

Abstract
Tumor-associated macrophages can promote growth of cancers. In neuroblastoma, tumor-associated macrophages have greater frequency in metastatic versus loco-regional tumors, and higher expression of genes associated with macrophages helps to predict poor prognosis in the 60% of high-risk patients who have MYCN-non-amplified disease. The contribution of cytotoxic T-lymphocytes to anti-neuroblastoma immune responses may be limited by low MHC class I expression and low exonic mutation frequency. Therefore, we modelled human neuroblastoma in T-cell deficient mice to examine whether depletion of monocytes/macrophages from the neuroblastoma microenvironment by blockade of CSF-1R can improve the response to chemotherapy. In vitro, CSF-1 was released by neuroblastoma cells, and topotecan increased this release. In vivo, neuroblastomas formed by subcutaneous co-injection of human neuroblastoma cells and human monocytes into immunodeficient NOD/SCID mice had fewer human CD14+ and CD163+ cells and mouse F4/80+ cells following CSF-1R blockade. In subcutaneous or intra-renal models in immunodeficient NSG or NOD/SCID mice, CSF-1R blockade alone did not affect tumor growth or mouse survival. However, when combined with cyclophosphamide plus topotecan, the CSF-1R inhibitor BLZ945, either without or with anti-human and anti-mouse CSF-1 mAbs, inhibited neuroblastoma growth and synergistically improved mouse survival. These findings indicate that depletion of tumor-associated macrophages from neuroblastomas can be associated with increased chemotherapeutic efficacy without requiring a contribution from T-lymphocytes, suggesting the possibility that combination of CSF-1R blockade with chemotherapy might be effective in patients who have limited anti-tumor T-cell responses.
Keywords: Neuroblastoma, tumor-associated macrophage, BLZ945, MCS110, 5A1
Introduction
Monocytes differentiate into macrophages, a cell type that can regulate normal tissue repair but also tumor growth. Tumor-associated macrophages (TAMs) have been reported to contribute to tumor progression and resistance to therapy.1, 2 A high frequency of TAMs is associated with worse prognosis in lung, breast, prostate, thyroid, and hepatocellular carcinoma, follicular lymphoma, and neuroblastoma (NB).3–7 Metastatic NBs have higher infiltration of TAMs than loco-regional NBs, and 25% of the accuracy of a novel 14-gene NB classification score was contributed by expression of genes associated with TAMs (CD16, CD33, IL6R, and IL10).7 Experimental studies indicate that TAMs promote tumor progression by multiple mechanisms including promotion of invasion, metastases, angiogenesis, survival signaling, chemotherapy resistance, and immune suppression.1 In a mouse model of NB, TAMs promote tumor growth via STAT3 phosphorylation and c-Myc up-regulation.8 These data suggest the general importance of interactions between tumor cells and TAMs, and the possibility that TAMs might be a therapeutic target in NB.7 To date, however, it has not been determined if targeted depletion of monocyte-derived cells or TAMs can enhance chemotherapy of human NB.
Monocytes and TAMs, as well as other monocyte-derived cells such as osteoclasts, microglia, and Langerhan cells, express colony stimulating factor 1 receptor (CSF-1R/M-CSFR/CD115);9, 10 in contrast, other cells such as granulocytes and lymphocytes do not. CSF-1, a well characterized ligand of CSF-1R, is released by many types of cancer cells and promotes monocyte and macrophage recruitment to and survival in the tumor microenvironment.11–13 CSF-1 expression is known to be associated with poor prognosis in breast, ovarian and prostate carcinoma.14–16 Therefore, targeting CSF-1R+ cells could prevent their recruitment to and survival in tumor microenvironments.1, 2
Most previous studies combining CSF-1R blockade with chemotherapy examined the relationship of murine tumors and macrophages in immune competent mice, and much of the efficacy of CSF-1R inhibition has been attributed to infiltrating T lymphocytes. For example, in immunocompetent murine cervical and breast cancer models, inhibition of CSF-1R with BLZ945 has been shown to increase infiltration by CD8+ lymphocytes with concurrent reduction in TAMs and tumor growth.17 In immunocompetent MMTV-PyMT mice, the CSF-1R inhibitor PLX3397 in combination with paclitaxel resulted in increased infiltration by both CD4+ and CD8+ T cells, increased intra-tumoral perforin, granzyme B, and IFN-γ mRNA, and decreased murine mammary tumor growth and pulmonary metastases in a CD8+ T lymphocyte-dependent manner.18 In an immunocompetent pancreatic cancer model, CSF-1R inhibition by another CSF-1R inhibitor PLX6134/GW2580 enhanced gemcitabine efficacy and reduced metastasis in a CD8+ T cell-dependent manner.19 Similar to these studies, it has been reported that BLZ945 in combination with immune checkpoint inhibition (anti-PD-1 and anti-PD-L1 antibodies) increased the frequency and activation of infiltrating CD8+ T cells and enabled control of spontaneously arising transgenic TH-MYCN NBs in immunocompetent mice,20 a model in which murine NB cells constitutively express a low but detectible level of MHC class I (H-2Db).21
To date, no studies have evaluated NB tumors in mice without T or NK cells but including human (and murine) monocyte-derived cells to examine the role of TAM:tumor interactions in modulation of therapeutic efficacy. Such an evaluation is of interest because, as in some other cancer types whose natural or inducible anti-cancer T lymphocyte responses are minimal or absent,22, 23 the anti-tumor activity of T lymphocytes in NB patients may be limited. Human NB tumors generally express no detectable MHC class I molecules (HLA-ABC), the lack of which impedes recognition of tumor cells by cytotoxic T cells.24, 25 In addition, newly diagnosed, stage 4 metastatic human NBs (> 18 months of age) have been shown to exhibit relatively few somatic mutations and are therefore likely to express few immunogenic molecules.26 Similarly, a meta-analysis has also indicated a relatively low frequency of mutations in high-risk primary NBs compared to multiple adult tumors.27 Finally, anti-NB functions of T lymphocytes may be suppressed by high levels of T regulatory cells in NB patients.28
To examine whether depletion of human and murine monocyte-derived cells can enhance chemotherapy without contribution from T or NK cells, we used a model of co-injection of human NB cells with human monocytes into NOD scid gamma (NSG) mice which lack T, B, and NK cells, and determined whether a CSF-1R inhibitor, BLZ945, and/or anti-CSF-1 neutralizing mAbs deplete TAMs from the NB microenvironment and enhance the response of NBs to chemotherapy.
Materials and Methods
Cell lines and patient-derived xenograft
CHLA-13629–31 and CHLA-25531–33 human NB cell lines and human NB patient-derived xenograft (PDX) COG-N-415x cells34 were derived from patients with progressive disease and were provided by the Children’s Oncology Group (COG) Cell Culture and Xenograft Repository (www.COGcell.org). CHLA-136 cells express a medium level of GD2, are chemoresistant, and have genomic amplification of MYCN as previously described.29 CHLA-255 cells express a high level of GD2, are moderately chemo-sensitive, and overexpress c-MYC mRNA and protein (Dr. Shahab Asgharzadeh, personal communication), thus representing the approximately 65% of patients with metastatic disease whose tumors lack amplification of the MYCN proto-oncogene7, 35 and the approximately 11% of undifferentiated NBs with augmented expression of c-MYC protein rather than amplified MYCN.36 Notably, patients expressing MYCN protein or c-MYC protein detected by immunofluorescence have similarly and significantly low survival.36 COG-N-415x PDX cells express a high level of GD2, have amplification of MYCN genes, and have mutated ALK (F1174L) (Dr. C. Patrick Reynolds, www.COGcell.org). We transduced the firefly luciferase (Fluc) gene into CHLA-136 (CHLA-136-Fluc) cells, CHLA-255 (CHLA-255-Fluc) cells, and freshly isolated human monocytes using a lentivirus vector, as previously described.31, 37 For specified experiments, CHLA-255 cells were transduced with the renilla luciferase gene (CHLA-255-hRL). Cells were tested for mycoplasma using MycoAlert (Lonza, Allendale, NJ, USA) and for correct identity using AmpFLSTR Identifiler PCR Amplification Kits (Applied Biosystems, Foster City, CA, USA), and were last tested at study completion. Cell lines were maintained in Iscove’s modified Dulbecco’s medium (IMDM) with 15% certified, heat-inactivated fetal bovine serum (Omega Scientific, Tarzana, CA, USA) at 37° C in a humidified incubator with 5% CO2.
Human monocyte isolation
Leukocytes were obtained as a by-product from a Trima Accel® instrument (TerumoBCT, Lakewood, CO) used to collect platelets from healthy human donors at the CHLA Blood Donor Center in accordance with a protocol approved by the Committee on Clinical Investigation. Peripheral blood mononuclear cells (PBMC) were isolated from these leukocytes or from blood using Histopaque-1077 for density centrifugation separation (Sigma-Aldrich, St. Louis, MO, USA). Monocytes were isolated from the PBMC using EasySep® negative selection monocyte isolation kits (StemCell Technologies, Seattle, WA, USA). Purity was 95-96% as determined by CD14 surface staining measured by flow cytometry.
Bioluminescent quantification of NB cell numbers
NB cell numbers were measured by adding D-luciferin potassium salt solution to co-cultures (0.035 mg/well) and immediately measuring tumor cell flux using the GloMax®-Multi detection system (Promega, Madison, WI, USA) with a one second read per well using the manufacturer’s protocol. Topotecan was tested without cyclophosphamide in vitro because cyclophosphamide requires in vivo conversion to achieve its active form.38
Cytokine assay
Conditioned media were assayed using the pre-mixed multiplex “Human Cytokine/Chemokine Panel I” (EMD Millipore, Billerica, MA, USA). Since CSF-1 was not available in the multiplex, a custom-made singleplex for CSF-1 was purchased (EMD Millipore). Assays were run according to the manufacturer’s protocols. For CSF-1 analysis, a seventh standard dilution was added below the suggested six. Data for CSF-1 was acquired on a Bio-Plex 200 instrument (BioRad Corporation, Hercules, CA, USA) and data from panel I was acquired on a Milliplex Analyzer (EMD Millipore). BioPlex software for CSF-1 (BioRad Corporation) and xPonent software for all other factors were used to fit standard curves to data obtained from analyte standards and to calculate absolute concentration (expressed as pg/mL) from respective standard curves.
Flow cytometry
Single cell suspensions were prepared from cultured cells using Puck’s EDTA, and from tumors using a gentleMACS Tissue Dissociator (Miltenyi Biotec, San Diego, CA, USA). Cells were stained with mAbs shown in Supplementary Table S1 in the dark for 45 minutes at 4° C, washed twice with PBS (without Ca++ and Mg++) containing 0.2% bovine serum albumin and 0.1% NaN3, and filtered through 50 μm mesh to remove clumps. 50,000 and 300,000 events were acquired per sample for cultured and tumor cells, respectively. Dead cells were excluded according to positive staining for DAPI. Debris was excluded by gating on forward and side scatter parameters. Doublets were excluded by gating on forward scatter area versus height. Data were acquired on an LSRII instrument with ultraviolet laser excitation of DAPI and analyzed using DIVA software version 6 (BD Biosciences, San Jose, CA, USA).
In Vivo NB models and treatment
For the intra-renal model, IL2-receptor gammanull NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice were bred in-house. On day 0, 1×106 NB cells were co-injected with 0.5×106 human monocytes (2:1 ratio) into left kidneys.39 Non-Obese Diabetic/Severe Combined Immunodeficiency (NOD/SCID) mice were bred in-house. On day -1, NSG and NOD/SCID mice were injected intraperitoneally with rat anti-mouse CD122 (200 μg/mouse) to deplete residual murine NK cells and total body irradiated (150 cGy) to deplete other hematopoietic cells. Tumor growth was assessed weekly by bioluminescence imaging using a Xenogen IVIS-200 system (Caliper Life Sciences, Hopkinton, MA, USA) 15 minutes after intraperitoneal injection of D-luciferin potassium salt solution (1.5 mg/mouse) as a substrate for firefly luciferase. Emitted photons in the range of 550-570 nm were quantified using Living Image 3.0 software (Caliper Life Sciences). Renilla luciferase was detected after intravenous injection of the substrate coelenterzine (100 μg/mouse), and blue light (480 nm) was collected. Animal experiments were performed in accordance with a protocol approved by the CHLA Institutional Animal Care and Usage Committee. Anti-CSF-1 mAbs MCS110 and 5A1 and small molecule inhibitor of CSF-1R BLZ945 were provided as a gift from Novartis (Basel, Switzerland). MCS110 is being tested in a phase II clinical trial with carboplatin and gemcitabine in triple negative breast cancer (NCT02435680) and in a phase 1b/II trial with the anti-PD-1 antibody PDR001 in advanced malignancies of adults (NCT02807844). Similarly, BLZ945 is being tested in a phase I/II clinical trial with PDR001 in advanced solid tumors of adults (NCT02829723). BLZ945 is a highly specific inhibitor of human CSF-1R that has 3200-fold selectivity relative to its closest off-target kinase c-Kit40 and also inhibits murine CSF-1R.20 BLZ945 was administered daily for 73 days, obviating rebound hematopoiesis during the treatment regimen. Cyclophosphamide and topotecan were administered i.p. at indicated times.
RNA preparation, cDNA synthesis, and Taqman® Low Density Array (TLDA) assay
Tumors were excised and cut into halves, one-half for TLDA and one-half for immunohistochemistry. The halves for TLDA were placed immediately into gentleMACS M Tubes™ containing 5 mL of TRIzol® (Invitrogen, Grand Island, NY, USA) and homogenized using the RNA_01 protocol on a gentleMACS Tissue Dissociator (Miltenyi Biotec, Auburn, CA, USA). Total RNA was prepared using TRIzol®, and RNA processed using an RNeasy® Mini Kit (Qiagen, Venlo, Netherlands). RNA Integrity Number (RIN) was obtained using an Agilent Bioanalyzer, and all specimens had a RIN ≥ 9. Reverse transcription of 2,500 ng total RNA was performed using Oligo-dT (ThermoFisher, Waltham, MA, USA) and M-MLV Reverse Transcriptase (ThermoFisher). Next, using the cDNA obtained from reverse transcription, the TLDA assay quantified the expression of 48 genes (5 NB-associated, 39 microenvironment, 4 housekeeping) in one reservoir of a custom-designed microfluidic card (Applied Biosystems, Carlsbad, CA, USA), enabling assays for all genes to be run simultaneously on a single TLDA card using a 7900HT instrument (ThermoFisher). Probe and primer sets for macrophage-associated genes are listed in Supplementary Table S2. Values for Ct represent the amplification cycle number at which fluorescence passed the threshold; ΔCt values are the Ct of the target gene minus the Ct of the mean of four housekeeping genes.
Immunohistochemistry and tissue preparation
Two or more mice from each group were euthanized by CO2 inhalation on specified days. Half of each tumor was fixed in 10% neutral buffered formalin for 24 hours at 4°C. Samples were embedded in paraffin blocks from which 5 μm sections were prepared and stained with hematoxylin, eosin and anti-CD163 mAb clone MRQ-26 (Leica Biosystems, Buffalo Grove, IL, USA) for human monocyte-derived cells or anti-F4/80 mAb clone CI:A3-1 (Abcam, Cambridge, MA, USA) for mouse macrophages.
Statistical analysis
In general, data points with intervals are presented as means ± standard error of the mean (SEM). In the subcutaneous model, mouse survival time was defined as the number of days from tumor injection until the end of study (survivors) or until euthanized due to weight loss (> 15%), inactivity/inability to move, hunched back, limping/abnormal gait, or tumor-mediated abdominal distension. In the intra-renal model, mouse survival was defined as number of days from tumor injection until euthanized for reasons above. Deaths not satisfying these criteria (e.g., due to imaging anesthesia) were right censored in analysis. For bioluminescence, flux values were normalized to baseline flux, and area under the curve (AUC) was computed, with AUC values right censored for mice that died before the end of observation period. Distribution of survival times and AUC are adequately described by a lognormal distribution, hence both endpoints were log10 transformed for analysis. Ordinary least square regression or interval regression (when censoring was present) of log10 survival and AUC on a grouping factor variable was used. Computations were performed using Stata 11 (StataCorp 2009, software release 11, College Station, TX, USA).
Results
Cultures of NB cells with or without monocytes release pro-tumor cytokines
We have previously shown that TAMs are strongly associated with poor survival of patients with NB that lack MYCN amplification7 and that TAMs promote NB via STAT3 phosphorylation and c-Myc up-regulation.8 To evaluate other potential mechanisms through which monocytes might affect NB cells, we determined the release of cytokines and chemokines induced by addition of monocytes to cultures of NB cells, thereby evaluating the potential efficacy of targeting one molecule released by monocytes or TAMs. Interaction of monocytes with CHLA-255 or CHLA-136 NB cell lines resulted in large increases in IL-6, IL-8/CXCL8, and GRO/CXCL1 compared to either cell type alone (Fig. 1a). IL-6 is known to promote NB cell survival,32 IL-8/CXCL8 can promote angiogenesis,40 and GRO/CXCL1 can form a paracrine network linking chemoresistance and metastasis.41, 42 There were apparent increases in anti-tumor cytokines IL-12p40, IL-12p70, IL-15, and TNFα; however, their concentrations remained relatively low (Fig. 1a, right columns). Similarly, there was an apparent increase in G-CSF, but the concentration was high for only one of the two NB cell lines. We also determined if CSF-1 is released from human NB cells, human monocytes, or their co-cultures. CSF-1 protein was released at a baseline of approximately 40-50 pg/ml by CHLA-255-Fluc and CHLA-136-Fluc cell lines, whether alone or in co-cultures with monocytes (1:1 ratio) (Fig. 1b). We found that human NB cells lack surface CSF-1R expression on their surface, whereas human monocytes expressed CSF-1R as expected (Supporting Information Fig. 1a, 1b). Interestingly, addition of topotecan (10 ng/ml) to NB cells resulted in 2-3-fold increases in release of CSF-1, regardless of whether the CSF-1R inhibitor BLZ945 (200 nM) or monocytes were added. Monocytes cultured alone did not increase their release of CSF-1 in response to topotecan. The effect of the anti-human CSF-1 mAb MCS110 was not examined in this experiment because MCS110 inhibited the ability of the Luminex assay to detect CSF-1. These experiments demonstrated that multiple pro-tumor factors are released in co-cultures of NB cells and monocytes, suggesting that targeting individual factors may not be feasible whereas targeting monocytes/macrophages themselves may be an alternative strategy. These experiments also demonstrated that CSF-1 can be released by NB cells and that NB release of CSF-1 is increased by topotecan.
Figure 1.
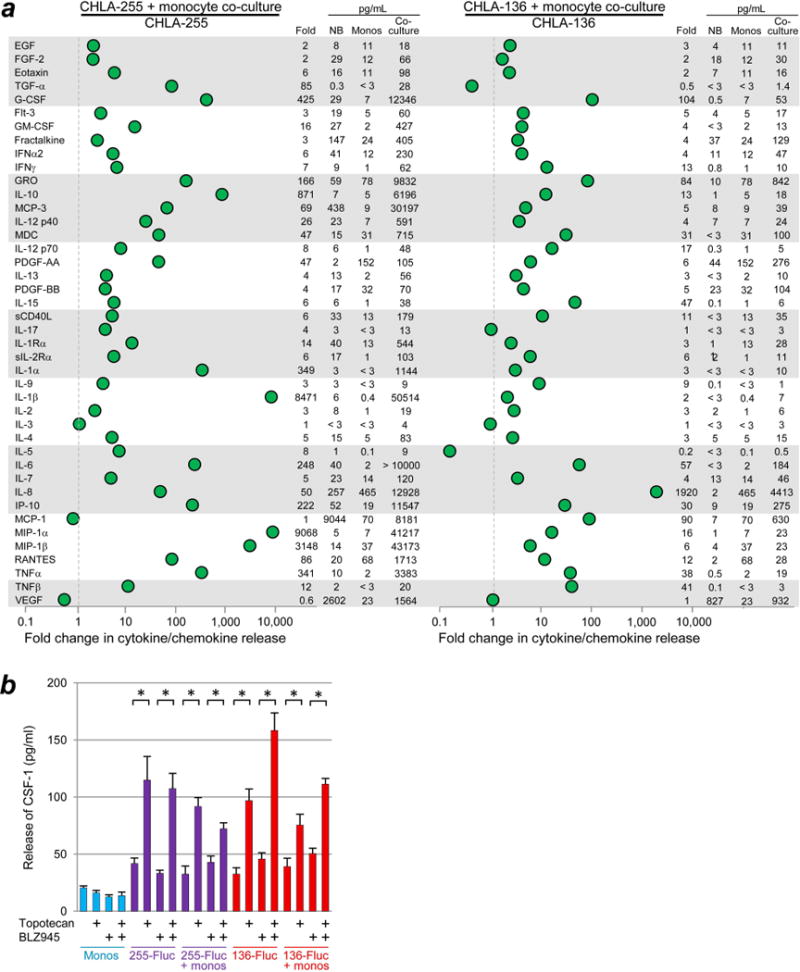
Release of cytokines and chemokines from co-cultures of human monocytes and NB cells. (a) Monocytes and NB cell line CHLA-255 or CHLA-136 were co-cultured for 72 hours at 1:1 ratio (106 of each cell type) and their medium was analyzed using a multiplex Luminex assay. Dotted lines are positioned at 1-fold to indicate no change in concentration. Fold change of cytokines and chemokines secreted from co-cultures is shown numerically on the right and was calculated as follows: (concentration in co-cultures)/(concentration in cultures of NB cells alone). For the purpose of this calculation, values above or below the range of the assay were assigned the value of the upper or lower limit. The concentrations (pg/mL) of cytokines or chemokines secreted from NB cells, monocytes, and their co-culture are also shown on the right of each panel. (b) Effect of topotecan on release of CSF-1 from NB cell lines ± monocytes. NB cells were cultured without or with monocytes (direct contact, 1×104 per well for both cell types, 1:1 ratio) ± topotecan (10 ng/ml) ± CSF-1R inhibitor BLZ945 (200 nM) in 96-well plates for 72 hours, and then CSF-1 in supernatants was quantified using a singleplex Luminex assay. Significance bars represent SEM; * denotes p < 0.02.
Based on the experiments above, we chose not to target individual pro-tumor cytokines but to target monocytes and macrophages themselves, exploiting their reliance on CSF-1/CSF-1R for recruitment and survival. In co-cultures of human monocytes with CHLA-255-Fluc cells (plated at a 1:1 ratio for 120 hours), monocytes persisted at a low frequency and this frequency was decreased by treatment with MCS110 (10 μg/mL) plus BLZ945 (200 nM); similarly, in co-cultures with CHLA-136-Fluc cells, monocytes persisted at a relatively higher frequency that was also decreased by CSF-1 blockade (Supporting Information Fig. 2a). Topotecan, which is active against neuroblastoma,43, 44 was added (10 ng/ml) 48 hours after beginning the co-cultures but had only modest effects on the frequency of monocytes.
Having found that CSF-1R blockade could reduce the frequency of monocytes, we examined if it could affect NB cell numbers. In co-cultures with monocytes, the combination of MCS110 plus BLZ945 reduced the number of CHLA-255-Fluc and CHLA-136-Fluc NB cells, both in the absence of topotecan and in the presence of a low dose of topotecan (1 ng/ml) (Supporting Information Fig. 2b).
Treatment of subcutaneous CHLA-255-Fluc NBs in NOD/SCID mice with anti-human CSF-1 mAb MCS110 decreases human monocyte-derived CD163+ cells but does not improve chemotherapy
We hypothesized that depletion of human monocyte-derived cells from subcutaneous tumors using MCS110 alone might enhance chemotherapy with cyclophosphamide plus topotecan, drugs used in induction therapy by the Children’s Oncology Group.43 As a control, the effect of MCS110 on monocytes was determined after subcutaneous injection of a 2:1 mixture of renilla luciferase-labeled CHLA-255 cells and firefly luciferase-labeled human monocytes into the shoulders of NOD/SCID mice. MCS110 administered alone once weekly reduced intra-tumor human monocyte-derived cells as demonstrated by three endpoints. First, Fluc-labeled human monocytes exhibited decreased flux in the MCS110-treated group as compared to untreated control (Supporting Information Fig. 3a). Second, immunohistochemistry staining showed a decrease in intra-tumor human CD163+ cells in the MCS110-treated group (Supporting Information Fig. 3b). Third, mRNA associated with human macrophage-associated marker genes CD86, CD163, CD40L, CD16, CSF1, and CSF1R showed lower expression in tumors of the MCS110-treated group (Supporting Information Fig. 3c). Unexpectedly though, as measured by bioluminescent imaging or mouse survival, MCS110 did not significantly improve chemotherapy of subcutaneous CHLA-255-Fluc tumors (Fig. 2a, 2b) even though it had reduced the number of human monocyte-derived cells and was detectible in serum before and during chemotherapy (Supporting Information Fig. 4).
Figure 2.
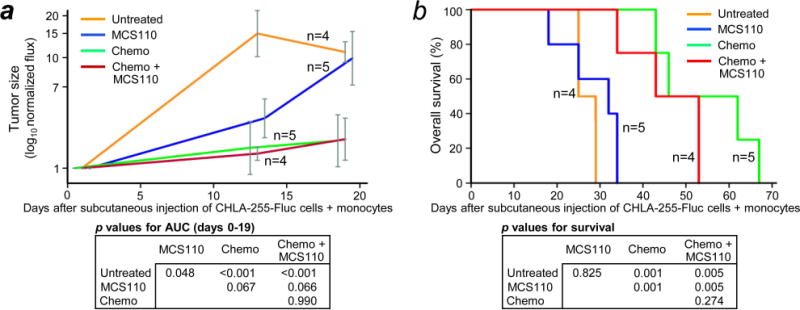
Effect of anti-human CSF-1 mAb MCS110 on NB response to chemotherapy in vivo. (a) Effect of mAb MCS110 and chemotherapy on tumors formed by co-injection of unlabeled human monocytes and firefly luciferase-expressing CHLA-255-Fluc NB cells subcutaneously in both left and right shoulders (enabling two tumor measurements per mouse). 4×106 CHLA-255-Fluc NB cells and 2×106 human monocytes (2:1 ratio) were injected on day 0. MCS110 (10 mg/kg injected intraperitoneally) was given twice a week, starting on day 1 until sacrificed due to symptomatic tumor progression. Chemotherapy (25 mg/kg/day cyclophosphamide plus 0.1 mg/kg/day topotecan intraperitoneally) was given on days 8-12, 29-33, and 48-52. Tumor size (log normalized flux) was determined at the indicated time points and plotted as mean ± SEM. P values for pairwise comparisons of area under the growth curve (AUC) for each group are shown below. (b) Kaplan-Meier survival curves for mice shown in (a).
Treatment of subcutaneous CHLA-255-Fluc NBs in NOD/SCID mice with anti-CSF-1 mAbs MCS110 and 5A1 combined with human/mouse CSF-1R inhibitor BLZ945 decreases the frequency of human CD14+ and CD163+ cells as well as mouse F4/80+ TAMs, and improves chemotherapy
Given that depleting human monocyte-derived cells with anti-human CSF-1 mAb MCS110 did not improve chemotherapy, we determined if mouse TAMs were present in NBs and if targeting them might improve chemotherapy. Human NBs growing in nu/nu mice have been reported to include infiltrating mouse TAMs,45 and consistent with this observation, we demonstrated with flow cytometry that subcutaneous CHLA-255-Fluc tumors growing in NOD/SCID mice included murine macrophages (Ly6G−CD11b+F4/80+) as well as human monocyte-derived cells (human CD45+CD14+) cells, both of which lacked GD2 expression (GD2−) (Fig. 3a, 3b, 3c). The frequency of these F4/80+ mouse cells and CD14+ human cells was reduced 3.4-fold and 2.8-fold (p = 0.01 and 0.001, respectively) by adding the inhibitor of human and mouse20 CSF-1R BLZ945 plus the anti-mouse CSF-1 mAb 5A1 to MCS110 treatment (triple agent CSF-1R blockade) (Fig. 3d). Most human CD14+ cells expressed CD163, consistent with a pro-tumor phenotype (Fig. 3e). Immunohistochemistry similarly showed that these tumors contained F4/80+ mouse TAMs whose frequency could be decreased by triple agent blockade (Fig. 3f). Similarly, treatment of these NBs with MCS110 alone or with triple agent blockade reduced CD163+ human monocyte-derived cells (Fig. 3f). Combining triple agent blockade with cyclophosphamide and topotecan was well tolerated and, importantly, it increased suppression of NB growth compared to chemotherapy alone as measured by bioluminescence (Fig. 3g and 3h) (p < 0.001).
Figure 3.
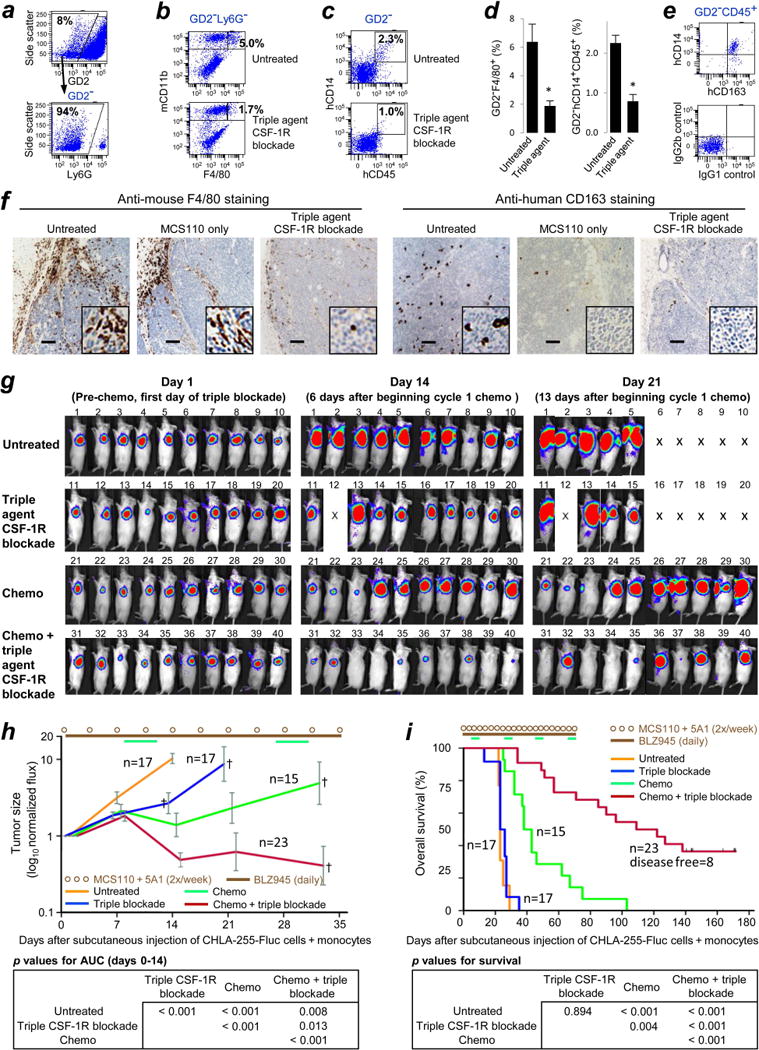
Effect of triple agent CSF-1R blockade on intra-tumor mouse F4/80+ TAMs, on human CD14+ and CD163+ monocyte-derived cells, and on response of CHLA-255-Fluc NBs to chemotherapy. 4×106 NB cells and 2×106 human monocytes (2:1) were co-injected subcutaneously into shoulders of irradiated NOD/SCID mice on day 0. Commencing from day 1, anti-mouse CSF-1 mAb 5A1 (10 mg/kg) and anti-human CSF-1 mAb MCS110 (10 mg/kg) were co-administered intraperitoneally 2×/week, and the CSF-1R inhibitor BLZ945 was administered daily at 160 mg/kg by oral gavage. (a-e) Analysis of subcutaneous CHLA-255-Fluc tumors on day 21 by eight-color flow cytometry. All antibodies are described in Supporting Information Table 1. (a) Cells from dissociated tumors were gated on the subpopulation lacking the NB antigen GD2 (upper panel). For analyses of murine components in tumors, GD2− cells were gated on Ly6G− cells to exclude mouse granulocytes (lower panel). (b) GD2−Ly6G− cells were examined for co-expression of mouse CD11b (mCD11b) and F4/80. The percentages shown represent frequencies of Ly6G−F4/80+CD11b+ murine TAMs among all GD2− cells, and were calculated as follows: (frequency of cells in the Ly6G−F4/80+CD11b+ gate) – (frequency of cells in the same gate when applied to the corresponding isotype-matched negative control). The strategy for positioning the F4/80 gate is shown in Supporting Information Fig. 5. (c) GD2− cells were examined for co-expression of human CD45 (hCD45) and hCD14. Percentages of hCD45+hCD14+ monocyte-derived cells were calculated as above. The absence of outgrowth of non-monocytic cells among hCD45+ cells shows that the purity of MACS-enriched monocytes was maintained in vivo. (d) Frequencies of murine TAMs (left) and human monocyte-derived cells (right) among all GD2− cells, as gated in A-C. Means + SEM are shown; n = 4 tumors examined per treatment group; * denotes ≤ p 0.01. (e) Co-expression of hCD14 and the M2 marker hCD163 on GD2−hCD45+ cells. The corresponding isotype-matched negative control is shown in the lower panel. (f) The presence of F4/80+ and CD163+ cells in CHLA-255-Fluc tumors from mice treated with MCS110 alone or with MCS110 plus 5A1 and BLZ945 (triple agent CSF-1R blockade) as determined on day 15 by immunohistochemistry. Images are representative of three tumors per group. No chemotherapy was administered. Note that human CD163+ cells were at a low frequency even in untreated tumors, as human monocytes did not persist for many weeks in the mouse model. Insets in the lower right of each image depict 5× digital enlargement processed using a sharpen filter in ImageJ software to enhance contrast. Scale bars represent 100 μm. (g) Bioluminescent images showing the effect of triple agent CSF-1R blockade on cyclophosphamide plus topotecan chemotherapy. Mice from two of four replicate experiments are shown. Chemotherapy doses were as in Fig. 2a and were given on days 8-12, 27-31, 48-52 and 69-73. MCS110 and 5A1 were co-administered intraperitoneally 2×/week, and BLZ945 was administered daily by gavage. Mouse 12 died of unknown causes on day 7 and other “X”s indicate mice that were sacrificed due to progressive tumor growth. (h) The sizes of tumors in mice shown in panel G as well as in mice from two additional experiments were measured and plotted as means ± SEM. At the top is the schedule for administration 2×/weekly of MCS110 plus 5A1 (brown circles) and daily BLZ945 (brown line), as described by brown lettering in the inset legend. Similarly shown is the schedule of chemotherapy (green lines on top). Crosses (†) indicate tumor-related death of one or more mice at indicated time points. Area under the curve (AUC) was measured from day 0 to 14, since day 14 was the last imaged time point in which all mice (except mouse 12) remained alive; differences at every time point beyond day 14 were significant. Curves for untreated and the triple blockade groups terminate at day 14 and day 21, respectively, owing to death of mice before the next measurement. (i) Kaplan-Meier survival curves summarizing four replicate experiments. All therapy (chemotherapy and triple agent blockade) was stopped after day 73.
Combining triple agent blockade with chemotherapy also resulted in greater mouse survival compared to chemotherapy alone, with mean survival times of 125 and 53 days, respectively (p < 0.001). Eight of 23 mice (35%) were disease-free when sacrificed on days 164-171 (Fig. 3i). The Wald statistical test indicated interaction (synergy) between chemotherapy and triple agent blockade (p < 0.001). Chemotherapy alone increased survival compared to either untreated control or triple agent alone groups (p < 0.001) (Fig. 3i lower panel). The untreated control and triple agent CSF-1R blockade alone groups were not statistically different (p = 0.894), and had mean survivals of 36 and 38 days, respectively. These experiments show that triple agent blockade of mouse F4/80+ and human CD14+ and CD163+ cells improves chemotherapy.
Having found that triple agent CSF-1R blockade can enhance chemotherapy against subcutaneous CHLA-255-Fluc tumors in NOD/SCID mice which have impaired T and B cell development and deficient natural killer (NK) cell function, we examined whether it might enhance chemotherapy against intra-renal NB cells growing in NSG mice which completely lack T, NK, and B lymphocytes. CHLA-136-Fluc cells were co-injected with human monocytes into the left kidney. Similar to the subcutaneous NB model, triple agent CSF-1R blockade alone had no effect on the growth of intra-renal NBs, and chemotherapy alone significantly reduced tumor growth by day 21 after tumor cell injection compared to untreated controls (Supporting Information Fig. 6a, 6b). Combining chemotherapy with triple agent CSF-1R blockade further reduced growth and increased survival as compared to chemotherapy alone with mean survival times of 93 and 72 days, respectively (Supporting Information Fig. 6c).
Treatment of intra-renal CHLA-255-Fluc NBs in NSG mice with CSF-1R inhibitor BLZ945 alone improves chemotherapy
Next, we sought to identify the most active single agent for potential clinical translation. NSG mice co-injected in the left kidney with CHLA-255-Fluc cells and human monocytes were treated with chemotherapy plus one of the following: i) triple agent blockade, ii) BLZ945 alone, iii) the two anti-CSF-1 mAbs (MCS110 and 5A1), iv) neither BLZ945 nor the mAbs. The combination of triple agent blockade with chemotherapy was superior to chemotherapy alone in inhibiting growth (p = 0.009), as was the combination of BLZ945 with chemotherapy (p = 0.003) (Fig. 4a). Unexpectedly, the combination of chemotherapy with MCS110 plus 5A1 was not significantly different from chemotherapy alone (p = 0.890), and was less effective than chemotherapy combined with either triple agent blockade (p = 0.006) or BLZ945 (p = 0.002).
Figure 4.
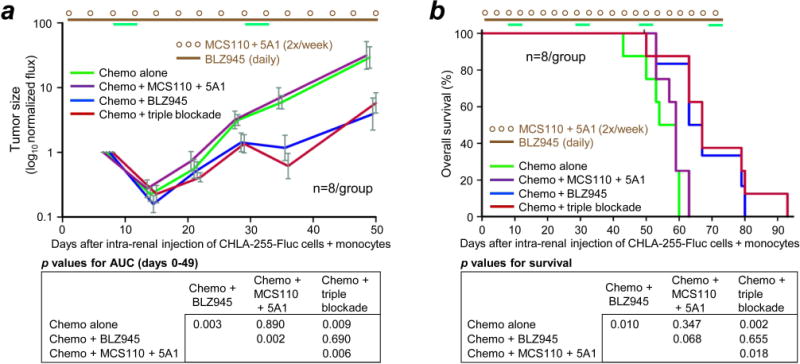
Effect of BLZ945 on chemotherapy of NB tumors formed by co-injection of CHLA-255-Fluc cells with monocytes into the kidney of NSG mice. (a and b) CHLA-255-Fluc cells and human monocytes (2:1) were co-injected into the left kidney, all mice received chemotherapy, and specified mice received anti-CSF-1 mAbs MCS110 and 5A1 together, CSF-1R inhibitor BLZ945 alone, or the combination of all three (triple blockade). Dosing and schedule of mAbs and BLZ945 was as in Fig. 3. Chemotherapy dosing was as in Fig. 2a and administered on days 8-12, 29-33, 48-52 and 69-73. All therapy stopped after day 73. One mouse from the chemotherapy group and two from the chemotherapy + BLZ945 group died accidentally during anesthesia and were censored.
As with tumor growth, mouse survival was increased by the combination of chemotherapy with BLZ945 (p = 0.010) or with triple agent blockade (p = 0.002) compared to chemotherapy alone (Fig. 4b). The combination of chemotherapy with BLZ945 was not different from chemotherapy with triple agent blockade (p = 0.655), and the combination of chemotherapy with anti-CSF-1 mAbs MCS110 plus 5A1 was not different from chemotherapy alone (p = 0.347). Taken together, these data suggest that BLZ945 as a single agent improves the ability of chemotherapy to inhibit NB growth and prolong survival.
Treatment of intra-renal PDX COG-N-415x NB in NSG mice with CSF-1R inhibitor BLZ945 alone improves chemotherapy
Having identified BLZ945 as an active single agent in combination with chemotherapy in the CHLA-255-Fluc model, we sought to extend the above findings in a model involving a NB patient-derived xenograft. COG-N-415x PDX cells (passage 4) and human monocytes were co-injected into the left kidney of NSG mice. Survival was the only endpoint, since PDX cells are not cultured and therefore are not labeled with luciferase. The mean survival of the untreated group and the BLZ945 alone group was 34 days, that of the chemotherapy alone group was 55 days, and that of the BLZ945 plus chemotherapy group was 92 days, the latter representing a significant enhancement compared to the chemotherapy alone group and other groups (p < 0.001) (Fig. 5). The Wald test demonstrated interaction between BLZ945 and chemotherapy (p < 0.001). Thus, BLZ945 alone synergistically improved chemotherapy of the PDX in the absence of T, NK, and B cells.
Figure 5.
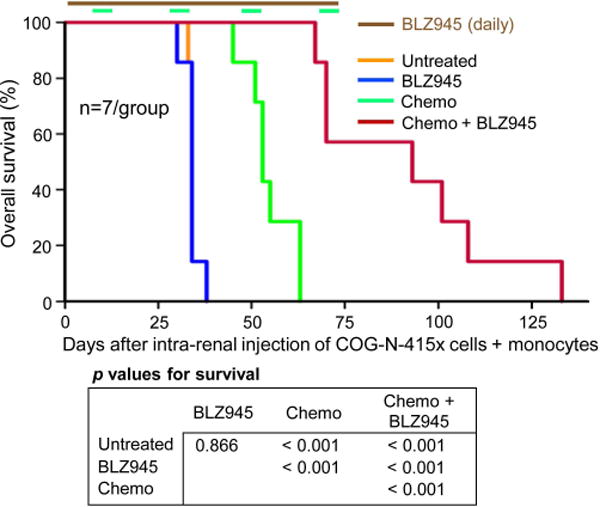
Effect of BLZ945 on chemotherapy of NB tumors formed by co-injection of patient-derived xenograft COG-N-415x cells with monocytes into the kidney of NSG mice. Kaplan-Meier curves for mice with PDX COG-N-415x NBs treated ± BLZ945 (160 mg/kg/day, oral gavage) without or with chemotherapy. Dosing and schedule of mAbs and BLZ945 was as in Fig. 3. Chemotherapy dosing was as in Fig. 2a and administered on days 8-12, 29-33, 48-52 and 69-73. All therapy stopped after day 73.
Discussion
Gene expression and immunohistochemical analyses have shown an association between macrophage infiltration and both extent of disease and progression-free survival of NB patients with MYCN-non-amplified disease.7 Macrophage migration and survival is regulated by CSF-1,12–14 and we demonstrate that human NB cells can release CSF-1 protein in vitro. Furthermore, we show that topotecan, which is clinically active against NB,43, 44, 46 can significantly increase the release of CSF-1 protein from NB cells, consistent with reports of increased CSF-1 mRNA expression in a breast cancer model after treatment with paclitaxel18 and in a prostate cancer model after ionizing radiation.47 In contrast with evidence for TAMs associating with outcome in NB, there is no published clinical evidence for an adaptive T lymphocyte response against human NBs.48 T lymphocyte responses in NB patients may be limited by absent expression of MHC class I molecules and by a low frequency of somatic mutations in the tumor cells.24–27 We therefore used NSG and NOD/SCID mice as models for deficient T lymphocyte responses. Co-injecting these mice with human monocytes and human NB cell lines or a PDX, we demonstrate that inhibition of CSF-1R by combination of BLZ945 with anti-CSF-1 mAbs MCS110 and 5A1 decreases the intra-tumor presence of human CD14+ and CD163+ cells as well as mouse F4/80+ cells. Importantly, we show that BLZ945 monotherapy can also improve chemotherapy of NB, without requiring T lymphocytes.
Multiple mechanisms may be operative whereby TAMs promote NB growth and resistance to chemotherapy. Experimentally, we have shown that TAMs promote NB growth via STAT3 phosphorylation and c-Myc up-regulation.8 This suggests that depletion of TAMs from the NB microenvironment may reduce pSTAT3 and c-Myc in NB cells. We show here that co-culturing monocytes with NB cells results in many-fold increases in IL-6, IL-8, and GRO, and thus depletion of TAMs may reduce these pro-tumor cytokines/chemokines in the tumor milieu. Together, these effects may sensitize NBs to chemotherapy, independent of anti-tumor functions of T lymphocytes. Additional benefit from CSF-1R blockade beyond what is observed in our experimental models might potentially occur in patients who mount anti-NB T lymphocyte responses.
Although CSF-1R blockade without chemotherapy was insufficient to extend survival of NB-bearing immunodeficient mice, others have found that CSF-1R blockade in immune competent mice bearing tumors was effective without chemotherapy. One such example is found in the immunocompetent TH-MYCN transgenic NB model in which murine NB cells, unlike patient NBs, constitutively express a low level of MHC class I molecules (MHC class Ilow),21 potentially enabling anti-NB responses by T lymphocytes. In this model, BLZ945 as a single agent was shown to decrease tumor growth, and this effect was reported to be larger when combined with immune checkpoint blockade.20 Similarly, single agent BLZ945 reduced tumor growth in immune competent mouse models of mammary and cervical carcinoma and of glioblastoma multiforme.17, 20, 49, 50 In the immune competent models of mammary and cervical carcinoma17 and MHC class Ilow NB,20 single agent BLZ945 was associated with increased CD8+ T cell infiltration, potentially explaining the anti-tumor effect. However, given the unclear contribution of T lymphocytes in the clinical therapy of high-risk NB patients488 as well as the undetectable expression of MHC class I molecules and low frequency of somatic mutations in tumors of NB patients,24–27 there may be advantages to identifying treatment modalities for NB that do not rely on T lymphocyte responses. Our findings suggest that NB patients who have ineffective anti-NB T lymphocyte responses might benefit from combination of chemotherapy with BLZ945.
We unexpectedly observed that neutralization of both human and mouse CSF-1 using anti-CSF1 mAbs MCS110 plus 5A1 combined with chemotherapy had no effect on intra-renal tumor growth or survival of NSG mice (Fig. 4a, 4b). It is possible that CSF-1R was bound not only by CSF-1 but also by murine IL-34,50 rendering neutralization of CSF-1 alone insufficient to inhibit signaling through CSF-1R in the NSG mouse model. It was beyond the scope of our study to evaluate a combination of an anti-IL-34 mAb with MCS110 and 5A1. An alternative explanation could be insufficient depletion of continuously infiltrating murine macrophages by anti-mouse CSF-1 mAb 5A1 administered twice weekly. Given these alternate explanations, our findings do not exclude the possibility that MCS110 might have a beneficial effect in humans.
Our findings provide direct evidence that monocyte-derived cells in the NB microenvironment can be important determinants of sensitivity to chemotherapy. CSF-1R blockade that included daily administration of BLZ945 significantly depleted human and mouse monocyte-derived cells from NBs and improved the efficacy of chemotherapy against MYCN-non-amplified and MYCN-amplified NBs in immune deficient mice lacking T lymphocytes. These findings may have implications for other cancers whose natural or inducible anti-cancer T lymphocyte responses are limited.22, 23
Supplementary Material
What’s New.
Monocytes and macrophages rely on CSF-1/CSF-1R for recruitment and survival. This work shows that neuroblastoma cells increase their release of CSF-1 after treatment with topotecan, and using models of co-injection of human neuroblastoma cells with human monocytes into immunodeficient mice, demonstrates that BLZ945, a small molecule inhibitor of CSF-1R, enhances chemotherapy in the absence of T-lymphocytes. These findings have implications for therapy of neuroblastoma and other cancers in which anti-cancer T-lymphocyte responses are limited.
Acknowledgments
Grant Support: This work was supported in part by grants P01 CA81403 (R.C. Seeger) and R01 CA182633 (R.C. Seeger) from the National Cancer Institute, and from the T.J. Martell Foundation (R.C. Seeger).
Footnotes
Conflict of Interest: Dylan Daniel worked previously for Novartis, the manufacturer and supplier of MCS110, 5A1, and BLZ945, but now has no potential conflict of interest. All other authors declare no potential conflict of interest.
References
- 1.Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27:462–72. doi: 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017 doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, Tsai MF, Chen CH, Yang PC. Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol. 2005;23:953–64. doi: 10.1200/JCO.2005.12.172. [DOI] [PubMed] [Google Scholar]
- 4.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–65. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 5.Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ, Wu WZ, Wang L, Tang ZY, Sun HC. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol. 2008;26:2707–16. doi: 10.1200/JCO.2007.15.6521. [DOI] [PubMed] [Google Scholar]
- 6.Farinha P, Masoudi H, Skinnider BF, Shumansky K, Spinelli JJ, Gill K, Klasa R, Voss N, Connors JM, Gascoyne RD. Analysis of multiple biomarkers shows that lymphoma-associated macrophage (LAM) content is an independent predictor of survival in follicular lymphoma (FL) Blood. 2005;106:2169–74. doi: 10.1182/blood-2005-04-1565. [DOI] [PubMed] [Google Scholar]
- 7.Asgharzadeh S, Salo JA, Ji L, Oberthuer A, Fischer M, Berthold F, Hadjidaniel M, Liu CW, Metelitsa LS, Pique-Regi R, Wakamatsu P, Villablanca JG, et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J Clin Oncol. 2012;30:3525–32. doi: 10.1200/JCO.2011.40.9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hadjidaniel MD, Muthugounder S, Hung LT, Sheard MA, Shirinbak S, Chan RY, Nakata R, Borriello L, Malvar J, Kennedy RJ, Iwakura H, Akamizu T, et al. Tumor-associated macrophages promote neuroblastoma via STAT3 phosphorylation and up-regulation of c-MYC. Oncotarget. 2017;8:91516–29. doi: 10.18632/oncotarget.21066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–44. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 10.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–83. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 11.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–9. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 12.Tang R, Beuvon F, Ojeda M, Mosseri V, Pouillart P, Scholl S. M-CSF (monocyte colony stimulating factor) and M-CSF receptor expression by breast tumour cells: M-CSF mediated recruitment of tumour infiltrating monocytes? J Cell Biochem. 1992;50:350–6. doi: 10.1002/jcb.240500403. [DOI] [PubMed] [Google Scholar]
- 13.Hume DA, MacDonald KP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119:1810–20. doi: 10.1182/blood-2011-09-379214. [DOI] [PubMed] [Google Scholar]
- 14.Aharinejad S, Salama M, Paulus P, Zins K, Berger A, Singer CF. Elevated CSF1 serum concentration predicts poor overall survival in women with early breast cancer. Endocrine-related cancer. 2013;20:777–83. doi: 10.1530/ERC-13-0198. [DOI] [PubMed] [Google Scholar]
- 15.Chambers SK, Kacinski BM, Ivins CM, Carcangiu ML. Overexpression of epithelial macrophage colony-stimulating factor (CSF-1) and CSF-1 receptor: a poor prognostic factor in epithelial ovarian cancer, contrasted with a protective effect of stromal CSF-1. Clin Cancer Res. 1997;3:999–1007. [PubMed] [Google Scholar]
- 16.Groblewska M, Mroczko B, Wereszczynska-Siemiatkowska U, Mysliwiec P, Kedra B, Szmitkowski M. Serum levels of granulocyte colony-stimulating factor (G-CSF) and macrophage colony-stimulating factor (M-CSF) in pancreatic cancer patients. Clin Chem Lab Med. 2007;45:30–4. doi: 10.1515/CCLM.2007.025. [DOI] [PubMed] [Google Scholar]
- 17.Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, Daniel D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8+ T cells. Oncoimmunology. 2013;2:e26968. doi: 10.4161/onci.26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, Rugo HS, Hwang ES, et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, Hewitt S, Udupi GM, et al. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Res. 2013;73:1128–41. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mao Y, Eissler N, Le Blanc K, Johnsen JI, Kogner P, Kiessling R. Targeting suppressive myeloid cells potentiates checkpoint inhibitors to control spontaneous neuroblastoma. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-1912. [DOI] [PubMed] [Google Scholar]
- 21.Carlson LM, Rasmuson A, Idborg H, Segerstrom L, Jakobsson PJ, Sveinbjornsson B, Kogner P. Low-dose aspirin delays an inflammatory tumor progression in vivo in a transgenic mouse model of neuroblastoma. Carcinogenesis. 2013;34:1081–8. doi: 10.1093/carcin/bgt009. [DOI] [PubMed] [Google Scholar]
- 22.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, Palaskas N, Rodriguez GA, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017;7:188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–30. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 24.Seeger RC. Immunology and immunotherapy of neuroblastoma. Seminars in cancer biology. 2011;21:229–37. doi: 10.1016/j.semcancer.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prigione I, Corrias MV, Airoldi I, Raffaghello L, Morandi F, Bocca P, Cocco C, Ferrone S, Pistoia V. Immunogenicity of human neuroblastoma. Ann N Y Acad Sci. 2004;1028:69–80. doi: 10.1196/annals.1322.008. [DOI] [PubMed] [Google Scholar]
- 26.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J, Lawrence MS, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–84. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morandi F, Croce M, Cangemi G, Barco S, Rigo V, Carlini B, Amoroso L, Pistoia V, Ferrini S, Corrias MV. IL-10 and ARG-1 concentrations in bone marrow and peripheral blood of metastatic neuroblastoma patients do not associate with clinical outcome. Journal of immunology research. 2015;2015:718975. doi: 10.1155/2015/718975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keshelava N, Zuo JJ, Chen P, Waidyaratne SN, Luna MC, Gomer CJ, Triche TJ, Reynolds CP. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001;61:6185–93. [PubMed] [Google Scholar]
- 30.Keshelava N, Davicioni E, Wan Z, Ji L, Sposto R, Triche TJ, Reynolds CP. Histone deacetylase 1 gene expression and sensitization of multidrug-resistant neuroblastoma cell lines to cytotoxic agents by depsipeptide. J Natl Cancer Inst. 2007;99:1107–19. doi: 10.1093/jnci/djm044. [DOI] [PubMed] [Google Scholar]
- 31.Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, Xu Y, Wang L, Sposto R, Kim ES, Asgharzadeh S, Seeger RC. TGFbetaR1 Blockade with Galunisertib (LY2157299) Enhances Anti-Neuroblastoma Activity of the Anti-GD2 Antibody Dinutuximab (ch14.18) with Natural Killer Cells. Clin Cancer Res. 2017;23:804–13. doi: 10.1158/1078-0432.CCR-16-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ara T, Nakata R, Sheard MA, Shimada H, Buettner R, Groshen SG, Ji L, Yu H, Jove R, Seeger RC, DeClerck YA. Critical role of STAT3 in IL-6-mediated drug resistance in human neuroblastoma. Cancer Res. 2013;73:3852–64. doi: 10.1158/0008-5472.CAN-12-2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, Malvar J, Blavier L, Shimada H, Asgharzadeh S, Seeger RC, DeClerck YA. Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res. 2017;77:5142–57. doi: 10.1158/0008-5472.CAN-16-2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopez-Barcons L, Maurer BJ, Kang MH, Reynolds CP. P450 inhibitor ketoconazole increased the intratumor drug levels and antitumor activity of fenretinide in human neuroblastoma xenograft models. Int J Cancer. 2017;141:405–13. doi: 10.1002/ijc.30706. [DOI] [PubMed] [Google Scholar]
- 35.Asgharzadeh S, Pique-Regi R, Sposto R, Wang H, Yang Y, Shimada H, Matthay K, Buckley J, Ortega A, Seeger RC. Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification. J Natl Cancer Inst. 2006;98:1193–203. doi: 10.1093/jnci/djj330. [DOI] [PubMed] [Google Scholar]
- 36.Wang LL, Teshiba R, Ikegaki N, Tang XX, Naranjo A, London WB, Hogarty MD, Gastier-Foster JM, Look AT, Park JR, Maris JM, Cohn SL, et al. Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children’s Oncology Group study. Br J Cancer. 2015;113:57–63. doi: 10.1038/bjc.2015.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Wu HW, Sheard MA, Sposto R, Somanchi SS, Cooper LJ, Lee DA, Seeger RC. Growth and activation of natural killer cells ex vivo from children with neuroblastoma for adoptive cell therapy. Clin Cancer Res. 2013;19:2132–43. doi: 10.1158/1078-0432.CCR-12-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siegel MM, Chung HS, Rucker N, Siegel SE, Seeger RC, Isaacs H, Jr, Benedict WF. In vitro and in vivo preclinical chemotherapy studies of human neuroblastoma. Cancer TreatRep. 1980;64:975–9. [PubMed] [Google Scholar]
- 39.Patterson DM, Shohet JM, Kim ES. Preclinical models of pediatric solid tumors (neuroblastoma) and their use in drug discovery. Current protocols in pharmacology/editorial board, SJ Enna. 2011:7. doi: 10.1002/0471141755.ph1417s52. Chapter 14: Unit 14. [DOI] [PubMed] [Google Scholar]
- 40.Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170:3369–76. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 41.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, Norton L, Brogi E, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165–78. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bandapalli OR, Ehrmann F, Ehemann V, Gaida M, Macher-Goeppinger S, Wente M, Schirmacher P, Brand K. Down-regulation of CXCL1 inhibits tumor growth in colorectal liver metastasis. Cytokine. 2012;57:46–53. doi: 10.1016/j.cyto.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 43.London WB, Frantz CN, Campbell LA, Seeger RC, Brumback BA, Cohn SL, Matthay KK, Castleberry RP, Diller L. Phase II randomized comparison of topotecan plus cyclophosphamide versus topotecan alone in children with recurrent or refractory neuroblastoma: a Children’s Oncology Group study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:3808–15. doi: 10.1200/JCO.2009.27.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park JR, Scott JR, Stewart CF, London WB, Naranjo A, Santana VM, Shaw PJ, Cohn SL, Matthay KK. Pilot induction regimen incorporating pharmacokinetically guided topotecan for treatment of newly diagnosed high-risk neuroblastoma: a Children’s Oncology Group study. J Clin Oncol. 2011;29:4351–7. doi: 10.1200/JCO.2010.34.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abraham D, Zins K, Sioud M, Lucas T, Schafer R, Stanley ER, Aharinejad S. Stromal cell-derived CSF-1 blockade prolongs xenograft survival of CSF-1-negative neuroblastoma. Int J Cancer. 2010;126:1339–52. doi: 10.1002/ijc.24859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santana VM, Furman WL, Billups CA, Hoffer F, Davidoff AM, Houghton PJ, Stewart CF. Improved response in high-risk neuroblastoma with protracted topotecan administration using a pharmacokinetically guided dosing approach. J Clin Oncol. 2005;23:4039–47. doi: 10.1200/JCO.2005.02.097. [DOI] [PubMed] [Google Scholar]
- 47.Xu J, Escamilla J, Mok S, David J, Priceman S, West B, Bollag G, McBride W, Wu L. CSF1R Signaling Blockade Stanches Tumor-Infiltrating Myeloid Cells and Improves the Efficacy of Radiotherapy in Prostate Cancer. Cancer Res. 2013;73:2782–94. doi: 10.1158/0008-5472.CAN-12-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Majzner RG, Heitzeneder S, Mackall CL. Harnessing the Immunotherapy Revolution for the Treatment of Childhood Cancers. Cancer Cell. 2017;31:476–85. doi: 10.1016/j.ccell.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–72. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, Holland EC, Sutton JC, Joyce JA. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352:aad3018. doi: 10.1126/science.aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.