

Abstract
Objective
Mucinous ovarian cancer (MOC) is a rare type of epithelial ovarian cancer resistant to standard chemotherapy regimens. We sought to characterize the repertoire of somatic mutations in MOCs and to define the contribution of massively parallel sequencing to the classification of tumors diagnosed as primary MOCs.
Methods
Following gynecologic pathology and chart review, DNA samples obtained from primary MOCs and matched normal tissues/blood were subjected to whole-exome (n=9) or massively parallel sequencing targeting 341 cancer genes (n=15). Immunohistochemical analysis of estrogen receptor, progesterone receptor, PTEN, ARID1A/BAF250a, and the DNA mismatch (MMR) proteins MSH6 and PMS2 was performed for all cases. Mutational frequencies of MOCs were compared to those of high-grade serous ovarian cancers (HGSOCs) and mucinous tumors from other sites.
Results
MOCs were heterogeneous at the genetic level, frequently harboring TP53 (75%), KRAS (71%) mutations and/or CDKN2A/B homozygous deletions/mutations (33%). Although established criteria for diagnosis were employed, four cases harbored mutational and immunohistochemical profiles similar to those of endometrioid carcinomas, and one case for colorectal or endometrioid carcinoma. Significant differences in the frequencies of KRAS, TP53, CDKN2A, FBXW7, PIK3CA and/or APC mutations between the confirmed primary MOCs (n=19) and HGSOCs, mucinous gastric and/or mucinous colorectal carcinomas were found, whereas no differences in the 341 genes studied between MOCs and mucinous pancreatic carcinomas were identified.
Conclusions
Our findings suggest that the assessment of mutations affecting TP53, KRAS, PIK3CA, ARID1A and POLE, and DNA MMR protein expression may be used to further aid the diagnosis and treatment decision-making of primary MOC.
Keywords: mucinous ovarian cancer, massively parallel sequencing, immunohistochemistry, classification, diagnosis
1. INTRODUCTION
Mucinous adenocarcinoma of the ovary is a rare type of epithelial ovarian cancer, representing approximately 3-4% of all epithelial ovarian malignancies [1]. These tumors are distinct at the biological, clinical and genetic levels from the common high-grade serous ovarian cancers (HGSOCs). Contrary to HGSOCs, mucinous ovarian cancers (MOCs) frequently present with early stage ovarian-confined disease associated with an overall favorable prognosis (stage I, 5 year survival rate 91%) [2,3]. In advanced stage disease, however, MOC is associated with poor outcome and overall survival rates lower than those reported for advanced stage HGSOC (5-year survival 11% MOC vs 26% HGSOC) [2,4–6]. MOCs are less sensitive to standard platinum/taxane chemotherapy regimens than HGSOCs, which may contribute to the observed poor outcome when diagnosed in advanced stages [4,7]. Previous studies employing targeted and whole-exome massively parallel sequencing have revealed that primary MOCs are heterogeneous at the genomic level, with TP53 (52-57%), KRAS (45-65%), BRAF (23%), ERBB2 (23%) and CDKN2A (60%) being the genes most commonly altered [8,9], and that these tumors may be distinct from other ovarian cancer subtypes at the genomic level.
The diagnosis of primary MOC and the differentiation from metastatic mucinous tumors originating in extraovarian primary sites, most commonly from the colorectum, especially appendiceal, is challenging [10,11]. Pathology review of 44 presumed MOCs as part of a prospective Gynecological Oncology Group phase 3 trial led to the reclassification of 61% of cases as ovarian metastases from tumors originating in other primary sites [6]. Therefore, the integration of clinical history and pathologic features has been shown to be essential for the diagnosis of primary MOC and to discriminate these tumors from their metastatic mimics [6,10]. Laterality and size provide important information as primary MOCs typically present as unilateral tumors measuring >10cm as compared to metastatic lesions [10]. In addition, immunohistochemistry has been used as an ancillary diagnostic test for the differentiation between MOCs and adenocarcinomas from other anatomical sites, in particular those demonstrating lower intestinal differentiation [11–16]. In advanced stage mucinous ovarian disease, upper and lower endoscopy, CT or PET imaging and/or serum tumor markers are warranted to rule out the presence of an extra-ovarian primary cancer (NCCN guidelines, https://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf).
In this study, we sought to characterize the repertoire of somatic genetic alterations focusing on key cancer genes in MOCs and to define the contribution of massively parallel sequencing to the classification of tumors diagnosed as MOCs based on current clinicopathologic criteria. To achieve these aims, we subjected a series of 24 MOCs to whole-exome sequencing (n=9) or massively parallel sequencing targeting all exons of 341 key cancer genes (MSK-IMPACT; n=15).
2. MATERIAL AND METHODS
Case selection
All primary MOCs diagnosed between July 2001–July 2013 with available tissue slides and blocks were retrieved from the files of the Department of Pathology at Memorial Sloan Kettering Cancer Center (MSKCC). This study was approved by the Institutional Review Board (IRB) of MSKCC, and patient consent was obtained where appropriate. Representative sections of each case diagnosed as primary MOC were re-reviewed by gynecologic pathologists (YRH, RAS, DD), and clinical information, including age, stage, laterality, therapy, endoscopy and follow-up, was obtained from the medical records. Invasive MOC was defined as the presence of confluent tumor cells with intracytoplasmic mucin, measuring >10mm2 and at least 5mm in one linear dimension [3]. Unilateral tumor size ≥10cm and expression of CK7 with or without CK20 expression were used to confirm the diagnosis of primary MOC along with normal upper and lower endoscopy and prolonged clinical follow-up without evidence of gastrointestinal involvement in patients with advanced stage disease, and the absence of the development of a second primary tumor during follow-up; in addition, cases with a unilateral tumor size <10cm or bilateral disease with a tumor size ≥10cm were acceptable if PAX8 was expressed [10,12,13,15,17]. The ovarian origin of the stage I carcinomas as opposed to a gastrointestinal carcinoma metastatic to the ovary was further confirmed by the prolonged survival, as 71% (12/17) of the stage I tumors had no evidence of disease at a median follow-up of 82 months (range 41-185 months), 12% (2/17) were alive with disease (40 and 326 months follow-up), and 18% (3/17) died of unknown cause (62, 98 and 102 months follow-up; Table 1). Following this review, 24 primary MOCs (15 formalin-fixed paraffin-embedded (FFPE), 9 fresh frozen) were included in this study.
Table 1.
Clinicopathologic information of the 24 primary mucinous ovarian cancers included in this study.
| ID | Age at diagnosis (years) |
FIGO Stage |
Tumor size (cm) |
Laterality | CK7 | CK20 | PAX8 | Adjuvant chemo- therapy |
Follow-up (months, status) |
Sequen cing type |
|---|---|---|---|---|---|---|---|---|---|---|
| MOC01 | 38 | IA | 13 | Unilateral (right) | Strong positive | Positive | Positive | No | 150, NED | WES |
| MOC04 | 68 | IIIA | 20 | Unilateral (right) | Strong positive | Positive | Focal positive | Yes | 4.2, DOD | IMPACT |
| MOC05 | 76 | IA | 15 | Unilateral (left) | Strong positive | Positive | Negative | Yes | 101.5, DUC | WES |
| MOC09 | 64 | IA | 12 | Unilateral (right) | Strong positive | Positive | Positive | No | 185.4, NED | WES |
| MOC10 | 59 | IIC | 11 | Unilateral (left) | Strong positive | Negative | Positive | Yes | 91.3, NED | WES |
| MOC17 | 77 | IA | 20 | Unilateral (left) | Strong positive | Negative | Focal positive | No | 98.1, DUC | IMPACT |
| MOC19* | 62 | IIC | 9 | Unilateral (right) | Strong positive | Positive | Positive | Yes | 244.4, DUC | IMPACT |
| MOC21* | 38 | IC | 11 | Unilateral (NA) | Strong positive | Negative | Positive | Yes | 326.1, AWD | IMPACT |
| MOC25 | 20 | IA | 21 | Unilateral (left) | Strong positive | Positive | Focal positive | No | 83.7, NED | IMPACT |
| MOC30 | 59 | IC | 30 | Unilateral (right) | Strong positive | Positive | Focal positive | No | 133.6, NED | IMPACT |
| MOC31 | 37 | IA | 20 | Unilateral (right) | Strong positive | Positive | Focal positive | No | 53.3, NED | IMPACT |
| MOC32 | 60 | IIIC | 13, 3 | Bilateral | Strong positive | Negative | Positive | Yes | 96.6, DOD | WES |
| MOC37 | 46 | IC | 11 | Unilateral (left) | Strong positive | Negative | Focal positive | Yes | 117.6, NED | WES |
| MOC38 | 86 | IC | 7 | Unilateral (right) | Strong positive | Positive | Positive | No | 61.6, DUC | IMPACT |
| MOC46 | 46 | IC | 25 | Unilateral (right) | Strong positive | Positive | Positive | Yes | 90.8, NED | IMPACT |
| MOC48 | 78 | IA | 8 | Unilateral (left) | Strong positive | Focal positive | Positive | No | 76.7, NED | IMPACT |
| MOC51 | 70 | IA | 16 | Unilateral (left) | Strong positive | Focal positive | Positive | No | 41.3, NED | IMPACT |
| MOC52 | 45 | IA | 29 | Unilateral (right) | Strong positive | Negative | Focal positive | No | 79.5, NED | IMPACT |
| MOC59 | 54 | IIIC | 30 | Unilateral (right) | Negative | Strong positive | Negative | Yes | 35.0, AWD | WES |
| MOC60 | 37 | IA | 15 | Unilateral (right) | Strong positive | Negative | Positive | No | 54.3, NED | IMPACT |
| MOC62 | 53 | IA | 12 | Unilateral (left) | Strong positive | Focal positive | Negative | Yes | 61.0, NED | IMPACT |
| MOC67* | 59 | IIB | 12 | Unilateral (left) | Weak positive | Positive | Positive | Yes | 31.6, DOD | IMPACT |
| MOC72 | 50 | IA | 32 | Unilateral (right) | Strong positive | Focal positive | Positive | No | 39.8, AWD | IMPACT |
| MOC73 | 41 | IIIB | 11 | Unilateral (right) | Strong positive | Negative | Focal positive | No | 47.9, DOD | WES |
AWD, alive with disease; CK, cytokeratin; DOD, dead of disease; DUC, dead of unknown cause; NA, not available; NED, no evidence of disease; WES, whole-exome sequencing;
recurrences.
Immunohistochemistry
Immunohistochemistry (IHC) for CK7, CK20 and PAX8, was performed in the diagnostic work-up of the tumors, following previously validated protocols [15,18]. For CK7 and CK20 only cytoplasmic staining was considered positive, and for PAX8 only nuclear staining was considered positive [15,18]. After sequencing, additional immunohistochemical analysis was performed for estrogen receptor (ER) and progesterone receptor (PR), PTEN, ARID1A/BAF250a and the DNA mismatch repair proteins MSH6 and PMS2 using previously described protocol [19–22]. ER and PR expression was defined as positive when >1% of the tumor nuclei showed immunoreactivity, following the ASCO/CAP guidelines for breast cancer [23]. Loss of MSH6, PMS2 and ARID1A expression was defined as complete absence of protein expression in unequivocal tumor cell nuclei [24]. Normal epithelium and stroma were used as internal controls for PTEN, MSH6, PMS2 and ARID1A expression; PTEN expression was defined as lost if tumor cells displayed no immunoreactivity or less than the internal control.
DNA extraction
Tumor sections were reviewed by two gynecologic pathologists (YRH, RAS) to ensure >20% neoplastic cells. Matched normal DNA was extracted from peripheral blood lymphocytes or normal tissue sections (benign lymph node), confirmed to be devoid of any neoplastic cells. Genomic DNA from tumor- and matched normal samples was extracted using the DNeasy Blood & Tissue kit (Qiagen).
Whole-exome and targeted massively parallel sequencing
Tumor and matched normal DNA samples were subjected to whole-exome sequencing (n=9) or massively parallel sequencing (n=15) using the Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) assay targeting all exons, selected intronic and regulatory regions of 341 key cancer genes, as previously described [25,26]. Sequencing data were analyzed as previously described (Supplementary Methods) [24,26]. Cancer cell fractions of each mutation were inferred using ABSOLUTE (v1.0.6) [27], as previously described [24,26]. Mutational signatures were defined for cases with at least 20 somatic mutations using deconstructSigs [28].
Comparison with high-grade serous ovarian, colorectal, gastric and pancreatic carcinomas
The mutational frequencies of the 341 genes in our targeted sequencing panel of MOCs were compared to those of HGSOCs from The Cancer Genome Atlas (TCGA; n=316) [29], mucinous colorectal carcinomas (TCGA; n=32) [30], mucinous gastric adenocarcinomas (TCGA; n=18)[31], pancreatic adenocarcinomas from the International Cancer Genome Consortium (ICGC; mucinous cystadenocarcinoma/intraductal papillary mucinous neoplasm with invasion, n=11; pancreatic ductal adenocarcinoma, n=177) [32] and to MOCs described by Ryland et al (n=12) [9]. The whole-exome sequencing-derived mutational data of the mucinous colorectal carcinomas and mucinous gastric adenocarcinomas were retrieved from GDAC Firehose (https://gdac.broadinstitute.org/; Mutation Annotation File) and of the HGSOCs and pancreatic adenocarcinomas from cBioPortal (http://www.cbioportal.org/) [33]. We restricted the comparison to the 341 genes targeted by our sequencing panel. Comparisons were performed using Fisher’s exact tests, and p-values <0.01 were deemed statistically significant.
3. RESULTS
The repertoire somatic mutations and copy number alterations of mucinous ovarian cancers
After secondary specialist gynecologic pathology and chart review, a final diagnosis of primary MOC was rendered in 24 cases (Table 1, Fig. 1). Whole-exome sequencing was performed at a median depth of coverage of 127x (range 92x-141x) for tumor and 119x (81x-156x) for normal samples, respectively, and targeted massively parallel sequencing of 266x (range 27x-620x) and 180x (range 27x-485x) for tumor and normal samples, respectively (Supplementary Table S1). When focusing on the 341 cancer-related genes, the 24 MOCs studied here harbored a median of 3.5 (range 1-189) non-synonymous somatic mutations; the mutational burden affecting these 341 genes that was significantly higher than that of HGSOCs (2, range 0-9 non-synonymous mutations; p<0.001, Mann-Whitney U test) but significantly lower than that of mucinous colorectal carcinomas (12, range 4-206 non-synonymous mutations; p<0.001, Mann-Whitney U test) from the TCGA datasets [29,30]. One of the MOCs analyzed here (MOC62) harbored a pathogenic POLE A456P exonuclease domain mutation, and a high mutational burden with 189 non-synonymous somatic mutations in the 341 genes tested (Supplementary Table S2).
Figure 1. Histologic features of primary mucinous ovarian carcinomas.
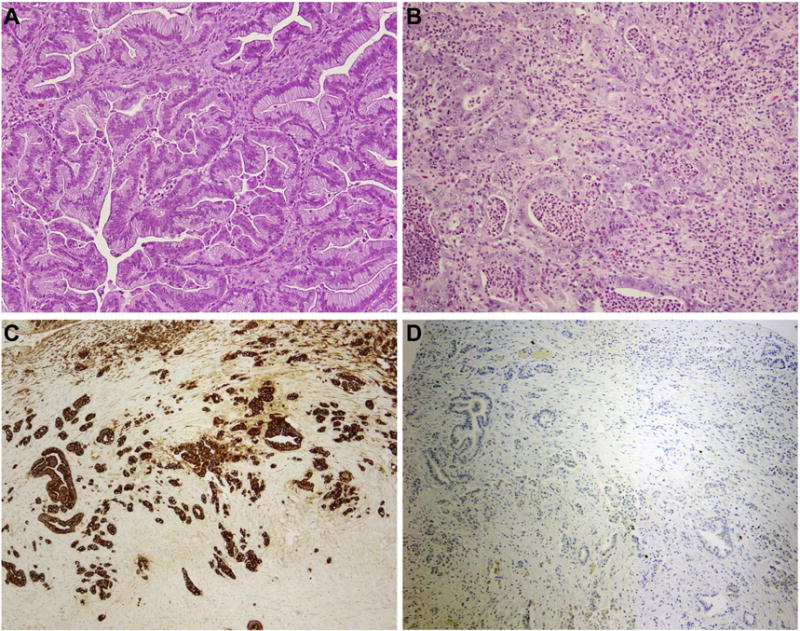
Representative hematoxylin and eosin stained sections of (A) a mucinous adenocarcinoma with an expansile pattern of growth and (B) a poorly differentiated mucinous adenocarcinoma with destructive stromal invasion. (C) Poorly differentiated mucinous adenocarcinoma showing strong CK7 expression. (D) Poorly differentiated mucinous adenocarcinoma displaying lack of CK20 expression. Magnification 400x.
The genes most frequently affected by somatic mutations were TP53 (18/24; 75%) and KRAS (17/24; 71%), which co-occurred in 13 MOCs (54%; Fig. 2), consistent with previously reported results [8,9,34]. Of the 18 TP53 mutations, 14 were found to be clonal (i.e. to be present in all cancer cells within a sample), 14 affected TP53 hotspot codons (58%), and 12 (50%) TP53 mutations displayed loss of heterozygosity (LOH) of the wild-type allele. All KRAS mutations affected the hotspot codons G12 (13/17), G13 (1/17) or Q61 (3/17), and all but three KRAS mutation were clonal (Fig. 2). Additional recurrent somatic mutations found in these tumors were SETD2 (4/24), GNAS (3/24) and ERBB3 (3/24), amongst others.
Figure 2. Recurrent non-synonymous somatic genetic alterations detected by massively parallel sequencing in primary mucinous ovarian cancers.

(A) Recurrent (n≥2) non-synonymous somatic mutations (top) and amplifications and homozygous deletions of interest (bottom) identified in 24 tumors initially diagnosed as primary mucinous ovarian cancer in 341 cancer-related genes. Mutation types, gene copy number alterations and sequencing type are color-coded according to the legend. Loss of heterozygosity of the wild-type allele in association with a somatic mutation is depicted by a diagonal bar. EDM, exonuclease domain; Indel, small insertion/deletion; Seq, sequencing; SNV, single nucleotide variant. (B) Cancer cell fractions of non-synonymous somatic mutations as defined by ABSOLUTE [27] in tumors initially diagnosed as primary mucinous ovarian cancer. Cancer cell fractions and sequencing type are color-coded according to the legend, and clonal mutations are depicted by an orange box. Loss of heterozygosity of the wild-type allele in association with a somatic mutation is depicted by a diagonal bar. Seq, sequencing.
MOCs displayed a heterogeneous pattern of copy number alterations, with 25% (6/24) cases harboring CDKN2A/B homozygous deletions (Fig. 2; Supplementary Fig. S1). Of note, two cases lacking CDKN2A/B homozygous deletions had a CDKN2A hotspot mutations coupled with LOH of the wild-type allele (MOC1) or a CDKN2A in-frame indel (MOC67). In addition, single cases harbored amplification of ERBB2 (MOC05), MYC (MOC04) or AKT2 (MOC25), or homozygous deletions of RB1 (MOC31) or SMAD4 (MOC67, Fig. 2; Supplementary Fig. S1).
Mutational signature analysis for the cases with at least 20 somatic mutations (n=11) revealed that all but two MOCs displayed a dominant signature 1 associated with aging (Fig. 3A) [35]. We further noted that of the nine MOCs with a signature 1, seven displayed an underlying mutational signature 3 associated with defective homologous recombination DNA repair (Fig. 3A) [35]. MOC62, the case with a pathogenic POLE A456P exonuclease domain mutation, had a POLE mutational signature (signature 10) and MOC38 a mutational signature associated with defective DNA MMR (mutational signature 6; Fig. 3B).
Figure 3. Mutational signatures in mucinous ovarian cancers.
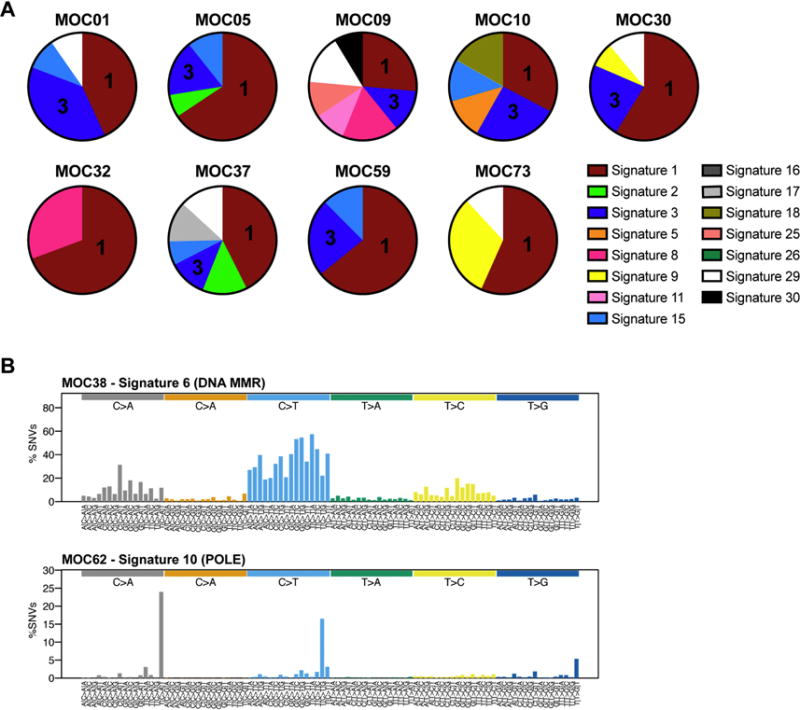
(A) Mutational signatures of all somatic single nucleotide variants (SNVs) identified in the nine mucinous ovarian cancers subjected to whole-exome sequencing using deconstructSigs [28]. Signature 1 associated with aging is the dominant signature in the MOCs studied. Signatures are color-coded according to the legend, including signature 1 (aging), signature 2 (APOBEC), signature 3 (defective homologous recombination DNA repair), and signatures 15 and 26 (defective DNA mismatch repair). (B) Mutational signatures of all SNVs in MOC38 and MOC62 subjected to MSK-IMPACT sequencing, displayed according to the 96 substitution classification defined by the substitution classes (C>A, C>G, C>T, T>A, T>C, and T>G bins), the 5’ and 3’ sequence context, normalized to the trinucleotide frequency of the human genome. MMR, mismatch repair.
Re-classification of presumed primary MOCs using ancillary markers
Whilst the majority of MOCs included in this study harbored alterations affecting the most common drivers of these lesions (i.e. TP53, KRAS and/or CDKN2A), we did identify six MOCs either lacking TP53, KRAS or CDKN2A mutations/homozygous deletions and/or harboring mutations in genes reported to be commonly mutated in endometrioid ovarian or endometrioid endometrial cancers [36,37], including PIK3CA or CTNNB1 hotspot mutations and/or ARID1A truncating/frameshift mutations (i.e. MOC10, MOC17, MOC19, MOC38, MOC46, MOC62; Fig. 2). To define whether these MOCs diagnosed upon re-review were truly primary MOCs, we performed a detailed analysis of the mutational profiles of these six tumors, and subjected them to immunohistochemical analysis using ER, PR, DNA MMR proteins, PTEN and ARID1A.
MOC46, which harbored KRAS and CTNNB1 hotspot mutations, was ER- and PR-negative and retained DNA MMR, PTEN and ARID1A expression; these features are consistent with a diagnosis of primary MOC. MOC62, however, a 12cm unilateral ER-negative, PR-negative, PAX8-negative tumor, harbored a POLE exonuclease domain mutation, a POLE mutational signature (signature 10) and also one truncating hotspot (p.R1114*) and two missense mutations affecting the tumor suppressor gene APC (Table 1; Figs. 2 and 3; Supplementary Table S2), suggesting that this tumor is unlikely to constitute a primary MOC. The differential diagnosis includes a primary ovarian carcinoma that lacks PAX8 expression or a metastasis from an occult extraovarian carcinoma (e.g. mucinous carcinoma of colorectal origin), however the patient did not develop an extraovarian carcinoma during surveillance following oophorectomy, and was alive without evidence of disease at 61 months follow-up (Table 1). Primary MOCs frequently lack PAX8 expression [18], as do many adenocarcinomas that arise in teratomas. Rare endometrioid carcinomas with or without mucinous differentiation may also, on occasion, be PAX8-negative.
Four MOCs displayed features consistent with a diagnosis of an endometrioid ovarian carcinoma with mucinous differentiation: 1) MOC38, a 7cm unilateral tumor, which harbored 28 non-synonymous mutations in the 341 genes tested, including a TP53, PIK3CA hotspot and ARID1A frameshift mutation, and the mutational signature 6 associated with defective DNA MMR and microsatellite instability (Fig. 2), was found to be ER-positive and to lack ARID1A and MLH1/PMS2 protein expression. The uterine pathology of this case revealed a complex atypical hyperplasia, however no invasive component was found. 2) MOC17, a 20cm unilateral lesion, which lacked KRAS or TP53 mutations presented with a PIK3CA hotspot mutation, was ER- and PR-positive and showed PTEN loss of expression (Fig. 2; Table 2). 3) MOC10, a 11cm unilateral tumor, harbored a KRAS mutation but also an ARID1A truncating mutation, a PTEN missense mutation and a PTEN frameshift insertion, expressed ER and PR, and showed equivocal PTEN expression by immunohistochemistry. 4) MOC19, which harbored a KRAS hotspot and ARID1A truncating mutation coupled with loss of ARID1A protein expression and was PR-positive.
Table 2.
Results of immunohistochemical analysis and mutation status of key genes to guide the diagnosis of primary mucinous ovarian cancer.
| ID | Immunohistochemical markers | Mutation status | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ER | PR | DNA MMR | PTEN | ARID1A | TP53 | KRAS | PIK3CA | ARID1A | POLE EDM | |
| MOC01 | Negative | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
| MOC04 | Negative | Negative | Retained | Retained | Retained | MUT | WT | WT | WT | WT |
| MOC05 | Negative | Negative | Retained | Retained | Retained | MUT | WT | WT | WT | WT |
| MOC09 | Negative | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
| MOC10 | Positive | Positive | Retained | Equivocal | Retained | WT | MUT | WT | MUT | WT |
| MOC17 | Positive | Positive | Retained | Loss | Retained | WT | WT | MUT | WT | WT |
| MOC19 | Negative | Positive | Retained | Retained | Loss | WT | MUT | MUT | MUT | WT |
| MOC21 | Negative | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
| MOC25 | Negative | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
| MOC30 | Negative | Negative | Retained | Retained | Retained | WT | MUT | WT | WT | WT |
| MOC31 | Positive | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
| MOC32 | Negative | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
| MOC37 | Positive | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
| MOC38 | Positive | Negative | PMS2/MLH1 loss | Retained | Loss | MUT | WT | MUT | MUT | WT |
| MOC46 | Negative | Negative | Retained | Retained | Retained | WT | MUT | WT | WT | WT |
| MOC48 | Negative | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
| MOC51 | Negative | Negative | Retained | Retained | Retained | WT | MUT | WT | WT | WT |
| MOC52 | Negative | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
| MOC59 | Negative | Negative | Retained | Retained | Retained | MUT | WT | WT | WT | WT |
| MOC60 | Negative | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
| MOC62 | Negative | Negative | Retained | Retained | Loss | WT | WT | MUT | MUT | MUT |
| MOC67 | Negative | Negative | Retained | Retained | Retained | MUT | WT | WT | WT | WT |
| MOC72 | Positive | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
| MOC73 | Negative | Negative | Retained | Retained | Retained | MUT | MUT | WT | WT | WT |
EDM, exonuclease domain; ER, estrogen receptor; MMR, mismatch repair; MUT, mutant; PR, progesterone receptor; WT, wild-type.
Our findings indicate that in addition to tumor size, laterality, CK7, CK20, ER and PR expression, the repertoire of mutations affecting a small panel of genes, including TP53, KRAS, PIK3CA, ARID1A and POLE, coupled with the assessment of DNA MMR protein expression may provide useful information for the differential diagnosis of mucinous tumors affecting the ovary.
Comparison of the mutational repertoire of MOCs with mucinous carcinomas from other sites
We next compared the repertoire of somatic mutations affecting the 341 genes included in the MSK-IMPACT assay in the MOCs studied here and in HGSOCs and mucinous cancers from other sites. For this analysis, only the tumors with a mutational/immunohistochemical profile suggestive of bona fide primary MOC were included in the comparisons (i.e. MOC10, MOC17, MOC19, MOC38 and MOC62 were excluded), whose pattern of mutations affecting the 341 cancer-related genes studied here did not significantly differ from those of the invasive MOCs reported in Ryland et al [9] (Supplementary Table S3). Compared to HGSOCs (n=316; TCGA)[29], the 19 bona fide MOCs significantly more frequently harbored mutations affecting KRAS and CDKN2A (KRAS, 79.0% vs 0.6%, p<0.001; CDKN2A, 10.5% vs 0%, p=0.003, Fisher’s exact test; Fig. 4). In addition, MOCs significantly more frequently harbored TP53 and KRAS mutations than mucinous gastric carcinomas (n=18; TCGA)[31] (TP53, 89.5% vs 16.7%, p<0.001; KRAS, 79.0% vs 11.1%, p<0.001, Fisher’s exact test). Compared to mucinous colorectal cancers (n=32; TCGA)[30], MOCs more frequently had TP53 mutations (89.5% vs 28.1%, p<0.001) but less frequently APC, PIK3CA and FBXW7 mutations (APC, 0% vs 65.6%, p<0.001; PIK3CA and FBXW7, 0% vs 31.3%, p=0.008, Fisher’s exact test; Fig. 4). Also pancreatic ductal carcinomas (n=177; ICGC)[32] were found to less frequently harbor TP53 and ERBB2 mutations than MOCs (TP53, 55.4% vs 89.5%, p=0.006; ERBB2, 0% vs 10.5%, p=0.009, Fisher’s exact test), whilst no statistically significant differences in the mutational repertoire of the 341 genes examined were found between the bona fide primary MOCs studied here and pancreatic mucinous carcinomas (n=11; Fig. 4).
Figure 4. Comparison of the mutational profiles of mucinous ovarian cancers with high-grade serous ovarian cancers and mucinous carcinomas from other sites.
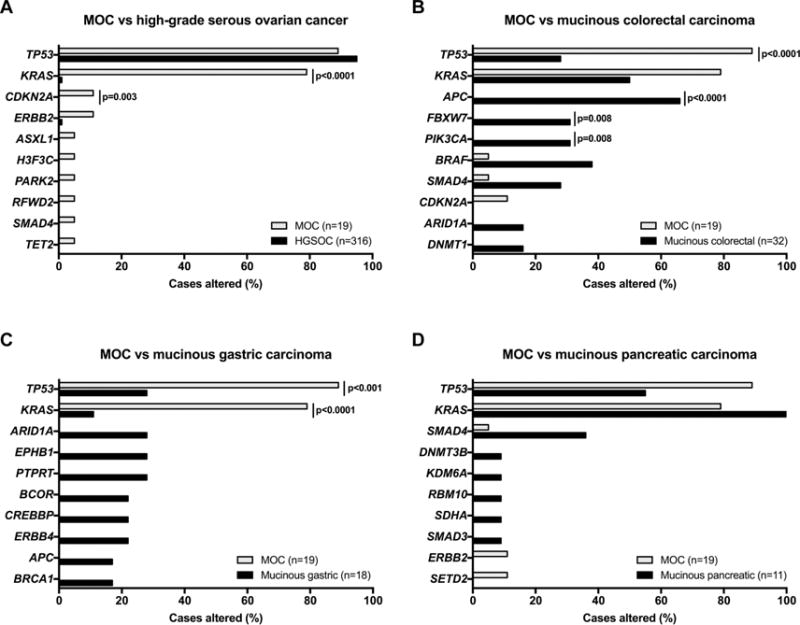
The repertoire of somatic mutations affecting the 341 genes included in the MSK-IMPACT assay in the bone fide mucinous ovarian cancers (MOCs) studied here (n=19) and in (A) high-grade serous ovarian cancers (HGSOCs) from The Cancer Genome Atlas (TCGA; n=316)[29], (B) mucinous colorectal carcinomas (TCGA; n=32)[30], (C) mucinous gastric carcinomas (TCGA; n=18)[31] and (D) mucinous pancreatic carcinomas from the International Cancer Genome Consortium (ICGC; n=11)[32]. The top most frequently mutated genes of a given cancer type and TP53 and KRAS are shown. Comparisons were performed using Fisher’s exact test and statistically significant p-values are shown.
4. DISCUSSION
Here we demonstrate that massively parallel sequencing resulted in the reclassification of a subset of tumors initially classified as MOCs based on clinicopathologic parameters. Furthermore, we show that MOCs display a heterogeneous repertoire of somatic genetic alterations, and confirm that these tumors are underpinned by TP53 mutations, KRAS mutations and/or CDKN2A alterations [8,9,34]. Similar frequencies of mutations affecting the 341 genes studied here were found between MOCs and pancreatic mucinous carcinomas, however at the mutational level, MOCs are an entity distinct from common-type epithelial ovarian cancers and mucinous colorectal or gastric cancers. This notion is further corroborated by the observation that HGSOCs and primary MOCs are characterized by the presence of recurrent TP53 mutations, however whilst HGSOCs lack KRAS mutations and harbor high numbers of copy number alterations, MOCs frequently harbor KRAS mutations and fewer gene copy number changes (Fig. 1, Supplementary Fig. S1). In addition, it has been reported that MOCs display risk factor profiles distinct from that of other ovarian cancers [38], providing evidence to suggest that MOCs have a different etiology than other types of ovarian cancer.
The heterogeneity of primary MOC is further reflected by the prevalence of ERBB2 amplifications. Primary MOCs have been reported to harbor ERBB2 amplification in up to 20% of cases [39], however only one of the MOCs studied here was ERBB2 amplified (4.2%). It should be noted that one other case harbored a hotspot ERBB2 p.R678Q mutation. Further studies are warranted to define whether ERBB2 altered primary MOCs constitute a subgroup of these lesions associated with distinct clinico-pathologic characteristics, stage at presentation, response to treatment and/or outcome. In addition, Ryland and colleagues reported that deleterious somatic mutations of the tumor suppressor gene RNF43 would play a role in a subset of primary MOCs [9], however none of the cases studied here harbored mutations or homozygous deletions affecting RNF43. Hence, the clinical and biological relevance of alterations affecting RNF43 in MOCs remains to be determined.
Despite pathologic and clinical re-review, 21% of the cases diagnosed as primary MOCs here are, based on their immunohistochemical and mutational profiles, more consistent with a diagnosis of endometrioid ovarian cancers with mucinous differentiation or a mucinous carcinoma of colorectal type. The clinical outcome of these patients, however, is consistent with the diagnosis of primary ovarian cancers. Given the challenges associated with the diagnosis of primary MOC and their distinction from other types of ovarian cancer, such endometrioid carcinoma with mucinous differentiation, and metastatic mucinous tumors of the colorectum, the implementation of a small gene panel and/or immunohistochemistry set of markers, including TP53/p53, KRAS, PIK3CA, ARID1A/ARID1A, and DNA MMR proteins, may be useful to guide the diagnosis of these lesions. Such an approach would not only ensure an accurate diagnosis, but also facilitate the delivery of precision medicine-based treatments for patients with MOCs. In fact, based upon current practice, primary MOCs deemed eligible for chemotherapy due either to advanced stage or high-grade histology are treated using a standard epithelial ovarian cancer regimen using carboplatin plus paclitaxel or oxaliplatin containing GI type regimens depending on the clinician preference (NCCN guidelines, https://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf). Importantly, however, by subjecting MOCs to a targeted capture sequencing approach, patients whose tumors display a hypermutator phenotype, such as the DNA MMR-deficient MOC38 or the POLE-mutant MOC62, would potentially be eligible for checkpoint blockage immunotherapy [40]. Also, we identified MOCs with ERBB2 amplification or mutations, which may benefit from anti-HER2 agents, and a case with EGFR amplification (MOC59).
Our study has several limitations. The sequencing coverage was low for a few FFPE samples, which may have affected the mutation and/or copy number analyses performed here. The frequency of cancer genes mutated in the MOCs studied here is, however, similar to that previously reported [9]. In addition, with the exception of the tumors harboring POLE exonuclease domain mutations or DNA MMR alterations, the number of somatic mutations in the MOCs subjected to targeted massively parallel sequencing (MSK-IMPACT) was too low to assess the mutational signatures in these lesions.
Despite these limitations, our data demonstrate that primary MOCs are heterogeneous at the genetic level and suggest that the assessment of mutations affecting TP53, KRAS, PIK3CA, ARID1A and POLE, and DNA MMR protein expression may be used in conjunction with current established criteria to guide the diagnosis and, potentially, the treatment of patients with primary MOCs. In addition, our genomic analysis of tumors diagnosed as primary MOCs based on current clinico-pathologic guidelines may in fact constitute endometrioid carcinomas with mucinous differentiation, given their repertoire of somatic genetic alterations and immunohistochemical profiles.
Supplementary Material
HIGHLIGHTS.
MOCs are heterogeneous at the mutational level
Mucinous ovarian cancers (MOCs) frequently harbor TP53 and KRAS mutations
The current pathologic criteria to diagnose MOCs may result in misclassifications
Mutation analysis of a small gene panel may help improve the accuracy of MOC diagnosis
Acknowledgments
Funding support: This work was funded in part by the Marie-Josée and Henry R. Kravis Center for Molecular Oncology, the National Cancer Institute Cancer Center Core Grant No. P30-CA008748, and Linda and David Yoon. The funders had no role in the design of the study, the collection, analysis or interpretation of the data, the writing of the manuscript or the decision to submit the manuscript for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
J.J.M. and B.A.S.: methodology, investigation, formal analysis, project administration, writing - original draft. R. K.: formal analysis (bioinformatics), software, data curation, investigation, visualization. N.O.: data curation, formal analysis, investigation, methodology. F.D.: data curation, formal analysis, investigation, methodology, software, visualization. N.A-R. and C.A.: project administration, supervision. D.D., Y.R.H. and R.A.S: investigation, formal analysis, data curation, supervision. D.A.L.: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, writing - original draft. B.W.: conceptualization, formal analysis, data curation, visualization, project administration, resources, supervision, writing - original draft. All authors: writing/review and editing of the final draft.
CONFLICT OF INTEREST
CA reports consultancy fees from Tesaro, Mateon Therapeutics, Clovis, Cerulean, Bayer and VentiRx, outside the submitted work. RAS reports grants from NIH, grants from DOD, personal fees from Springer Publishing, and personal fees from Cambridge Univ Press, outside the submitted work. The remaining authors have no conflicts of interest to declare.
References
- 1.Kobel M, Kalloger SE, Huntsman DG, Santos JL, Swenerton KD, Seidman JD, et al. Differences in tumor type in low-stage versus high-stage ovarian carcinomas. Int J Gynecol Pathol. 2010;29:203–211. doi: 10.1097/PGP.0b013e3181c042b6. [DOI] [PubMed] [Google Scholar]
- 2.Brown J, Frumovitz M. Mucinous tumors of the ovary: current thoughts on diagnosis and management. Curr Oncol Rep. 2014;16:389. doi: 10.1007/s11912-014-0389-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riopel MA, Ronnett BM, Kurman RJ. Evaluation of diagnostic criteria and behavior of ovarian intestinal-type mucinous tumors: atypical proliferative (borderline) tumors and intraepithelial, microinvasive, invasive, and metastatic carcinomas. Am J Surg Pathol. 1999;23:617–635. doi: 10.1097/00000478-199906000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Hess V, A’Hern R, Nasiri N, King DM, Blake PR, Barton DP, et al. Mucinous epithelial ovarian cancer: a separate entity requiring specific treatment. J Clin Oncol. 2004;22:1040–1044. doi: 10.1200/JCO.2004.08.078. [DOI] [PubMed] [Google Scholar]
- 5.Simons M, Ezendam N, Bulten J, Nagtegaal I, Massuger L. Survival of Patients With Mucinous Ovarian Carcinoma and Ovarian Metastases: A Population-Based Cancer Registry Study. Int J Gynecol Cancer. 2015;25:1208–1215. doi: 10.1097/IGC.0000000000000473. [DOI] [PubMed] [Google Scholar]
- 6.Zaino RJ, Brady MF, Lele SM, Michael H, Greer B, Bookman MA. Advanced stage mucinous adenocarcinoma of the ovary is both rare and highly lethal: a Gynecologic Oncology Group study. Cancer. 2011;117:554–562. doi: 10.1002/cncr.25460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu W, Rush J, Rickett K, Coward JI. Mucinous ovarian cancer: A therapeutic review. Crit Rev Oncol Hematol. 2016;102:26–36. doi: 10.1016/j.critrevonc.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 8.Mackenzie R, Kommoss S, Winterhoff BJ, Kipp BR, Garcia JJ, Voss J, et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer. 2015;15:415. doi: 10.1186/s12885-015-1421-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryland GL, Hunter SM, Doyle MA, Caramia F, Li J, Rowley SM, et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Med. 2015;7:87. doi: 10.1186/s13073-015-0210-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee KR, Young RH. The distinction between primary and metastatic mucinous carcinomas of the ovary: gross and histologic findings in 50 cases. Am J Surg Pathol. 2003;27:281–292. doi: 10.1097/00000478-200303000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Kelemen LE, Kobel M. Mucinous carcinomas of the ovary and colorectum: different organ, same dilemma. Lancet Oncol. 2011;12:1071–1080. doi: 10.1016/S1470-2045(11)70058-4. [DOI] [PubMed] [Google Scholar]
- 12.Kobel M, Kalloger SE, Boyd N, McKinney S, Mehl E, Palmer C, et al. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med. 2008;5:e232. doi: 10.1371/journal.pmed.0050232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCluggage WG. Immunohistochemistry in the distinction between primary and metastatic ovarian mucinous neoplasms. J Clin Pathol. 2012;65:596–600. doi: 10.1136/jcp.2010.085688. [DOI] [PubMed] [Google Scholar]
- 14.Vang R, Gown AM, Wu LS, Barry TS, Wheeler DT, Yemelyanova A, et al. Immunohistochemical expression of CDX2 in primary ovarian mucinous tumors and metastatic mucinous carcinomas involving the ovary: comparison with CK20 and correlation with coordinate expression of CK7. Mod Pathol. 2006;19:1421–1428. doi: 10.1038/modpathol.3800698. [DOI] [PubMed] [Google Scholar]
- 15.Park KJ, Bramlage MP, Ellenson LH, Pirog EC. Immunoprofile of adenocarcinomas of the endometrium, endocervix, and ovary with mucinous differentiation. Appl Immunohistochem Mol Morphol. 2009;17:8–11. doi: 10.1097/PAI.0b013e318174f012. [DOI] [PubMed] [Google Scholar]
- 16.Moh M, Krings G, Ates D, Aysal A, Kim GE, Rabban JT. SATB2 Expression Distinguishes Ovarian Metastases of Colorectal and Appendiceal Origin From Primary Ovarian Tumors of Mucinous or Endometrioid Type. Am J Surg Pathol. 2016;40:419–432. doi: 10.1097/PAS.0000000000000553. [DOI] [PubMed] [Google Scholar]
- 17.Seidman JD, Kurman RJ, Ronnett BM. Primary and metastatic mucinous adenocarcinomas in the ovaries: incidence in routine practice with a new approach to improve intraoperative diagnosis. Am J Surg Pathol. 2003;27:985–993. doi: 10.1097/00000478-200307000-00014. [DOI] [PubMed] [Google Scholar]
- 18.Nonaka D, Chiriboga L, Soslow RA. Expression of pax8 as a useful marker in distinguishing ovarian carcinomas from mammary carcinomas. Am J Surg Pathol. 2008;32:1566–1571. doi: 10.1097/PAS.0b013e31816d71ad. [DOI] [PubMed] [Google Scholar]
- 19.Leitao MM, Jr, Hensley ML, Barakat RR, Aghajanian C, Gardner GJ, Jewell EL, et al. Immunohistochemical expression of estrogen and progesterone receptors and outcomes in patients with newly diagnosed uterine leiomyosarcoma. Gynecol Oncol. 2012;124:558–562. doi: 10.1016/j.ygyno.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 20.Garg K, Leitao MM, Jr, Kauff ND, Hansen J, Kosarin K, Shia J, et al. Selection of endometrial carcinomas for DNA mismatch repair protein immunohistochemistry using patient age and tumor morphology enhances detection of mismatch repair abnormalities. Am J Surg Pathol. 2009;33:925–933. doi: 10.1097/PAS.0b013e318197a046. [DOI] [PubMed] [Google Scholar]
- 21.Ye J, Zhou Y, Weiser MR, Gonen M, Zhang L, Samdani T, et al. Immunohistochemical detection of ARID1A in colorectal carcinoma: loss of staining is associated with sporadic microsatellite unstable tumors with medullary histology and high TNM stage. Hum Pathol. 2014;45:2430–2436. doi: 10.1016/j.humpath.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Garg K, Broaddus RR, Soslow RA, Urbauer DL, Levine DA, Djordjevic B. Pathologic scoring of PTEN immunohistochemistry in endometrial carcinoma is highly reproducible. Int J Gynecol Pathol. 2012;31:48–56. doi: 10.1097/PGP.0b013e3182230d00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784–2795. doi: 10.1200/JCO.2009.25.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeLair DF, Burke KA, Selenica P, Lim RS, Scott SN, Middha S, et al. The genetic landscape of endometrial clear cell carcinomas. J Pathol. 2017;243:230–241. doi: 10.1002/path.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng CKY, Piscuoglio S, Geyer FC, Burke KA, Pareja F, Eberle CA, et al. The Landscape of Somatic Genetic Alterations in Metaplastic Breast Carcinomas. Clin Cancer Res. 2017;23:3859–3870. doi: 10.1158/1078-0432.CCR-16-2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 33.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rechsteiner M, Zimmermann AK, Wild PJ, Caduff R, von Teichman A, Fink D, et al. TP53 mutations are common in all subtypes of epithelial ovarian cancer and occur concomitantly with KRAS mutations in the mucinous type. Exp Mol Pathol. 2013;95:235–241. doi: 10.1016/j.yexmp.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 35.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McConechy MK, Ding J, Senz J, Yang W, Melnyk N, Tone AA, et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod Pathol. 2014;27:128–134. doi: 10.1038/modpathol.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas Research Network. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soegaard M, Jensen A, Hogdall E, Christensen L, Hogdall C, Blaakaer J, et al. Different risk factor profiles for mucinous and nonmucinous ovarian cancer: results from the Danish MALOVA study. Cancer Epidemiol Biomarkers Prev. 2007;16:1160–1166. doi: 10.1158/1055-9965.EPI-07-0089. [DOI] [PubMed] [Google Scholar]
- 39.Anglesio MS, Kommoss S, Tolcher MC, Clarke B, Galletta L, Porter H, et al. Molecular characterization of mucinous ovarian tumours supports a stratified treatment approach with HER2 targeting in 19% of carcinomas. J Pathol. 2013;229:111–120. doi: 10.1002/path.4088. [DOI] [PubMed] [Google Scholar]
- 40.Heong V, Ngoi N, Tan DS. Update on immune checkpoint inhibitors in gynecological cancers. J Gynecol Oncol. 2017;28:e20. doi: 10.3802/jgo.2017.28.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.