

Abstract
ADAM metalloproteases have been shown to play critical roles during development. In this review we will describe functional evidence that implicates ADAM proteins during the genesis, migration and differentiation of neural crest cells. We will restrict our analysis to the transmembrane ADAMs as other reviews have addressed the role of extracellular metalloproteases (L. Christian, Bahudhanapati, & Wei, 2013). This review will describe advances that have been obtained mainly through the use of two vertebrate model systems, the frog and avian embryos. The role of the principal substrates of ADAMs, the cadherins, has been extensively described in other reviews, most recently in (Cousin, 2017; Taneyhill & Schiffmacher, 2017). The function of ADAMs in the migration of other cell types, including the immune system, wound healing and cancer has been described previously in (Dreymueller, Theodorou, Donners, & Ludwig, 2017). Our goal is to illustrate both the importance of ADAMs in controlling neural crest behavior and how neural crest cells have helped us understand the molecular interactions, substrates and functions of ADAM proteins in vivo.
Keywords: Disintegrin, metalloprotease, neural crest, development, Xenopus, avian
Introduction
ADAMs are transmembrane proteins that contain A Disintegrin and A Metalloprotease domain. While they were originally identified in sperm and shown to play a role in fertilization (Blobel, 2000; Cho, 2012; Evans, 2001; Evans, Schultz, & Kopf, 1995), they have now been demonstrated to be critical for most biological processes. In particular two ADAM proteins (ADAM10 and ADAM17) are essential for development as they control Notch, EGF and Ephrin signaling as well as the epithelial-to-mesenchymal transition (EMT) through cleavage of cadherins. Most other ADAMs appear to have either partially redundant roles or function as modulators, rather than an on/off switch, so that the phenotypes associated with their loss are subtle.
The domain organization of ADAM proteins is similar to snake venom metalloprotease, a family of proteins in which the disintegrin domain binds to and inhibits platelet integrins while the metalloprotease domain cleaves multiple proteins contributing to blood vessel leakage (Gutierrez, Rucavado, Escalante, & Diaz, 2005). ADAMs have a conserved domain organization, starting from the N-terminus, which possesses a pro-domain that associates with the metalloprotease domain to keep it inactive and help with folding. The presence of key Histidines (H) and one Glutamic acid (E) (HExxHxGxxHD) within the metalloprotease domain determines whether the ADAM is an active protease. Only about half of the more than thirty known ADAMs are predicted to be active proteases, suggesting that the other domains are also critical for function. The metalloprotease domain in the ADAM is then followed by the disintegrin and cysteine rich domains. These two domains have been implicated in protein-protein interactions that control cell adhesion and endow substrate recognition. Some ADAMs also have a single EGF repeat before the transmembrane and cytoplasmic domains. For ADAM13, we have shown that the EGF repeat is important to position the protease active site at the proper distance from the plasma membrane to allow for cleavage of Cadherin-11 (Abbruzzese, Becker, Kashef, & Alfandari, 2016). The cytoplasmic domain is the most divergent between ADAMs in both length and sequence. While sequence similarity is always weak, the cytoplasmic domains of multiple ADAMs share a basic isoelectric point and large number of Prolines that have been shown to form SH3 (Src Homology) docking sites (Cousin, Gaultier, Bleux, Darribere, & Alfandari, 2000).
Many excellent reviews have described the role of ADAMs in multiple models (Atapattu, Lackmann, & Janes, 2014; L. M. Christian, 2012; Dreymueller, Pruessmeyer, Groth, & Ludwig, 2012; Giebeler & Zigrino, 2016; Hartmann, Herrlich, & Herrlich, 2013; Pollheimer, Fock, & Knofler, 2014; van der Vorst, Keijbeck, de Winther, & Donners, 2012). Experimental studies over the years have shown that ADAM cleavage of a transmembrane protein can serve to 1) remove the active protein (e.g. ADAM10 cleavage of Ephrin), 2) activate a latent protein (e.g. ADAM10 cleavage of Notch), 3) release a protein from the plasma membrane (ADAM10 and ADAM17 cleavage of pro-EGF) and 4) produce a substrate for subsequent cleavage by γ-secretase, leading to the release of a biologically active C-terminal fragment in the cytoplasm and/or the nucleus (e.g. ADAM10 cleavage of Notch and cadherins).
The roles of ADAMs in cellular migration are extremely complex in part because they can act at the surface of migrating cells or within the environment in which the cells are migrating. For example, ADAM10 cleavage of E-cadherin contributes to EMT so that a cell can leave the epithelium and migrate (Maretzky et al., 2005), but the same function is also used to make an epithelium more permeable to allow invasion of immune cells toward a site of inflammation (Schulz et al., 2008). ADAM proteins also cleave and release chemokines that will attract cells toward the source of production (Hundhausen et al., 2003). In addition, ADAMs can transform a cell adhesion molecule into a signaling protein. For example, cleavage of N- and E-cadherin in cancer cells produces an extracellular fragment that promotes cell migration by binding to the EGF receptor 2 (Her2) (Brouxhon, Kyrkanides, Teng, Athar, et al., 2014; Brouxhon, Kyrkanides, Teng, O’Banion, et al., 2014). Ultimately, these various functions only describe the role of the metalloprotease domain; however, the other domains of ADAM proteins are involved in cell adhesion as well as transcriptional control, adding additional layers of complexity. While the progress to clearly define the role of ADAM proteins in vivo has been slow and subtle, the neural crest has been instrumental to that progress and, in turn, has advanced our understanding of ADAM function during development and disease.
The original neural crest ADAM: Xenopus ADAM13/33
The first ADAM identified with a specific expression in neural crest cells was ADAM13/33 (Alfandari, Wolfsberg, White, & DeSimone, 1997). In Xenopus laevis, it is first expressed in most dorsal structures including dorsal mesoderm and ectoderm during gastrulation. It is then restricted to the neural folds and later the cranial neural crest (CNC) and somitic mesoderm. Studies in chick and quail have also shown that ADAM13/33 is expressed dorsally including the neural crest but its expression is not restricted or particularly enriched in the CNC (Lin, Redies, & Luo, 2007). In Xenopus tropicalis, ADAM13 is expressed in the neural crest but is less restricted than in Xenopus laevis (Wei et al., 2010). Given its obvious expression in the migrating neural crest and the complete absence of data indicating a role for ADAMs in cell motility at the time, ADAM13 was the target of many studies to understand its contribution to neural crest cell biology. The structure and proteolytic processing of ADAM13 is represented in Figure 1.
Figure 1. ADAM13 processing in neural crest cells.
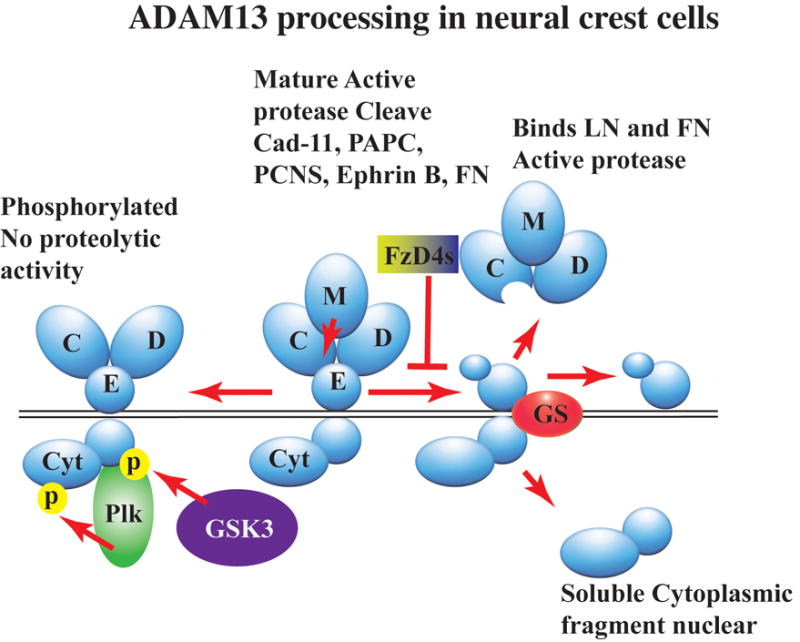
ADAMs are proteins that contain a Disintegrin (D) and Metalloprotease (M) domain. They are synthesized as pro-enzymes that get cleaved by Furin during the transit to the cell surface (elimination of the pro-domain not presented here). At the surface, ADAM13 cleaves both transmembrane proteins such as Cadherins, Protocadherins and Ephrin-B as well as extracellular matrix proteins such as fibronectin (FN). ADAM13 undergoes additional processing steps including self-proteolysis (right) within the cysteine (C) rich domain that releases the metalloprotease (M) domain associated with the disintegrin (D) and part of the cysteine (C) rich domain in the extracellular compartment. This protein remains proteolytically active and can bind to FN and Laminin (LN). The EGF repeat (E) remains associated with the transmembrane and cytoplasmic (Cyt) domains and becomes a substrate for γ-secretase (GS), which can cleave the cytoplasmic domain to allow it to translocate to the nucleus. Alternatively (left), the metalloprotease (M) domain can be cleaved to produce a non-proteolytic ADAM, which is the substrate of two kinases, GSK3 and Polo like kinase (Plk). The function of that form of ADAM13 is unknown, but the phosphorylation is critical for CNC migration.
Phenotypical analysis
The first functional study used a version of ADAM13 with a point mutation of the Glutamic acid that resides in the catalytic active site, E341/A (E/A-ADAM13), and this mutant was thought to act as a dominant negative. Expression of this protein in the CNC perturbed the position of neural crest cells in vivo as shown by in situ hybridization for Twist and assessment of migration using a graft of GFP-expressing CNC (Alfandari et al., 2001). The neural crest cells from hyoid and branchial segments remained dorsal near the original position while those from the mandibular segment where not affected by the mutant suggesting that migration in the most anterior pathway does not depend on ADAM13 proteolytic activity. The notion that the E/A-ADAM13 mutant could act as a dominant negative was based on studies of ADAM10 in Drosophila showing that a proteolytically dead ADAM10 could block Notch cleavage and signaling (Pan & Rubin, 1997). The rationale was that the mutant ADAMs could still associate with their substrates but no longer process them, thereby acting as a competitive inhibitor rather than a true dominant negative. With the advent of morpholino antisense oligonucleotides (MO), the knockdown (KD) of various ADAMs became possible in Xenopus but surprisingly, despite a nearly complete KD of ADAM13, the migration of grafted neural crest cells was not significantly affected, suggesting that the dominant negative could perturb multiple ADAMs and that some redundancy between other ADAMs may occur (Cousin, Abbruzzese, McCusker, & Alfandari, 2012). The most obvious candidate was the ADAM13S/MDC13 homolog (duplicated copy), but KD of ADAM13S together with ADAM13L did not inhibit CNC migration but instead affected cell viability (Alfandari and Cousin, unpublished). Given the extremely low expression of ADAM13S, it is not surprising that it may not compensate for the loss of ADAM13L, unless the expression level was drastically increased in the embryo lacking ADAM13L (Session et al., 2016). In addition, the ADAM13S protein does not appear to be processed into a mature protease (Alfandari, unpublished observation). This phenomenon has been observed in both human and mouse in which the 3′ UTR of the ADAM33 gene is missing (Umland et al., 2003; Umland et al., 2004).
Two major findings came out of these experiments, the first of which was that ADAM13 function could be partially rescued by physically separating the neural crest from the underlying mesoderm, a step necessary to perform the graft (Cousin et al., 2012). In line with this observation, CNC explants placed on fibronectin (FN) migrate well even in the absence of ADAM13, confirming that ADAM13 does not control the intrinsic motility of cells. In addition, the function of ADAM13 was shown to be critical in the leading cells because cells lacking ADAM13 activity could follow those expressing the protein (Cousin et al., 2012). This is particularly interesting as in the chick, the leading neural crest cells, called the “trailblazer” cells, also strongly express ADAM13/33, suggesting a conserved role (McLennan et al., 2015). The second finding was that simultaneous KD of ADAM19 and ADAM13 efficiently blocked the migration of grafted neural crest cells, confirming the redundancy hypothesis (Cousin, Abbruzzese, Kerdavid, Gaultier, & Alfandari, 2011; Cousin et al., 2012). In Xenopus tropicalis, ADAM13 KD also leads to a defect in CNC migration but the molecular mechanism is different (see below). Surprisingly, ADAM33 KO in mice does not cause any obvious craniofacial defects (Chen, Huang, & Sheppard, 2006). In mammals, the ADAM33 cytoplasmic domain, which is critical in frogs, is not conserved, due most likely to a recombination event that has truncated this domain. In contrast, this domain is well conserved from the worm Caenorhabditis elegans to the marsupial Monodelphis domestica, and these isolated cytoplasmic domains can functionally compensate for the absence of the ADAM13 cytoplasmic domain in Xenopus laevis CNC (Cousin et al., 2011). Given the partial redundancy of ADAM9, ADAM13 and ADAM19, it would be interesting to observe the phenotypes after multiple ADAM KD and KO in chick, zebrafish and mouse.
Molecular analysis of ADAM13
Proteolysis
ADAM13/33 appears to have multiple functions during neural crest cell development. In Xenopus tropicalis, ADAM13 cleaves Ephrin-B to stop forward signaling, which attenuates the Wnt/β-catenin signaling pathway. As a result, loss of ADAM13 increases Ephrin-B forward signaling and decreases Wnt signaling, preventing normal Snail2 expression in neural crest cells (Wei et al., 2010). In Xenopus laevis, loss of ADAM13L does not decrease Snail2, Sox9 or Sox10, but does inhibit the expression of Tfap2α, a transcription factor that controls neural plate border formation and subsequently neural crest cell development (Cousin et al., 2011; Vikram Khedgikar, 2017).
Does this mean that ADAM13 controls neural crest cell induction in Xenopus tropicalis but not in Xenopus laevis?
In Xenopus laevis, loss of both ADAM13L and ADAM13S does decrease Snail2 expression and affects β-catenin levels during gastrulation (Alfandari, unpublished), suggesting that combined they may control CNC induction as well. On the other hand, CNC lacking both ADAM13 copies do migrate when grafted into host embryos or when placed in vitro on FN. This ability to migrate even in the presence of reduced Snail2 is not surprising given that Snail2 appears to be dispensable for migration, at least in some neural crest cell populations, such as the chick trunk, where migratory neural crest cells do not express Snail2 (del Barrio & Nieto, 2002; Sela-Donenfeld & Kalcheim, 1999), Furthermore, Snail2 controls many other cellular processes, including EMT, survival, immune regulation, and cell fate (Barrallo-Gimeno & Nieto, 2005; Cobaleda, Perez-Caro, Vicente-Duenas, & Sanchez-Garcia, 2007; Wu & Zhou, 2010). Which markers truly define neural crest cells? In Xenopus laevis, the fact that the explanted neural crest cells migrate in vivo (shown by grafting) and in vitro suggest that induction is not affected. In fact, perhaps the best test to determine if neural crest cells have been induced is to explant them on FN, as neither the neural plate nor the non-neural ectoderm migrate on this substrate (Alfandari, Cousin, Gaultier, Hoffstrom, & DeSimone, 2003). Experiments that test specifically the requirement of each of the transcription factors that define the neural crest in migration assays in vitro have not been done and would greatly improve our understanding of this key feature of neural crest cells.
In Xenopus laevis, ADAM13 binds, via its cysteine rich domain, multiple substrates including Cadherin-11 (McCusker, Cousin, Neuner, & Alfandari, 2009), the protocadherins Pcdh8l (PCNS) and Pcdh8 (PAPC) (Abbruzzese, Cousin, Salicioni, & Alfandari, 2014; Vikram Khedgikar, 2017) and FN (Alfandari et al., 2001). For FN the exact site of the interaction is the second heparin binding site (Hep2), and mutations of FN that abolish Syndecan-4 binding to this domain also prevent ADAM13 binding to FN (Gaultier, Cousin, Darribere, & Alfandari, 2002). Using modeling tools, a groove within the cysteine rich domain was discovered that aligns with the metalloprotease active site. In vivo, experiments using chimeras between ADAM10 and ADAM13 have shown that the cysteine rich domain of ADAM13 can endow ADAM10 with the ability to produce an ectopic and expanded cement gland, suggesting that it controls ADAM13 specificity (Smith et al., 2002). The signaling pathway that is triggered by ADAM13 in this assay is still unknown. The role of ADAM13 cleavage of FN in the context of CNC migration has not been resolved and the substrate(s) cleaved by ADAM13 that links the CNC to the underlying mesoderm remains to be identified.
Cleavage of Cadherin-11 by ADAM13 generates an extracellular fragment (EC1-3) containing the first three cadherin repeats including the homophilic binding site (Fig. 2). This fragment promotes cell migration in embryos lacking ADAM13 or overexpressing Cadherin-11 (McCusker et al., 2009). Interestingly, this function does not require the presence of the homophilic binding site (Abbruzzese et al., 2016). This appears to be a relatively non-specific function as the extracellular fragment from two other substrates of ADAM13, PAPC and PCNS, can also rescue CNC migration albeit much less efficiently (Vikram Khedgikar, 2017). Although the exact molecular pathway is not known, the Cadherin-11 extracellular fragment can bind to multiple growth factor receptors and can induce AKT phosphorylation upon binding to ErbB2 (EGF-receptor 2). ErbB2 and AKT are also important for CNC migration and while exposing CNC explants to EC1-3 increases AKT phosphorylation and persistence of CNC migration, inhibition of ErbB2 reduces AKT phosphorylation and CNC migration, suggesting a direct link. In vitro, the Cadherin-11 EC1-3 fragment appears to increase the association of the ErbB2/ErbB3 heterodimer with PI3K, one of the kinases upstream of AKT phosphorylation. Inhibition of PI3K impedes CNC migration in vivo and in vitro, demonstrating the requirement for each of the partners of this signaling pathway (Mathavan, Khedgikar, Bartolo, & Alfandari, 2017).
Figure 2. ADAM molecular functions in neural crest cells.
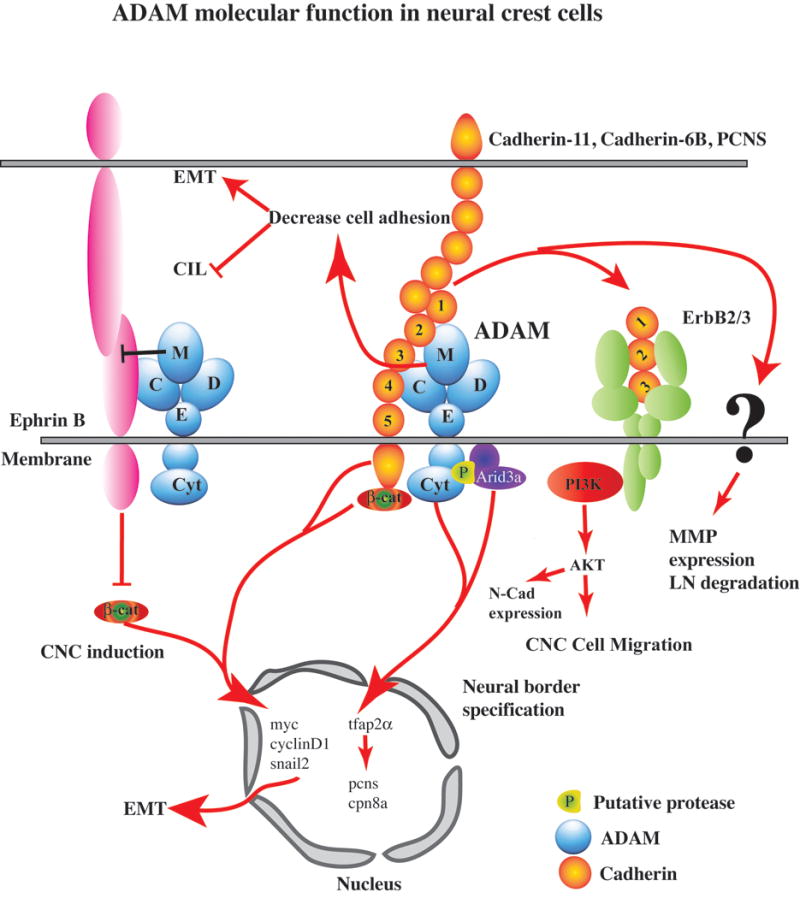
ADAMs have been shown to regulate the Wnt/β-catenin signaling pathway in two ways. ADAM13 can cleave the Ephrin B ligand, thereby blocking signaling that represses β-catenin. Thus, ADAM13 cleavage of Ephrin B increases β-catenin signaling, resulting in the robust expression of β-catenin target genes such as Snail2, CyclinD1 and myc. Second, by cleaving cadherins that associate with β-catenin at the membrane, ADAMs can initiate a series of proteolytic processing events, leading to the release of the cadherin cytoplasmic domain/C-terminal fragment (CTF2) still associated with β-catenin and thus stimulating this pathway. By cleaving cell adhesion molecules such as cadherins, ADAMs positively control the epithelial-to-mesenchymal transition (EMT) as well as negatively affect contact inhibition of locomotion (CIL). EMT is also stimulated by the activation of β-catenin target genes, which can occur through the formation of a CTF2/β-catenin complex. In the quail trunk neural crest, ADAM10 processes N-cadherin, forming a CTF2 that increases expression of β-catenin and CyclinD1, while chick cranial neural crest cells use ADAM10 and ADAM19 to cleave Cadherin-6B, liberating a CTF2/β-catenin complex that associates with chromatin and activates these and other EMT effector genes, including Snail2. This pro-EMT role for a cadherin cleavage fragment is also observed for the Cadherin-6B NTF, which furthers EMT by enhancing Laminin (LN) degradation, likely through activation of MMPs. ADAM13 cleavage of Cadherin-11 produces an extracellular fragment, EC1-3, that binds to the ErbB2/3 complex and stimulates the phosphorylation of AKT in CNC and eventual CNC migration. EC1-3 increases the persistence of migration while AKT phosphorylation has been shown to increase N-cadherin, a protein critical for CIL and controlling the onset of CNC migration. ADAM13 also controls the nuclear translocation of Arid3a and its ability to induce Tfap2α to define the neural plate border and, subsequently, the neural crest. At least two genes, PCNS and Calpain8a, which lie downstream of ADAM13/Arid3a/Tfap2α, are critical for CNC migration.
Other studies have shown the importance of AKT phosphorylation and CNC migration (Bahm et al., 2017; Figueiredo et al., 2017). In particular, the Mayor group revealed that PDGFR stimulation results in increased AKT phosphorylation and N-cadherin expression, which in turn contributes to the transition from pre- to post-migratory CNC (Bahm et al., 2017). Given that Cadherin-11 EC1-3 also binds to PDGFR, it is possible that the cleaved cadherin fragment contributes to multiple signals that modulate neural crest cell migration. Other groups have shown that the cytoplasmic domain of Cadherin-11 is critical for neural crest specification by regulating β-catenin directly (Koehler et al., 2013) and filopodia formation through an interaction with the GEF-trio (Kashef et al., 2009). In addition, Cadherin-11 is found in nascent focal adhesions where it interacts with Syndecan-4 (Langhe et al., 2016). Thus, the complex of ADAM13, Cadherin-11 and Syndecan-4 could be present in these focal adhesions, interacting and possibly competing for Hep2 on FN. This is a particularly intriguing possibility as we have shown that the Hep2 domain of FN contributes to normal CNC migration in vitro and that, when given a choice, CNC migrate preferentially on FN with available Hep2 binding sites (Alfandari et al., 2003). By cleaving Cadherin-11, ADAM13 may be able to initiate simultaneously an extracellular signaling cascade involving growth factor receptors (FGFR1, EGFRs, PDGFR) and a nuclear signaling event if γ-secretase preferentially cleaves the Cadherin-11 membrane stump to release the cytoplasmic domain associated with β-catenin, as has been demonstrated for Cadherin-6B in chick CNC (Schiffmacher et al., 2014; see below). It is important to note that the level of expression of Cadherin-11 is much lower than any of the other cadherins (Huang, Kratzer, Wedlich, & Kashef, 2016), suggesting that its role as a signaling protein may be more important than its function as a classical cell adhesion molecule.
Non-proteolytic regulation of gene expression
In addition to these proteolytic functions, ADAM13 was shown to regulate gene expression via its cytoplasmic domain. The process is controlled at multiple steps. First, ADAM13 cleaves its own cysteine rich domain, producing a soluble active metalloprotease linked to the disintegrin and part of the cysteine rich domain (Gaultier et al., 2002). The remaining protein in the membrane contains a portion of the cysteine rich domain and one EGF repeat in addition to the cytoplasmic domain. This membrane stump is then cleaved by γ-secretase to release the cytoplasmic domain, which translocates into the nucleus (Fig. 1). Adding a nuclear export signal to the cytoplasmic domain is sufficient to block its ability to promote CNC migration in vivo. Using complementation assays we showed that the cytoplasmic domain of ADAM13 could be provided as a soluble GFP-fusion (GFP-C13) to complement ADAM13 lacking a cytoplasmic domain (A13ΔCyto). Through these experiments we also demonstrated that the cytoplasmic domains of ADAM13 homologues from the worm Caenorhabditis elegans to marsupial Monodelphis domestica were functional while the mouse ADAM13 cytoplasmic domain was not. Genes transcriptionally regulated by the cytoplasmic domain of ADAM13, such as the cytoplasmic protease calpain8a, can also complement in this assay. Interestingly, the cytoplasmic domains of mouse and Xenopus ADAM19 can complement while those from ADAM9 and ADAM10 cannot (Cousin et al., 2011).
The regulation of transcription by ADAM13 is not direct (Fig. 2). The cytoplasmic domain does not possess DNA binding capacity and does not activate genes even when fused to the VP16 activator domain (Alfandari, unpublished). Instead, ADAM13 associates with the transcription factor Arid3a at the membrane where it induces a proteolytic cleavage of Arid3a. While Arid3a is found at the membrane, cytoplasm and nucleus, the cleaved form of Arid3a is mostly restricted to the nucleus. ADAM13 association with Arid3a drives the expression of Tfap2α, a transcription factor that is in turn responsible for the expression of calpain8a and the protocadherin pcdh8l (PCNS), among others (Vikram Khedgikar, 2017).
The “nuclear” function of ADAM13 is regulated both at the membrane as well as in the cytoplasm. Association of ADAM13 with the Wnt receptor Frizzled 4 (Fzd4), and/or its secreted form Fzd4-v1, inhibits ADAM13 self-cleavage within its cysteine rich domain, and consequently the production of the cleaved cytoplasmic domain (Abbruzzese et al., 2015). KD of Fzd4 in embryos results in early processing of the cytoplasmic domain of ADAM13 and increased expression of Tfap2α, while overexpression of Fzd4 produces the opposite effect. In HEK293T cells, expression of Fzd4-v1 abrogates activation of a Tfap2α-luciferase reporter by ADAM13. In the cytoplasm, two kinases control the nuclear activity of ADAM13. First, GSK3 phosphorylates the ADAM13 cytoplasmic domain creating a docking site for Polo like kinase (Plk). Phosphorylation of ADAM13 by Plk is essential for CNC migration but does not regulate ADAM13 proteolytic activity, the cleavage of the cytoplasmic domain or its translocation into the nucleus. It is unclear at the moment how Plk phosphorylation controls ADAM13 function (Abbruzzese et al., 2014).
ADAM19
ADAM19 is critical for cardiac neural crest cell development in mouse. ADAM19 KO mice have a ventricular septal defect, a structure that is produced by cardiac neural crest cells. Using a lineage tracer, the authors showed that the cardiac neural crest cells populate the proper region of the heart in the KO, suggesting that induction, proliferation and migration of neural crest cells is not perturbed. By re-expressing ADAM19 specifically in cardiac neural crest cells using the PO-Cre promoter, the author were able to rescue the cardiac defect, implying that cardiac neural crest cells require ADAM19 at later stages during differentiation to mediate the proper signals (e.g. EGF) to form the ventricular septum (Komatsu et al., 2007).
In Xenopus laevis, ADAM19 KD reduces the expression of several neural crest cell markers including Snail2 and Sox8 while increasing the expression of Sox2, suggesting that ADAM19 is important in the induction of the CNC at the neural plate border. Using targeted injection, the protein was also shown to be crucial for CNC migration in vivo (Neuner, Cousin, McCusker, Coyne, & Alfandari, 2009). Interestingly, ADAM19 KD, similar to ADAM13 KD, did not affect CNC migration in vitro or in grafts (Cousin et al., 2012). At the molecular level, ADAM19 KD decreased phosphorylation of AKT, a key component of the EGF signaling pathway that is critical for CNC migration (see above). It is interesting to note that the ADAM19 KD phenotype is similar to the ErbB2 KD and that, at least during gastrulation, ADAM19 KD can be partially rescued by expressing a cleaved extracellular fragment of Neuregulin (Neuner and Alfandari, unpublished). In Xenopus tropicalis, ADAM19 KD produces phenotypes that are very similar to ADAM13 KD. In fact, loss of ADAM19 results in a dramatic decrease in the ADAM13 protein. This is also true in Xenopus laevis although less dramatic. The control of ADAM13 protein levels by ADAM19 depends on the presence of the ADAM13 cytoplasmic domain but is independent of ADAM19 or ADAM13 proteolytic activity. Indeed, KD of ADAM13 can be rescued by a proteolytically dead ADAM19 (E/A-ADAM19). In the absence of ADAM19, ADAM13 is ubiquitinated on a conserved lysine (K911 in Xenopus tropicalis) in the cytoplasmic domain, resulting in its degradation by the proteasome (Jiejing Li1, accepted). This is consistent with observations in Xenopus laevis that ADAM13 lacking its cytoplasmic domain is expressed at a much higher level (8-fold) than the intact protein (Alfandari et al., 2001; Cousin et al., 2000). ADAM13 and ADAM19 form a complex mediated by the cysteine rich domain that can be immunoprecipitated, suggesting that this physical interaction may compete with an ubiquitin ligase. It will be interesting to determine if the ADAM13/ADAM19 complex has a different activity than the individual proteins or if this interaction controls the subcellular localization of the proteins in the neural crest. Given that the ADAM19 cytoplasmic domain can regulate gene expression in a similar way to ADAM13 but no evidence of self-processing of ADAM19 currently exists, it will be interesting to see if other ADAMs (ADAM9 or ADAM13) are involved in the extracellular cleavage of ADAM19 to produce a substrate for γ-secretease-mediated cleavage. Since both ADAM13 and ADAM19 can induce a Tfap2α reporter in HEK293T cells, it will be important to determine if they can both bind to the Arid3a transcription factor and contribute to the activating complex at the membrane.
This exquisite interaction between ADAM proteins, and the ability of ADAMs to regulate each other (initially found in the sperm ADAM (ADAM1 and 2)), may be the key to understanding the non-proteolytic functions of ADAM proteins. For example, ADAM11 is also expressed in Xenopus laevis CNC (H. Cai, Kratzschmar, Alfandari, Hunnicutt, & Blobel, 1998) where it binds to ADAM13 (Alfandari, unpublished). It is unknown at present if ADAM11 is critical for CNC development and if it may stabilize or inhibit ADAM13 function. Similar to ADAM13, ADAM11 was also found in “trailblazer” neural crest cells in chick, suggesting that in this model system the two proteins could also functionally interact. The identification of ADAM function in multiple vertebrate species during neural crest cell migration is critical to help us understand key conserved mechanism. Similarly, the analysis from the two closely related Xenopus have taken advantage of the ploidy (Xenopus laevis tetraploid, Xenopus tropicalis diploid) to uncover multiple facets of ADAM protein function that could not have been identified otherwise. In the second part of this review we will focus on recent advances in the avian model system.
ADAM proteins in avian neural crest
ADAM10 expression and function in avian trunk neural crest cells
ADAM expression and function in avian neural crest has been studied over the past 15 years. Initially, ADAM10 expression and function was described in chick neural crest cells cultured in vitro (Hall & Erickson, 2003). This group also noted ADAM10 protein throughout the ectoderm, including the basement membrane, as well as in the basement membrane that surrounds the dorsal neural tube, all within the chick trunk at later stages of embryogenesis (>HH13). Although functional studies to alter ADAM10 expression were carried out by introducing a dominant negative ADAM10 construct or MO to ADAM10 into the embryo ectoderm, the role of ADAM10 in the avian trunk neural crest remained unexplored until a few years later. Work by Shoval and colleagues suggested that ADAM10 proteolytic processing of N-cadherin mediates quail trunk neural crest cell EMT (Shoval, Ludwig, & Kalcheim, 2007). These authors proposed that BMP4 signaling (through a reduction in noggin transcripts) leads to activation of ADAM10 within premigratory trunk neural crest cells, followed by a subsequent decrease in N-cadherin and the onset of EMT. Explants of quail trunk premigratory neural crest cells cultured ex vivo in the presence of GI254023X, a chemical inhibitor that targets ADAM10, revealed maintenance of membrane-bound N-cadherin through immunohistochemistry, unlike the typical loss of N-cadherin observed in control cultures, thus negatively impacting EMT and migration (Shoval et al., 2007).
Because ADAM10-mediated extracellular processing of cadherins is typically followed by cleavage within the cadherin cytoplasmic tail by γ-secretase to liberate an intracellular C-terminal fragment (CTF2) (Marambaud et al., 2002; Marambaud et al., 2003), additional experiments were carried out to examine the function of N-cadherin CTF2. In situ hybridization demonstrated an increase in CyclinD1 and β-catenin transcripts in the neural tube and in neural crest cells following long-term (>16 hours) N-cadherin CTF2 overexpression in trunk premigratory neural crest cells, although the number of delaminating neural crest cells was only modestly increased. It is important to note, however, that NC markers such as Sox2, Sox10, FoxD3 and Snail2 were not analyzed in these experiments, making it difficult to evaluate effects on NC identity, EMT and migration. N-cadherin CTF2 was also localized to trunk neural crest cell nuclei within ex vivo neural crest cell explants, implying a potential transcriptional role for this cadherin CTF2. Furthermore, N-cadherin CTF2 overexpression rescued the loss of delamination and EMT noted upon treatment with GI254023X, further underscoring the epistatic relationship between ADAM10 and N-cadherin in quail trunk neural crest cells. The fact that overexpression of N-cadherin CTF2 can overcome the loss of neural crest cell delamination and EMT observed upon inhibition of ADAM10-mediated N-cadherin cleavage is striking and points to a role for N-cadherin CTF2 in regulating trunk neural crest cell delamination and EMT that is independent of a function in cell-cell adhesion, as was recently delineated for Cadherin-6B in the chick CNC (Schiffmacher et al., 2016; see below). Interestingly, in the mouse ADAM10 is also required for the development of the maxillofacial bone, a structure derived from the CNC, suggesting an important functional conservation of the protein (Tan et al., 2016).
More recent studies in the chick have shed further light on the cadherin repertoire expressed (at both the mRNA and protein levels) during neurulation and NC delamination and migration (Dady, Blavet, & Duband, 2012; Dady & Duband, 2017). Specifically, immunohistochemical analysis of chick sections taken at the level of the head and anterior trunk (cervical region) revealed that N-cadherin protein is not readily detectable in the NC (from specification up through delamination and migration) until the cessation of NC delamination at this axial level, but is instead observed in the more posterior trunk, which undergoes secondary neurulation. Shoval and colleagues also noted this difference in N-cadherin protein distribution, particularly within the dorsal neural tube (Shoval et al., 2007), and used these findings to design experiments to test how N-cadherin proteolysis impacts NC delamination (described above). Intriguingly, N-cadherin transcripts are present in the dorsal neural tube (Dady et al., 2012; Shoval et al., 2007), although possibly at reduced levels (Dady & Duband, 2017), particularly prior to NC delamination, implying some level of post-transcriptional control over N-cadherin. These data collectively imply that N-cadherin protein clearance must be an ongoing process in the premigratory NC (due to the absence of N-cad immunoreactivity, see below), and that other molecular events likely occur to trigger NC delamination, particularly in the case of the anterior trunk.
Recent findings now suggest that the absence of N-cadherin protein in the anterior trunk may not be attributed to extracellular cleavage of N-cadherin due to the identical pattern of N-cadherin immunostaining observed (i.e., little to no N-cadherin detected) using two antibodies raised to different regions of the N-cadherin extracellular domain (at the N-terminus and close to the plasma membrane of the extracellular domain; (Dady & Duband, 2017)). It is possible, however, that any proteolytic fragments of N-cadherin are short-lived and therefore difficult to detect in a static section by immunohistochemistry. Indeed, Shoval et al. were only able to visualize loss of membrane N-cadherin, along with apparent differential immunoreactivity and distribution for N-cadherin (using the same N-terminal antibody described above and a second antibody to the N-cadherin intracellular domain), in NC explants (Shoval et al., 2007). These explant cultures were maintained for 15 hours prior to imaging, though, so they may not entirely recapitulate what is happening in vivo (i.e., the absence of N-cadherin immunoreactivity in premigratory and migratory NC). Higher magnification images of in vivo sections using confocal microscopy together with antibodies that specifically recognize epitopes in the N-cadherin extracellular and intracellular regions, and/or live imaging of NC possessing fluorescently modified N-cadherin at both its extracellular and intracellular domains (using CRISPR, see (Gandhi, Piacentino, Vieceli, & Bronner, 2017)), would aid in detection of potential cleavage products and help resolve the degree and timing of proteolysis that may be occurring in the anterior trunk. Nevertheless, the evidence provided by these publications strongly indicates differential expression of N-cadherin, and perhaps function, in trunk NC dependent upon axial level of origin.
Studies by other groups have established the importance of additional cadherins besides N-cadherin, such as E-cadherin and Cadherin-6B, in premigratory and migratory NC throughout the cranial and trunk axial levels of the chick (Coles, Taneyhill, & Bronner-Fraser, 2007; Dady et al., 2012; Dady & Duband, 2017; Lee et al., 2013; S Nakagawa & Takeichi, 1995; S. Nakagawa & Takeichi, 1998; Taneyhill, Coles, & Bronner-Fraser, 2007). Through immunohistochemical and in situ hybridization analyses, Dady and Duband (Dady et al., 2012; Dady & Duband, 2017) demonstrated that E-cadherin and Cadherin-6B are expressed in the proper spatio-temporal pattern to play a role in the specification of the NC and defining the premigratory NC domain (Dady & Duband, 2017), including in the head and anterior and mid trunk. Furthermore, these authors demonstrate that Cadherin-6B expression, but not N-cadherin, defines the premigratory NC domain in these truncal regions, with delaminating cells here devoid of Cadherin-6B, N-cadherin, and E-cadherin, but later expressing Cadherin-7 during migration. Based on their careful analysis of Cadherin-6B spatio-temporal distribution in the chick head and trunk, the authors propose that Cadherin-6B expression is important to sort neural crest cells from neural tube and epidermal cells at the neural plate border, leading to NC specification, a hypothesis initially proposed 20 years ago (S Nakagawa & Takeichi, 1995). On the other hand, expression of E- and N-cadherin occurs later and is further refined to allow for the segregation of non-neural and neural ectoderm, respectively. A second function of Cadherin-6B is the induction of NC delamination and subsequent emigration of NC from the neural tube. In light of these new data highlighting a role for Cadherin-6B (as opposed to N-cadherin) in specifying neural crest cells and determining the premigratory NC domain in the anterior trunk, it is important to now revisit the prior proteolysis study for N-cadherin to determine how this molecular mechanism controls NC delamination and whether the identity, adhesion and other properties of the NC may, in fact, be altered upon molecular perturbation of N-cadherin.
ADAM10, ADAM19 and γ-secretase expression and function in chick CNC
The localization of ADAM10 in the chick head region is comparable to that noted in the chick trunk, with ADAM10 also observed in premigratory CNC (Schiffmacher, Padmanabhan, Jhingory, & Taneyhill, 2014). In chick premigratory and migratory CNC, however, an additional ADAM, ADAM19, is expressed (Schiffmacher et al., 2014). The spatio-temporal distribution of ADAM19 was initially described at later stages of chick embryogenesis, with expression noted in neural crest-derived structures of the peripheral nervous system (Lewis, Farlie, & Newgreen, 2004; Yan, Lin, Markus, Rolfs, & Luo, 2011). Importantly, both the premigratory and migratory CNC populations also express membrane-localized presenilin-1 (Schiffmacher et al., 2014), the catalytic subunit of the γ-secretase complex, whose activity is required to cleave at the cadherin juxtamembrane domain and release cadherin CTF2 (Marambaud et al., 2002; Marambaud et al., 2003). Thus, like their trunk counterparts, chick premigratory and migratory CNC possess the appropriate protease machinery to process cadherins during EMT and delamination.
Proteolysis
While chick premigratory CNC express multiple and at times overlapping (in space and time) cadherin proteins (e.g., N-cadherin, E-cadherin, Cadherin-6B), Cadherin-6B is the sole cadherin whose expression is substantially reduced in premigratory neural crest cells during EMT and subsequently lost from the migratory neural crest cell population (Cousin, 2017; Dady et al., 2012; Dady & Duband, 2017; Lee et al., 2013; Schiffmacher et al., 2014; Taneyhill et al., 2007; Taneyhill & Schiffmacher, 2017). While transcriptional repression of Cadherin-6B is achieved during EMT through a complex consisting of Snail2 and other chromatin remodeling proteins (Strobl-Mazzulla & Bronner, 2012; Taneyhill et al., 2007), this cannot account for the rapid loss of Cadherin-6B protein over a 90 minute time period within premigratory CNC (and the eventual absence of Cadherin-6B in migratory CNC), particularly since Cadherin-6B possesses a half-life typical of other cadherins (~5.5 hours; see (Schiffmacher et al., 2014)). This result paved the way to examine whether ADAM-mediated proteolytic cleavage of Cadherin-6B could clear existing membrane-bound pools of Cadherin-6B during EMT.
With this hypothesis in mind, we identified that both ADAM10 and ADAM19, together with γ-secretase, cleave Cadherin-6B during EMT to liberate a shed Cad6B extracellular domain or N-terminal fragment (NTF) and an intracellular CTF2 (Schiffmacher et al., 2014). Using Concanavalin A pull-down from whole cell lysates prepared from isolated premigratory neural crest cells at stages prior to and during EMT (5-8 somite stage, ss) coupled with immunoblotting, we demonstrated loss of full-length Cadherin-6B protein and the appearance of a Cadherin-6B NTF as neural crest cells undergo EMT. Using a C-terminally-tagged Cadherin-6B due to the lack of a commercially available antibody to the Cadherin-6B C-terminus, the presence of the Cadherin-6B CTF2 was also validated, with high levels observed during active stages of EMT (8ss) (Schiffmacher et al., 2014). Additional studies later showed that Cadherin-6B CTF2 is in fact observed prior to EMT, indicating that Cadherin-6B proteolysis is likely an ongoing process that commences before EMT to generate the Cadherin-6B NTF and CTF2 (Schiffmacher, Xie, & Taneyhill, 2016); see below). This proteolysis of Cadherin-6B in vivo was in fact blocked using GM6001, a broad-spectrum inhibitor of both ADAMs and matrix metalloproteases (MMPs), providing additional evidence that Cad6B proteolysis occurs during cranial neural crest cell EMT.
To define the protease(s) involved in Cadherin-6B processing, an in vitro transfection approach was adopted to narrow down candidates (Schiffmacher et al., 2014). Through this method, both ADAM10 and ADAM19 were identified, as transfection of the wild-type protein could produce the typical Cadherin-6B fragments while the catalytically inactive mutants of either ADAM led to a loss of Cadherin-6B processing in vitro. The inclusion of a γ-secretase inhibitor (L-685,458) also implicated this complex in cleaving the Cadherin-6B intracellular domain. While other ADAMs were tested (ADAM12, ADAM13/33, ADAM17), they were unable to cleave Cadherin-6B, at least in this in vitro assay, providing evidence for potential cadherin substrate specificity within the ADAM family. Notably, ADAM10 and ADAM19 are expressed in premigratory and migratory CNC (described above), indicating that they possess the proper spatio-temporal distribution to process Cadherin-6B in vivo.
Phenotypic analysis
To address ADAM functionality in premigratory CNC, both overexpression and knockdown assays were conducted in the chick embryo (Schiffmacher et al., 2014). We demonstrated that overexpression of either ADAM in chick premigratory CNC prior to EMT results in precocious clearance of Cadherin-6B protein. Conversely, introduction of a MO to either ADAM (singly or together) into chick premigratory CNC before EMT leads to the maintenance of Cadherin-6B (46% or 25% increase in dorsal neural tube/premigratory neural crest cell Cadherin-6B upon ADAM10 or ADAM19 KD, respectively, as assessed by immunoblotting). In the embryo, this translates to a 1.4-fold or 1.2-fold increase in the Cadherin-6B-positive premigratory CNC domain after MO-mediated depletion of ADAM10 or ADAM19, respectively, while a 1.4-fold increase is observed when MOs to both ADAMs are introduced. Cadherin-6B has been hypothesized to serve in segregating premigratory neural crest cells from other neural tube cells to create this specific domain in the dorsal neural tube, as described above and in (Coles et al., 2007; Dady & Duband, 2017; S Nakagawa & Takeichi, 1995; Schiffmacher et al., 2014). As such, proteases like ADAM10 may play a key role in establishing and/or maintaining the proper size of the premigratory neural crest cell domain. Interestingly, depletion of either ADAM (or both simultaneously) does not alter the size of the migratory neural crest cell domain (based upon the number of migratory neural crest cells), at least at the stage examined in this report (Schiffmacher et al., 2014).
To gain insight into the function of Cadherin-6B CTF2, a recombinant protein corresponding to the proteolytic fragment was expressed in premigratory CNC, resulting in the surprising loss of Cadherin-6B protein (29% reduction in the size of the Cadherin 6B-positive premigratory neural crest cell domain) and thereby phenocopying ADAM overexpression. This latter result provided the foundation for additional studies (described below) to address a potential pro-EMT function for Cadherin-6B CTF2 in the CNC, as loss of Cadherin-6B in neural crest cells is a hallmark of chick CNC EMT. Collectively, these data indicate that, like in Xenopus, multiple ADAMs are used to regulate cadherin protein levels in the chick CNC. It is likely, however, that other proteases that function in chick CNC, such as MMPs (Duong & Erickson, 2004; Monsonego-Ornan et al., 2012; Roth, Kalev-Altman, Monsonego-Ornan, & Sela-Donenfeld, 2017), may also contribute to Cadherin-6B proteolytic processing during EMT, as given by our new preliminary data (see below, Schiffmacher and Taneyhill, submitted).
ADAM- and γ-secretase-mediated proteolysis of Cadherin-6B facilitates CNC EMT through both clearance of full-length Cadherin-6B protein (and loss of intercellular adhesion among premigratory neural crest cells) and the generation of Cadherin-6B CTF2, which further promotes EMT by activating EMT effector genes in vivo (Schiffmacher et al., 2016). The data supporting the existence of this positive, feed-forward network is described in the next section.
Using sub-phenotypic electroporation of C-terminally-tagged full-length Cadherin-6B (due to the absence of a commercially available antibody to allow for labeling of the Cadherin-6B C-terminus; see (Schiffmacher et al., 2014)) into premigratory CNC, we tracked the appearance of Cadherin-6B CTF2 over developmental time via immunoblotting (Schiffmacher et al., 2016). We noted that Cadherin-6B CTF2 is observed both prior to and during neural crest cell EMT, indicating that Cadherin-6B is subjected to continuous proteolytic processing in the CNC (Schiffmacher et al., 2016). Similarly to what is observed in cell culture (Ferber et al., 2008; Marambaud et al., 2002; Sadot, Simcha, Shtutman, Ben-Ze’ev, & Geiger, 1998; Uemura et al., 2006), Cadherin-6B CTF2 and β-catenin can be co-immunoprecipitated both in vitro and in vivo in the neural crest. Moreover, binding of CTF2 to β-catenin generates a complex that stabilizes both proteins, resulting in a decreased rate of degradation for wild-type Cadherin-6B CTF2 (170 minutes) compared to that observed for a mutant version of Cadherin-6B CTF2 that possesses an almost 90% reduction in its ability to bind β-catenin (MUT9; 59.9 minutes). Importantly, both wild-type Cadherin-6B CTF2 and MUT9 are expressed at comparable levels in vivo, implying that their stability can be directly correlated to β-catenin binding. In keeping with this finding, wild-type Cadherin-6B CTF2 is turned over more rapidly (1.6x faster) upon depletion of β-catenin from cultured cells, whereas the degradation rate of MUT9 is unaffected by β-catenin knockdown. Importantly, Cadherin-6B CTF2 overexpression in premigratory CNC leads to the re-distribution of β-catenin from the cytosol to the nucleus and the importation of both CTF2 and β-catenin into the nucleus, both in vivo and in ex vivo neural crest cell explant cultures (Fig. 2). Taken together, these biochemical and localization results point to a potential transcriptional role for Cadherin-6B CTF2 in the CNC, as had been proposed previously, but not definitively proven, for N-cadherin CTF2 in trunk neural crest cells (Shoval et al., 2007).
To investigate a transcriptional role for the Cad6B CTF2/β-catenin complex in the CNC, either Cadherin-6B CTF2 or MUT9 was overexpressed in premigratory CNC for only five hours (the minimum time required for CTF2 expression), followed by quantitative PCR (QPCR) for known EMT effector genes, including β-catenin, CyclinD1 and Snail2 (Schiffmacher et al., 2016). We noted that all three genes showed a statistically significant up-regulation in the presence of Cadherin-6B CTF2 but not MUT9, revealing that only Cadherin-6B CTF2 possessing an intact β-catenin binding domain is able to transactivate gene expression. The finding that Cadherin-6B CTF2 can enhance Snail2 expression was particularly remarkable in light of the ability of Snail2 to repress Cadherin-6B transcription and facilitate loss of full-length Cadherin-6B (Strobl-Mazzulla & Bronner, 2012; Taneyhill et al., 2007). In keeping with this result, Cadherin-6B transcripts were also decreased by 20% in this QPCR assay. The mechanism underlying the up-regulation of Snail2 by Cadherin-6B CTF2 was further refined by performing similar QPCR experiments but instead examining expression of a GFP reporter controlled by a minimal Snail2 promoter (1.2 kb upstream of the Snail2 translational start site (Sakai et al., 2005)) that was introduced into premigratory neural crest cells along with either Cadherin-6B CTF2 or MUT9. In this assay, only Cadherin-6B CTF2, but not MUT9, could induce GFP expression. Collectively, these data reveal yet an additional means by which Cadherin-6B levels are reduced to promote CNC EMT and, importantly, shed light on how Cadherin-6B CTF2 can impact the gene regulatory network that controls EMT at the level of transcription (Schiffmacher et al., 2016).
The preceding GFP expression assay provided critical information with regards to the potential position(s) within the Snail2 promoter that may mediate Snail2 expression in the presence of Cadherin-6B CTF2. To demonstrate an association between Cadherin-6B CTF2 and this region of the Snail2 promoter, chromatin immunoprecipitation assays coupled with QPCR were conducted in chick premigratory neural crest cells expressing Cadherin-6B CTF2 or MUT9 (Schiffmacher et al., 2016). In this experiment, Cadherin-6B CTF2- or MUT9-bound chromatin was immunoprecipitated from neural crest, followed by QPCR using sets of overlapping primers spanning the 1.2 kb region of the endogenous Snail2 promoter that was demonstrated to be responsive to Cadherin-6B CTF2 in the GFP reporter QPCR experiment. While MUT9 did not associate to any of the tested chromatin regions, we showed that Cadherin-6B CTF2 was enriched in two areas of chromatin within the endogenous Snail2 promoter, indicating that an intact β-catenin binding domain within Cadherin-6B CTF2 is required to mediate this association. These regions of chromatin exhibiting CTF2 association lacked any putative Lef/TCF sites to which β-catenin could bind in a complex with Lef/TCF, implying that regulation occurs through a complex consisting of CTF2, β-catenin and potentially other protein(s), such as DNA binding proteins, to regulate Snail2. Indeed, this finding is not surprising given that β-catenin binding to a cadherin CTF2 and a Lef/TCF family member is mutually exclusive due to the overlap in the binding sites for CTF2 and Lef/TCF proteins on β-catenin (Ferber et al., 2008; Sadot et al., 1998). Moreover, Cadherin-6B CTF2 lacks an apparent importin-α nuclear localization sequence that is observed in other catenins, although the presence of an undiscovered cryptic nuclear localization sequence cannot be ruled out (Schiffmacher et al., 2016). As such, Cadherin-6B CTF2/β-catenin complexes in the CNC regulate transcription through at least two mechanisms: shuttling β-catenin to the nucleus where it can interact with other proteins (e.g. Lef/TCF family members) and associating with chromatin, as this bipartite complex or with other proteins, such as those providing a DNA binding domain (e.g. transcriptional scaffold). These data now highlight, for the first time, that a cadherin CTF2 can interact with chromatin in vivo and control transcription of key genes in the neural crest to facilitate EMT (Schiffmacher et al., 2016). Notably, the use of ADAMs and other proteases to generate shed cadherin NTFs and intracellular CTF2s, the latter of which in turn possess novel functions in gene regulation, may be directly translatable to other developmental- and disease-related EMTs.
In addition to the novel transcriptional function acquired by Cadherin-6B CTF2 upon proteolysis of Cadherin-6B, a potential unique function for the Cadherin-6B NTF is now being explored. Our preliminary results in cell culture and in vivo indicate that multiple metalloproteases can cleave Cadherin-6B. This gives rise to NTFs of distinct molecular weights that are observed after immunoblotting with an antibody to the Cadherin-6B N-terminus. When fragments of these latter sizes are cloned and expressed in CHO cells, which lack endogenous cadherins, the Cadherin-6B NTFs that are produced are in fact secreted, N-glycosylated, and their relative electrophoretic mobilities mimic the size of NTFs generated by proteolysis of full-length Cadherin-6B (Schiffmacher and Taneyhill, submitted).
To gain insight into a potential function for these distinct Cadherin-6B NTFs, the NTFs were expressed in chick premigratory CNC, followed by immunostaining to evaluate effects on EMT. Strikingly, Cadherin-6B NTF-positive neural crest cells were surrounded by less laminin, with some neural crest cells prematurely emerging through the basement membrane, leading to an overall effect of precocious delamination. Given that the absence of laminin from the dorsal neural tube basement membrane is critical to allow chick CNC to emerge from the neural tube, we investigated whether Cadherin-6B NTF overexpression increased levels of proteases for which laminin would be a substrate, such as MMPs. Through an in-gel zymography assay, a two-fold increase in protease activity was noted in supernatants of CNC cultured ex vivo that overexpress Cadherin-6B NTF over control. Intriguingly, this activity was observed at 72 kDa and 62 kDa, which respectively correspond to the molecular weights of pro-MMP2 and activated MMP2, a protease expressed in neural crest cells (D. H. Cai, Vollberg, Hahn-Dantona, Quigley, & Brauer, 2000; Duong & Erickson, 2004) that can also degrade laminin. These data point to a potential mechanism by which the shed Cadherin-6B NTF from proteolysis of full-length Cadherin-6B enhances laminin degradation, and thus promotes delamination and EMT, by up-regulating MMP2 levels and/or activity (Schiffmacher and Taneyhill, submitted). Collectively, these findings outlining the novel pro-EMT functions for Cadherin-6B NTFs and CTF2s in the chick reveal that, like in Xenopus, N- and C-terminal fragments of cadherins are biologically active and possess crucial roles in development (Fig. 2).
Conclusion
The study of ADAMs in the neural crest has uncovered multiple levels of regulation of proteins that are essential for neural crest cell EMT and migration. Notably, it has forced us to look beyond transcriptional control within gene regulatory networks to understand protein function at the plasma membrane. In return, the neural crest model has provided a fantastic in vivo system to probe ADAM function in live cells in their natural environment. Many more questions have been raised by these studies that will continue to collectively advance the two fields of research. In particular what is the role of non-proteolytic ADAMs in neural crest development? Can we use proteomics-based methods to provide an unbiased, exhaustive list of ADAM proteolytic substrates in the neural crest? Is there an evolutionary conserved role of the ADAM cytoplasmic domain at the membrane and in the nucleus? How do ADAMs acquire substrate specificity versus possessing redundant roles in cleaving substrates within the neural crest? Investigating these questions in multiple vertebrate models, such as frog and avian species, will provide insight into both conserved and unique mechanisms by which ADAM-mediated proteolysis controls neural crest development, and, in turn, will shed light on the roles of ADAMs and their substrates in human development and disease.
Acknowledgments
This work was supported by the National Institutes of Health, U.S. Public Health Service, Grant R01-DE016289 and R24OD021485 (D.A.), R01-DE024217 (L.A.T.), and an American Cancer Society Research Scholars Grant RSG-15-023-01 – CSM (L.A.T.).
References
- Abbruzzese G, Becker SF, Kashef J, Alfandari D. ADAM13 cleavage of cadherin-11 promotes CNC migration independently of the homophilic binding site. Dev Biol. 2016;415(2):383–390. doi: 10.1016/j.ydbio.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbruzzese G, Cousin H, Salicioni AM, Alfandari D. GSK3 and Polo-like kinase regulate ADAM13 function during cranial neural crest cell migration. Mol Biol Cell. 2014;25(25):4072–4082. doi: 10.1091/mbc.E14-05-0970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbruzzese G, Gorny AK, Kaufmann LT, Cousin H, Kleino I, Steinbeisser H, Alfandari D. The Wnt receptor Frizzled-4 modulates ADAM13 metalloprotease activity. J Cell Sci. 2015;128(6):1139–1149. doi: 10.1242/jcs.163063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfandari D, Cousin H, Gaultier A, Hoffstrom BG, DeSimone DW. Integrin alpha5beta1 supports the migration of Xenopus cranial neural crest on fibronectin. Dev Biol. 2003;260(2):449–464. doi: 10.1016/s0012-1606(03)00277-x. [DOI] [PubMed] [Google Scholar]
- Alfandari D, Cousin H, Gaultier A, Smith K, White JM, Darribere T, DeSimone DW. Xenopus ADAM 13 is a metalloprotease required for cranial neural crest-cell migration. Curr Biol. 2001;11(12):918–930. doi: 10.1016/s0960-9822(01)00263-9. [DOI] [PubMed] [Google Scholar]
- Alfandari D, Wolfsberg TG, White JM, DeSimone DW. ADAM 13: a novel ADAM expressed in somitic mesoderm and neural crest cells during Xenopus laevis development. Dev Biol. 1997;182(2):314–330. doi: 10.1006/dbio.1996.8458. [DOI] [PubMed] [Google Scholar]
- Atapattu L, Lackmann M, Janes PW. The role of proteases in regulating Eph/ephrin signaling. Cell Adh Migr. 2014;8(4):294–307. doi: 10.4161/19336918.2014.970026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahm I, Barriga EH, Frolov A, Theveneau E, Frankel P, Mayor R. PDGF controls contact inhibition of locomotion by regulating N-cadherin during neural crest migration. Development. 2017;144(13):2456–2468. doi: 10.1242/dev.147926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132(14):3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- Blobel CP. Functional processing of fertilin: evidence for a critical role of proteolysis in sperm maturation and activation. Rev Reprod. 2000;5(2):75–83. doi: 10.1530/ror.0.0050075. [DOI] [PubMed] [Google Scholar]
- Brouxhon SM, Kyrkanides S, Teng X, Athar M, Ghazizadeh S, Simon M, Ma L. Soluble E-cadherin: a critical oncogene modulating receptor tyrosine kinases, MAPK and PI3K/Akt/mTOR signaling. Oncogene. 2014;33(2):225–235. doi: 10.1038/onc.2012.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouxhon SM, Kyrkanides S, Teng X, O’Banion MK, Clarke R, Byers S, Ma L. Soluble-E-cadherin activates HER and IAP family members in HER2+ and TNBC human breast cancers. Mol Carcinog. 2014;53(11):893–906. doi: 10.1002/mc.22048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai DH, Vollberg TM, Sr, Hahn-Dantona E, Quigley JP, Brauer PR. MMP-2 expression during early avian cardiac and neural crest morphogenesis. Anat Rec. 2000;259(2):168–179. doi: 10.1002/(SICI)1097-0185(20000601)259:2<168::AID-AR7>3.0.CO;2-U. doi:10.1002/(SICI)1097-0185(20000601)259:2<168::AID-AR7=3.0.CO;2-U [pii] [DOI] [PubMed] [Google Scholar]
- Cai H, Kratzschmar J, Alfandari D, Hunnicutt G, Blobel CP. Neural crest-specific and general expression of distinct metalloprotease-disintegrins in early Xenopus laevis development. Dev Biol. 1998;204(2):508–524. doi: 10.1006/dbio.1998.9017. [DOI] [PubMed] [Google Scholar]
- Chen C, Huang X, Sheppard D. ADAM33 is not essential for growth and development and does not modulate allergic asthma in mice. Mol Cell Biol. 2006;26(18):6950–6956. doi: 10.1128/MCB.00646-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C. Testicular and epididymal ADAMs: expression and function during fertilization. Nat Rev Urol. 2012;9(10):550–560. doi: 10.1038/nrurol.2012.167. [DOI] [PubMed] [Google Scholar]
- Christian L, Bahudhanapati H, Wei S. Extracellular metalloproteinases in neural crest development and craniofacial morphogenesis. Crit Rev Biochem Mol Biol. 2013;48(6):544–560. doi: 10.3109/10409238.2013.838203. [DOI] [PubMed] [Google Scholar]
- Christian LM. The ADAM family: Insights into Notch proteolysis. Fly (Austin) 2012;6(1):30–34. doi: 10.4161/fly.18823. [DOI] [PubMed] [Google Scholar]
- Cobaleda C, Perez-Caro M, Vicente-Duenas C, Sanchez-Garcia I. Function of the zinc-finger transcription factor SNAI2 in cancer and development. Annu Rev Genet. 2007;41:41–61. doi: 10.1146/annurev.genet.41.110306.130146. [DOI] [PubMed] [Google Scholar]
- Coles EG, Taneyhill LA, Bronner-Fraser M. A critical role for Cadherin6B in regulating avian neural crest emigration. Dev Biol. 2007;312(2):533–544. doi: 10.1016/j.ydbio.2007.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin H. Cadherins function during the collective cell migration of Xenopus Cranial Neural Crest cells: revisiting the role of E-cadherin. Mech Dev. 2017;148:79–88. doi: 10.1016/j.mod.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin H, Abbruzzese G, Kerdavid E, Gaultier A, Alfandari D. Translocation of the cytoplasmic domain of ADAM13 to the nucleus is essential for Calpain8-a expression and cranial neural crest cell migration. Dev Cell. 2011;20(2):256–263. doi: 10.1016/j.devcel.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin H, Abbruzzese G, McCusker C, Alfandari D. ADAM13 function is required in the 3 dimensional context of the embryo during cranial neural crest cell migration in Xenopus laevis. Dev Biol. 2012;368(2):335–344. doi: 10.1016/j.ydbio.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin H, Gaultier A, Bleux C, Darribere T, Alfandari D. PACSIN2 is a regulator of the metalloprotease/disintegrin ADAM13. Dev Biol. 2000;227(1):197–210. doi: 10.1006/dbio.2000.9871. [DOI] [PubMed] [Google Scholar]
- Dady A, Blavet C, Duband JL. Timing and kinetics of E- to N-cadherin switch during neurulation in the avian embryo. Dev Dyn. 2012;241(8):1333–1349. doi: 10.1002/dvdy.23813. [DOI] [PubMed] [Google Scholar]
- Dady A, Duband JL. Cadherin interplay during neural crest segregation from the non-neural ectoderm and neural tube in the early chick embryo. Dev Dyn. 2017;246(7):550–565. doi: 10.1002/dvdy.24517. [DOI] [PubMed] [Google Scholar]
- del Barrio MG, Nieto MA. Overexpression of Snail family members highlights their ability to promote chick neural crest formation. Development. 2002;129(7):1583–1593. doi: 10.1242/dev.129.7.1583. [DOI] [PubMed] [Google Scholar]
- Dreymueller D, Pruessmeyer J, Groth E, Ludwig A. The role of ADAM-mediated shedding in vascular biology. Eur J Cell Biol. 2012;91(6-7):472–485. doi: 10.1016/j.ejcb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Dreymueller D, Theodorou K, Donners M, Ludwig A. Fine Tuning Cell Migration by a Disintegrin and Metalloproteinases. Mediators Inflamm. 2017;2017:9621724. doi: 10.1155/2017/9621724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong TD, Erickson CA. MMP-2 plays an essential role in producing epithelial-mesenchymal transformations in the avian embryo. Dev Dyn. 2004;229(1):42–53. doi: 10.1002/dvdy.10465. [DOI] [PubMed] [Google Scholar]
- Evans JP. Fertilin beta and other ADAMs as integrin ligands: insights into cell adhesion and fertilization. Bioessays. 2001;23(7):628–639. doi: 10.1002/bies.1088. [DOI] [PubMed] [Google Scholar]
- Evans JP, Schultz RM, Kopf GS. Mouse sperm-egg plasma membrane interactions: analysis of roles of egg integrins and the mouse sperm homologue of PH-30 (fertilin) beta. J Cell Sci. 1995;108(Pt 10):3267–3278. doi: 10.1242/jcs.108.10.3267. [DOI] [PubMed] [Google Scholar]
- Ferber EC, Kajita M, Wadlow A, Tobiansky L, Niessen C, Ariga H, Fujita Y. A role for the cleaved cytoplasmic domain of E-cadherin in the nucleus. J Biol Chem. 2008;283(19):12691–12700. doi: 10.1074/jbc.M708887200. doi:M708887200 [pii] 10.1074/jbc.M708887200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo AL, Maczkowiak F, Borday C, Pla P, Sittewelle M, Pegoraro C, Monsoro-Burq AH. PFKFB4 control of AKT signaling is essential for premigratory and migratory neural crest formation. Development. 2017;144(22):4183–4194. doi: 10.1242/dev.157644. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Piacentino ML, Vieceli FM, Bronner ME. Optimization of CRISPR/Cas9 genome editing for loss-of-function in the early chick embryo. Dev Biol. 2017;432(1):86–97. doi: 10.1016/j.ydbio.2017.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaultier A, Cousin H, Darribere T, Alfandari D. ADAM13 disintegrin and cysteine-rich domains bind to the second heparin-binding domain of fibronectin. J Biol Chem. 2002;277(26):23336–23344. doi: 10.1074/jbc.M201792200. [DOI] [PubMed] [Google Scholar]
- Giebeler N, Zigrino P. A Disintegrin and Metalloprotease (ADAM): Historical Overview of Their Functions. Toxins (Basel) 2016;8(4):122. doi: 10.3390/toxins8040122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez JM, Rucavado A, Escalante T, Diaz C. Hemorrhage induced by snake venom metalloproteinases: biochemical and biophysical mechanisms involved in microvessel damage. Toxicon. 2005;45(8):997–1011. doi: 10.1016/j.toxicon.2005.02.029. [DOI] [PubMed] [Google Scholar]
- Hall RJ, Erickson CA. ADAM 10: an active metalloprotease expressed during avian epithelial morphogenesis. Dev Biol. 2003;256(1):146–159. doi: 10.1016/s0012-1606(02)00133-1. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Herrlich A, Herrlich P. Who decides when to cleave an ectodomain? Trends Biochem Sci. 2013;38(3):111–120. doi: 10.1016/j.tibs.2012.12.002. [DOI] [PubMed] [Google Scholar]
- Huang C, Kratzer MC, Wedlich D, Kashef J. E-cadherin is required for cranial neural crest migration in Xenopus laevis. Dev Biol. 2016;411(2):159–171. doi: 10.1016/j.ydbio.2016.02.007. [DOI] [PubMed] [Google Scholar]
- Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Ludwig A. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102(4):1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- Li Jiejing1, Perfetto Mark1 2§, Neuner Russell3, Bahudhanapati Harinath1, Christian Laura1, Mathavan Ketan3, Bridges Lance C4, Alfandari Dominique R3, Wei Shuo1 2* Xenopus ADAM19 regulates Wnt signaling and neural crest specification through nonproteolytic stabilization of ADAM13. Development. doi: 10.1242/dev.158154. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashef J, Kohler A, Kuriyama S, Alfandari D, Mayor R, Wedlich D. Cadherin-11 regulates protrusive activity in Xenopus cranial neural crest cells upstream of Trio and the small GTPases. Genes Dev. 2009;23(12):1393–1398. doi: 10.1101/gad.519409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler A, Schlupf J, Schneider M, Kraft B, Winter C, Kashef J. Loss of Xenopus cadherin-11 leads to increased Wnt/beta-catenin signaling and up-regulation of target genes c-myc and cyclin D1 in neural crest. Dev Biol. 2013;383(1):132–145. doi: 10.1016/j.ydbio.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Komatsu K, Wakatsuki S, Yamada S, Yamamura K, Miyazaki J, Sehara-Fujisawa A. Meltrin beta expressed in cardiac neural crest cells is required for ventricular septum formation of the heart. Dev Biol. 2007;303(1):82–92. doi: 10.1016/j.ydbio.2006.10.037. [DOI] [PubMed] [Google Scholar]
- Langhe RP, Gudzenko T, Bachmann M, Becker SF, Gonnermann C, Winter C, Kashef J. Cadherin-11 localizes to focal adhesions and promotes cell-substrate adhesion. Nat Commun. 2016;7:10909. doi: 10.1038/ncomms10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RT, Nagai H, Nakaya Y, Sheng G, Trainor PA, Weston JA, Thiery JP. Cell delamination in the mesencephalic neural fold and its implication for the origin of ectomesenchyme. Development. 2013;140(24):4890–4902. doi: 10.1242/dev.094680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SL, Farlie PG, Newgreen DF. Isolation and embryonic expression of avian ADAM 12 and ADAM 19. Gene Expr Patterns. 2004;5(1):75–79. doi: 10.1016/j.modgep.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Lin J, Redies C, Luo J. Regionalized expression of ADAM13 during chicken embryonic development. Dev Dyn. 2007;236(3):862–870. doi: 10.1002/dvdy.21071. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Robakis NK. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002;21(8):1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1FAD mutations. Cell. 2003;114(5):635–645. doi: 10.1016/j.cell.2003.08.008. doi:S0092867403006512 [pii] [DOI] [PubMed] [Google Scholar]
- Maretzky T, Reiss K, Ludwig A, Buchholz J, Scholz F, Proksch E, Saftig P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc Natl Acad Sci U S A. 2005;102(26):9182–9187. doi: 10.1073/pnas.0500918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathavan K, Khedgikar V, Bartolo V, Alfandari D. The ectodomain of cadherin-11 binds to erbB2 and stimulates Akt phosphorylation to promote cranial neural crest cell migration. PLoS One. 2017;12(11):e0188963. doi: 10.1371/journal.pone.0188963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker C, Cousin H, Neuner R, Alfandari D. Extracellular cleavage of cadherin-11 by ADAM metalloproteases is essential for Xenopus cranial neural crest cell migration. Mol Biol Cell. 2009;20(1):78–89. doi: 10.1091/mbc.E08-05-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan R, Schumacher LJ, Morrison JA, Teddy JM, Ridenour DA, Box AC, Kulesa PM. VEGF signals induce trailblazer cell identity that drives neural crest migration. Dev Biol. 2015;407(1):12–25. doi: 10.1016/j.ydbio.2015.08.011. [DOI] [PubMed] [Google Scholar]
- Monsonego-Ornan E, Kosonovsky J, Bar A, Roth L, Fraggi-Rankis V, Simsa S, Sela-Donenfeld D. Matrix metalloproteinase 9/gelatinase B is required for neural crest cell migration. Dev Biol. 2012;364(2):162–177. doi: 10.1016/j.ydbio.2012.01.028. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Takeichi M. Neural crest cell-cell adhesion controlled by sequential and subpopulation-specific expression of novel cadherins. Development. 1995;121:1321–1332. doi: 10.1242/dev.121.5.1321. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Takeichi M. Neural crest emigration from the neural tube depends on regulated cadherin expression. Development. 1998;125(15):2963–2971. doi: 10.1242/dev.125.15.2963. [DOI] [PubMed] [Google Scholar]
- Neuner R, Cousin H, McCusker C, Coyne M, Alfandari D. Xenopus ADAM19 is involved in neural, neural crest and muscle development. Mech Dev. 2009;126(3-4):240–255. doi: 10.1016/j.mod.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Rubin GM. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 1997;90(2):271–280. doi: 10.1016/s0092-8674(00)80335-9. [DOI] [PubMed] [Google Scholar]
- Pollheimer J, Fock V, Knofler M. Review: the ADAM metalloproteinases - novel regulators of trophoblast invasion? Placenta. 2014;35(Suppl):S57–63. doi: 10.1016/j.placenta.2013.10.012. [DOI] [PubMed] [Google Scholar]
- Roth L, Kalev-Altman R, Monsonego-Ornan E, Sela-Donenfeld D. A new role of the membrane-type matrix metalloproteinase 16 (MMP16/MT3-MMP) in neural crest cell migration. Int J Dev Biol. 2017;61(3-4-5):245–256. doi: 10.1387/ijdb.160286ds. [DOI] [PubMed] [Google Scholar]
- Sadot E, Simcha I, Shtutman M, Ben-Ze’ev A, Geiger B. Inhibition of beta-catenin-mediated transactivation by cadherin derivatives. Proc Natl Acad Sci U S A. 1998;95(26):15339–15344. doi: 10.1073/pnas.95.26.15339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai D, Tanaka Y, Endo Y, Osumi N, Okamoto H, Wakamatsu Y. Regulation of Slug transcription in embryonic ectoderm by b-catenin-Lef/TCF and BMP-Smad signaling. Develop Growth Differ. 2005;47:471–482. doi: 10.1111/j.1440-169X.2005.00821.x. [DOI] [PubMed] [Google Scholar]
- Schiffmacher AT, Padmanabhan R, Jhingory S, Taneyhill LA. Cadherin-6B is proteolytically processed during epithelial-to-mesenchymal transitions of the cranial neural crest. Mol Biol Cell. 2014;25(1):41–54. doi: 10.1091/mbc.E13-08-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmacher AT, Xie V, Taneyhill LA. Cadherin-6B proteolysis promotes the neural crest cell epithelial-to-mesenchymal transition through transcriptional regulation. J Cell Biol. 2016;215(5):735–747. doi: 10.1083/jcb.201604006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P, Reiss K. ADAM10 regulates endothelial permeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res. 2008;102(10):1192–1201. doi: 10.1161/CIRCRESAHA.107.169805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela-Donenfeld D, Kalcheim C. Regulation of the onset of neural crest migration by coordinated activity of BMP4 and Noggin in the dorsal neural tube. Development. 1999;126(21):4749–4762. doi: 10.1242/dev.126.21.4749. [DOI] [PubMed] [Google Scholar]
- Session AM, Uno Y, Kwon T, Chapman JA, Toyoda A, Takahashi S, Rokhsar DS. Genome evolution in the allotetraploid frog Xenopus laevis. Nature. 2016;538(7625):336–343. doi: 10.1038/nature19840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoval I, Ludwig A, Kalcheim C. Antagonistic roles of full-length N-cadherin and its soluble BMP cleavage product in neural crest delamination. Development. 2007;134(3):491–501. doi: 10.1242/dev.02742. [DOI] [PubMed] [Google Scholar]
- Smith KM, Gaultier A, Cousin H, Alfandari D, White JM, DeSimone DW. The cysteine-rich domain regulates ADAM protease function in vivo. J Cell Biol. 2002;159(5):893–902. doi: 10.1083/jcb.200206023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl-Mazzulla PH, Bronner ME. A PHD12-Snail2 repressive complex epigenetically mediates neural crest epithelial-to-mesenchymal transition. J Cell Biol. 2012;198(6):999–1010. doi: 10.1083/jcb.201203098. doi:jcb.201203098 [pii] 10.1083/jcb.201203098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Fu R, Liu J, Wu Y, Wang B, Jiang N, Fang B. ADAM10 is essential for cranial neural crest-derived maxillofacial bone development. Biochem Biophys Res Commun. 2016;475(4):308–314. doi: 10.1016/j.bbrc.2016.05.101. [DOI] [PubMed] [Google Scholar]
- Taneyhill LA, Coles EG, Bronner-Fraser M. Snail2 directly represses cadherin6B during epithelial-to-mesenchymal transitions of the neural crest. Development. 2007;134(8):1481–1490. doi: 10.1242/dev.02834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneyhill LA, Schiffmacher AT. Should I stay or should I go? Cadherin function and regulation in the neural crest. Genesis. 2017;55(6) doi: 10.1002/dvg.23028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura K, Kihara T, Kuzuya A, Okawa K, Nishimoto T, Bito H, Shimohama S. Activity-dependent regulation of beta-catenin via epsilon-cleavage of N-cadherin. Biochem Biophys Res Commun. 2006;345(3):951–958. doi: 10.1016/j.bbrc.2006.04.157. doi:S0006-291X(06)00977-6 [pii] 10.1016/j.bbrc.2006.04.157. [DOI] [PubMed] [Google Scholar]
- Umland SP, Garlisi CG, Shah H, Wan Y, Zou J, Devito KE, Ralston R. Human ADAM33 messenger RNA expression profile and post-transcriptional regulation. Am J Respir Cell Mol Biol. 2003;29(5):571–582. doi: 10.1165/rcmb.2003-0028OC. [DOI] [PubMed] [Google Scholar]
- Umland SP, Wan Y, Shah H, Garlisi CG, Devito KE, Braunschweiger K, Del Mastro R. Mouse ADAM33: two splice variants differ in protein maturation and localization. Am J Respir Cell Mol Biol. 2004;30(4):530–539. doi: 10.1165/rcmb.2003-0220OC. [DOI] [PubMed] [Google Scholar]
- van der Vorst EP, Keijbeck AA, de Winther MP, Donners MM. A disintegrin and metalloproteases: molecular scissors in angiogenesis, inflammation and atherosclerosis. Atherosclerosis. 2012;224(2):302–308. doi: 10.1016/j.atherosclerosis.2012.04.023. [DOI] [PubMed] [Google Scholar]
- Khedgikar Vikram, Ketan Mathavan GA2, Szydlo Hannah, Cousin Hélène, Alfandari Dominique#. Dual control of pcdh8l/PCNS expression and function by adam13/33 via tfap2α and arid3a. elife. 2017 doi: 10.7554/eLife.26898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Xu G, Bridges LC, Williams P, White JM, DeSimone DW. ADAM13 induces cranial neural crest by cleaving class B Ephrins and regulating Wnt signaling. Dev Cell. 2010;19(2):345–352. doi: 10.1016/j.devcel.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zhou BP. Snail: More than EMT. Cell Adh Migr. 2010;4(2):199–203. doi: 10.4161/cam.4.2.10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Lin J, Markus A, Rolfs A, Luo J. Regional expression of ADAM19 during chicken embryonic development. Dev Growth Differ. 2011;53(3):333–346. doi: 10.1111/j.1440-169X.2010.01238.x. [DOI] [PubMed] [Google Scholar]