation, human ASM cells (Lonza, Switzerland) ± nicotine (50 μg/ml, 72 h) and human ASM cells isolated from healthy and asthmatic donors were cultured in serum-free media. Primary antibodies employed included polyclonal rabbit anti-MLC (1:1000, Cell Signaling Technologies), polyclonal rabbit anti-phospho-MLC (1:500, Cell Signaling Technologies), and polyclonal rabbit anti-GAPDH (1:10,000, Sigma). Secondary antibodies used were IRDye 680LT conjugated polyclonal donkey anti-mouse IgG (1:20,000, LI-COR Biosciences) and IRDye 800CW conjugated polyclonal goat anti-rabbit IgG (1: 20,000, LI-COR Biosciences). Quantification of protein expression was performed by measuring integrated intensity using the Odyssey Infrared Imaging System (LI-COR Biosciences). Densitometry was used to quantify relative protein expression levels using Image J (National Institutes of Health). The band of the protein of interest was normalized to the appropriate loading control.
Antibody blocking
ASM cells were incubated with monoclonal antibodies selective for α5β1 (MAB1969, Millipore), αvβ5 (MAB1961, Millipore) and αvβ3 (MAB1976, Millipore) at 10 μg/ml for 30 min. Additionally, 10 μg of antibody was added to each functionalized surface for full inhibition of α5β1, αvβ5 and αvβ3 integrins.
Protein engineering and dye labeling
I27 protein sensors expressing p-azidophenylalanine were designed with N-terminal ligand (either TVYAVTGRGDSPASSAA or TVYAVTGRGESPASSAA) and two C-terminal cysteines for attachment onto AuNPs. For kinetic studies, we used a I27 variant protein in which Cys47 and Cys63 were mutated to Ala. Also, Gly32 and Ala75 were mutated to cysteines to form disulfide bond as previously described.[17] The proteins were expressed in BL21(DE3) E. coli cells, purified by Ni2+ affinity chromatography and kept at −80 °C in 0.1 M potassium phosphate buffer (pH 7.4). To label the protein sensors with the dye, I27 protein constructs were incubated with either DBCO-Cy3 or DIBO-A647 for 1 h at 37 °C, followed by overnight incubation at room temperature. To purify the dye-labelled protein sensors, P2 gel size exclusion beads were used and the labeling ratio was determined by UV-Vis absorption (NanoDrop).
Protein expression with UAA incorporation
The pET22b plasmid encoding MTFM with a TAG codon was co-transformed with pEVOL-pAzF plasmid into electrocompetent BL21(DE3) E. coli cells.[37] Cells were grown at 37 °C in the presence of ampicillin, chloramphenicol, and 0.2% glucose to an optical density (OD) of 0.2, at which 1 mM of 4-azido-L-phenylalanine was added. At an OD of 0.4, L-arabinose was added to a final concentration of 0.02% (w/v) and at an OD of 0.8, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM. Cells were shaken for 16 h at 30 °C and purified using Ni2+ affinity chromatography.
Fabrication of protein-AuNP surfaces
Glass coverslips (number 2, 25 mm diameter; VWR) were rinsed with Nanopure water (18.2 mΩ), dried at 80 °C, piranha etched in a 3:1 mixture of sulfuric acid and 30% hydrogen peroxide for 30 min (Caution: Piranha can be explosive if mixed with organic solvent!), and then functionalized with an APTMS solution in acetone for 1 h. The samples were then rinsed in acetone three times and baked in an oven at 80 °C for 30 min. After cooling, the substrates were incubated with a solution of 5% (w/v) mPEG-NHS and 0.5% (w/v) lipoic acid-PEG-NHS in 0.1 M fresh sodium bicarbonate overnight at 4 °C. Subsequently, 12 nM of tannic acid stabilized AuNPs (diameter = 9 nm) were added onto the lipoic acid coated surfaces and incubated for 20 min. The AuNP surfaces were then rinsed with water and incubated with the binary mixture of mPEG, SH(CH2-CH2-O)8COOH and the protein sensor in 0.1 M potassium phosphate buffer (pH 7.4) at 1:5 molar stoichiometry for 1 h at RT. The protein surfaces were rinsed with 0.1 M potassium phosphate buffer (pH 7.4) to remove unbound protein sensors. Functionalized coverslips were then assembled into a custom-made chamber and filled with cell media containing 0.5% FBS. The surfaces were used within the same day.
Fluorescence immunostaining
ASM cells were gently rinsed with 1 X phosphate buffered saline (PBS) in the imaging chamber, fixed with 4% paraformaldehyde for 10 min, washed with 1 X PBS and then treated with 0.1% Triton X-100 for 10 min. Cells were next rinsed with 1 X PBS and immunostained with 1: 220 dilution of α-Bungarotoxin-tetramethylrhodamine for 2 h at RT. Before imaging, cell samples were rinsed with 1 X PBS.
Drug treatment
ASM cells were plated at ~50% density and treated chronically with 50 μg/ml (1 dose/day) of nicotine for 72 h. A day before cell experiment, smooth muscle cells were incubated with 4 μg/ml of doxycycline to prevent matrix metalloprotease release from cells. To obtain EC50, ASM cells were allowed to spread on the protein sensor functionalized surfaces for 2 h. Next, increasing amounts of Salbutamol (albuterol, 0.01 nM–1 mM) were added every 5 min and fluorescence-dose response curves were generated, from which the EC50 values were determined using Origin dose response fitting function.
Live cell fluorescence microscopy imaging
For live cell imaging at 37 °C, a Nikon Eclipse Ti microscope with Elements software was used, along with the warming apparatus consisting of an objective warmer and sample warmer. The microscope features a TIRF launcher with three laser lines: 488 nm (10 mW), 561 (50 mW), and 647 nm (20 mW), an Intensilight epifluorescence source (Nikon), an Evolve electron multiplying charge coupled device (EMCCD camera, Photometrics), and the Nikon Perfect Focus system that prevents losing focus during imaging. The microscope is equipped with TIRF 488, TIRF 640, FITC, TRITC, and RICM (reflection interference contrast microscopy) filter cubes that were purchased from Chroma (Bellows Falls, VT).
Kinetic unclamping experiment
Kinetic measurements of S-S reduction were performed as previously described.[17] Briefly, normal and human ASM cells were cultured on a RGD-Cy3-I27G32C-A75C for 2 h. DTT at different concentrations (0, 0.25 mM, 2.5 mM, 5 mM, 12.5 mM, and 25 mM) was added to the chamber (t = 0) and the time lapse video of the fluorescence increase was recorded at the 300 ms exposure time. Next, single-exponential fits were used to determine rates of protein unfolding for each DTT concentration.
Data analysis and statistics
We first generated background subtracted fluorescent images to eliminate signals from quenched probes. Masks of tension signal from individual spread cells before drug treatment were then isolated using Fiji ImageJ (Image → Adjust → Threshold). Raw intensities of the same cells before and after drug treatment were isolated and measured using the masks. Percent inhibition by dividing the raw tension signals of an untreated cell to those of cells after drug incubation (Idrug/Iuntreated). For EC50 measurement, we normalized tension intensity after incubation of varying concentrations of drug to the raw tension intensity before treatment. EC50 was defined as the drug concentration that leads to half-maximal intensities of the tension signal.
All data were analyzed using built-in analysis methods in GraphPad Prism 6.0 (Graphpad Software. San Diego, CA) or Origin 9.0 (OriginLab, Northampton, MA). Two tailed Student’s t-test, Wilcoxon signed-rank test and Mann-Whitney test were used for statistical comparisons. P values were used to access the significance of the data, where *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. All data with error bars were presented as mean ± standard error of the mean (SEM).
Supplementary Material
Acknowledgments
K.S. acknowledges support from National Science Foundation (CAREER-1553344) and National Institutes of Health (R01GM124472). V.P.-Y.M. is a recipient of the National Cacner Institutes Predoctoral to Postdoctoral Fellow Transition Award (F99CA223074). This study was supported in part by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000454. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Kornelia Galior, Department of Chemistry, Emory University, Atlanta, GA 30322, USA
Victor Pui-Yan Ma, Department of Chemistry, Emory University, Atlanta, GA 30322, USA.
Dr. Yang Liu, Department of Chemistry, Emory University, Atlanta, GA 30322, USA
Hanquan Su, Department of Chemistry, Emory University, Atlanta, GA 30322, USA.
Nusaiba Baker, Emory University School of Medicine, Emory University, Atlanta, GA 30307, USA. Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332, USA.
Prof. Dr. Reynold Panettieri, Jr., Rutgers Institute for Translational Medicine and Science, Rutgers, The State University of New Jersey, New Brunswick, NJ 08901, USA
Dr. Cherry Wongtrakool, Emory University School of Medicine, Emory University, Atlanta, GA 30307, USA. Atlanta Veterans Affairs Medical Center, Decatur, GA 30033, USA
Prof. Dr. Khalid Salaita, Department of Chemistry, Emory University, Atlanta, GA 30322, USA. Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332, USA. Winship Cancer Institute, Emory University, Atlanta, GA 30322, USA
References
- 1.a) McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Dev Cell. 2004;6:483. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]; b) Judokusumo E, Tabdanov E, Kumari S, Dustin ML, Kam LC. Biophys J. 2012;102:L5. doi: 10.1016/j.bpj.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Brugues A, Anon E, Conte V, Veldhuis JH, Gupta M, Colombelli J, Munoz JJ, Brodland GW, Ladoux B, Trepat X. Nat phys. 2014;10:683. doi: 10.1038/nphys3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Janmey PA, Miller RT. J Cell Sci. 2011;124:9. doi: 10.1242/jcs.071001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Konen J, Summerbell E, Dwivedi B, Galior K, Hou Y, Rusnak L, Chen A, Saltz J, Zhou W, Boise LH, Vertino P, Cooper L, Salaita K, Kowalski J, Marcus AI. Nature Commun. 2017;8:15078. doi: 10.1038/ncomms15078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Park CY, Zhou EH, Tambe D, Chen B, Lavoie T, Dowell M, Simeonov A, Maloney DJ, Marinkovic A, Tschumperlin DJ, Burger S, Frykenberg M, Butler JP, Stamer WD, Johnson M, Solway J, Fredberg JJ, Krishnan R. Integr Biol. 2015;7:1318. doi: 10.1039/c5ib00054h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Coppola S, Carnevale I, Danen EHJ, Peters GJ, Schmidt T, Assaraf YG, Giovannetti E. Drug Resist Updat. 2017;31:43. doi: 10.1016/j.drup.2017.07.001. [DOI] [PubMed] [Google Scholar]; c) Hu J, Gondarenko AA, Dang AP, Bashour KT, O’Connor RS, Lee S, Liapis A, Ghassemi S, Milone MC, Sheetz MP, Dustin ML, Kam LC, Hone JC. Nano Lett. 2016;16:2198. doi: 10.1021/acs.nanolett.5b04364. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Krishnan R, Park JA, Seow CY, Lee PVS, Stewart AG. Trends Pharmacol Sci. 2016;37:87. doi: 10.1016/j.tips.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Huang C, Holfeld J, Schaden W, Orgill D, Ogawa R. Trends Mol Med. 2013;19:555. doi: 10.1016/j.molmed.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Effron MB, Bhatnagar GM, Spurgeon HA, Ruaño-Arroyo G, Lakatta EG. Circ Res. 1987;60:238. doi: 10.1161/01.res.60.2.238. [DOI] [PubMed] [Google Scholar]
- 5.Grosberg A, Nesmith AP, Goss JA, Brigham MD, McCain ML, Parker KK. J Pharmacol Toxicol Methods. 2012;65:126. doi: 10.1016/j.vascn.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Wahlquist C, Jeong D, Rojas-Munoz A, Kho C, Lee A, Mitsuyama S, van Mil A, Jin Park W, Sluijter JPG, Doevendans PAF, Hajjar RJ, Mercola M. Nature. 2014;508:531. doi: 10.1038/nature13073. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kijlstra Jan D, Hu D, Mittal N, Kausel E, van der Meer P, Garakani A, Domian Ibrahim J. Stem Cell Rep. 2015;5:1226. doi: 10.1016/j.stemcr.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith BA, Tolloczko B, Martin JG, Grütter P. Biophys J. 2005;88:2994. doi: 10.1529/biophysj.104.046649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang H, Kamm RD, So PTC, Lee RT. Hypertension. 2001;38:1158. doi: 10.1161/hy1101.096456. [DOI] [PubMed] [Google Scholar]
- 9.a) An SS, Laudadio RE, Lai J, Rogers RA, Fredberg JJ. Am J Physiol Cell Physiol. 2002;283:C792. doi: 10.1152/ajpcell.00425.2001. [DOI] [PubMed] [Google Scholar]; b) An SS, Askovich PS, Zarembinski TI, Ahn K, Peltier JM, von Rechenberg M, Sahasrabudhe S, Fredberg JJ. Respir Res. 2011;12:8. doi: 10.1186/1465-9921-12-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Tse HTK, Gossett DR, Moon YS, Masaeli M, Sohsman M, Ying Y, Mislick K, Adams RP, Rao J, Di Carlo D. Sci Transl Med. 2013;5:212ra163. doi: 10.1126/scitranslmed.3006559. [DOI] [PubMed] [Google Scholar]; b) Otto O, Rosendahl P, Mietke A, Golfier S, Herold C, Klaue D, Girardo S, Pagliara S, Ekpenyong A, Jacobi A, Wobus M, Töpfner N, Keyser UF, Mansfeld J, Fischer-Friedrich E, Guck J. Nat Methods. 2015;12:199. doi: 10.1038/nmeth.3281. [DOI] [PubMed] [Google Scholar]
- 11.a) Oliver T, Jacobson K, Dembo M. Cell Motil Cytoskeleton. 1995;31:225. doi: 10.1002/cm.970310306. [DOI] [PubMed] [Google Scholar]; b) Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Proc Natl Acad Sci USA. 2003;100:1484. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Plotnikov SV, Sabass B, Schwarz US, Waterman CM. In: Methods in Cell Biology. Waters JC, Wittman T, editors. Vol. 123. Academic Press; 2014. p. 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Bashour KT, Gondarenko A, Chen H, Shen K, Liu X, Huse M, Hone JC, Kam LC. Proc Natl Acad Sci USA. 2014;111:2241. doi: 10.1073/pnas.1315606111. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Basu R, Whitlock BM, Husson J, Le Floc’h A, Jin W, Oyler-Yaniv A, Dotiwala F, Giannone G, Hivroz C, Biais N, Lieberman J, Kam LC, Huse M. Cell. 2016;165:100. doi: 10.1016/j.cell.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Park CY, Zhou EH, Tambe D, Chen B, Lavoie T, Dowell M, Simeonov A, Maloney DJ, Marinkovic A, Tschumperlin DJ, Burger S, Frykenberg M, Butler JP, Stamer WD, Johnson M, Solway J, Fredberg JJ, Krishnan R. Integr Biol. 2015;7:1318. doi: 10.1039/c5ib00054h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Park C, Burger S, Watts E, Frykenberg M, Tambe D, Zhou E, Krishinan R, Marinkovic A, Tschumperlin D, Butler J, Solway J, Fredberg J. FASEB J. 2016;30:1013.4. [Google Scholar]; c) Yoshie H, Koushki N, Kaviani R, Rajendran K, Dang Q, Husain A, Yao S, Li C, Sullivan JK, Saint-Geniez M, Krishnan R, Ehrlicher A. bioRxiv. 2017 doi: 10.1101/162206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Stabley DR, Jurchenko C, Marshall SS, Salaita KS. Nat Methods. 2012;9:64. doi: 10.1038/nmeth.1747. [DOI] [PubMed] [Google Scholar]; b) Jurchenko C, Salaita K. Mol Cell Biol. 2015;35:2570. doi: 10.1128/MCB.00195-15. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu Y, Galior K, Ma VPY, Salaita K. Acc Chem Res. 2017;50:2915. doi: 10.1021/acs.accounts.7b00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Zhang Y, Ge C, Zhu C, Salaita K. Nature Commun. 2014;5:5167. doi: 10.1038/ncomms6167. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blakely BL, Dumelin CE, Trappmann B, McGregor LM, Choi CK, Anthony PC, Duesterberg VK, Baker BM, Block SM, Liu DR, Chen CS. Nat Methods. 2014;11:1229. doi: 10.1038/nmeth.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu Y, Blanchfield L, Ma VPY, Andargachew R, Galior K, Liu Z, Evavold B, Salaita K. Proc Natl Acad Sci USA. 2016;113:5610. doi: 10.1073/pnas.1600163113. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ma VPY, Liu Y, Blanchfield L, Su H, Evavold BD, Salaita K. Nano Lett. 2016;16:4552. doi: 10.1021/acs.nanolett.6b01817. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ma VPY, Liu Y, Yehl K, Galior K, Zhang Y, Salaita K. Angewandte Chemie International Edition. 2016;55:5488. doi: 10.1002/anie.201600351. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zhang Y, Qiu Y, Blanchard AT, Chang Y, Brockman JM, Ma VPY, Lam WA, Salaita K. Proc Natl Acad Sci USA. 2017 doi: 10.1073/pnas.1710828115. [DOI] [Google Scholar]; g) Brockman JM, Blanchard AT, Ma VPY, Derricotte WD, Zhang Y, Fay ME, Lam WA, Evangelista FA, Mattheyses AL, Salaita K. Nat Methods. 2018;15:115. doi: 10.1038/nmeth.4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang Y, Liu Z, Zhang Y, Galior K, Yang J, Salaita K. J Am Chem Soc. 2016;138:2901. doi: 10.1021/jacs.5b11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galior K, Liu Y, Yehl K, Vivek S, Salaita K. Nano Lett. 2016;16:341. doi: 10.1021/acs.nanolett.5b03888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Humphrey JD, Dufresne ER, Schwartz MA. Nat Rev Mol Cell Biol. 2014;15:802. doi: 10.1038/nrm3896. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yan J, Yao M, Goult BT, Sheetz MP. Cell Mol Bioeng. 2015;8:151. doi: 10.1007/s12195-014-0364-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Freyer AM, Johnson SR, Hall IP. Am J Respir Cell Mol Biol. 2001;25:569. doi: 10.1165/ajrcmb.25.5.4605. [DOI] [PubMed] [Google Scholar]; b) Nguyen TT, Ward JP, Hirst SJ. Am J Respir Crit Care Med. 2005;171:217. doi: 10.1164/rccm.200408-1046OC. [DOI] [PubMed] [Google Scholar]; c) Peng Q, Lai D, Nguyen TT, Chan V, Matsuda T, Hirst SJ. J Immunol. 2005;174:2258. doi: 10.4049/jimmunol.174.4.2258. [DOI] [PubMed] [Google Scholar]; d) Tatler AL, John AE, Jolly L, Habgood A, Porte J, Brightling C, Knox AJ, Pang L, Sheppard D, Huang X, Jenkins G. J Immunol. 2011;187:6094. doi: 10.4049/jimmunol.1003507. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chen C, Kudo M, Rutaganira F, Takano H, Lee C, Atakilit A, Robinett KS, Uede T, Wolters PJ, Shokat KM, Huang X, Sheppard D. J Clin Invest. 2012;122:2916. doi: 10.1172/JCI60387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.An SS, Bai TR, Bates JH, Black JL, Brown RH, Brusasco V, Chitano P, Deng L, Dowell M, Eidelman DH, Fabry B, Fairbank NJ, Ford LE, Fredberg JJ, Gerthoffer WT, Gilbert SH, Gosens R, Gunst SJ, Halayko AJ, Ingram RH, Irvin CG, James AL, Janssen LJ, King GG, Knight DA, Lauzon AM, Lakser OJ, Ludwig MS, Lutchen KR, Maksym GN, Martin JG, Mauad T, McParland BE, Mijailovich SM, Mitchell HW, Mitchell RW, Mitzner W, Murphy TM, Pare PD, Pellegrino R, Sanderson MJ, Schellenberg RR, Seow CY, Silveira PS, Smith PG, Solway J, Stephens NL, Sterk PJ, Stewart AG, Tang DD, Tepper RS, Tran T, Wang L. Eur Respir J. 2007;29:834. doi: 10.1183/09031936.00112606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cazzola M, Page CP, Calzetta L, Matera MG. Pharmacol Rev. 2012;64:450. doi: 10.1124/pr.111.004580. [DOI] [PubMed] [Google Scholar]
- 22.Ma X, Cheng Z, Kong H, Wang Y, Unruh H, Stephens NL, Laviolette M. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1181. doi: 10.1152/ajplung.00389.2001. [DOI] [PubMed] [Google Scholar]
- 23.a) Labeit S, Kolmerer B, Linke WA. Circ Res. 1997;80:290. doi: 10.1161/01.res.80.2.290. [DOI] [PubMed] [Google Scholar]; b) Lu H, Isralewitz B, Krammer A, Vogel V, Schulten K. Biophys J. 1998;75:662. doi: 10.1016/S0006-3495(98)77556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Carrion-Vazquez M, Marszalek PE, Oberhauser AF, Fernandez JM. Proc Natl Acad Sci USA. 1999;96:11288. doi: 10.1073/pnas.96.20.11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Liu Y, Yehl K, Narui Y, Salaita K. J Am Chem Soc. 2013;135:5320. doi: 10.1021/ja401494e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu Y, Medda R, Liu Z, Galior K, Yehl K, Spatz JP, Cavalcanti-Adam EA, Salaita K. Nano Lett. 2014;14:5539. doi: 10.1021/nl501912g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a) Leguillette R, Laviolette M, Bergeron C, Zitouni N, Kogut P, Solway J, Kachmar L, Hamid Q, Lauzon AM. Am J Respir Crit Care Med. 2009;179:194. doi: 10.1164/rccm.200609-1367OC. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mahn K, Hirst SJ, Ying S, Holt MR, Lavender P, Ojo OO, Siew L, Simcock DE, McVicker CG, Kanabar V, Snetkov VA, O’Connor BJ, Karner C, Cousins DJ, Macedo P, Chung KF, Corrigan CJ, Ward JP, Lee TH. Proc Natl Acad Sci USA. 2009;106:10775. doi: 10.1073/pnas.0902295106. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mahn K, Ojo OO, Chadwick G, Aaronson PI, Ward JP, Lee TH. Thorax. 2010;65:547. doi: 10.1136/thx.2009.129296. [DOI] [PubMed] [Google Scholar]
- 26.a) Carroll N, Elliot J, Morton A, James A. Am Rev Respir Dis. 1993;147:405. doi: 10.1164/ajrccm/147.2.405. [DOI] [PubMed] [Google Scholar]; b) Trian T, Benard G, Begueret H, Rossignol R, Girodet PO, Ghosh D, Ousova O, Vernejoux JM, Marthan R, Tunon-de-Lara JM, Berger P. J Exp Med. 2007;204:3173. doi: 10.1084/jem.20070956. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bara I, Ozier A, Tunon de Lara JM, Marthan R, Berger P. Eur Respir J. 2010;36:1174. doi: 10.1183/09031936.00019810. [DOI] [PubMed] [Google Scholar]
- 27.Ma X, Cheng Z, Kong H, Wang Y, Unruh H, Stephens NL, Laviolette M. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1181. doi: 10.1152/ajplung.00389.2001. [DOI] [PubMed] [Google Scholar]
- 28.a) Takayanagi I, Kizawa Y, Sone H. Gen Pharmacol. 1984;15:349. doi: 10.1016/0306-3623(84)90013-2. [DOI] [PubMed] [Google Scholar]; b) Streck E, Jorres RA, Huber RM, Bergner A. J Investig Allergol Clin Immunol. 2010;20:324. [PubMed] [Google Scholar]; c) Wongtrakool C, Grooms K, Bijli KM, Crothers K, Fitzpatrick AM, Hart CM. PLoS One. 2014;9:e109602. doi: 10.1371/journal.pone.0109602. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jiang Y, Dai A, Zhou Y, Peng G, Hu G, Li B, Sham JS, Ran P. Cell Physiol Biochem. 2014;33:389. doi: 10.1159/000356678. [DOI] [PubMed] [Google Scholar]; e) He F, Li B, Zhao Z, Zhou Y, Hu G, Zou W, Hong W, Zou Y, Jiang C, Zhao D, Ran P. PLoS One. 2014;9:e93508. doi: 10.1371/journal.pone.0093508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a) Wang Y, McNiven MA. J Cell Biol. 2012;196:375. doi: 10.1083/jcb.201105153. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gu Z, Fonseca V, Hai CM. Vascul Pharmacol. 2013;58:87. doi: 10.1016/j.vph.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li ZZ, Guo ZZ, Zhang Z, Cao QA, Zhu YJ, Yao HL, Wu LL, Dai QY. Mol Cell Biochem. 2015;399:49. doi: 10.1007/s11010-014-2231-z. [DOI] [PubMed] [Google Scholar]
- 30.Sundaram A, Chen C, Khalifeh-Soltani A, Atakilit A, Ren X, Qiu W, Jo H, DeGrado W, Huang X, Sheppard D. J Clin Invest. 2017;127:365. doi: 10.1172/JCI88555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J, Xiong W, Baca-Regen L, Nagase H, Baxter BT. J Vasc Surg. 2003;38:1376. doi: 10.1016/s0741-5214(03)01022-x. [DOI] [PubMed] [Google Scholar]
- 32.Dom AM, Buckley AW, Brown KC, Egleton RD, Marcelo AJ, Proper NA, Weller DE, Shah YH, Lau JK, Dasgupta P. Invest Ophthalmol Vis Sci. 2011;52:4428. doi: 10.1167/iovs.10-5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hallsworth MP, Twort CH, Lee TH, Hirst SJ. Br J Pharmacol. 2001;132:729. doi: 10.1038/sj.bjp.0703866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott MG, Swan C, Jobson TM, Rees S, Hall IP. Br J Pharmacol. 1999;128:721. doi: 10.1038/sj.bjp.0702829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ward JE, Harris T, Bamford T, Mast A, Pain MCF, Robertson C, Smallwood D, Tran T, Wilson J, Stewart AG. Eur Respir J. 2008;32:362. doi: 10.1183/09031936.00119307. [DOI] [PubMed] [Google Scholar]
- 36.Vargas A, Peltier A, Dubé J, Lefebvre-Lavoie J, Moulin V, Goulet F, Lavoie J-P. Am J Vet Res. 2017;78:359. doi: 10.2460/ajvr.78.3.359. [DOI] [PubMed] [Google Scholar]
- 37.Young TS, Ahmad I, Yin JA, Schultz PG. J Mol Biol. 2010;395:361. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.