

Abstract
Ischemic stroke caused by an embolus or local thrombosis results in neural tissue damage (an infarct) in the territory of the occluded cerebral artery. Decades of studies have increased our understanding of the molecular events during cerebral infarction; however, translation of these discoveries to druggable targets for ischemic stroke treatment has been largely disappointing. Interleukin-4 (IL-4) is a multifunctional cytokine that exerts its cellular activities via the interleukin-4 receptor α (IL-4Rα). This cytokine-receptor complex is associated with diverse immune and inflammatory responses. Recent studies have suggested a role of the cytokine IL-4 in long-term ischemic stroke recovery, involving immune cell activity. By contrast, the role of the receptor, IL-4Rα especially in the acute phase of infarction is unclear. In this study, we determined that IL-4Rα is expressed on neurons and that during the early phases of cerebral infarction (24 hours) levels of this receptor are increased to regulate cellular apoptosis factors through activation of STAT6. In this context, we show a neuroprotective role for IL-4Rα in an in vivo surgical model of cerebral ischemia and in ex vivo brain slice explants, using both genetic knockout of this receptor and RNAi-mediated gene knockdown. IL-4Rα may therefore represent a novel target and pathway for therapeutic development in ischemic stroke.
Keywords: Interleukin-4 receptor alpha (Il4rα), Ischemic stroke, Neuroprotection
Graphical abstract
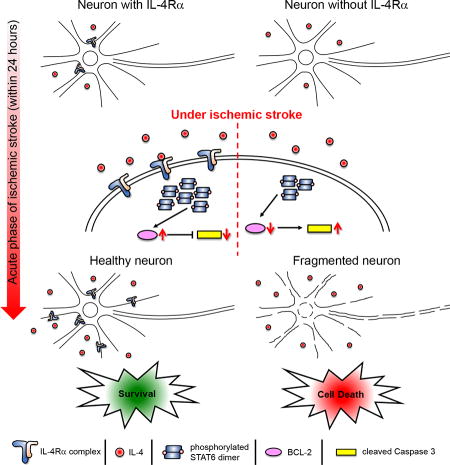
In this study, we report that neurons in the mouse brain express IL-4Rα and that during the early phase of ischemic stroke (within 24 hours), levels of this receptor are increased to regulate cellular apoptotic factors through activation of STAT6. Therefore, IL-4Rα plays a cell autonomous role in neuroprotection in the acute phase of ischemic stroke.
Introduction
Stroke is the fourth-leading cause of death in the US, with almost 800,000 new cases occurring each year [1]. The majority (over 80%) of strokes are of ischemic origin caused by disrupted blood flow within the territory of an occluded blood vessel, resulting in irreversible death of brain tissue (infarction). Total health care costs for stroke are estimated at $40 billion per year and with the aging population in the coming decades stroke burden is expected to increase substantially. Despite decades of research, the only treatment option approved by the FDA for stroke therapy remains intravenous recombinant tissue plasminogen activator (tPA), currently given to only 2–3% of stroke patients in the US because of its limited time window for drug administration of <4.5 h beyond stroke [2]. Moreover, tPA does not provide protection to neural tissues damaged by stroke. Thus, alternative strategies for brain protection and/or neuroprotection are urgently needed for stroke treatment.
IL-4 is a multifunctional cytokine that exerts its cellular activities via the IL-4Rα. This cytokine-receptor complex is associated with diverse immune and inflammatory responses [3–5], but recent studies have suggested critical roles for this cytokine-receptor complex in the brain. Diminished IL-4 levels in the aging brain may lead to cognitive decline and increased risk of Alzheimer’s disease [6], and in zebrafish brain displaying an Alzheimer’s disease-like phenotype, activation of IL-4 coupled with its receptor IL-4Rα promotes neural stem cell proliferation and neurogenesis [7]. Furthermore, several clinical and animal studies have demonstrated that IL-4 can function as a protective modulator, improving tissue recovery after ischemic stroke onset. Specifically, under a chronic stroke condition, macrophages expressing IL-4Rα are transformed into the alternatively activated M2 polarized phenotype that is essential for tissue preservation and repair [8–10].
Here, we report that neurons in the mouse brain express IL-4Rα and that during the acute phase of ischemic stroke (within 24 h), neurons are autonomously activated to modulate neuronal apoptosis through the IL-4/IL-4Rα signaling pathway, including STAT6 activation. This neuroprotective effect of IL-4Rα activation alters the extent of ischemic infarction (tissue death). These data suggest a new pathway for neural protection in ischemic stroke.
Results
Absence of Il4rα modulates both cerebral collateral vessel anatomy and ischemic infarct volume
Exploiting variation in infarct volume after middle cerebral artery occlusion (MCAO) across different inbred mouse strains, we genetically mapped a locus on distal chromosome 7 (Civq1, cerebral infarct volume QTL 1) that robustly modulates infarct volume [11]. Subsequent work demonstrated that this locus controls cerebral collateral blood vessel anatomy, where inbred strains with extensive collateral vessel connections exhibit smaller infarcts due to their ability to rapidly re-perfuse the ischemic territory [12,13]. Additional work suggested that the locus may be more complex, containing multiple genes that independently modulate infarct volume by either vascular-dependent and/or vascular-independent mechanisms [13,14]. Among the candidate genes mapping within this locus, one gene, Il4rα, showed more than a 2-fold expression difference in P1 brain cortex between two strains that differ widely in infarct volume, C57BL/6J (B6) and BALB/cJ [14]. The expression difference in brain tissue suggested a potential vascular-independent role in the infarct phenotype. To investigate whether Il4rα modulates ischemic infarct volume and/or collateral vessel phenotypes, we employed an Il4rα KO mouse strain. As these two phenotypes vary widely across inbred mouse strains, it was critical that we consider the genetic background of the animals in order to distinguish any effects of the loss of the Il4rα gene from effects contributed by the rest of the genome. The Il4rα KO mouse was originally generated and maintained in the BALB/cJ strain background. After confirming that this KO allele is fully contained in the BALB/cJ background by whole genome SNP genotyping, we examined effects of the loss of this gene on pial collateral vessel density and infarct volume after MCAO.
We first measured the number of collateral vessel connections between the anterior cerebral artery (ACA) and middle cerebral artery (MCA). The number of vessel connections in genotypes of Il4rα KO allele mice did not differ among the genotypes (WT (~2.3), Het (~2.5), and KO (~2.0)), nor when compared to the parental background strain (BALB/cJ (~3.1)) (Fig. 1A). We next measured infarct volume after MCAO for each genotype of the Il4rα KO mice. Infarct volumes among genotypes also did not differ between WT (45.1 mm3), Het (47.5 mm3), and KO (47.3 mm3), nor when compared to the parental background strain (BALB/cJ (41.7 mm3)) (Fig. 1B).
Figure 1. Collateral vessel anatomy and infarct volume after MCAO in the Il4rα KO mice.

(A) The graph indicates the average number of collateral vessel connections in the brain and total number of animals for 129S1, B6, BALB/cJ, Il4rα-BALB/c WT, Il4rα-BALB/c Het, and Il4rα-BALB/c KO are 31, 37, 30, 16, 28, and 17 animals, respectively. (B) The graph shows the infarct volume for 129S1, B6, BALB/cJ, Il4rα-BALB/c WT, Il4rα-BALB/c Het, and Il4rα-BALB/c KO are 32, 32, 31, 21, 21, and 27 animals, respectively. (C, E, and G) Representative images are shown for Il4rα, wild-type (C), heterozygous KO (E), and homozygous KO strains (G). Scale bar: 1 mm. (D, F, and H) D, F, and H are three times magnified from C, E and G, respectively, and red arrowheads indicate vessel connections between the ACA and MCA. (I) The graph indicates the average number of collateral vessel connections in the brain. The total number of animals for Il4rα WT, -Het, and -KO are 19, 12, and 10 animals, respectively. (J – L) Serial brain sections (1 mm) for each genotype of Il4rα WT (J), Het (K), and KO (L) 24 h after MCAO. The infarct appears as white tissue after 2% TTC staining. Scale bar: 5 mm. (M) The graph shows the infarct volume for Il4rα WT, -Het, and -KO; 12, 20, and 14 animals, respectively. Data represent the mean ± SEM. Statistical analysis, 2-tailed Student’s t test (*** p<.001 vs. 129S1 and B6 (A and B), Il4rα WT and -Het (I and M); ### p<.001 vs. Il4rα WT (M)).
However, the BALB/cJ inbred strain is a phenotypic outlier among most inbred mouse strains [13,14], having very few collateral vessel connections (Fig. 1A) and consequently exhibiting extremely large infarct volumes following unilateral MCAO (Fig. 1B). Therefore, the effects of genetic loss of Il4rα could have been masked by the already extreme phenotype of the BALB/cJ strain. To test this, we backcrossed the Il4rα KO allele to a different inbred strain that lies on the opposite extreme of the phenotypic spectrum [11,13,14]. Strain 129S1/SvlmJ (129S1) exhibits a high number of collateral vessel connections and after MCAO, small infarct volumes (Fig. 1A and B) as the B6 mouse strain [13,14].
In the 129S1 background (backcrossed to N5 and validated the genetic background by whole genome SNP genotyping), we examined the effects of the loss of Il4rα on pial collateral vessel density and infarct volume. The number of vessel connections in the Il4rα KO (~15.5) was slightly reduced compared with both WT (~21.1) and Het (~22.1) (Fig. 1C – I). We then examined the effects of this gene deletion on ischemic infarct volume in the permanent occlusion model. Infarct volume in Il4rα KO mice (38.6 mm3) was ~4.9-fold larger than observed in WT (7.8 mm3) (Fig. 1J – M). Interestingly, unlike with the collateral phenotype, infarct volume after MCAO showed a significant gene dosage effect. Il4rα Het mice (18.8 mm3) showed ~2.4-fold increased infarct volume compared to WT mice (Fig. 1M). The lack of complete inverse correlation [12–14] between collateral phenotype and infarct volume suggests that Il4rα displays both collateral-dependent and –independent effects on cerebral infarct volume. In all of the different inbred strain backgrounds and the different genotypes, we found no differences between males and females in any of our phenotypic measures, and thus, males and females were analyzed together.
Absence of Il4rα alters neuronal protection in the brain after ischemic stroke induction
We thus hypothesized that in addition to a minor effect on collateral vasculature, upon focal cerebral ischemia Il4rα might modulate a neuroprotective pathway. IL-4Rα is thought to be expressed in endothelial, epithelial, fibroblast, hematopoietic, hepatocyte, muscle, and brain tissue; and its ligand IL-4 a multifunctional cytokine is mainly secreted by T-helper 2 cells, mast cells, eosinophils, basophils, and stromal cells [15,16]. Recent studies reported that IL-4 is expressed on neurons in both mouse and zebra fish brain tissues [7–9], but expression of the IL-4Rα by neurons is not clear. To address this, we performed immunohistochemistry using antibodies to IL-4 and IL-4Rα along with a neuronal marker, NeuroD2, to identify the source of both IL-4 and IL-4Rα in mouse brain tissue. Both IL-4 and IL-4Rα were clearly expressed on neurons in the brain tissue (Fig. 2A and B). To further investigate which cell type in the brain expresses the IL-4Rα during ischemia, we induced ischemic stroke in Il4rα WT mice using the permanent occlusion model (MCAO) and then performed immunohistochemistry 4 h after occlusion, using antibodies to IL-4Rα and either a neuronal marker, NeuroD2, or a microglia/macrophage marker, Iba1. Although neurons showed weak expression of IL-4Rα in the contralateral hemisphere, the microglia/macrophages in this region exhibited much higher expression (Fig. 2C, C1, and C2). However, 4 h after ischemic stroke was induced, levels of IL-4Rα were greatly increased in the neurons of the penumbra (P) and surrounding the ischemic core (IC) (Fig. 2C, C3, and C4).
Figure 2. IL-4 and IL-4Rα are expressed on neurons in Il4rα WT mouse brain and neuronal IL-4Rα levels are increased during ischemia.

(A and B) Immunofluorescence staining for either IL-4 or IL-4Rα is indicated in green. Neurons stained with NeuroD2 are indicated in red. Merged images show that IL-4 and IL-4Rα are expressed on neurons that are located in layer 2/3 of brain cortex. Scale bar: 10 µm. (C) A schematic illustration shows the regions of the infarct (penumbra and ischemic core) after permanent MCAO in the mouse brain. Images were obtained from both the contralateral and ipsilateral regions of the mouse brain 4 h after occlusion. Immunofluorescence of IL-4Rα is shown in green and either NeuroD2 or Iba1 is shown in red. Merged images from the contralateral region of the brain (boxed regions are magnified in C1 and C2) show that IL-4Rα is predominantly expressed on microglia/macrophages but also expressed at lower levels on neurons. Merged images from the ipsilateral region of the brain (boxed regions are magnified in C3 and C4) show that 4 h after MCAO, neurons show dramatically increased levels of IL-4Rα as shown by staining with NeuroD2. Scale bar: 100 µm for most images and 30 µm for the magnified boxed regions.
The expression of both IL-4 and IL-4Rα in neurons allowed us to use a brain slice explant model to determine whether Il4rα modulates intrinsic neuroprotection via a collateral-independent mechanism. This ex vivo brain slice assay model maintains the complex multicellular architecture of the tissue while obviating any effects of reperfusion of the tissue by the cerebrovasculature including by collateral circulation. Using this assay, we examined the degree of neuronal cell death induced by oxygen-glucose deprivation (OGD) in brain slice explants prepared from the Il4rα KO mouse strains.
We first showed that there were no differences in YFP transfection efficiency (used to delineate neurons) and cell viability in non-OGD treated Il4rα WT and KO brain slices (Fig. 3A, B, and E). As expected, transient OGD of both Il4rα WT and KO brain slices resulted in the degeneration and clearance of a large proportion of cortical pyramidal neurons within 24 h (Fig. 3A – E). However, after OGD, Il4rα KO brain slices showed a >40% increase in neuronal cell death when compared to Il4rα WT brain slices (Fig. 3E and F). To investigate whether neuronal cell death is actively induced in Il4rα KO brain slices, we monitored cleaved Caspase 3 (c-Caspase 3) activity in the neurons 12 hours after transient OGD (Fig. 3G – L). c-Caspase 3 intensity in neurons without Il4rα was ~7.7-fold higher than that in neurons with Il4rα (Fig. 3M), consistent with the observed increase in neuronal cell death being due to the loss of Il4rα. Together, these results suggest that Il4rα plays a protective role in ischemic brain damage, independent of tissue reperfusion through the collateral circulation.
Figure 3. Il4rα KO mice show increased neuronal cell death in the cortical brain slice ischemia assay.

(A – D) Cortical brain slices from Il4rα WT and KO mice biolistically transfected with an YFP expression plasmid under normal conditions and 24 h after 5.5 min of OGD. YFP expressed in cortical pyramidal neurons appears dark in these contrast-inverted fluorescence micrographs. Scale bar: 100 µm. (E) The graph indicates the average cell viability of Il4rα WT and KO before and 24 h after OGD. The OGD dramatically induced neuronal cell death for both Il4rα WT and KO compared their non-OGD control. Experiments were performed three times using three to four mice per genotype for each experiment (Total number of animals: WT (n=10) and KO (n=11)). The number of brain slices analyzed for Il4rα WT(non-OGD), WT (OGD), KO (non-OGD), and KO (OGD) were 55, 58, 62, and 60, respectively. (F) The graph indicates the average cell viability of Il4rα WT and KO 24 h after OGD. Il4rα KO increases neuronal cell death by 40% normalized to Il4rα WT. (G – L) Brain slices for Il4rα WT (G, H, and I) and KO (J, K, and L) were stained with a neuronal-specific MAP2 antibody (G and J) and c-Caspase 3 antibody (H and K). (I) and (L) capture the image of a single neuron located in layer 2/3 of the brain cortex for Il4rα WT (G) and KO (J), and white dots in the outline of the cell represent c-Caspase 3 normalized to the same level of both Il4rα WT and KO neurons using Image J. Scale bar: 5 µm. (M) The graph indicates the average intensity of c-Caspase 3. Il4rα KO neurons showed a 7.7-fold increase in c-Caspase 3 intensity compared to WT neurons. The number of neurons analyzed for Il4rα WT and KO were 10 and 15, respectively. Data represent the mean ± SEM. Statistical analysis, 2-tailed Student’s t test (*** p<.001 vs. Il4rα WT).
Absence of Il4ra affects levels of Il4 mRNA
IL-4Rα forms a heterodimeric receptor complex with the common cytokine receptor γ chain (γc) and through this receptor complex (termed IL-4 receptor, type I) IL-4 signaling is initiated [16–18]. IL-4 signal transduction and functional activity is impaired but not completely eliminated in Il4rα KO mice [19]. Consistent with previously published data [19], we found that Il4 mRNA expression was diminished in Il4rα knockout animals (Fig. 4A) and appeared unresponsive to OGD. By contrast, levels of both Il4 and Il4rα increased 12 h and 24 h after OGD treatment in wild-type mice (Fig. 4A).
Figure 4. Il4rα KO mice show increased c-Caspase 3 after ischemic stroke stimulation in an ex vivo brain slice stroke model.

(A) mRNA levels of Il4, Il4rα, Bcl-2, and Caspase 3 from Il4rα WT and KO were determined by qRT-PCR 12 h and 24 h after 5.5 min OGD. Gapdh was used for normalization. Experiments were performed three times using three to four mice per genotype for each experiment. (B) Western blots were performed to detect both BCL-2 and c-Caspase 3 in explanted brain slices of Il4rα WT and KO 12 h and 24 h after 5.5 min OGD. (C and D) Levels of BCL-2 (C) and c-Caspase 3 (D) protein were determined. GAPDH was used for normalization. Experiments were performed four times using three to four mice per genotype for each experiment. Data represent the mean ± SEM. Statistical analysis, two-way ANOVA followed by Bonferroni’s multiple comparison test (* p<.05; ** p<.01; *** p<.001).
Absence of Il4rα affects apoptosis
To determine whether the observed increased neuronal cell death in Il4rα KO brain tissue after OGD treatment was due to increased cellular apoptosis we determined mRNA transcription levels of the anti- and pro-apoptotic factors Bcl-2 and Caspase 3, respectively. Transcript levels of Bcl-2 in Il4rα KO brain slices were significantly reduced at 12 h after OGD treatment compared to Il4rα WT brain slices (Fig. 4A). Furthermore, at the protein level, Il4rα KO brain slices showed significantly reduced BCL-2 levels compared to Il4rα WT brain slices after OGD treatment (Fig. 4B and C). Caspase-3 mRNA expression levels in Il4rα KO brain slices were significantly increased 12 h after OGD treatment compared to Il4rα WT brain slices (Fig. 4A) and c-Caspase 3 protein levels in Il4rα KO brain slices were significantly increased after OGD treatment (Fig. 4B and D). These results indicate that Il4rα regulates cellular apoptosis pathways after OGD.
Il4rα modulates apoptotic factors through STAT6 phosphorylation
IL-4 interaction with type I receptor IL-4Rα results in tyrosine phosphorylation of Janus Kinase 1 and 3 (JAK1 and JAK3), and JAK1 induces signal transducer and activator of transcription 6 (STAT6) phosphorylation [20,21]. Phosphorylated STAT6 (pSTAT6) homo-dimerizes and translocates to the nucleus where it serves as a transcription factor to promote transcription of target genes, including Il4rα [22–24]. Recent studies have implicated STAT6 activity in cellular apoptosis across many cell types [25–27]. To investigate whether IL-4/IL-4Rα signals through STAT6 during apoptosis due to cerebral ischemia, we determined Stat6 mRNA transcription levels from both Il4rα WT and KO brain slices after transient OGD treatment. Although Stat6 mRNA expression levels were elevated in both Il4rα WT and KO brain slices after OGD treatment, this effect was much reduced in Il4ra KO brain tissue (Fig. 5A). This pattern was also reflected in the active form of the protein, pSTAT6 especially at 12 h post ischemia (Fig. 5B and C). These results suggest that IL-4/IL-4Rα may signal through the JAK/STAT pathway to modulate neuronal apoptosis within the first 24 h after ischemia.
Figure 5. Il4rα KO mice show reduced levels of STAT6 transcript and phosphorylated STAT6 protein after ischemic stroke stimulation in an ex vivo brain slice stroke model.

(A) mRNA levels of Stat6 from Il4rα WT and KO were determined by qRT-PCR 12 h and 24 h after 5.5 min OGD. Gapdh was used for normalization. Experiments were performed three times using three to four mice per genotype for each experiment. (B) Western blots were performed to detect both pSTAT6 and STAT6 in explanted brain slices of Il4rα WT and KO 12 h and 24 h after 5.5 min OGD. (C) Levels of pSTAT6 protein were determined using total STAT6 protein levels for normalization. Experiments were performed three times using three to four mice per genotype for each experiment. Data represent the mean ± SEM. Statistical analysis, two-way ANOVA followed by Bonferroni’s multiple comparison test (* p<.05; ** p<.01; *** p<.001).
Acute knockdown of Il4rα gene expression by siRNA also affects neuronal apoptosis
Finally, we sought to exclude a significant neurodevelopmental role in the effects of Il4rα gene deletion by employing acute knock down of Il4rα gene expression in wild-type brain tissue explants using Il4rα-specific siRNAs. We first evaluated the efficiency of siRNA delivery system in the mouse brain slice cultures. Il4rα mRNA transcript levels in Il4rα WT brain slices showed an approximately 55% reduction after transfection of Il4rα-specific siRNA compared to that observed after transfection of non-specific siRNA (Fig. 6A). As predicted from previously published data documenting the regulatory cross talk between receptor and cytokine [3,5,16] Il4 mRNA expression levels were also affected by knockdown of its receptor, showing ~50% reduction (Fig. 6A). IL-4Rα protein levels showed a concomitant 40% reduction (Fig. 6B and C).
Figure 6. Cellular apoptosis is induced through pSTAT6 by reduction of IL-4Rα expression level using Il4rα-specific siRNA in the cortical brain slice ischemia assay.

(A) Non-specific (control) or Il4rα-specific siRNA were transfected into Il4rα WT cortical brain slices for 48 h. mRNA levels of Il4, Il4rα from brain slices transfected either control or Il4rα-specific siRNA were determined by qRT-PCR 24 h after 30 min OD. Gapdh was used for normalization. Experiments were performed three times using three mice per genotype for each experiment. (B) Western blots were performed to detect IL-4Rα in explanted brain slices of Il4rα WT brain slices transfected with either control siRNA or Il4rα-specific siRNA in both control and OD conditions. (C) Levels of IL-4Rα protein were normalizing to GAPDH protein levels. Experiments were performed three times using three mice per genotype for each experiment. Data represent the mean ± SEM. Statistical analysis, 2-tailed Student’s t test (* p<.05 vs. non-specific siRNA). (D) Non-specific (control) or Il4rα-specific siRNA were transfected into wild-type cortical brain slices. 48 h later mRNA levels of Il4, Il4rα, Stat6, Bcl-2, and Caspase 3 from Il4rα WT were determined by qRT-PCR 24 h after 30 min OD. Gapdh was used for normalization. Experiments were performed three times using three mice per genotype for each experiment. (E) Western blots were performed to detect both pSTAT6 and STAT6 in explanted brain slices of Il4rα WT mice transfected with either control siRNA or Il4rα-specific siRNA for both control and OD conditions. (F) Levels of pSTAT6 protein were determined using total STAT6 protein levels for normalization. (G) Western blot were performed to detect both BCL-2 and c-Caspase 3 in explanted brain slices of Il4rα WT and KO transfected with either control siRNA or Il4rα-specific siRNA for both control and OD conditions. (H and I) Levels of BCL-2 (H) and c-Caspase 3 (I) proteins were determined using normalizing to GAPDH protein levels. Experiments were performed four times using three mice per genotype for each experiment. Data represent the mean ± SEM. Statistical analysis, two-way ANOVA followed by Bonferroni’s multiple comparison test (* p<.05; ** p<.01; *** p<.001).
Next, to apply Il4rα siRNA knockdown in the brain slice ischemia assay, we had to slightly modify the OGD model which is incompatible with siRNA transfection due to technical issues [14]. Instead, we employed an ex vivo oxygen deprivation (OD) model [14]. This OD model enabled the delivery of Il4rα-specific siRNA into cultured mouse brain slices, and provided sufficient time for RNAi-mediated knockdown at both the mRNA and protein levels before application of OD to the brain slice. We have previously validated this approach and shown similar cell viability when compared with the OGD assay [14].
Using this OD assay in brain slice cultures, we determined the transcript and protein levels of apoptotic factors after transfection of Il4rα-specific siRNA. Consistent with the results observed with Il4rα KO brain slices after OGD treatment (Fig. 4A), caspase-3 mRNA transcript levels in Il4rα-specific siRNA transfected brain slices were markedly increased after OD treatment when compared to scrambled siRNA controls (Fig. 6D). Bcl-2 and Stat6 transcript levels were also decreased by Il4rα-specific siRNA compared to scrambled siRNA controls, again consistent with the data using Il4rα KO brain slices. Protein levels of pSTAT6, BCL-2, and c-Caspase 3 showed concomitant and similar changes as were observed for their mRNA transcript counterparts (Fig. 6E – I). Taken together, these data support an ongoing role for Il4rα signaling in modulating levels of apoptotic factors in brain tissue after acute ischemic insult.
Discussion
Although epidemiologic studies have estimated that two-thirds of the population-attributable risk for ischemic stroke is due to genetic factors, a significant genetic component to ischemic stroke still remains elusive [28–30]. To identify some of the more recalcitrant genetic factors, especially those that modulate the extent (size) of the infarcted tissue, we employed permanent middle cerebral artery occlusion across various inbred strains of mice and performed QTL mapping of loci that modulate infarct volume [11,13,14,31]. Using this approach, we and others have determined that some genetic loci modulate the size of the infarct via a collateral vessel-dependent mechanism, thereby enabling reperfusion of the ischemic territory. Other loci influence infarct volume via a collateral vessel-independent mechanism [13,14,31] and are more likely to reflect innate neuroprotective mechanisms. The strongest of the modifier loci identified to date maps to distal chromosome 7 [11,13,14]. Further fine-mapping of the relatively broad genomic interval using ancestral SNP haplotype analysis identified 12 candidate genes [14], a short list that includes Il4rα, the receptor for the cytokine IL-4.
The role of the cytokine IL-4 in ischemic stroke recovery has been well established [8,9] and has focused on long-term recovery from ischemia, involving the activity of immune cells. In particular, these long-term effects of the IL-4/IL-4Rα complex involve macrophages and are associated with both detrimental- and beneficial effects [32–34]. Cytokine IL-4 together with its receptor IL-4Rα induce host defense responses including anti-inflammatory and tissue repair phenotypes in macrophages [17,35,36]. However, the effects of this immune cascade mechanism occur later in the process.
By contrast, the role of the receptor, IL-4Rα, especially in the acute phase of infarction, has been less studied. We sought to investigate the role of Il4rα during infarction, focusing on the acute phase, that is, within 24 hours of the ischemic insult. We first postulated that if the Il4rα modulated the extent of cell death in the acute phases of infarction, and had a role in innate neuroprotection, the receptor might be expressed on neurons. Previously published work has shown that the cytokine, IL-4 is indeed expressed on neurons [8,9] but its receptor, IL-4Rα is not clear. We showed that IL-4Rα is also expressed on neurons albeit at lower levels than in microglia/macrophages (Fig. 2) but importantly, the levels of neuronal IL-4Rα are dramatically increased upon induction of ischemic stroke (Fig. 2).
We next sought to generate a complete loss of function (homozygous KO) of the Il4rα gene in multiple inbred mouse strains. The phenotypic effects of a gene knockout can vary depending on genetic background, and the effects of the knockout allele of a gene can be masked if these effects would drive the phenotype in the direction where the genetic strain background is already at the extreme of the phenotypic range. The well-studied Il4rα KO allele has been maintained in BALB/cJ, a strain exhibiting very large infarct volumes due primarily to a lack of collateral vessels [13,14]. We sought to move the allele into the 129S1 background that shows ~5-fold smaller infarcts. We found that even with minimal (or none in the case of the heterozygous KO) differences in collateral vessel number, infarct volume after MCAO is dramatically increased with both reduced (Het) and full loss (KO) of Il4rα. These results suggest that Il4rα modulates ischemia-induced tissue death in large part via a vascular-independent mechanism.
We next verified the non-vascular neuroprotective effect of Il4rα using an ex vivo brain slice explant ischemia assay in which the vascular circulation is entirely absent. We further showed that Il4rα induces apoptosis via activation of STAT6 signaling, regulating the anti-apoptotic factor, BCL-2 and the pro-apoptotic factor, c-Caspase 3. These same apoptotic factors were shown to be Il4rα-dependent whether a genetic knockout of the receptor was used or when acutely introducing Il4rα-specific siRNA into wild-type brain slices, further supporting an ongoing neuroprotective, anti-apoptotic effect of Il4rα signaling. Importantly, it should be stressed that we were measuring early effects (within 24 hours) of the loss of Il4rα on ischemia-induced neuronal death. Our data support a neuronal, cell-autonomous effect of Il4rα during the early events of ischemia-induced cell death.
Ischemic stroke causes brain tissue damage (infarction) leading to disability and/or death. The only treatment option currently approved by the FDA is tPA, which by dissolving the blood clot, re-establishes blood flow to the affected territory. However, even this treatment does not modulate intrinsic neuroprotective mechanisms of the brain tissue. The identification of Il4rα as a vascular-independent modulator of infarct volume suggests an endogenous neuroprotective pathway that could be targeted for therapy, especially during the early phases of ischemic stroke.
Materials and methods
Animals
All inbred mouse strains and Il4rα KO mice (BALB/c-Il4rαtm1Sz/J) were obtained from the Jackson Laboratory (Bar Harbor, ME) or bred locally from breeding pairs of each strain. The Il4rα KO mice in the BALB/cJ mouse background were backcrossed a minimum of five generations with 129S1, or BALB/cJ for Il4rα-129S1, or Il4rα-BALB/c, respectively. The genetic background of each Il4rα KO line was confirmed by whole genome SNP genotyping (OpenArray Technology). Mice (both male and female animals) were age matched (P10 for brain slice culture, P21 for collateral vessel perfusion, and 12 ± 1 week for MCAO) for all experiments. All animal procedures were conducted under protocols approved by the Duke University IACUC in accordance with NIH guidelines.
Collateral vessel density measurement
As collateral vessel traits are determined by 3 weeks of age and remain constant for many months [37], the collateral vessel phenotype was measured at P21. Mice were anesthetized with ketamine (100 mg/kg) and xylazine (2.5 mg/kg), and the ascending thoracic aorta was cannulated. The animals were perfused with freshly made buffer (1 mg/ml adenosine, 40 g/ml papaverine, and 25 mg/ml heparin in PBS) to remove the blood. The pial circulation was then exposed after removal of the dorsal calvarium and adherent dura mater. The cardiac left ventricle was cannulated and a polyurethane solution with a viscosity sufficient to minimize capillary transit (1:1 resin to 2-butanone, PU4ii, VasQtec) was slowly infused; cerebral circulation was visualized under a stereomicroscope during infusion. The brain surface was topically rinsed with 10% PBS-buffered formalin and the dye solidified for 20 min. After post-fixation with 10% PBS-buffered formalin, pial circulation was imaged. All collaterals interconnecting the anterior- and middle cerebral artery trees of both hemispheres were counted.
Permanent MCAO
Focal cerebral ischemia was induced by direct permanent occlusion of the distal MCA as previously described [14]. Briefly, adult mice were anesthetized with ketamine (100 mg/kg) and xylazine (2.5 mg/kg). The right MCA was exposed by a 0.5 cm vertical skin incision midway between the right eye and ear under a dissecting microscope. After the temporalis muscle was split, a 2-mm burr hole was made with a high-speed microdrill at the junction of the zygomatic arch and the squamous bone through the outer surface of the semi-translucent skull. The MCA was clearly visible at the level of the inferior cerebral vein. The inner layer of the skull was removed with fine forceps, and the dura was opened with a 32-gauge needle. While visualizing under an operating microscope, the right MCA was electrocauterized. The cauterized MCA segment was then transected with microscissors to verify permanent occlusion. The surgical site was closed with 6-0 sterile nylon sutures, and 0.25% bupivacaine was applied. The temperature of each mouse was maintained at 37°C with a heating pad during the surgery until the animal was fully recovered from the anesthetic. Mice were then returned to their cages and allowed free access to food and water in an air-ventilated room with the ambient temperature set to 25°C.
Infarct volume measurement
Cerebral infarct volumes were measured 24 h after surgery because the size of the cortical infarct is largest and stable at 24 h after distal permanent MCA occlusion [38]. Twenty-four hours after MCAO surgery, the animals were euthanized by decapitation, and the brains were carefully removed. The brains were placed in a brain matrix and sliced into 1 mm coronal sections after being chilled at −80°C for 4 min to slightly harden the tissue. Each brain slice was placed in 1 well of a 24-well plate and incubated for 20 min in a solution of 2% 2,3,5-triphenyltetrazolium chloride (TTC) in PBS at 37°C in the dark. The sections were then washed once with PBS and fixed with 10% PBS-buffered formalin at 4°C. Then, 24 h after fixation, the caudal face of each section was scanned using a flatbed color scanner. The scanned images were used to determine infarct volume [39]. Image-Pro software (Media Cybernetics) was used to calculate the infarcted area of each slice by subtracting the infarcted area of the hemisphere from the non-infarcted area of the hemisphere to minimize error introduced by edema. The total infarct volume was calculated by summing the individual slices from each animal.
Preparation of brain slices
Preparation of cortical brain slice explants was performed as previously described [13,14]. Coronal brain slices containing neocortex were prepared from P10 mice. Under sterile conditions, brains were dissected and cut into 250 µm coronal slices on a vibratome in chilled culture medium containing 15% heat-inactivated horse serum, 10 mM KCl, 10 mM HEPES, 100 U/ml penicillin/streptomycin, 1 mM MEM sodium pyruvate, and 1 mM l-glutamine in Neurobasal A supplemented with NMDA receptor inhibitor (1 µM MK-801).
Oxygen Glucose Deprivation (OGD) and Oxygen Deprivation (OD)
Brains were divided into “hemi-coronal” slices. For OGD, slices were suspended at 34°C for 5.5 min in glucose-free, N2-bubbled artificial CSF containing 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 24 mM D-glucose, and 10 mM HEPES. Control and OGD-treated brain slices were plated into 12-well plates in interface configuration atop solid culture medium made by the addition of 0.5% agarose. After explanting the brain slices, plates were placed for recovery at 37°C for 30 min in a humidified incubator under 5% CO2. Gold particle (1.6 µm) coated with plasmids expressing yellow fluorescent protein (YFP) were introduced into the brain slices by biolistic transfection using a Helios Gene Gun (Bio-Rad). Slice cultures were maintained at 37°C for 24 h in humidified incubator under 5% CO2. For OD, we previously modified from the established OGD model [13,14]. Brain slices were placed onto the solid culture media and incubated for recovery at 37°C for 30 min in a humidified incubator under 5% CO2. Gold particles coated with plasmids encoding YFP were introduced into the brain slices by biolistic transfection using a Helios Gene Gun (BioRad). After 24 h of incubation, each slice was submerged for 30 min at 37°C in N2-bubbled artificial CSF containing 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 24 mM D-glucose, and 10 mM HEPES to induce ischemic injury. The slices were carefully washed with pre-warmed PBS and incubated for an additional 24 h under normoxic conditions.
Il4rα-specific siRNA knock-down experiments
To reduce Il4rα mRNA expression by RNA interference, the Il4rα-specific siRNA was purchased from Dharmacon (GE Healthcare, IL) and used as recommended. For the ex vivo stroke experiments, siRNA was delivered to the cortical brain slices before the OD. Briefly, after explanting brain slices, plates were placed for recovery at 37°C for 30 min in a humidified incubator under 5% CO2. 5 mM of non-target pooled (D-001910-10-05) or Il4rα-specific pooled (E-043730-00-0005) siRNAs were introduced onto the brain slices and the slices were maintained at 37°C in a humidified incubator under 5% CO2. Forty-eight hours after introducing the siRNA, brain slices were deprived of oxygen using glucose-free, N2-bubbled artificial CSF. The slices were then incubated for an additional 24 h.
qRT-PCR
mRNA levels were measured by qRT-PCR as previously described [14]. To determine transcript levels for each genotype, brain slices were obtained from each genotype. To quantify mRNA levels of Il4, Il4rα, Stat6, Bcl-2, and Caspase 3, brain slices from Il4rα WT and KO mice were used for either OGD treatment or OD treatment after Il4rα-specific siRNA transfection. All samples were run in triplicate, and an additional assay for endogenous Gapdh was performed to control for input cDNA template quantity.
Western blot analysis
To quantify protein expression levels, brain slices were collected either after OGD or OD treatment after Il4rα-specific siRNA transfection. Apoptosis was determined by measuring apoptotic factor c-Caspase 3 and anti-apoptotic factor BCL-2. Brain slices were homogenized in cold lysis buffer (50 mM Tris-HCl [pH 7.8], 150 mM NaCl, 0.2% Triton X-100) containing protease and phosphatase inhibitor cocktail (Thermo Scientific). Protein samples (50–100 µg) were electrophoresed on a 10% to 15% polyacrylamide gel and then transferred on PVDF membrane for 90 min on ice. Membranes were incubated with primary and secondary antibodies, and the level of protein was visualized via chemiluminescence (ECL Detection Kit, Thermo Fisher Scientific). The following primary antibodies were used: anti-IL-4Rα (H-4) (1:1,500, Santa Cruz Biotechnology Inc., sc-28361), anti–phospho-STAT6 (Tyr641) (1:1,500, BD Pharmingen, 558241), anti-STAT6 (M-20) (1:2,000, Santa Cruz Biotechnology Inc., sc-981), anti-cleaved Caspase 3 (Asp175) (1:1,500, Cell Signaling Technology, 9661), anti-BCL-2 (50E3) (1:2,000, Cell Signaling Technology, 2870), and anti-GAPDH (1:4,000, Santa Cruz Biotechnology, sc-32233). HRP-conjugated anti-mouse and anti-rabbit (Santa Cruz Biotechnology Inc.) secondary antibodies were used to detect proteins.
Immunohistochemistry
Tissue from both in vivo MCAO and ex vivo brain slice cultures were fixed overnight in 4% PFA and 4% sucrose at 4°C. Brain slices were permeabilized and blocked with blocking solution containing 3% BSA and 0.2% Triton X-100 in PBS overnight at 4°C. Brain slices were incubated for 72 h with primary antibodies at 4°C and then incubated in secondary antibodies for 12 h at 4°C after washing with PBS. The following primary antibodies were used: anti-IL-4 (HIL41) (1:500, Santa Cruz Biotechnology Inc., sc-12723), anti-IL-4Rα (H-4) (1:500, Santa Cruz Biotechnology Inc., sc-28361), anti-NeuroD2 (1:500, Abcam, ab104430), anti-Iba1 (1:500, Waco, 019-19741), anti-cleaved Caspase 3 (Asp175) (1:1,500, Cell Signaling Technology, 9661), and anti-MAP2 (1:1,000, Sigma-Aldrich, M1406). The following secondary antibodies were used: Alexa Fluor 488 and 594 (1:1,000, Molecular Probes). Images were captured using a LSM 710 confocal microscope (Zeiss).
Statistics
Statistical analysis was performed either with SigmaPlot Version 10 (Systat Software Inc) or with Prism 7 (GraphPad Software). Statistical significance was evaluated using either a 2-tailed Student’s t test when comparing two groups or two-way ANOVA followed by Bonferroni’s multiple comparison test, according to the following definition: p > .05, not significant; p = 0.01–0.05, significant (*); p = 0.001–0.01, very significant (**); and p < .001, highly significant (***). All values from quantitative data are reported as mean ± SEM.
Acknowledgments
The authors thank Dr. Il Hwan Kim for helpful discussion concerning image analysis. This work was supported by a grant from the Edna and Fred L. Mandel Jr. Foundation, a Holland-Trice Scholar Award, and NIH grant 5R01HL097281.
Abbreviations
- 129S1
129S1/SvlmJ
- ACA
anterior cerebral artery
- B6
C57BL/6J
- c-Caspase 3
cleaved Caspase 3
- Civq1
cerebral infarct volume QTL 1
- Il4
interleukin-4
- Il4rα
interleukin-4 receptor alpha
- MCA
middle cerebral artery
- MCAO
middle cerebral artery occlusion
- OD
oxygen deprivation
- OGD
oxygen-glucose deprivation
- STAT6
signal transducer and activator of transcription 6
- tPA
tissue plasminogen activator
Footnotes
The authors have declared that no conflict of interest exists.
Author Contributions
HKL, DCL, and DAM designed research. HKL and SK performed research. HKL and DAM analyzed data. HKL, DCL, and DAM wrote the paper.
References
- 1.Roger VL, et al. Heart disease and stroke statistics - 2011 update: a report from the American Heart Association. Circulation. 2011;123(4):e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suwanwela N, Koroshetz WJ. Acute Ischemic Stroke: Overview of Recent Therapeutic Developments. Annual Review of Medicine. 2007;58:89–106. doi: 10.1146/annurev.med.58.070605.115306. [DOI] [PubMed] [Google Scholar]
- 3.Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat. Rev. Immunol. 2015;15:271–282. doi: 10.1038/nri3831. [DOI] [PubMed] [Google Scholar]
- 4.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 5.Luzina IG, Keegan AD, Heller NM, Rook GA, Shea-Donohue T, Atamas SP. Regulation of inflammation by interleukin-4: a review of "alternatives". J Leukoc Biol. 2012;92(4):753–64. doi: 10.1189/jlb.0412214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nolan Y, Maher FO, Martin DS, Clarke RM, Brady MT, Bolton AE, Mills KH, Lynch MA. Role of interleukin-4 in regulation of age-related inflammatory changes in the hippocampus. J Biol Chem. 2005;280(10):9354–9362. doi: 10.1074/jbc.M412170200. [DOI] [PubMed] [Google Scholar]
- 7.Bhattarai P, Thomas AK, Cosacak MI, Papadimitriou C, Mashkaryan V, Froc C, Reinhardt S, Kurth T, Dahl A, Zhang Y, Kizil C. IL4/STAT6 Signaling Activates Neural Stem Cell Proliferation and Neurogenesis upon Amyloid-β42 Aggregation in Adult Zebrafish Brain. Cell Rep. 2016;17(4):941–948. doi: 10.1016/j.celrep.2016.09.075. [DOI] [PubMed] [Google Scholar]
- 8.Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J. Neuronal Interleukin-4 as a Modulator of Microglial Pathways and Ischemic Brain Damage. J Neurosci. 2015;35(32):11281–91. doi: 10.1523/JNEUROSCI.1685-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu X, Liu J, Zhao S, Zhang H, Cai W, Cai M, Ji X, Leak RK, Gao Y, Chen J, Hu X. Interleukin-4 Is Essential for Microglia/Macrophage M2 Polarization and Long-Term Recovery After Cerebral Ischemia. Stroke. 2016;47(2):498–504. doi: 10.1161/STROKEAHA.115.012079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJM, Ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16(9):1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keum S, Marchuk DA. A locus mapping to mouse chromosome 7 determines infarct volume in a mouse model of ischemic stroke. Circ Cardiovasc Genet. 2009;2(6):591–8. doi: 10.1161/CIRCGENETICS.109.883231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Prabhakar P, Sealock R, Faber JE. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J Cereb Blood Flow Metab. 2010;30(5):923–34. doi: 10.1038/jcbfm.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keum S, Lee HK, Chu PL, Kan MJ, Huang MN, Gallione CJ, Gunn MD, Lo DC, Marchuk DA. Natural genetic variation of integrin alpha L (Itgal) modulates ischemic brain injury in stroke. PLoS Genet. 2013;9(10):e1003807. doi: 10.1371/journal.pgen.1003807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee HK, Keum S, Sheng H, Warner DS, Lo DC, Marchuk DA. Natural allelic variation of the IL-21 receptor modulates ischemic stroke infarct volume. J Clin Invest. 2016;126(8):2827–38. doi: 10.1172/JCI84491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gadani SP, Cronk JC, Norris GT, Kipnis J. IL-4 in the brain: a cytokine to remember. J Immunol. 2012;189(9):4213–9. doi: 10.4049/jimmunol.1202246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–38. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- 17.Van Dyken SJ, Locksley RM. Interleukin-4- and Interleukin-13-Mediated Alternatively Activated Macrophages: Roles in Homeostasis and Disease. Annu Rev Immunol. 2013;31:317–43. doi: 10.1146/annurev-immunol-032712-095906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hage T, Sebald W, Reinemer P. Crystal structure of the interleukin-4/receptor alpha chain complex reveals a mosaic binding interface. Cell. 1999;97(2):271–81. doi: 10.1016/s0092-8674(00)80736-9. [DOI] [PubMed] [Google Scholar]
- 19.Noben-Trauth N, Shultz LD, Brombacher F, Urban JF, Jr, Gu H, Paul WE. An interleukin 4 (IL-4)-independent pathway for CD4+ T cell IL-4 production is revealed in IL-4 receptor-deficient mice. Proc Natl Acad Sci U S A. 1997;94(20):10838–43. doi: 10.1073/pnas.94.20.10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mudter J, Neurath MF. The role of signal transducers and activators of transcription in T inflammatory bowel diseases. Inflammatory bowel diseases. Inflamm Bowel Dis. 2003;9(5):332–7. doi: 10.1097/00054725-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Cetkovic-Cvrlje M, Uckun FM. Targeting Janus kinase 3 in the treatment of leukemia and inflammatory diseases. Arch Immunol Ther Exp (Warsz) 2004;52(2):69–82. [PubMed] [Google Scholar]
- 22.Kuperman DA, Schleimer RP. Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med. 2008;8(5):384–92. doi: 10.2174/156652408785161032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller-Ladner U, Judex M, Ballhorn W, Kullmann F, Distler O, Schlottmann K, Gay RE, Scholmerich J, Gay S. Activation of the IL-4 STAT pathway in rheumatoid synovium. J Immunol. 2000;164(7):3894–901. doi: 10.4049/jimmunol.164.7.3894. [DOI] [PubMed] [Google Scholar]
- 24.Acacia de Sa Pinheiro A, Morrot A, Chakravarty S, Overstreet M, Bream JH, Irusta PM, Zavala F. IL-4 induces a wide-spectrum intracellular signaling cascade in CD8+ T cells. J Leukoc Biol. 2007;81(4):1102–10. doi: 10.1189/jlb.0906583. [DOI] [PubMed] [Google Scholar]
- 25.Galka E, Thompson JL, Zhang WJ, Poritz LS, Koltun WA. Stat6 (null phenotype) human lymphocytes exhibit increased apoptosis. J. Surg. Res. 2004;122(1):14–20. doi: 10.1016/j.jss.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M, Zhou Y, Xie C, Zhou F, Chen Y, Han G, Zhang WJ. STAT6 specific shRNA inhibits proliferation and induces apoptosis in colon cancer HT-29 cells. Cancer Lett. 2006;243(1):38–46. doi: 10.1016/j.canlet.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 27.Zhang WJ, Li BH, Yang XZ, Li PD, Yuan Q, Liu XH, Xu SB, Zhang Y, Yuan J, Gerhard GS, Masker KK, Dong C, Koltun WA, Chorney MJ. IL-4-induced Stat6 activities affect apoptosis and gene expression in breast cancer cells. Cytokine. 2008;42(1):39–47. doi: 10.1016/j.cyto.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Elkind MS. Why now? Moving from stroke risk factors to stroke triggers. Curr Opin Neurol. 2007;20(1):51–7. doi: 10.1097/WCO.0b013e328012da75. [DOI] [PubMed] [Google Scholar]
- 29.Boehme AK, Esenwa C, Elkind MS. Stroke Risk Factors, Genetics, and Prevention. Circ Res. 2017;120(3):472–495. doi: 10.1161/CIRCRESAHA.116.308398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bevan S, Traylor M, Adib-Samii P, Malik R, Paul NL, Jackson C, Farrall M, Rothwell PM, Sudlow C, Dichgans M, Markus HS. Genetic heritability of ischemic stroke and the contribution of previously reported candidate gene and genomewide associations. Stroke. 2012;43(12):3161–7. doi: 10.1161/STROKEAHA.112.665760. [DOI] [PubMed] [Google Scholar]
- 31.Chu PL, Keum S, Marchuk DA. A novel genetic locus modulates infarct volume independently of the extent of collateral circulation. Physiol Genomics. 2013;45(17):751–63. doi: 10.1152/physiolgenomics.00063.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu B, Gao HM, Wang JY, Jeohn GH, Cooper CL, Hong JS. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann N Y Acad Sci. 2002;962:318–31. doi: 10.1111/j.1749-6632.2002.tb04077.x. [DOI] [PubMed] [Google Scholar]
- 33.Moss DW, Bates TE. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. Eur J Neurosci. 2001;13(3):529–38. doi: 10.1046/j.1460-9568.2001.01418.x. [DOI] [PubMed] [Google Scholar]
- 34.Liang J, Takeuchi H, Jin S, Noda M, Li H, Doi Y, Kawanokuchi J, Sonobe Y, Mizuno T, Suzumura A. Glutamate induces neurotrophic factor production from microglia via protein kinase C pathway. Brain Res. 2010;1322:8–23. doi: 10.1016/j.brainres.2010.01.083. [DOI] [PubMed] [Google Scholar]
- 35.Minutti CM, Jackson-Jones LH, García-Fojeda B, Knipper JA, Sutherland TE, Logan N, Rinqvist E, Guillamat-Prats R, Ferenbach DA, Artigas A, Stamme C, Chroneos ZC, Zaiss DM, Casals C, Allen JE. Local amplifiers of IL-4Rα-mediated macrophage activation promote repair in lung and liver. Science. 2017;356(6342):1076–1080. doi: 10.1126/science.aaj2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, Weinstein JS, Licona-Limon P, Schmid ET, Pelorosso F, Gagliani N, Craft JE, Flavell RA, Ghosh S, Rothlin CV. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science. 2017;356(6342):1072–1076. doi: 10.1126/science.aai8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clayton JA, Chalothorn D, Faber JE. Vascular Endothelial Growth Factor-A Specifies Formation of Native Collaterals and Regulates Collateral Growth in Ischemia. Circ Res. 2008;103(9):1027–36. doi: 10.1161/CIRCRESAHA.108.181115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lambertsen KL, Meldgaard M, Ladeby R, Finsen B. A quantitative study of microglial-macrophage synthesis of tumor necrosis factor during acute and late focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005;25(1):119–35. doi: 10.1038/sj.jcbfm.9600014. [DOI] [PubMed] [Google Scholar]
- 39.Wexler EJ, Peters EE, Gonzales A, Gonzales ML, Slee AM, Kerr JS. An objective procedure for ischemic area evaluation of the stroke intraluminal thread model in the mouse and rat. J Neurosci Methods. 2002;113(1):51–8. doi: 10.1016/s0165-0270(01)00476-9. [DOI] [PubMed] [Google Scholar]