

Abstract
Hematopoietic stem and progenitor cells maintain blood formation throughout our lifetime by undergoing long and short-term self-renewal, respectively. As progenitor cells progress through the hematopoiesis process, their differentiation capabilities narrow, and at the point where potential limits the fate, and the precursors become committed to only one or two lineages. This review focuses on recent advances in the identification and characterization of bipotent megakaryocytic-erythroid progenitors (MEP), the cell that can further produce two completely different functional outputs: platelets and red blood cells. Potentially contradicting data have been published on the presence of MEP. Additionally, studies describing the requirement for this intermediate progenitor stage prior to commitment to the erythroid and megakaryocytic lineages can be contradictory. Interpretation of these studies is complicated by the variety of species, cell sources, and analytical approaches used along with the inherent challenge in the continuum of hematopoiesis, where hematopoietic progenitors do not stop at discrete steps on single paths as classically drawn in hematopoietic hierarchy models. With the goal of improving our understanding of human hematopoiesis, we discuss findings in both human and murine cells. Based on these data, MEP clearly represents a transitional stage of differentiation in at least one route to the generation of both megakaryocytes and erythroid cells.
Graphical Abstract
Hematopoietic Stem and Progenitor Cells
In humans, the bone marrow generates between one and three hundred billion blood cells every day, of which platelets and red blood cells comprise 99% of the cellular components produced. One of the most intriguing and fundamental phenomena in cell biology is the mechanism by which stem cells give rise to daughter cells committed to different lineages. The hematopoietic systems is an excellent model for studying this process as the hematopoietic stem cell (HSC) can self-renew and differentiate down multiple different lineages to provide hematopoietic function from fetal through adult life.
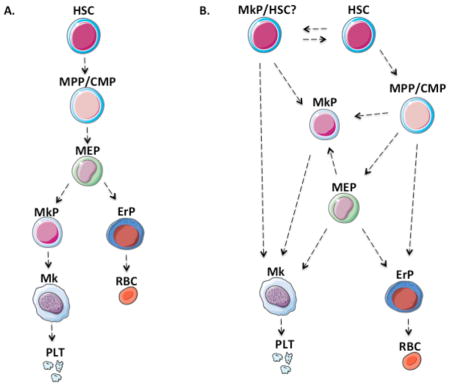
The ‘classical’ paradigm of hematopoiesis is represented as a hierarchy or tree, as shown in Figure 1A. with progressive discrete steps. Historically, this model was established based on bone marrow transplantation, flow cytometric analyses, fluorescence activated cell sorting (FACS), and single cell colony forming unit (CFU) assays including in vivo CFU-spleen and in vitro CFU-c. These early studies identified colonies [1] and sorted cell populations that had both erythroid and megakaryocytic potential [2], while lacking myeloid potential. Ultimately, these cells were thus termed megakaryocytic erythroid progenitors (MEP).
Fig. 1. Models of hematopoiesis.

(A) The classical hematopoiesis “tree,” which depicts a strictly hierarchical stepwise process, with each step representing a unique well defined population. (B) The adapted model of hematopoiesis with multiples routes to unilineage committed cells such as megakaryocyte progenitors. HSC, hematopoietic stem cell; MPP, multi-potent progenitor; CMP, common myeloid progenitor; MEP, megakaryocyte-erythroid progenitor; GMP, granulocyte-monocyte progenitor; MkP, megakaryocyte progenitor; Gran, granulocyte; Mono, monocyte; ErP, erythroid progenitor; RBC, red blood cell, Mk, megakaryocyte; Plt, platelets.
Although the stepwise hierarchical model is extremely useful for visualization of hematopoiesis as well as for prospective isolation of stem and progenitor cell subpopulations by FACS, inherently such schematics cannot accurately represent the complexity of individual cells as they undergo gradual hematopoietic maturation changes at the genetic and epigenetic levels.
Recent advances in single cell technologies have led to a deeper understanding of the continuum of hematopoiesis. A more complex network of hematopoietic progenitors is being revealed with novel approaches for studying hematopoietic clones in their native environment, as well as index sorting, and single cell genomics (Figure 1B). While it is not yet clear how all cells of the megakaryocytic and erythroid lineages are derived, recent findings have revealed the possibility that the megakaryocytic (Mk) and erythroid lineages may arise via multiple different routes including MEP, as well as the potentially more directly from hematopoietic stem cells.
Megakaryopoiesis and Erythropoiesis
Megakaryocytes are rare polyploid cells whose main function is to produce platelets. However, megakaryocytes also play significant roles in the HSPC niche in the bone marrow by secreting pleotropic factors such as PF4 [3] and TGF-B.[4] The phenotypic surface proteins that are most commonly used for identification of the megakaryocytic lineage are the integrins CD41 (GPIIb) and CD61 (GPIIIa), which form heterodimers on the platelet surface. The GPIIb/IIIa complex functions mainly as a receptor for fibrinogen during platelet aggregation.[5] Von Willebrand factor (vWF) is highly expressed in platelets and endothelial cells.[6] During maturation, a unipotent CFU-Mk, which is also CD41+, matures ultimately through polyploidization creating megakaryocytes, which undergo complex cytoplasmic cytoskeletal rearrangement to release platelets. Recently, surface expression of CD41 and vWF have also been reported in a subpopulation of phenotypic HSC that may represent Mk-biased HSC[7–9], which are addressed in further detail below. Many observations have been made over the years indicating significant overlaps of megakaryocyte markers (CD41, CD61, vWF and PF4) in Mk and HSC.[5] Both Mk and HSC are dependent on thrombopoietin (TPO) for survival. While not widely appreciated, all hematopoietic stem cells and most hematopoietic progenitor cells (including multi potent progenitor (MPP) and common myeloid progenitor (CMP), but excluding committed erythroid progenitors) express MPL, the receptor for TPO, on their cell membranes.[5, 10–13]
Erythroid progenitors play a critical role in erythropoiesis as the source of exponential expansion of the erythroid lineage, which then become enucleated red blood cells that are responsible for oxygen delivery throughout the body. RBCs are derived from unipotent BFU-E and its descendent, the CFU-E. It is estimated that thousands of RBCs are generated from each erythroid committed progenitor.[14] The surface protein used most commonly to identify committed erythroid cells is glycophorin A (Gly A) for human cells and Ter119 for murine cells, as well as the transferrin receptor, CD71. Of note, CD71 is expressed on all dividing cells. However, its surface expression increases by at least 10-fold in erythroid committed cells. The immunophenotype of mouse and human erythroid committed cells was recently clarified and includes co-expression of high levels of CD71 and endoglin (Table 1).[15, 16]
Table 1.
Current gating strategies to identify MEPs by flow cytometry and the respective in vitro assays to evaluate their lineage potential.
| Species | Name | Immunophenotype | Assay | Lineage Potential | Ref |
|---|---|---|---|---|---|
| Mouse | MEP | IL-7Ra-Lin-Sca-1-c-Kit+ FcgRloCD34− | CFC in methylcellulose | 20% of Mk/E, 30% of Mk and 10% E and 40% N/A | [2] |
| Mouse | PreMegE | Lin-cKit+Sca-FcgRII/III-CD41−CD150+CD105− | CFC in methylcellulose | 10% of Mk/E, 50% of Mk and 40% E | [16] |
| Human | MEP | CD34+CD38+IL3RA- CD45RA- | CFC in methylcellulose | 10% Mk/E, 5% Mk, 70% E and 15% N/A | [43] |
| Human | MEP | CD34+CD38+Thy-1−CD45RA−CD135−CD7−CD10− | MS-5 co-culture and CFC in methylcellulose | No colonies in co-culture. E-only colonies in CFC assays | [57] |
| Human | MEP | CD19-CD34+IL-3Ralphalo/-CD45RA-BAH1+ | CFU-Mk/E in megacult and methocult | Less than 3% Mk/E, 3% Mk, 37%E, 55% N/A in megault. E-only in methylcellulose | [42] |
| Human | MEP (F1) | CD34+CD38+CD10−FLT3CD45RA–CD71-BAH1- | MS-5 co-culture followed by flow cytometry | 60% myeloid (GlyA−CD41− CD45+CD11b+), 20% E and 10% Mix (myeloid and E or Mk) | [21] |
| Human | MEP (F3) | CD34+CD38+CD10−FLT3CD45RA–CD71+BAH1+ | MS-5 co-culture followed by flow cytometry | 90% E-only | [21] |
| Human | MEP | CD34+Lin-CD135-CD45Ra-CD38midCD110+CD36-CD41- | Dual CFU-Mk/E in megacult | 50% Mk/E, 20% Mk, 25% E in megacult. 90% E and 10%G/M in methylcellulose | [10] |
An important caveat to studies of output of an MEP arises from the differences in how these two lineages (Mk and erythroid) mature. In early work, Debili et al found that in liquid culture dual potential progenitors quickly made lower numbers of MK than erythroid cells. These Mks then disappeared after making platelets followed by a much greater numerical expansion of erythroid cells.[17] According to our unpublished observations using single cell live imaging and the literature, erythroid-committed cells undergo considerably more cell divisions than megakaryocyte-committed cells [18, 19] and mature megakaryocytes rarely survive for 3 weeks in culture. [20] Thus, analysis of a cell’s potential in culture by flow cytometry could mask the yield of rare megakaryocytes, especially when there are large numbers of erythroid cells, and miss megakaryocyte potential no longer evident at later timepoints used for erythroid and myeloid progeny. Furthermore, the kinetics of the acquisition of lineage specific surface markers is not consistent between lineages although they all may be deemed “early” markers. For example, when evaluating the fate decision of the MEP towards the Mk vs. erythroid lineages, one could misunderstand the output lineages if evaluating the expression of CD41 (Mk specific) and GlyA (erythroid specific) at early time-points. Surface expression of CD41 occurs 4–5 days before, which could lead to the conclusion that the starting progenitor was already Mk committed. However, if assessed at later time-points (around 10 days), the GlyA expression could be observed in some cases without any CD41+ cells remaining (Scanlon and Krause, manuscript in preparation).
Challenges in isolating MEP
The controversy related to the existence of MEPs may be attributed in large part to misconceptions sustained by hematopoietic hierarchy models that include immunophenotypes, but which have ignored the fact that classic murine and human MEP gating strategies actually greatly enrich for unipotent erythroid progenitors, as shown in Table 1 [14, 21]. These erythroid-committed cells far outnumber the bipotent MEP within the classic immunophenotypically-defined populations referred to as “MEP.” In the murine system, Akashi et al. identified the CD34-Lin-c-Kit+Sca- FcγRlowIL-7Ra- fraction of mouse bone marrow cells as MEP as the progeny of these cells in vitro lacked monocytes and granulocytes. However, the CFU assay data showed that the majority of these cells were already erythroid or Mk committed (CFU-E and CFU-Mk, respectively).[2] When transplanted into irradiated mice, cells from this ‘MEP’ gate reconstituted only Ter119+ cells, consistent with enrichment for erythroid-committed cells, which correlates with the finding that they expressed high levels of EpoR.[2] In subsequent studies using this sorting strategy, an average of 80% of the cells were erythroid-committed based on CFU assays. [16] Note that it is well established that erythroid committed cells express Kit (reviewed in [22]).
Despite the knowledge that the CD34−Lin-c-Kit+Sca-FcγRlowIL-7Ra- bone marrow subpopulation is composed predominantly of erythroid committed cells, this immunophenotyping strategy is still broadly used to quantitate and isolate cells that investigators incorrectly refer to as murine ‘MEPs,’ including in recent studies using single cell RNAseq, which, not surprisingly, showed erythroid enriched genes that clustered together and almost no Mk-related genes or recognition of a unique bipotent progenitor in this population.[23–26]
In one such paper, Paul et al. [24] proposed that multipotent myeloid progenitors commit very early to unilineage committed progenitors without going through stages of progenitor differentiation with gradually decreasing potency (e.g. CMP, MEP). They reached this conclusion because no tripotent or bipotent progenitors were identified among the thousands of Lin-Sca-c-Kit+ cells on which they performed single cell mRNA-seq. The investigators then determined which subpopulations (based on indexed FACS analysis of conventional surface markers) were associated with patterns of lineage-specific gene expression. [2] They found very few cells that co-expressed transcription factors known to regulate different lineages and observed a lack of discriminative power of CD34 and FcgR, commonly used markers to distinguish cells between CMP, MEP and GMP populations. None of the cells within the “MEP” gate as defined by Akashi et al. [2] had expression of Mk specific genes, which is consistent with the high content of erythroid-committed cells within this population.
Two assumptions may have further hampered discovery of rare MEPs in this single cell RNA sequencing study. First, it was assumed that the frequency of multi- and bi-potent cells would be high enough to detect within a population of ~3,000 single Kit+ cells, which would include highly proliferative erythroid progenitors. Secondly, they assumed that multi- or bi-potent progenitors would share gene expression patterns predictive of the lineages into which they could potentially differentiate. But, it is possible that a bipotent progenitor does not co-express lineage specific genes of the lineages into which it can differentiate at levels that would be detected by this approach. If functional MEP express transcription factors of both the Mk and E lineages, it is highly likely that these genes would be expressed at very low levels that may require gene specific amplification to detect.
Inaccurate use of the term “MEP” to define a phenotype that lacks functional MEP has also contributed to confusion in studies of human MEPs.[14, 21, 27, 28] For human cells, the classic ‘MEP’ gate usually includes all CD34+CD38+ cells because CD38+ is used to distinguish HSC, which are CD38-, from progenitor cells. However, early experiments by Debili et al. found dual-lineage (Mk and erythroid) colonies composed of 100 to 1,000 erythroblasts intermixed with a few MK that were enriched in the CD34+CD38+/− and CD34+CD38− cell fractions. Even though CD34+CD38high human cells have been published as being greatly enriched for erythroid committed cells,[10] papers still use this gate to isolate what they refer to as human “MEP.” Consequently, single cell RNA sequencing data from this population shows that many of these cells are erythroid committed, which would lead investigators to question the existence of human MEPs. [27, 28]
Recently, Notta et al. used a 3-week culture assay to test the fate decision of different human hematopoietic subsets within bone marrow, cord blood and fetal liver[21]. Using a FACS panel containing antibodies against CD34, CD38, CD7, CD10, Thy1, CD45Ra, FLT3, and CD49f as well as the BAH-1 monoclonal antibody, they co-cultured single cells with the MS5 murine stromal cell line and evaluated the progeny of these single cells by flow cytometry after 2–3 weeks. The BAH-1 antibody is not specific to MPL (the TPO-R) as well demonstrated by Abbot et al. [29] Because these populations were sorted based on staining with the BAH-1 monoclonal antibody known to be non-specific, it is not clear what these cell types actually were. Combining the described in vitro assay with single cell RNA-seq and in vivo assays, Notta et al. [21] identified Mk progenitor activity only in MPP and HSC populations upstream of what has traditionally been gated as an MEP population.
Although it is claimed that flow cytometric analysis 3 weeks after culturing single cells in liquid media provides conditions for the differentiated progeny to survive, propagate, and expand to permit detection, it is not clear what percentage of progenitors expanded enough to reach an adequate cell number for flow cytometric analysis. It certainly is possible that the only progenitors that would still produce Mk after 2–3 weeks in culture would be those further up the hierarchy than MEP or CFU-Mk.
A similar observation was made by Velter et al. [30] using an analogous approach of combining single cell index sort, RNAseq and in vitro studies. Like Notta et al,[21] they analyzed the progeny of single cells after 3 weeks in culture by flow cytometry. Their data suggested that hematopoietic stem and progenitor cells gradually acquire lineage priming; and at the CD34+CD38− stage, the major branches are still multipotent but have committed to the lymphoid or myeloid lineages. However, Lin-CD34+CD38+ cells, which were characterized by CD38 upregulation and cell cycle activation, were unipotent. [30] While these studies have valuable insights on progenitor hierarchy, conclusions about bipotent MEP must be tempered by the caveats of the immunophenotype and the lengthy culturing times used.
Alternatives to the megakaryocytic-erythroid progenitor (MEP) intermediate
While issues with immunophenotyping and optimal assays have complicated interpretation of ex vivo assays of Mk potential, recent studies using transplantation of a variety of cells have indicated there may be diversity in the pathways that can yield megakaryocytes.
Mk Biased-HSC and long-lived MkP may coexist and be difficult to discriminate
The past few years have been marked by the growing discussion about the origin of megakaryocytes and the existence of intermediate progenitors in the hematopoiesis process. The concept of an Mk/platelet biased HSC was initially based on the many similarities between HSC and megakaryocytes.[5] For example, Mk and HSC share dependency on thrombopoietin for survival.[12] While not widely appreciated, all hematopoietic stem cells and most hematopoietic progenitor cells (excluding committed erythroid progenitors) express MPL, the receptor for thrombopoietin, on their cell membranes.[10, 31, 32]
Gekas and Graf [7] identified a CD41+ subpopulation of adult HSC as being myeloid-biased. Through competitive bone marrow transplantation, they observed that the CD41+ HSC were more quiescent than CD41− HSC, and that CD41 knockout led to a transplantable pancytopenia as the animals showed a reduction not only in platelet numbers, but also in red blood cells and leukocytes [7]. Similarly, vWF+HSC were shown to be more quiescent, and when transplanted, led to engraftment of all lineages but with higher platelet production than vWF- HSCs. These cells also exhibited significant enrichment of megakaryocyte-associated genes and were affected by knocking out the Tpo gene. Thus, as seen with CD41+ HSC, these findings may suggest a subset of HSC with a more general myeloid bias (with enhanced Mk/erythroid/myeloid engraftment) or an Mk bias. The possible inclusion of a downstream MEP intermediate during maturation of this biased HSC is unclear.
Reconciling the two hypothesis, Yamamoto et al. [33] demonstrated both the presence of functional MEP in vivo and the possibility that an Mk committed progenitor could be derived directly from an HSC, using single cell transplantation of cells from reporter mice, which in addition to nucleated cells, also have fluorescently labeled RBCs and platelets.
In this approach, a longterm self-renewing cell was determined based on persistence over time, transplantability, and the lineages detected (platelets, erythrocytes, neutrophil/monocyte, B cell and T cell) at a level higher than 0.1% during primary and secondary transplantations. They observed that repopulating MEPs generated over 10% of the platelets, and 5% of the erythrocytes for up to 5 months after primary transplantation, but no more than 1% of platelets and approximately 0.1% of RBCs after secondary transplants. Mk committed progenitors gave rise to 1% of the platelets in primary transplants, and barely (<0.1%) detectable platelets in 1 mouse after secondary transplantation.
In order to functionally evaluate the immediate progeny of a single cell, they performed single cell sorting of candidate HSPCs and watched their division in vitro. After the first cell division, cells from each cell pair were transplanted into two different recipients. With this elegant approach, it was possible to observe that reconstituting HSC generated different types of daughter cells that included 1 LT-HSC and 1 Mk-committed progenitor and in another example, an HSC gave rise to 1 ST-HSC and 1 Mk-committed progenitor, strongly suggesting that Mk committed progenitors can be generated directly from HSC without further cell division. Importantly, the repopulating Mk progenitor generated in this case were rare compared to HSC/HSC pairs and reconstituted not more than 0.1% of platelets that were sustained after secondary transplantation.[33] Together, these data prove the existence of a potentially Mk-biased HSC, but this cell did not seem to serve as the major source of platelets in hematopoiesis when compared to the reconstitution of MEPs. One possibility is that this Mk biased HSC serves as an important source of Mk under specific physiological conditions.
Different from Yamamoto’s findings [33], a single cell transplantation approach was recently able to prove the long-term multilineage reconstituting potential of HSC subpopulations that were restricted to generating platelets for up to 44 weeks in the donor mice,[34] which is consistent with prior single cell transplantation studies of Yamamoto discussed above.[33] Another important finding of this study was the demonstration that the restricted platelet fate observed in vivo does not always correlate with the potential in vitro, as LSK cells derived from mice with robust and stable platelet-restricted reconstitution showed potential to grow granulocytes, monocytes/macrophages and T lymphocytes in vitro. [34] Thus, one must be cognizant of differences between hematopoietic fate and hematopoietic potential. Hopefully, future studies will focus on how fate vs. potential is regulated, as well as determining if there is a specific and consistent intermediate stage in the progenitor hierarchy that multipotency is lost.
It seems reasonable to assume that the commitment of individual cells within the HSPC compartment is linked to the blood demand in case of infection or bleeding, for example. Haas et al. [8] demonstrated that the immunophenotypically defined (Lin−cKit+CD150+CD48−) HSC pool contains committed (unilineage) Mk progenitors that, in response to LPS induced inflammation, upregulate CD41 and produce Mk directly without evidence that they pass though intermediate stages. While it is possible that these cells were derived directly from a multipotent HSC, this remains to be shown. Thus, further assays are needed to determine if this is an HSC now limited to Mk, or an Mk-progenitor gaining CD41 expression in response to stress signals induced by LPS. The possibility that Mk progenitors are not derived from HSCs but simply share an identical surface marker pattern was also raised by Roch et al. [35] who observed that 15% of single HSCs (LSK CD150+CD48−CD34−) directly differentiate to megakaryocytes without cell division.[35] In either case, these progenitors could be unique in their ability to serve as an emergency pool for platelet production in response to inflammatory insults, [36] and would be consistent with the hypothesis that platelets, which are extremely important in response to injury and inflammation, have more than one production route.
Studies focusing on steady state hematopoiesis used a transposon tagging strategy to clonally trace the fates of stem and progenitors cells. [37] In a subsequent study, this strategy revealed that half of the Mk lineage appeared to be produced independently from other lineages by the HSC compartment. [38] However, the authors suggested that the MEP stage could be too transient to be detected by their approach. [38]
Performing clonal analysis of human cells in vivo is challenging, especially if one wants to test potential differences between steady state and stress hematopoiesis. However, a study by Biasco et al. [39] followed Wiskott-Aldrich syndrome patients up to 4 years after transplantation with lentivirally transduced autologous CD34+ cells, and through the identification of the integration site of the lentiviral vector used, they tracked the output of the transplanted cells to reveal clonal dynamics during early and steady-state reconstitution phases.[39] The analysis of the input level that each lineage received from CD34+ cells at different time points using unsupervised heat maps showed that GlyA+ erythroid cells and CD61+ megakaryocytic cells clustered together, indicating a common progenitor. In addition, the greatest similarities in integration sites were found 4 years after the bone marrow transplantation, indicating that this is a result of steady state hematopoiesis. It is possible that this represents engraftment of longterm reconstituting Mk and erythroid biased HSC analogous to published murine studies discussed above. [33, 34]
Data supporting the existence of a bipotent megakaryocytic-erythroid progenitor
Genetic and physiological associations between Mk and E support the concept of an MEP. Megakaryocytes and erythroid cells share dependence on several critical transcription factors including GATA-1, FOG-1, GATA-2, and NF-E2 (reviewed in [40]). To explain the physiological thrombocytopenia that occurs concomitant with stimulated erythropoiesis under hypoxia conditions, McDonald and Sullivan speculated the competition between two potential fates of a common precursor of erythroid and Mk lineages that is affected by hypoxia [41]. Using two markers, CD105 and CD150, Pronk et al. [16] demonstrated the complexity of progenitors within the previous “MEP” gate, [2, 16] and improved upon previous enrichment strategies for murine MEP. Within the previously published “MEP” gate, they further fractionated CD150+CD105− cells, approximately 20% of which are bipotent Mk/E progenitors. The CFU-Mk were found in the CD150+CD41+ population, while BFU-E (or ‘Pre-CFU-E”) were in the CD150+CD105+ population. These results support the existence of a murine MEP and provide tools for its enrichment.
Our group developed an improved strategy for enrichment of primary human MEP.[10] Using a highly specific functional clonal assay, individual cells that are capable of both E and Mk differentiation (CFU-Mk/E) and lack potential to differentiate down the monocytic or granulocytic lineages were identified. This functional assay showed that previously published approaches for human MEP enrichment [27, 42, 43] yielded heterogeneous mixtures of granulocyte/monocyte (G/M) progenitors, Mk progenitors and erythroid progenitors with only low levels (~15%) of “true” MEP. Colonies were grown from sparsely plated single cells (100 cells/35 mm plate) or as single cells in 96 well plates in collagen based medium with appropriate cytokines for both lineages, and mature cells were identified based on dual immunostaining for CD41 (Mk specific) and GlyA (erythroid specific). The improved method for enriching MEP (lin-CD34+Flt3-CD45RA-CD38midMPLl+CD36-CD41−) yields 45–60% CFU-Mk/E, with ≤3% contaminating CFU-G/M. While the MEP population is not 100% pure, it results in highly reproducible relative compositions of 3 major cell populations: CFU-Mk/E, BFU-E, and CFU-Mk. In addition, it was optimized both Mk-progenitor (CD34+lin-Flt3-CD45RA-CD38midMPL+CD36-CD41+) and erythroid progenitor (CD34+lin-Flt3-CD45RA-CD38highMPL-) with >80% and >90% purity for CFU-Mk and BFU-E, respectively.
While our MEP strategy presents the highest purity in vitro of bipotent (Mk/E) cells published to date, it is still to be determined if the CFU-Mk or BFU-E only colonies that are still present may have already biased them toward one lineage or the other, but has kept the “MEP” immunophenotype or if the “MEP” gate also is contaminated with unipotent progenitors. Consistent with the first possibility, our unpublished single cell RNA sequencing data suggest that about 90% of cells within the MEP gate share a unique gene expression pattern that is intermediate between that of CMP and unilineage progenitors, and this gene expression pattern is quite unique from that of the sorted Mk and erythroid committed populations described above.
It is important to note that these cells are a reasonable model to study potential in vitro, but do not prove the existence of the human MEP in vivo nor do the in vitro data demonstrates that this cell is responsible for producing platelets and red blood cells in vivo.
Mechanisms underlying the bipotent MEP fate decision
Cell fate decisions require a change in the balance between different gene expression programs, mainly executed by transcriptional and epigenetic regulators in response to extracellular signals. The mechanisms underlying the fate decision of MEPs down the Mk versus erythroid lineages are not yet clear.
A misconception regarding MEP fate decisions is that TPO promotes Mk whereas EPO promotes the erythroid fate commitment. As mentioned before, TPO is necessary for HSC survival and stimulates proliferation of nearly all hematopoietic progenitor types.[13] In contrast, EPO is not necessary for erythroid fate commitment.[44] Rather, EPO is necessary for proliferation and maturation of already committed erythroid progenitors. Thus, TPO enhances the proliferation, survival and differentiation of HSPCs as well as Mks, and EPO is required for erythroid proliferation, survival and differentiation. These cytokines do not play an instructive role in the fate decision of uncommitted progenitors, but regulate maturation and yield [10, 45, 46].
Studies suggest that the MEP fate decision is governed by coordinated regulation of a precise balance of several transcription factors including MYB, KLF1 and FLI1 with higher levels of expression of KLF1 and FLI1 promoting E and Mk differentiation, respectively. [10, 40, 47–51]
Taking advantage of our dual human CFU-Mk/E assay, we reported that decreasing MYB expression specifically in primary human MEP results in an increased number of Mk colonies at the expense of erythroid and dual Mk/E colonies.[10] Studies related to the mechanisms upstream and downstream of Myb in the MEP fate decision show that, 1) Myb is a miR-150 target,[52] and that Myb promotes erythropoiesis via binding and activation of KLF1 and LMO2,[50] and via transactivation miR-486–3p. [49]
Upstream of transcription factors, GTPase activating proteins such as ARHGAP21 (Rho GAP [53]) and Rgs18 (GAP [54]) play a role in the MEP fate decision, potently supporting erythroid and suppressing Mk commitment. Acting as an effector for GFI1, RGS18 regulates the downstream signaling through ERK1 and ERK2 to modify the balance of the transcription factors Fli1 and Klf1.[54]
Ultimately, the mechanisms underlying erythroid and Mk fate decisions from bipotent or more multipotent progenitors will be revealed. The development of new single cell techniques including ChIP, next-generation sequencing and ATAC-seq are defining the full repertoire of genomic loci occupied by hematopoietic transcription factors or loci that are “open” for potential expression. [40, 55] In addition, improved methods will be developed to functionally evaluate the fate decision at the single cell level without perturbing other cell functions such as proliferation, cell cycle kinetics and apoptosis. [10, 21] Also, the field is still missing sensitive reporters to evaluate cell fate decisions in real time. In order to be accurate, these reporters most likely cannot be based on protein or gene expression because these events are a product of a fate decision that has already been made. Fate decision reporters will need to be based on earlier events such as methylation or chromatin accessibility to genes determined to reliably report commitment to different lineages.
Conclusion
Hematopoietic cells do not always appear to follow the hematopoietic hierarchy as defined by the dogma. Here, we have reviewed published data showing longterm self-renewal of hematopoietic stem/progenitor cells with limited differentiation potential, and that, while MEP clearly do exist in vivo in both mice and humans, there may be alternative pathways for hematopoietic cells to achieve fate commitment to the E and Mk lineages that do not require that they pass through this bipotent MEP stage.
The lack of uniform effective approaches to purify or even to consider the MEP as a player in hematopoiesis in different studies that have combined approaches including functional, single cell sequencing and lineage potential assays, suggests that the identification of MEPs and their subpopulations is highly dependent on the gating strategy [14]. Additionally, the more reliable MEP gating strategies published to date [10, 16, 56] demonstrate that these bipotent progenitors comprise a rare population (less than 1% of human CD34+ cells and murine Lin- cells) and are based on populations that are negative for different lineage markers rather than positive for others, as we are still lacking a positive and specific marker for MEP.
MEPs may represent a rare transitional state and are thus difficult to isolate in contrast to other more well-defined subpopulations of unipotent progenitors for megakaryocytes or erythroid cells. Since this stage could be transient, and may not always have a distinct FACS phenotype, it may be that even “HSC-like” cells that seem to undergo Mk fate commitment undergo fate transitions by passing through a currently undetectable transient epigenetic bipotent state. It is still unknown whether the contribution of MEP, MkP and/or Mk-biased HSC to the generation of Mk and platelets changes upon stress or in disease settings. These studies will have the potential to harness our new insights to better treat megakaryocytic diseases or improve platelet recovery following bone marrow transplantation.
The understanding of how hematopoietic fate decisions are made is rapidly developing. The vision of hematopoiesis as a dynamic process with multiple complementary routes to unilineage progenitors will benefit the diagnosis, prognosis and treatment of benign and malignant hematologic disorders.
Acknowledgments
We apologize to colleagues whose work was not cited due to space limitations.
The authors thank Kathleen McGrath, Nadia Ayala-Lopez and Vanessa Scanlon for critical input. JXF is the recipient of a National Blood Foundation Early Career award. Work was supported by NIHR01DK114031 and 1U54DK106857.
Footnotes
Author contributions: JXF wrote the manuscript, and created the table and figure. DSK oversaw the work and wrote the manuscript.
The authors have no relevant conflicts of interest.
References
- 1.Vannucchi AM, Paoletti F, Linari S, et al. Identification and characterization of a bipotent (erythroid and megakaryocytic) cell precursor from the spleen of phenylhydrazine-treated mice. Blood. 2000;95:2559–2568. [PubMed] [Google Scholar]
- 2.Akashi K, Traver D, Miyamoto T, et al. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 3.Bruns I, Lucas D, Pinho S, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20:1315–1320. doi: 10.1038/nm.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao M, Perry JM, Marshall H, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014;20:1321–1326. doi: 10.1038/nm.3706. [DOI] [PubMed] [Google Scholar]
- 5.Huang H, Cantor AB. Common features of megakaryocytes and hematopoietic stem cells: what’s the connection? J Cell Biochem. 2009;107:857–864. doi: 10.1002/jcb.22184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huck V, Schneider MF, Gorzelanny C, et al. The various states of von Willebrand factor and their function in physiology and pathophysiology. Thromb Haemost. 2014;111:598–609. doi: 10.1160/TH13-09-0800. [DOI] [PubMed] [Google Scholar]
- 7.Gekas C, Graf T. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood. 2013;121:4463–4472. doi: 10.1182/blood-2012-09-457929. [DOI] [PubMed] [Google Scholar]
- 8.Haas S, Hansson J, Klimmeck D, et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell. 2015;17:422–434. doi: 10.1016/j.stem.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Sanjuan-Pla A, Macaulay IC, Jensen CT, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 2013;502:232–236. doi: 10.1038/nature12495. [DOI] [PubMed] [Google Scholar]
- 10.Sanada C, Xavier-Ferrucio J, Lu YC, et al. Adult human megakaryocyte-erythroid progenitors are in the CD34+CD38mid fraction. Blood. 2016;128:923–933. doi: 10.1182/blood-2016-01-693705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woolthuis CM, Park CY. Hematopoietic stem/progenitor cell commitment to the megakaryocyte lineage. Blood. 2016;127:1242–1248. doi: 10.1182/blood-2015-07-607945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian H, Buza-Vidas N, Hyland CD, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell. 2007;1:671–684. doi: 10.1016/j.stem.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Kimura S, Roberts AW, Metcalf D, et al. Hematopoietic stem cell deficiencies in mice lacking c-Mpl, the receptor for thrombopoietin. Proc Natl Acad Sci U S A. 1998;95:1195–1200. doi: 10.1073/pnas.95.3.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dulmovits BM, Hom J, Narla A, et al. Characterization, regulation, and targeting of erythroid progenitors in normal and disordered human erythropoiesis. Curr Opin Hematol. 2017;24:159–166. doi: 10.1097/MOH.0000000000000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Hale J, Bhagia P, et al. Isolation and transcriptome analyses of human erythroid progenitors: BFU-E and CFU-E. Blood. 2014;124:3636–3645. doi: 10.1182/blood-2014-07-588806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pronk CJ, Rossi DJ, Mansson R, et al. Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell. 2007;1:428–442. doi: 10.1016/j.stem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Debili N, Hegyi E, Navarro S, et al. In vitro effects of hematopoietic growth factors on the proliferation, endoreplication, and maturation of human megakaryocytes. Blood. 1991;77:2326–2338. [PubMed] [Google Scholar]
- 18.Besancenot R, Chaligne R, Tonetti C, et al. A senescence-like cell-cycle arrest occurs during megakaryocytic maturation: implications for physiological and pathological megakaryocytic proliferation. PLoS Biol. 2010:8. doi: 10.1371/journal.pbio.1000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pop R, Shearstone JR, Shen Q, et al. A key commitment step in erythropoiesis is synchronized with the cell cycle clock through mutual inhibition between PU.1 and S-phase progression. PLoS Biol. 2010:8. doi: 10.1371/journal.pbio.1000484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sim X, Poncz M, Gadue P, et al. Understanding platelet generation from megakaryocytes: implications for in vitro-derived platelets. Blood. 2016;127:1227–1233. doi: 10.1182/blood-2015-08-607929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Notta F, Zandi S, Takayama N, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351:aab2116. doi: 10.1126/science.aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munugalavadla V, Kapur R. Role of c-Kit and erythropoietin receptor in erythropoiesis. Crit Rev Oncol Hematol. 2005;54:63–75. doi: 10.1016/j.critrevonc.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Wilson NK, Kent DG, Buettner F, et al. Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell. 2015;16:712–724. doi: 10.1016/j.stem.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paul F, Arkin Y, Giladi A, et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell. 2015;163:1663–1677. doi: 10.1016/j.cell.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 25.Hamey FK, Nestorowa S, Kinston SJ, et al. Reconstructing blood stem cell regulatory network models from single-cell molecular profiles. Proc Natl Acad Sci U S A. 2017;114:5822–5829. doi: 10.1073/pnas.1610609114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Graaf CA, Choi J, Baldwin TM, et al. Haemopedia: An Expression Atlas of Murine Hematopoietic Cells. Stem Cell Reports. 2016;7:571–582. doi: 10.1016/j.stemcr.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Psaila B, Barkas N, Iskander D, et al. Single-cell profiling of human megakaryocyte-erythroid progenitors identifies distinct megakaryocyte and erythroid differentiation pathways. Genome Biol. 2016;17:83. doi: 10.1186/s13059-016-0939-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyawaki K, Iwasaki H, Jiromaru T, et al. Identification of unipotent megakaryocyte progenitors in human hematopoiesis. Blood. 2017;129:3332–3343. doi: 10.1182/blood-2016-09-741611. [DOI] [PubMed] [Google Scholar]
- 29.Abbott C, Huang G, Ellison AR, et al. Mouse monoclonal antibodies against human c-Mpl and characterization for flow cytometry applications. Hybridoma (Larchmt) 2010;29:103–113. doi: 10.1089/hyb.2009.0095. [DOI] [PubMed] [Google Scholar]
- 30.Velten L, Haas SF, Raffel S, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. 2017;19:271–281. doi: 10.1038/ncb3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murone M, Carpenter DA, de Sauvage FJ. Hematopoietic deficiencies in c-mpl and TPO knockout mice. Stem Cells. 1998;16:1–6. doi: 10.1002/stem.160001. [DOI] [PubMed] [Google Scholar]
- 32.Alexander WS, Roberts AW, Nicola NA, et al. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood. 1996;87:2162–2170. [PubMed] [Google Scholar]
- 33.Yamamoto R, Morita Y, Ooehara J, et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. 2013;154:1112–1126. doi: 10.1016/j.cell.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 34.Carrelha J, Meng Y, Kettyle LM, et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature. 2018;554:106–111. doi: 10.1038/nature25455. [DOI] [PubMed] [Google Scholar]
- 35.Roch A, Trachsel V, Lutolf MP. Brief Report: Single-Cell Analysis Reveals Cell Division-Independent Emergence of Megakaryocytes From Phenotypic Hematopoietic Stem Cells. Stem Cells. 2015;33:3152–3157. doi: 10.1002/stem.2106. [DOI] [PubMed] [Google Scholar]
- 36.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11:264–274. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 37.Sun J, Ramos A, Chapman B, et al. Clonal dynamics of native haematopoiesis. Nature. 2014;514:322–327. doi: 10.1038/nature13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodriguez-Fraticelli Alejo ESLW, Weinreb Caleb S, Panero Riccardo, Patel Sachin H, Jankovic Maja, Sun Jianlong, Calogero Raffaele A, Klein Allon M, Camargo Fernando D. Clonal analysis of lineage fate in native haematopoiesis. Nature. 2018;553:212–216. doi: 10.1038/nature25168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biasco L, Pellin D, Scala S, et al. In Vivo Tracking of Human Hematopoiesis Reveals Patterns of Clonal Dynamics during Early and Steady-State Reconstitution Phases. Cell Stem Cell. 2016;19:107–119. doi: 10.1016/j.stem.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dore LC, Crispino JD. Transcription factor networks in erythroid cell and megakaryocyte development. Blood. 2011;118:231–239. doi: 10.1182/blood-2011-04-285981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonald TP, Sullivan PS. Megakaryocytic and erythrocytic cell lines share a common precursor cell. Exp Hematol. 1993;21:1316–1320. [PubMed] [Google Scholar]
- 42.Edvardsson L, Dykes J, Olofsson T. Isolation and characterization of human myeloid progenitor populations--TpoR as discriminator between common myeloid and megakaryocyte/erythroid progenitors. Exp Hematol. 2006;34:599–609. doi: 10.1016/j.exphem.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 43.Manz MG, Miyamoto T, Akashi K, et al. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci U S A. 2002;99:11872–11877. doi: 10.1073/pnas.172384399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu H, Liu X, Jaenisch R, et al. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83:59–67. doi: 10.1016/0092-8674(95)90234-1. [DOI] [PubMed] [Google Scholar]
- 45.Richmond TD, Chohan M, Barber DL. Turning cells red: signal transduction mediated by erythropoietin. Trends Cell Biol. 2005;15:146–155. doi: 10.1016/j.tcb.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 46.Deutsch VR, Tomer A. Megakaryocyte development and platelet production. Br J Haematol. 2006;134:453–466. doi: 10.1111/j.1365-2141.2006.06215.x. [DOI] [PubMed] [Google Scholar]
- 47.Starck J, Cohet N, Gonnet C, et al. Functional cross-antagonism between transcription factors FLI-1 and EKLF. Mol Cell Biol. 2003;23:1390–1402. doi: 10.1128/MCB.23.4.1390-1402.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuvardina ON, Herglotz J, Kolodziej S, et al. RUNX1 represses the erythroid gene expression program during megakaryocytic differentiation. Blood. 2015;125:3570–3579. doi: 10.1182/blood-2014-11-610519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bianchi E, Bulgarelli J, Ruberti S, et al. MYB controls erythroid versus megakaryocyte lineage fate decision through the miR-486–3p-mediated downregulation of MAF. Cell Death Differ. 2015;22:1906–1921. doi: 10.1038/cdd.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bianchi E, Zini R, Salati S, et al. c-myb supports erythropoiesis through the transactivation of KLF1 and LMO2 expression. Blood. 2010;116:e99–110. doi: 10.1182/blood-2009-08-238311. [DOI] [PubMed] [Google Scholar]
- 51.Bouilloux F, Juban G, Cohet N, et al. EKLF restricts megakaryocytic differentiation at the benefit of erythrocytic differentiation. Blood. 2008;112:576–584. doi: 10.1182/blood-2007-07-098996. [DOI] [PubMed] [Google Scholar]
- 52.Lu J, Guo S, Ebert BL, et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev Cell. 2008;14:843–853. doi: 10.1016/j.devcel.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xavier-Ferrucio J, Ricon L, Vieira K, et al. Hematopoietic defects in response to reduced Arhgap21. Stem Cell Res. 2017;26:17–27. doi: 10.1016/j.scr.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sengupta A, Upadhyay G, Sen S, et al. Reciprocal regulation of alternative lineages by Rgs18 and its transcriptional repressor Gfi1b. J Cell Sci. 2016;129:145–154. doi: 10.1242/jcs.177519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buenrostro JD, Wu B, Litzenburger UM, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–490. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klimchenko O, Mori M, Distefano A, et al. A common bipotent progenitor generates the erythroid and megakaryocyte lineages in embryonic stem cell-derived primitive hematopoiesis. Blood. 2009;114:1506–1517. doi: 10.1182/blood-2008-09-178863. [DOI] [PubMed] [Google Scholar]
- 57.Doulatov S, Notta F, Eppert K, et al. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol. 2010;11:585–593. doi: 10.1038/ni.1889. [DOI] [PubMed] [Google Scholar]