

Abstract
Prohibitin (PHB) is a ubiquitously expressed and evolutionarily conserved mitochondrial protein with multiple functions. We have recently shown that PHB upregulation offers robust protection against neuronal injury in models of cerebral ischemia in vitro and in vivo, but the mechanism by which PHB affords neuroprotection remains to be elucidated. Here, we manipulated PHB expression in PC12 neural cells to investigate its impact on mitochondrial function and the mechanisms whereby it protects cells exposed to oxidative stress. PHB overexpression promoted cell survival while PHB downregulation diminished cell viability. Functionally, manipulation of PHB levels did not affect basal mitochondrial respiration, but it increased spare respiratory capacity. Moreover, PHB overexpression preserved mitochondrial respiratory function of cells exposed to oxidative stress. Preserved respiratory capacity in differentiated PHB overexpressing cells exposed to oxidative stress was associated with an elongated mitochondrial morphology, while PHB downregulation enhanced fragmentation. Mitochondrial complex I oxidative degradation was attenuated by PHB overexpression and increased in PHB knockdown cells. Changes in complex I degradation were associated with alterations of respiratory chain supercomplexes. Furthermore, we showed that PHB directly interacts with cardiolipin and that downregulation of PHB results in loss of cardiolipin in mitochondria, which may contribute to destabilizing respiratory chain supercomplexes. Taken together, these data demonstrate that PHB modulates mitochondrial integrity and bioenergetics under oxidative stress, and suggest that the protective effect of PHB is mediated by stabilization of the mitochondrial respiratory machinery and its functional capacity, by regulation of cardiolipin content.
Keywords: Prohibitin, mitochondria, oxidative stress, PC12 cells, respiratory chain complexes
Graphical Abstract
The mitochondrial protein prohibitin (PHB) confers protection against a variety of cellular insults. In this study we manipulated expression of PHB in cells to demonstrate that PHB level regulates the viability of cells in an oxidative stress paradigm and maintains mitochondrial respiratory capacity, morphology, and respiratory complex stability. We show that PHB directly interacts with and maintains the content of the mitochondrial phospholipid cardiolipin (CL) that is required for respiratory chain complex and supercomplex stability and organelle morphology.
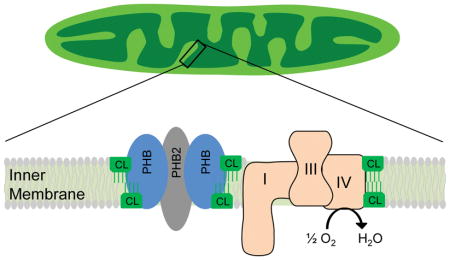
Introduction
Prohibitin (PHB) is a nuclear-encoded protein that localizes to mitochondria. PHB is highly conserved among species and is involved in wide variety of cellular functions (Thuaud et al. 2013, Merkwirth & Langer 2009), ranging from mitochondrial morphogenesis and formation of cristae (Merkwirth & Langer 2009) to stem cell rejuvenation in aged mice (Zhang et al. 2016). PHB forms a high molecular weight complex with PHB2 in the inner mitochondrial membrane that is required for its mitochondrial function (Tatsuta et al. 2005). PHB is also involved in mitochondrial dynamics regulation through stabilization of the dynamin-related GTPase OPA1 (Merkwirth & Langer 2009), a key protein in inner membrane fusion and cristae structure (Song et al. 2007).
Previously, we discovered that PHB is upregulated in ischemic preconditioning and demonstrated that PHB expression provides robust protection against different neuronal injury paradigms in vitro (Zhou et al. 2012) or cerebral ischemia in vivo (Kurinami et al. 2014, Kahl et al. 2017). Mitochondria play a critical role in the pathophysiology of ischemic brain injury (Sims & Anderson 2002, Fiskum et al. 2004, Chinopoulos & Adam-Vizi 2006, Sims & Muyderman 2010). In ischemia/reperfusion, mitochondria produce large amounts of reactive oxygen species (ROS) (Liu et al. 2002, Chan 2001, Turrens & Boveris 1980, Lesnefsky et al. 2001, Niatsetskaya et al. 2012) that can damage mitochondria and injure neural tissues. PHB has been implicated in the assembly or degradation of mitochondrial complex I (CI) (Bourges et al. 2004), and a recent report suggests that PHB stabilizes electron transfer chain (ETC) supercomplexes in mitochondria (Jian et al. 2017). However, the mechanisms whereby PHB protects mitochondria under oxidative stress conditions remain to be elucidated.
To gain new insight into the mechanisms of PHB-mediated neuroprotection we investigated its ability to protect neural cells in an oxidative stress paradigm. PHB levels were modulated in PC12 cells exposed to toxic levels of hydrogen peroxide (H2O2) and the effects on cell viability and mitochondrial functional and morphological properties were analyzed. We found that moderate changes in PHB expression levels had profound effects on cell viability, respiratory function, respiratory chain stability, and mitochondrial morphology. We also found that PHB binds cardiolipin and modulates its content in mitochondria. These findings identify PHB as a positive modulator of mitochondrial integrity and bioenergetic function in neural cells under oxidative stress, potentially via mediating the stability of cardiolipin, and provide a mechanistic link between PHB expression, mitochondrial health, and neuroprotection.
Materials and Methods
Materials
All routine laboratory chemicals including salts, detergents, and buffers were purchased from Sigma-Aldrich (St Louis, MO). Special chemicals including oligomycin (11342), FCCP (15218), antimycin A (19433) were from Cayman Chemicals (Ann Arbor, Michigan). All tissue culture reagents were purchased from Thermo-Fisher Scientific with the following catalog numbers: F-12K medium (21127030); heat-inactivated fetal bovine serum (16140071); heat inactivated horse serum (26050088); G418 (10131027); puromycin (A1113802); Lipofectamine LTX (15338500).
Cell culture and viability assays
The study was not pre-registered and used PC12, a cell line derived from rat adrenal gland pheochromocytoma that is not listed as a commonly misidentified cell line by the International Cell Line Authentication Committee (ICLAC) but not authenticated in this study. PC12 cells were purchased from ATCC (# CRL-1721; RRID:CVCL_0481) and cultured in F-12K medium containing 2.5% heat-inactivated fetal bovine serum and 15% heat inactivated horse serum at 37°C with 5% CO2, unless otherwise indicated. The culture medium was changed twice a week. Experiments were conducted in cell lines at the same passage number and lines were discarded after 20 passages. Cell viability was assayed with a commercially available MTS assay kit following the manufacture’s protocols (Promega).
No Institutional approval was required for this study other than primary neuronal culture. The experimental procedures for primary neuronal culture using timed pregnant mice were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medicine (protocol number 2009-0079). Timed pregnant mice (C57BL/6) at embryonic day 16 were sacrificed under deep anesthesia. Primary cortical neuronal cultures were obtained following procedures described previously (Zhou et al. 2012).
DNA transfection and stable cell line selection
The DNA construct for human PHB expression was described previously (Zhou et al. 2012). The shRNA plasmid (psi-U6) specific for rat PHB was purchased from GeneCopoeia. PC12 cells were transfected with linearized plasmid DNA using an electroporation apparatus (Nucleofector-2b, Amaxa-Lonza). Three days after transfection, cells were selected in 1mg/ml G418 (PHB expression plasmid) or 10μg/ml puromycin (PHB shRNA plasmid) for two weeks and sub-cloned in 96 wells. The lines were expanded and the expression levels of PHB assayed by immunoblotting using an anti-PHB1 antibody (LifeSpan Cat# LS-C85387-100, RRID:AB_1665767).
Oxygen consumption assay
Oxygen consumption in intact PC12 cells was performed according to the procedure described by Brand and Nicolls (Brand & Nicholls 2011). PC12 cells stably transfected with PHB or knockdown for PHB (shPHB) were assayed for O2 consumption together with their corresponding controls: vector for PHB and a scrambled shRNA (shSCR) for shPHB. Cells (2×106/assay) were suspended in warm fresh culture medium (with glucose) and placed in a sealed chamber equipped with a Clark-type O2 sensor (Oxygraphy Plus, Hansatech) at 37°C. After 3 min in the chamber to obtain a stable oxygen trace, oligomycin (1.5μM) was added to the cells to inhibit the mitochondrial ATPase. The oxygen consumption rate before oligomycin addition was used as basal respiration rate. The rate after oligomycin addition was used as a measure of proton leak. The cells were then treated with the protonophore carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP, 1μM) to fully uncouple mitochondria and stimulate maximum respiration. At the end of the measurement, a combination of rotenone and antimycin A (1μM each) was added to the cells to fully inhibit mitochondrial respiration and measure non-respiratory chain driven oxygen consumption, which was subtracted from all other values in the calculation of mitochondrial respiration.
Mitochondrial morphology measurements
For mitochondrial length measurements in neuritic processes, 0.35×106 PC12 cells were cultured on collagen coated glass cover slips (25mm, EMS) and co-transfected with mito-RFP to label mitochondria and GFP for cytosolic labeling using Lipofectamine LTX (Invitrogen). After transfection, cell differentiation was induced with recombinant human β-NGF (100ng/ml, R&D Systems, 256-GF/CF). After 6 days, differentiated PC12 cells were treated with 0.25mM H202 for 2hrs in order to induce oxidative stress and then fixed with 4% paraformaldehyde in PBS and mounted on imaging slides.
Mitochondria were visualized using a Leica TCS SP5 confocal laser-scanning microscope (Mannheim, Germany). Images were captured with a 63x oil objective and a zoom of 4. Confocal settings were kept unchanged for all experiments (z-step-size: 0.1μm, 600Hz, 1024×1024). To confirm cell integrity and localization of the mitochondria in the cell processes, only mito-RFP/GFP labeled cells were analyzed. Neurites from 5–10 cells were randomly selected and imaged per cover slip. Mitochondrial length was assessed using NIH Image J software by an operator blinded to treatment conditions.
For analysis of mitochondrial morphology in primary neurons, freshly dissociated cortical neurons were co-transfected with mouse PHB specific siRNA (si-PHB) and a mitochondria-targeted red fluorescent protein, dsRed2 (mt-dsRed). Control cells were transfected with a scrambled siRNA (si-Ctrl) and mt-dsRed. Neurons were seeded either on glass-bottom or plastic dishes. After five days in culture, cells in plastic dishes were used to make lysates for western blot analysis, while cells on glass bottom dishes were used to acquire mitochondrial morphology images using a confocal microscope (63 x objective lens) as described above. Ten random images containing mt-dsRed transfected neurons were taken for each dish by one person. Each image was coded with letter/number combination that revealed no information about the cells in the image. The images were analyzed in a blinded manner by another person who had no knowledge of the image coding and scored according to the morphology of mitochondria in neurite branches. Neurons were scored to a particular category when a majority of the imaged mitochondria were tubular, termed “elongated” or punctate, termed “fragmented.” Neurons that contain an intermediate proportion of elongated and fragmented mitochondrial in their neurites were labeled “mix.”
Mitochondrial Preparation and blue native (BN) gel electrophoresis
Crude mitochondrial preparations were made from cell lines as previously described (D’Aurelio et al., 2010). Cells were washed and harvested in a buffer containing 225mM mannitol, 75mM sucrose, 5mM HEPES, and 1mM EGTA before homogenization in a glass/Teflon homogenizer. Whole cells were centrifuged at 800xg for 5 minutes, supernatant collected and spun at 12,000xg for 10 minutes at 4°C. The crude mitochondrial pellet was washed once in the same buffer and centrifuged at 12,000xg for 10 minutes. The pellets were used for the BN gel electrophoresis performed as described previously (Wittig et al. 2006). Briefly, for assembled mitochondrial respiratory chain complex I detection using one-dimension BN gels, mitochondria were lysed with n-dodecyl β-d-maltoside (1.6g/g of protein) and 20μg of protein was loaded per lane in 4–12% Bis-Tris gradient gels and run at 4°C overnight. To detect respiratory chain supercomplexes, mitochondria were lysed in digitonin (8mg/ml) for 20 min followed by BN gel electrophoresis. The separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes for immunoblotting.
Mitochondrial Cardiolipin Content Measurement
Cardolipin content was measured in crude mitochondrial preparations using an enzyme-linked immunosorbent assay kit according to manufacturer’s instructions (EKU02951,Biomatik, Wilmington, DE). Mitochondria were isolated and 100μg were lysed in PBS by sonication on ice immediately before assaying.
Cardlolipin Binding Assay
Mitochondrial cardiolipin-binding proteins were pulled down using cardiolipin coated beads (P-BCLP, Echelon Biosciences, Salt Lake City, UT). Following crude mitochondrial isolation, 100μg of mitochondria were lysed in 1.0% NP-40, 150 mM NaCl, 10 mM HEPES, pH 7.4 on ice for 30 minutes. The resulting lysate was incubated overnight with beads coated in cardiolipin or uncoated control beads. Beads were washed 5 times with 0.5% NP-40, 150 mM NaCl, 10 mM HEPES, pH 7.4 and bound proteins were eluted in 2x SDS sample buffer. Eluted samples and 25μg of unbound mitochondrial input lysate were resolved by SDS-PAGE and immunoblotted as indicated.
Western blot analyses
For immunoblotting in whole cell lysates, cells were homogenized in RIPA (50mM Tris HCl, pH of 7.4, 150mM NaCl, 1.0% (v/v) NP-40, 0.5% (w/v) sodium deoxycholate, and 1.0mM EDTA, 0.1% (w/v) SDS) buffer plus 1% protease inhibitors (05892791001, Complete Mini; Roche, Mannheim, Germany). Equal amounts of proteins (25 – 50 μg) were separated by SDS-PAGE and transferred to PVDF membranes. For immunoblotting in mitochondria, equal amounts of proteins from crude mitochondrial preps were lysed in RIPA with 1% protease inhibitor for 30 minutes on ice before SDS-PAGE. Antibodies used were anti-PHB antibody (1:2000 dilution, clone # II-14-10, Thermo Scientific), anti-PHB2 (1:1000, Cell Signaling Technology), anti-HSP60 (1:1000, source), anti-SLP-2 (1:1000, ProteinTech), anti-β-actin (1:3000, Cell Signaling Technology). Representative subunits of CI were detected with anti-NDUFA9 (1:1000, Abcam) and anti-NDUFVI (1:500, Santa Cruz) antibodies. Complex III was detected with an anti-Core 2 subunit antibody (1:1000, Abcam). Protein bands were detected using a LI-COR Digital Imaging system and band intensities were quantified with Image Studio software.
Relative mitochondrial DNA copy number measurement by RT-PCR
Total DNA was extracted from undifferentiated PC12 cells using Wizard SV Genomic DNA Purification System (A2360, Promega, Madison, WI) and measured by quantitative real-time PCR analysis with Maxima SYBR Green/ROX qPCR Master Mix (K0221,Thermo Scientific, Waltham, MA). Amplifications were performed on a Chromo 4 Detector (Bio-Rad, Hercules, CA) using a two-step cycling protocol (50°C for 2 min and 95°C for 10 min followed by 45 cycles of 95°C for 15 sec, 60°C for 1 min). The following primers were used: mtDNA (F: 5′-AGAGAAAGTACCGCAAGGGAA-3′ and R: 5′-GTTTGGTTTCGGGGTTCTTAG-3′) and 28sRNA nuclear DNA (F: 5′-GGGTTTTAAGCAGGAGGTGTC-3′ and R: 5′-TGGCAACAACACATCATCAGT-3′). The data are shown as mean±SEM relative to control lines.
Statistical analyses
Data are presented as mean+/−SEM. No sample calculation and no randomization was performed. Statistical differences between two groups were evaluated by unpaired, two-tailed, t-test. ANOVA with Tukey’s post-hoc was used to compare differences across multiple samples. Kruskal-Wallis test was used for comparing mitochondrial size in different groups followed by Dunn’s for multiple comparison test using Graphpad Prism 7 software. Differences were considered significant when p<0.05.
Results
PHB expression modulates cell viability under oxidative stress
To evaluate the effect of PHB on mitochondrial function, we generated PC12 cell lines with stably overexpressed or downregulated PHB levels (Fig 1A, B). PHB overexpressing cells showed a 1-fold increase of PHB protein levels compared to vector control cells. The maximum stable shRNA-mediated knockdown we could achieve resulted in a 60% decrease of PHB level. This is likely because cells with lower PHB levels did not survive the selection process, as PHB is essential for cell viability (Artal-Sanz et al. 2003, He et al. 2008). This level of silencing is comparable to RNAi-mediated PHB knockdown in other studies (Jian et al. 2017). Under normal cell culture conditions, PHB overexpressing or knockdown cell lines exhibited morphological features, such as cell shape, size, growth rate, and viability, similar to control lines (Supplemental Figure 1).
Figure 1. PHB levels in PC12 cells affect cell viability in oxidative stress conditions.

(A) Western blots analyses for PHB. PC12 cells stably transfected with PHB overexpression, shRNA silencing, and vector control plasmids and grown in normal growth medium were harvested and lysed. Each lane was loaded with 20μg of cell lysate. β-actin was used as loading control. (B) Protein band intensity quantification. Mean band intensity was normalized to β-actin levels and expressed as a proportion of control lines. **p<0.01 compared to vector or shSCR lines, n=8 independent experiments. (C) Cell viability analyses. Cell viability was determined using MTS assay after cells were treated for 2hrs with H2O2 (0.2mM) or vehicle. Viability was calculated as percentage of vehicle treated cells for each line. ***p<0.001 compared to vector or shSCR, n=6 independent experiments. The data are shown as mean±SEM.
To determine if PHB modulates the viability of PC12 cells under oxidative stress, we treated cells with H2O2 (0.2mM for 2hrs). Consistent with our previous results in primary neuronal cultures (Zhou et al. 2012), PHB overexpression significantly improved cell survival of PC12 cells, while PHB downregulation caused decreased cell viability (Fig 1C). In order to control for off-target effects of the shRNA used to produce our knockdown line, we repeated this experiment using two independent lentiviral shRNAs constructs targeting PHB. We observed comparable decrease in both PHB level and viability under oxidative stress (Supplemental Fig 2A, B). These data support the hypothesis that PHB levels modulate cell viability under oxidative stress.
PHB expression modulates mitochondrial respiration
To gain insight into the potential mechanisms underlying the protective effect of PHB, we investigated how PHB expression modulates mitochondrial respiratory function in basal and oxidative stress conditions. We measured respiration in intact cells, in which the undisturbed cellular environment and the preserved interaction of mitochondria with other cellular components is useful in assessing the functional status of mitochondria (Brand & Nicholls 2011). In normal culture conditions, PHB expressing cells showed similar O2 consumption rates compared to control cells (Fig 2A). PHB knockdown did not result in a significant decline of O2 consumption in basal conditions, suggesting that endogenous PHB levels can be decreased up to 60% without causing overt deficits in respiration. After exposure to oxidative stress, O2 consumption decreased in all cell lines, indicating mitochondrial impairment. This impairment was ameliorated by PHB overexpression (Fig. 2A), while PHB knockdown resulted in a more severe respiratory decline, consistent with the hypothesis that PHB levels mediate mitochondrial susceptibility to oxidative stress. To determine if oxidative stress caused uncoupling of mitochondria we measured proton leak. Addition of oligomycin decreased O2 consumption with no significant differences among cell lines with or without H2O2 treatment (Fig 2B). These results indicate that PHB levels do not affect respiration by altering proton leak and that the oxidative stress used in our model does not cause uncoupling of mitochondria.
Figure 2. PHB levels modulate mitochondrial respiration.

PC12 cells treated with H2O2 (0.2 mM, 2 hrs) or vehicle were analyzed by oxygraph. (A) Basal respiration. (B) Proton leakage measurement after oligomycin (1μM) addition. (C) Maximum respiration induced by the protonophore FCCP (1μM). (D) Spare respiratory capacity obtained by subtracting basal respiration from maximum respiration. *p<0.05 compared to vehicle treated cells, #p<0.05 compared to corresponding shSCR group. Each bar represents between 6 and 9 samples from 3 independent experiments. (E) Relative mitochondrial DNA copy number measured by real-time RT-qPCR and normalized to 28sRNA nuclear DNA. The data are shown as mean±SEM.
In normal culture conditions, PHB overexpression did not affect the maximal respiration rate induced by the protonophore FCCP (Fig. 2C). However, under oxidative stress, control cells showed a significant decrease of the maximum respiration rate, which was ameliorated by PHB overexpression. Interestingly, the maximal respiration in PHB knockdown cells was significantly decreased in both normal and oxidative conditions. Consequently, spare respiratory capacity, which is the difference between the maximal and basal respiration, was preserved in PHB overexpressing cells under oxidative stress, but not in vector cells. In PHB knockdown cells, spare respiratory capacity was reduced in basal conditions and severely impaired after oxidative stress (Fig 2D). These data suggest that PHB modulates the function of the respiratory chain under oxidative stress.
To determine if differences in mitochondrial respiration could be attributed to altered mitochondrial content, we measured the amount of mitochondrial DNA relative to nuclear DNA by real-time PCR. No significant differences in relative mitochondrial copy number between control cells and PHB overexpressing or knockdown lines were observed (Fig 2E), thereby excluding that differences in cell respiration were due to changes in mitochondrial content.
PHB levels modulate mitochondrial dynamics
Next, we examined whether modulation of PHB expression was associated with changes in mitochondrial morphology. PC12 cells were transfected with a mitochondrially targeted RFP (mito-RFP) and differentiated into neuron-like cells with nerve growth factor (NGF) to facilitate the visualization of mitochondrial morphology in neural processes. We measured mitochondrial length in normal and oxidative stress conditions to assess the potential association between PHB protein level and mitochondrial size. In normal conditions, analysis of the relative frequency distribution of mitochondrial size revealed that PHB overexpression significantly shifted the size distribution toward longer mitochondria. Conversely, downregulation of PHB resulted in a shift towards shorter (fragmented) mitochondria (Fig. 3A, B, C). After exposure to H2O2, mitochondrial length distributions underwent dramatic changes. In PHB overexpressing cells, mitochondrial fragmentation was largely prevented, maintaining a length distribution similar to untreated control cells (Fig. 3A H2O2 panels, B, C). In contrast, PHB knockdown cells exhibited severe fragmentation (Fig. 3A H2O2 panels, B, C), with 95% of mitochondria having the major axis shorter than 0.6 μm (Fig. 3A, shPHB/H2O2 panel). These results demonstrate that PHB is an important regulator of mitochondrial morphology, and is able to prevent mitochondrial fragmentation in conditions of oxidative stress.
Figure 3. PHB levels modulate mitochondrial length in differentiated PC12 cells.

PC12 cells on glass bottom dishes were transfected with mito-RFP and induced to differentiate with NGF for 6 days. (A) Mitochondrial length distribution in normal (upper panels) and in oxidative stress conditions (lower panels). (B) Box-and-whisker diagrams showing mitochondrial length distribution derived from the Kruskal-Wallis test for the four cell lines in basal conditions (untreated) and under oxidative stress (H2O2 treatment). The box is interquartile range from the first to the third (midspread 50%); the data range (max and min values) is represented by the lines above and below the box; the bar inside the box represents the median value. Significant differences among groups revealed by the Dunn’s test for multiple comparisons are indicated (*p<0.05, total n=359–763 mitochondria per group in normal conditions and n=211–1096 mitochondria per group in H2O2 treated cells). (C) Representative images of mitochondrial network in PHB expressing and PHB knockdown cells in basal conditions and after oxidative stress. Data were obtained from three independent experiments measuring individual mitochondria.
To assess if these findings extend to neurons, we analyzed mitochondrial morphology in mouse primary cortical neurons, while manipulating PHB content. We knocked down endogenous PHB using siRNA electroporation and simultaneously labeled mitochondria with mt-dsRed. Neurons were cultured for five days in vitro (DIV), a time point we have previously reported to be sufficient for PHB knockdown but before any ensuing reduction in cell viability (Zhou et al. 2012). At 5 DIV, we lysed cells for PHB protein level assessment or imaged neurons to analyze mitochondrial morphology. On average, si-PHB transfection reduced PHB protein content by 70% relative to si-Ctrl transfected neurons (Fig. 4A, B). Mitochondria in processes of PHB knockdown neurons exhibited a dramatic shift from an elongated to a predominantly fragmented morphology (Fig. 4C). When PHB was downregulated, the fraction of neurons containing elongated mitochondria decreased by half, and the proportion of neurons with fragmented mitochondria doubled (Fig. 4D). Together with data obtained using PC12 cells, these results demonstrate that PHB is a potent modulator of mitochondria morphology also in neurons.
Figure 4. PHB knockdown in primary neurons causes mitochondrial fragmentation.

Primary neurons co-transfected with PHB siRNA and mt-dsRed were cultured for 5 days before PHB protein level and mitochondrial morphology analyses. (A) Western blot analysis of PHB protein level in si-PHB and si-Ctrl transfected neurons and β-actin as loading control. (B) Quantitative measurement of PHB protein band intensity normalized to β-actin. (C) Representative confocal images of mitochondrial morphology in the neurites of neurons transfected with mt-dsRed and either si-PHB or si-Ctrl as indicated. Images were taken with 63X oil objective. (D) Quantitative analyses of siRNA transfected neurons containing elongated tubular or fragmented mitochondria. Neurons containing a mix of elongated and fragmented mitochondria were grouped separately. *p<0.05, n=3 separate experiments totaling 224 cells for si-PHB group and 254 for si-Ctrl group. Data are shown as mean±SEM.
PHB expression promotes mitochondrial respiratory chain complex and supercomplex stability
In order to gain insight into how PHB expression modulates mitochondrial function, we examined the levels of individual subunits of the electron transfer chain complexes. We found that representative subunits of CI were significantly reduced in PHB knockdown cells (Fig. 5A–D), suggesting that PHB is needed in maintaining the levels of CI specifically, as no significant alteration in levels of CIII core 2 subunit was observed (Fig 5E, F). These results were confirmed using two independent lentiviral shRNAs targeting PHB in PC12 cells (Supplemental Fig 2 C).
Figure 5. PHB expression modulates mitochondrial complex I subunit levels and RSC formation.

Mitochondria from PHB overexpressing or knockdown cells and respective controls were partially purified and lysed with either RIPA buffer for SDS-PAGE or by digitonin for BN-PAGE analyses. (A, B) Mitochondrial complex CI level assessment with CI subunit NDUFA9 specific antibody and quantitative protein band analysis. HSP60 was used as loading control. (C, D) CI level assessment with a different antibody specific for CI subunit NDUFVI and protein band intensity measurements. (E, F) CIII level assessment with antibody specific for CIII subunit core 2 and protein band intensity measurements. (G) Supercomplex assembly assessment with antibodies for CI subunit A9. Mitochondrial preparations were solubilized using digitonin and separated by BN-PAGE. (H) Supercomplex assembly assessed by antibody specific for CIII core 2 subunit. Shown in each panel is one representative blot image from 3–4 separate experiments. *p<0.05, **p<0.01. The data are shown as mean±SEM.
Mitochondrial respiratory complexes exist in a dynamic arrangement of supercomplexes (RSCs), which facilitate efficient energy production (Genova & Lenaz 2014, Lapuente-Brun et al. 2013). RSC can be formed from combinations of either CIII with CIV (CIII2/CIV) or CI with CIII and CIV (CI/CIII2/CIV), among other variations (Acin-Perez et al. 2008, Moreno-Lastres et al. 2012). Because CI plays an essential role in RSC assembly (Moreno-Lastres et al. 2012, Lenaz et al. 2016) and CI level was modulated by PHB expression (Fig 5A–D), we further examined whether PHB expression also modulates CI in RSCs of PC12 cells.
RSCs can be visualized in digitonin permeabilized mitochondrial fractions by BN gel electrophoresis combined with western blotting (Acin-Perez et al. 2008, Schagger & Pfeiffer 2001, Dudkina et al. 2010). Strikingly, when digitonin solubilized mitochondria were resolved by BN gel and probed with CI and CIII specific antibodies, we observed more RSCs composed of CI, CIII2 and CIV in PHB overexpressing cells than in controls (Fig. 5G, two left lanes). Mitochondria from shPHB cells showed much lower RSC levels than sh-scrambled controls (Fig. 5G, two right lanes). Similar results were obtained when a CIII antibody was used to probe the RSC in the same set of samples (Fig. 5H), confirming the effect of PHB expression on RSC assembly.
Next, we treated cells with H2O2 in order to assess the stability of CI under oxidative stress conditions. We confirm previous reports that CI is sensitive to oxidative stress, as assembled CI detected by BN-PAGE in maltoside-purified mitochondria underwent a time- and dose-dependent degradation (Fig. 6A, B). PHB overexpression largely preserved the levels of CI subunit NDUFA9 from H2O2 induced degradation, while PHB knockdown led to the acceleration of its breakdown (Fig 6C, D). These data suggest that PHB not only contributes to the stability of CI subunits but also in complex stability, including supercomplex stability, under oxidative stress conditions.
Figure 6. PHB expression prevents oxidative breakdown of CI.

Oxidative breakdown of CI was assayed in mitochondrial preparations from WT PC12 cells. Intact CI was solubilized using maltoside from mitochondria of cells that were treated with increasing concentrations of H2O2 for 2 hrs (A) or with 0.5mM H2O2 for 0, 30, 60, 90, 120, or 180 minutes (B). Solubilized intact CI was resolved by BN-PAGE and probed with antibodies to CI subunit NDUFA9. To assess the effect of PHB on CI stability, PHB overexpressing or knockdown cells and a vector control line were treated with 0.5mM H2O2 for 4 hrs. Mitochondrial preparations were lysed and content of representative CI subunit NDUFA9 was assayed by SDS-PAGE and western blotting (C). CI NDUFA9 band intensity was normalized using TIM23 (D). *p<0.05, n=4 separate experiments; ns, not significant. The data are shown as mean±SEM.
PHB binds cardiolipin and modulates mitochondrial cardiolipin content
Increasing evidences suggest that phospholipids play a critical role in modulating mitochondrial respiratory complex activity and the assembly of RSC (Mileykovskaya & Dowhan 2014, Pfeiffer et al. 2003). However, whether mitochondrial lipids are involved in the effects of PHB expression on RSC assembly remains to be determined. Therefore, we first performed a cardiolipin binding assay to examine if native PHB and cardiolipin interact directly using cardiolipin coated beads to pull down cardiolipin-binding proteins from mitochondrial lysates. As shown in Fig 7A, cardiolipin coated beads bound and pulled down PHB and PHB2, indicating a direct interaction between PHB complex and cardiolipin. Because of the direct interaction between PHB and cardiolipin, we next sought to examine whether PHB expression could directly modulate cardiolipin content in our model. Using ELISA, we found a significant 40% decrease of cardiolipin content in mitochondria isolated from PHB knockdown cells compared to scrambled shRNA control cells (Fig 7B). This result is consistent with previous reports that PHB is required for the formation of mature cardiolipin in mitochondria (Osman et al. 2009, Richter-Dennerlein et al. 2014). Together, these data suggest that PHB may participate in the modulation of RSC assembly through its binding to cardiolipin.
Figure 7. PHB binds cardiolipin and modulates its content in mitochondria.

(A) Levels of PHB1 and SLP-2 were measured in whole cell lysates by immunoblot. HSP60 was used as a loading control. (B) Cardiolipin content was measured in mitochondria isolated from shSCR and shPHB PC12 cells. (C) Cardiolipin binding proteins were pulled down from mitochondrial lysates using cardiolipin coated beads and separated by SDS-PAGE for immunoblotting. Uncoated beads were used to control for non-specific binding and unbound lysate was used as an input control. *p<0.05, n=3 separate experiments. (D) The data are shown as mean±SEM.
To our surprise, contrary to a previous report (Christie et al. 2011), we did not observe interaction between cardiolipin and another mitochondrial inner membrane protein, stomatin like protein 2 (SLP-2). SLP-2 has been reported to interact with PHB, and SLP-2 knockdown by siRNA leads to the instability of PHB (Da Cruz et al. 2008). However, when we assessed SLP-2 protein levels in cells in which PHB was downregulated, we found a significant decrease in expression of SLP-2 (Fig 7C, D), suggesting that SLP-2 is not stable when PHB levels are decreased. Taken together, these data suggest that PHB and SLP-2 are mutually dependent in PC12 mitochondria but in our assay condition only PHB binds cardiolipin directly.
Discussion
We investigated the mechanistic aspects of PHB mediated neuroprotection, focusing on the effects of modulating PHB levels on mitochondrial function in normal and oxidative stress conditions. We found that PHB modules the mitochondrial response to oxidative stress by preserving mitochondrial morphology, respiratory capacity, preventing oxidative degradation of complex I, and stabilizing respiratory supercomplexes. PHB binds to cardiolipin and regulates its levels, which are known to be critical for complex and supercomplex assembly and stability (Pfeiffer et al. 2003, Mileykovskaya & Dowhan 2014). Collectively, these findings suggest that PHB is essential for stabilizing the lipid environment and respiratory machinery in the mitochondrial inner membrane and that, in conditions of oxidative stress, PHB overexpression exerts a protective effect by preserving the integrity of the respiratory machinery and bioenergetic capacity.
Surprisingly, PHB overexpression did not increase respiration in basal conditions (Fig. 2) despite increased levels of respiratory supercomplexes (Fig. 5). Therefore, it appears that endogenous PHB content is sufficient to achieve maximum basal respiratory capacity in normal conditions. However, PHB overexpression largely preserved both basal and spare respiratory capacity in oxidative stress conditions (Fig 2), suggesting that increased PHB levels are beneficial when cells are subjected to stress. On the other hand, decreasing PHB by 60% was sufficient to impair basal respiration after oxidative stress. Further, PHB knockdown significantly impaired maximal mitochondrial respiration and spare respiratory capacity both in basal and oxidative stress conditions (Fig. 2). These data demonstrate that PHB is necessary to maintain healthy mitochondrial function under basal and stressed conditions.
Maintaining a healthy mitochondrial spare respiratory capacity could prevent energy defects in pathological states. Impaired spare respiratory capacity, for example, was identified in fibroblasts derived from AD patients (Gray & Quinn 2015). Furthermore, it was shown that spare respiratory capacity regulates excitotoxicity mediated by rotenone-induced complex I inhibition in a neuronal culture model of Parkinson’s disease (Yadava & Nicholls 2007). A decline of spare respiratory capacity has also been linked to aging (Desler et al. 2012). Conversely, human adipocytes with higher spare respiratory capacity are better at maintaining ATP homeostasis under hypoglycemic conditions (Keuper et al. 2014). Furthermore, enhanced spare respiratory capacity in non-transformed fibroblasts is linked to increased cell survival (Nickens et al. 2013). Our results suggest that PHB modulates mitochondrial function by increasing spare respiratory capacity, although the detailed mechanisms responsible for this effect of PHB need to be further investigated.
Mitochondria are dynamic organelles that constantly undergo balanced fission and fusion that control shape, size, and number of the organelle. Balanced mitochondrial dynamics is critical for normal cell activity, as evidenced by the neuropathology in patients harboring loss-of-function mutations in the mitochondrial fusion genes, OPA1 and MFN2. OPA1 mutations cause autosomal dominant optic atrophy in retinal ganglion cells and optic nerve degeneration (Delettre et al. 2000) and mutations in MFN2 cause peripheral motor and sensory neuron loss (Zuchner et al. 2004). On the other hand, diminished fission is equally detrimental to cells. Impaired fission due to deletion of dynamin-related protein 1 (Drp1), a protein involved in the fission process, leads to the disruption of mitochondrial bioenergetics and synaptic function in CA1 hippocampal neurons (Shields et al. 2015). In response to stress, the balance tends to shift toward increased fission, leading to mitochondrial fragmentation and depolarization, ultrastructural damage, subsequent cytochrome c release and ROS production that contribute to cell death (Barsoum et al. 2006, Knott et al. 2008, Youle & van der Bliek 2012, Burte et al. 2015). Stress-induced mitochondrial fragmentation precedes neurite injury and neuronal cell death (Barsoum et al. 2006), highlighting a causative role of imbalanced mitochondrial fusion and fission in neuronal injury. Our results show that mitochondrial dynamics in the neurites of differentiated PC12 cells and in primary neurons is modulated by PHB. The preservation of mitochondrial length by PHB under oxidative stress (Fig. 3) may explain why PC12 cells overexpressing PHB exhibited increased viability, whereas cells with reduced PHB levels showed exacerbated cell death after H2O2 treatment. Further detailed biochemical studies are needed to elucidate how PHB modulates mitochondrial dynamics.
Complex I is degraded in damaged mitochondria (Guaras & Enriquez 2017, Pryde et al. 2016). Here we show for the first time that PHB overexpression preserves complex I in cells under oxidative stress. Furthermore, RSC formation was enhanced in PHB overexpressing cells, while it was diminished in PHB knockdown cells. Because of the critical role that CI plays in RSC assembly (Moreno-Lastres et al. 2012), and since PHB interacts with CI (Bourges et al. 2004), PHB may favor the association of the subunits of CI into a functional complex that serves as a foundation for RSC assembly together with CIII and CIV. PHB overexpression prevented the degradation of CI subunits under oxidative stress, while PHB knockdown exacerbated it (Fig. 6), indicating an important function of PHB in protecting CI from oxidative breakdown. Preserving assembled CI could contribute to the protection of CI-containing RSC and enhanced spare respiratory capacity.
Cardiolipin is an essential mitochondrial phospholipid. There is convincing evidence to support a role for cardiolipin in higher order organization of respiratory supercomplexes. Mitochondria from patients of Barth syndrome, a disease characterized by cardiomyopathy and skeletal myopathy caused by mutations in the gene for cardiolipin maturation, show lower levels of cardiolipin and decreased RSC formation (McKenzie et al. 2006). RSCs composed of CIII2CIV are unstable in the absence of cardiolipin (Pfeiffer et al. 2003, Zhang et al. 2002). Cardiolipin also contributes to mitochondrial inner membrane fusion, according to a recent study (Liu & Chan 2017, Ban et al. 2017). Our observation that PHB level modulates both RSC stability and cardiolipin content raises the possibility that the effect of PHB on CI and RSC is mediated through its ability to control cardiolipin content. Further, PHB expression may modulate both RSC formation and mitochondrial morphology by directly regulating cardiolipin content in mitochondria, although the detailed mechanisms by which PHB modulates cardiolipin level need to be further elucidated.
A role for the PHB in the regulation of cardiolipin has been previously reported by synthetic lethality screening in yeast (Osman et al. 2009) and in the maturation of cardiolipin in HEK cells (Richter-Dennerlein et al. 2014). Our data demonstrate the interaction of the PHB complex with cardiolipin and confirm that reduction of PHB decreases cardiolipin in mammalian mitochondria (Fig 7). Another SPFH-domain containing protein, SLP-2, has been proposed to complex with PHB, bind cardiolipin, and regulate ETC supercomplex stability (Da Cruz et al. 2008, Christie et al. 2011, Mitsopoulos et al. 2017). Further, it has been reported that SLP-2 stabilizes PHB in mitochondrial lipid microdomains (Christie et al. 2011). We found that loss of PHB leads to a reduction in SLP-2. The reciprocal regulation of PHB and SLP-2 is intriguing and increases the complexity of the role of PHB in regulating mitochondrial function. Taken together, these data suggest that the decrease of PHB and consequent reduction of cardiolipin drive mitochondrial dysfunction.
In summary, here we demonstrate that PHB plays an important role in the protection of PC12 cells subjected to oxidative stress by modulating mitochondrial respiratory machinery, dynamics, and supercomplexes stability. This study provides a deeper mechanistic understanding of how PHB may exert its protective effects. Our findings have potential implications for brain ischemia and other pathologies involving oxidative stress, as modulating the expression of PHB may have a significant impact on brain injury and recovery.
Supplementary Material
Acknowledgments
This work was supported by NIH grants NS067078 (PZ), NS034179 (CI), NS095692 (GM and CI), and a DFG fellowship KA 3810/1-1 to AK. The contribution of the Feil Family Foundation is gratefully acknowledged.
Abbreviation list
- PHB
prohibitin
- CI
complex I
- CIII
complex III
- ETC
electron transfer chain
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- shRNA
short hairpin ribonucleic acid
- SCR
scrambled
- FCCP
carbonyl cyanide-4-trifluoromethoxy)phenylhydrazone
- mito
mitochondria
- RFP
red fluorescence protein
- GFP
green fluorescence protein
- β-NGF
beta-nerve growth factor
- PBS
phosphate buffered saline
- siRNA
small interference RNA
- BN
blue native
- HEPES
hydroxyethyl piperazineethanesulfonic acid
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- PVDF
polyvinylidene difluoride
- SDS-PAGE
sodium dodecyl sulfate- polyacrylamide gel electrophoresis
- RIPA
Radioimmunoprecipitation assay
- HSP
heat shock protein
- SLP-2
Stomatin-like protein 2
- RT-PCR
real time polymerase chain reaction
- mtDNA
mitochondrial DNA
- DIV
days in vitro
- RSC
respiratory supercomplex
- ELISA
enzyme-linked immunosorbent assay
- ROS
reactive oxygen species
- HEK cells
Human Embryonic Kidney cells
Footnotes
Conflicts of interest
All authors claim no conflicts of interest.
Open Science Badges
This article has received a badge for *Open Materials* because it provided all relevant information to reproduce the study in the manuscript. The complete Open Science Disclosure form for this article can be found at the end of the article. More information about the Open Practices badges can be found at https://cos.io/our-services/open-science-badges/.
References
- Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Molecular cell. 2008;32:529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Artal-Sanz M, Tsang WY, Willems EM, Grivell LA, Lemire BD, van der Spek H, Nijtmans LG. The mitochondrial prohibitin complex is essential for embryonic viability and germline function in Caenorhabditis elegans. The Journal of biological chemistry. 2003;278:32091–32099. doi: 10.1074/jbc.M304877200. [DOI] [PubMed] [Google Scholar]
- Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, Oka T, Mihara K, Ishihara N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nature cell biology. 2017;19:856–863. doi: 10.1038/ncb3560. [DOI] [PubMed] [Google Scholar]
- Barsoum MJ, Yuan H, Gerencser AA, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. The EMBO journal. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourges I, Ramus C, Mousson de Camaret B, Beugnot R, Remacle C, Cardol P, Hofhaus G, Issartel JP. Structural organization of mitochondrial human complex I: role of the ND4 and ND5 mitochondria-encoded subunits and interaction with prohibitin. Biochem J. 2004;383:491–499. doi: 10.1042/BJ20040256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nature reviews. Neurology. 2015;11:11–24. doi: 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Adam-Vizi V. Calcium, mitochondria and oxidative stress in neuronal pathology. Novel aspects of an enduring theme. Febs J. 2006;273:433–450. doi: 10.1111/j.1742-4658.2005.05103.x. [DOI] [PubMed] [Google Scholar]
- Christie DA, Lemke CD, Elias IM, et al. Stomatin-like protein 2 binds cardiolipin and regulates mitochondrial biogenesis and function. Mol Cell Biol. 2011;31:3845–3856. doi: 10.1128/MCB.05393-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Cruz S, Parone PA, Gonzalo P, Bienvenut WV, Tondera D, Jourdain A, Quadroni M, Martinou JC. SLP-2 interacts with prohibitins in the mitochondrial inner membrane and contributes to their stability. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2008;1783:904–911. doi: 10.1016/j.bbamcr.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nature genetics. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- Desler C, Hansen TL, Frederiksen JB, Marcker ML, Singh KK, Juel Rasmussen L. Is There a Link between Mitochondrial Reserve Respiratory Capacity and Aging? Journal of aging research. 2012;2012:192503. doi: 10.1155/2012/192503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudkina NV, Kouril R, Peters K, Braun HP, Boekema EJ. Structure and function of mitochondrial supercomplexes. Biochimica et biophysica acta. 2010;1797:664–670. doi: 10.1016/j.bbabio.2009.12.013. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Rosenthal RE, Vereczki V, Martin E, Hoffman GE, Chinopoulos C, Kowaltowski A. Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. Journal of bioenergetics and biomembranes. 2004;36:347–352. doi: 10.1023/B:JOBB.0000041766.71376.81. [DOI] [PubMed] [Google Scholar]
- Genova ML, Lenaz G. Functional role of mitochondrial respiratory supercomplexes. Biochimica et biophysica acta. 2014;1837:427–443. doi: 10.1016/j.bbabio.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Gray NE, Quinn JF. Alterations in mitochondrial number and function in Alzheimer’s disease fibroblasts. Metabolic brain disease. 2015 doi: 10.1007/s11011-015-9667-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guaras AM, Enriquez JA. Building a Beautiful Beast: Mammalian Respiratory Complex I. Cell metabolism. 2017;25:4–5. doi: 10.1016/j.cmet.2016.12.019. [DOI] [PubMed] [Google Scholar]
- He B, Feng Q, Mukherjee A, Lonard DM, DeMayo FJ, Katzenellenbogen BS, Lydon JP, O’Malley BW. A repressive role for prohibitin in estrogen signaling. Molecular endocrinology. 2008;22:344–360. doi: 10.1210/me.2007-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian C, Xu F, Hou T, Sun T, Li J, Cheng H, Wang X. Deficiency of PHB complex impairs respiratory supercomplex formation and activates mitochondrial flashes. Journal of cell science. 2017;130:2620–2630. doi: 10.1242/jcs.198523. [DOI] [PubMed] [Google Scholar]
- Kahl A, Anderson CJ, Qian L, Voss H, Manfredi G, Iadecola C, Zhou P. Neuronal expression of the mitochondrial protein prohibitin confers profound neuroprotection in a mouse model of focal cerebral ischemia. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2017 doi: 10.1177/0271678X17720371. 271678X17720371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keuper M, Jastroch M, Yi CX, Fischer-Posovszky P, Wabitsch M, Tschop MH, Hofmann SM. Spare mitochondrial respiratory capacity permits human adipocytes to maintain ATP homeostasis under hypoglycemic conditions. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2014;28:761–770. doi: 10.1096/fj.13-238725. [DOI] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nature reviews. Neuroscience. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurinami H, Shimamura M, Ma T, et al. Prohibitin viral gene transfer protects hippocampal CA1 neurons from ischemia and ameliorates postischemic hippocampal dysfunction. Stroke; a journal of cerebral circulation. 2014;45:1131–1138. doi: 10.1161/STROKEAHA.113.003577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapuente-Brun E, Moreno-Loshuertos R, Acin-Perez R, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340:1567–1570. doi: 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- Lenaz G, Tioli G, Falasca AI, Genova ML. Complex I function in mitochondrial supercomplexes. Biochimica et biophysica acta. 2016;1857:991–1000. doi: 10.1016/j.bbabio.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Gudz TI, Migita CT, Ikeda-Saito M, Hassan MO, Turkaly PJ, Hoppel CL. Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron-sulfur protein subunit of electron transport complex III. Arch Biochem Biophys. 2001;385:117–128. doi: 10.1006/abbi.2000.2066. [DOI] [PubMed] [Google Scholar]
- Liu R, Chan DC. OPA1 and cardiolipin team up for mitochondrial fusion. Nature cell biology. 2017;19:760–762. doi: 10.1038/ncb3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. Journal of molecular biology. 2006;361:462–469. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Langer T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochimica et biophysica acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Mileykovskaya E, Dowhan W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chemistry and physics of lipids. 2014;179:42–48. doi: 10.1016/j.chemphyslip.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsopoulos P, Lapohos O, Weraarpachai W, Antonicka H, Chang YH, Madrenas J. Stomatin-like protein 2 deficiency results in impaired mitochondrial translation. PloS one. 2017;12:e0179967. doi: 10.1371/journal.pone.0179967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Lastres D, Fontanesi F, Garcia-Consuegra I, Martin MA, Arenas J, Barrientos A, Ugalde C. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell metabolism. 2012;15:324–335. doi: 10.1016/j.cmet.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niatsetskaya ZV, Sosunov SA, Matsiukevich D, Utkina-Sosunova IV, Ratner VI, Starkov AA, Ten VS. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia-ischemia in neonatal mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:3235–3244. doi: 10.1523/JNEUROSCI.6303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickens KP, Wikstrom JD, Shirihai OS, Patierno SR, Ceryak S. A bioenergetic profile of non-transformed fibroblasts uncovers a link between death-resistance and enhanced spare respiratory capacity. Mitochondrion. 2013;13:662–667. doi: 10.1016/j.mito.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Haag M, Potting C, Rodenfels J, Dip PV, Wieland FT, Brugger B, Westermann B, Langer T. The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. The Journal of cell biology. 2009;184:583–596. doi: 10.1083/jcb.200810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schagger H. Cardiolipin stabilizes respiratory chain supercomplexes. The Journal of biological chemistry. 2003;278:52873–52880. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- Pryde KR, Taanman JW, Schapira AH. A LON-ClpP Proteolytic Axis Degrades Complex I to Extinguish ROS Production in Depolarized Mitochondria. Cell reports. 2016;17:2522–2531. doi: 10.1016/j.celrep.2016.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Dennerlein R, Korwitz A, Haag M, et al. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell metabolism. 2014;20:158–171. doi: 10.1016/j.cmet.2014.04.016. [DOI] [PubMed] [Google Scholar]
- Schagger H, Pfeiffer K. The ratio of oxidative phosphorylation complexes I-V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. The Journal of biological chemistry. 2001;276:37861–37867. doi: 10.1074/jbc.M106474200. [DOI] [PubMed] [Google Scholar]
- Shields LY, Kim H, Zhu L, et al. Dynamin-related protein 1 is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell death & disease. 2015;6:e1725. doi: 10.1038/cddis.2015.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR, Anderson MF. Mitochondrial contributions to tissue damage in stroke. Neurochem Int. 2002;40:511–526. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- Sims NR, Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochimica et biophysica acta. 2010;1802:80–91. doi: 10.1016/j.bbadis.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. The Journal of cell biology. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T, Model K, Langer T. Formation of membrane-bound ring complexes by prohibitins in mitochondria. Molecular biology of the cell. 2005;16:248–259. doi: 10.1091/mbc.E04-09-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuaud F, Ribeiro N, Nebigil CG, Desaubry L. Prohibitin ligands in cell death and survival: mode of action and therapeutic potential. Chemistry & biology. 2013;20:316–331. doi: 10.1016/j.chembiol.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I, Braun HP, Schagger H. Blue native PAGE. Nature protocols. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- Yadava N, Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:7310–7317. doi: 10.1523/JNEUROSCI.0212-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016 doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- Zhang M, Mileykovskaya E, Dowhan W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. The Journal of biological chemistry. 2002;277:43553–43556. doi: 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- Zhou P, Qian L, D’Aurelio M, Cho S, Wang G, Manfredi G, Pickel V, Iadecola C. Prohibitin reduces mitochondrial free radical production and protects brain cells from different injury modalities. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:583–592. doi: 10.1523/JNEUROSCI.2849-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nature genetics. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.