

Abstract
Nucleophilic aromatic substitution (SNAr) is one of the most widely applied reaction classes in pharmaceutical and chemical research, providing a broadly useful platform for the modification of aromatic ring scaffolds. The generally accepted mechanism for SNAr reactions involves a two-step addition–elimination sequence via a discrete, non-aromatic Meisenheimer complex. Here we use 12C/13C kinetic isotope effect (KIE) studies and computational analyses to provide evidence that prototypical SNAr reactions in fact proceed through concerted mechanisms. The KIE measurements were made possible by a new technique that leverages the high sensitivity of 19F as an NMR nucleus to quantitate the degree of isotopic fractionation. This sensitive technique permits the measurement of KIEs on 10 mg of natural abundance material in one overnight acquisition. As a result, it provides a practical tool for performing detailed mechanistic analyses of reactions that form or break C–F bonds.
Graphical Abstract
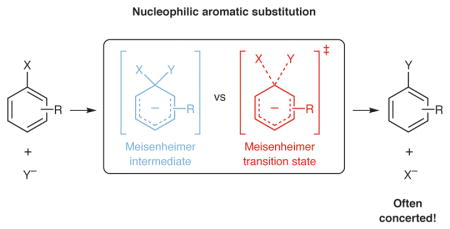
Whether a given chemical transformation proceeds through a stepwise or concerted mechanism is a question of both fundamental interest and practical importance, with direct implications for the stereospecificity, sensitivity to medium effects, and product distribution of the reaction.1 In general, stepwise pathways are favored when productive reactive intermediates are energetically accessible. For example, the isolation and characterization of the direct products of nucleophilic addition to aromatic rings, termed Meisenheimer complexes (Figure 1a), have led to the widely held notion that SNAr reactions2 generally proceed via stepwise, addition/elimination mechanisms.1,2
Figure 1.

Scope of study. (a) Previously observed Meisenheimer complexes have always been highly stabilized by poor leaving groups and electron-deficient rings. (b) The SNAr reactions studied here. Bold letters denote Meisenheimer complexes, which span a range of stabilities. (The term ‘complex’ does not necessarily indicate that the structure is a true intermediate.)
The participation of a discrete Meisenheimer complex as an intermediate in an SNAr mechanism requires that the anionic adduct be more thermodynamically stable than the transition state (TS) for the concerted pathway, as well as possess sufficient kinetic stability to have a significant lifetime prior to elimination. Nearly all known Meisenheimer complexes contain both strongly electron-withdrawing substituents, such as nitro groups, and poor leaving groups, such as fluoride, that are expected to retard elimination.3 In the SNAr reactions that are typically employed in pharmaceutical synthesis,4 which involve less stabilized anions or good leaving groups, the intermediates are not detectable but have nonetheless been assumed to exist.2
However, recent theoretical and experimental work, most notably from the Ritter group,5,6 has challenged this assumption. More broadly, concerted nucleophilic substitution reactions at sp2 carbons have long been established for reactions of vinyl halides,7–11 as well as acyl,12–24 phosphorus,25–29 and sulfur group transfer reactions.30–34 In the context of SNAr reactions, Williams et al. have shown that the quasi-symmetric1 addition of substituted phenoxides to phenoxytriazines is concerted,35,36 while the apparently analogous addition of pyridines to pyridyltriazines is stepwise.37,38 For many other reactions, computations39–50 predict concerted mechanisms, contradicting the textbook view that SNAr reactions proceed via stepwise addition-elimination mechanisms.51
We sought to use experiments to ascertain whether stepwise mechanisms are indeed generally operative in SNAr reactions by studying three transformations with putative intermediates that would be either strongly, moderately, or weakly stabilized (Figure 1b). Structure A is strongly stabilized by nitro group substituents and poor leaving groups (fluoride and methoxide), and related reactions have been demonstrated to be stepwise in nature.52 In contrast, structure B is only weakly stabilized by its nitrogen-containing heterocycle and ester substituent and contains a good leaving group (bromide), which are all factors that might be expected to favor a concerted reaction. Reaction C represents a borderline case, in which structure C is stabilized by strongly electron-withdrawing substituents, but destabilized by a good leaving group (chloride).
Kinetic isotope effects (KIEs) can provide direct insight into the bonding in transition states, and are therefore useful tools for the evaluation of reaction concertedness.1 In SNAr reactions, the primary 12C/13C KIE at the carbon undergoing substitution is expected to provide the most useful information. One powerful strategy for measuring such KIEs is to use quantitative 13C NMR spectroscopy, ideally at natural isotopic abundance.53 However, the high precision required for the determination of intrinsically small (<10%) heavy-atom KIEs, combined with the low natural abundance and gyromagnetic ratio of the of 13C nucleus, renders this type of experiment generally challenging to implement. When the carbons of interest are not bound to a proton, as is the case for the sites undergoing substitution in SNAr reactions, dipolar relaxation is particularly inefficient. This requires long delays (minutes) between scans and further reduces sensitivity.
Results
We envisioned an alternative approach for the quantification of 13C in the specific case of transformations involving C–F bond formation or cleavage, which are characteristic of many SNAr reactions. In 1H-decoupled 19F NMR spectra of organofluorine compounds at natural abundance, resonances appear as a large 12C–19F parent singlet flanked by a small 13C–19F satellite doublet (Figure 2a). In principle, integration of these satellite integrals could provide a highly sensitive means of determining 12C/13C isotope fractionation because the signal to noise ratio (S/N) of each 13C–19F satellite is theoretically 13.6 times greater (per fluorine) than that of the corresponding peak in a 13C spectrum. This increase in sensitivity translates to a potential 185-fold reduction in acquisition time.54
Figure 2.

Assessing 13C isotopic fractionation by suppressing NMR signals from fluorine atoms bound to 12C. (a) Standard 19F{1H} spectrum showing the parent 12C–19F peak flanked by two 13C–19F satellites. (b) The standard spectrum in (a), enlarged 125×. Accurate satellite integrals cannot be obtained directly due to overlap with the parent peak. (c) MQF 19F{1H} spectrum. Suppression of the parent peak allows accurate integration of the satellites.
Unfortunately, it is not possible to obtain accurate satellite integrals directly55 because of interference from the much larger parent peak, as well as possible overlapping impurities (Figure 2b). However, we found that accurate satellite integrals could be obtained from multiple-quantum-filtered (MQF) spectra in which the 12C signals are suppressed (Figure 2c).54 This technique was validated by determining the 12C/13C KIE for a simple nucleophilic substitution reaction using both the traditional 13C NMR and new MQF methods (Table 1). The MQF method was found to require far less material (as little as 10 mg with overnight acquisition; see entry 6) and acquisition time (under one hour with 50 mg of material; see entry 7). The consensus value of 1.059(3) determined by the new method agrees with the predicted SN2 KIE of 1.057 (M11/jun-cc-pVTZ/PCM). Consistent with prior studies,56,57 the predicted KIE is insensitive to both TS geometry and computational method (see Supplementary Information Section 4b).
Table 1.
Comparison of KIE Measurements
| ||
|---|---|---|
| method | time (h) | KIE (std. err.) |
| 1 Singleton (300 mg) | 9.9 | 1.058(6), 1.060(6) |
| 2 MQF (50 mg)a | 4.6 | 1.057(5), 1.065(6) |
| 3 MQF (50 mg) | 5.2 | 1.057(3), 1.062(4) |
| 4 MQF (50 mg)b | 5.2 | 1.059(4), 1.060(4) |
| 5 MQF (10 mg)c | 9.9 | 1.061(6) |
| 6 MQF (50 mg) | 0.7 | 1.055(10) |
| consensus (std. dev.): 1.059(3) | ||
Samples are pure unless otherwise noted. Acquisition times are given for each pair of partial and full conversion samples. Pairs of KIEs refer to independent chemical replicates. The error bars (in parentheses) refer to standard errors of the mean (t-distribution) and reflect technical variation due to errors in the measurement of conversion and satellite area. KIEs are referenced to 12C–19F unless otherwise noted.
KIEs are referenced to 13C-19F.
Unpurified sample.
Shigemi tube used.
The greatly increased sensitivity of the MQF method results from the larger gyromagnetic ratio and shorter T1 relaxation times of 19F over 13C nuclei. Sensitivity can be further increased by using the parent 12C–19F peak from a routine 19F{1H} spectrum, rather than a different 13C–19F satellite in the MQF spectrum, as an internal reference58 (entries 4–7). Additionally, because a fluorination or defluorination reaction generally involves the appearance or disappearance of a well-separated 19F resonance, unpurified material can be used (entry 5).
The accuracy and sensitivity of the MQF method allowed us to determine the 12C/13C KIEs for several SNAr fluorination and defluorination reactions. To provide a theoretical basis for interpreting these measurements, we assessed the accuracy of various computational methods in describing the potential energy surfaces of reactions A, B, and C. Using geometries spanning a range of carbon/nucleophile and carbon/leaving group distances, we found that B3LYP-D3BJ/jun-cc-pVTZ59 most closely reproduced the energies calculated using the benchmark coupled cluster method DLPNO-CCSD(T)60/aug-cc-pVTZ (see Supplementary Information Sections 4c–e).
The surface for reaction A in implicit solvent (PCM) clearly shows an intermediate (Figure 3a), and is consistent with the second step being rate-determining. The experimental KIE of 1.035(3) agrees closely with the calculated KIE of 1.033. This provides strong evidence for the validity of the stepwise mechanism in the case of reaction A.
Figure 3.

Computational analysis of the transition from stepwise to concerted behavior (B3LYP-D3BJ/jun-cc-pVTZ). (a) Potential energy surface for reaction A. An intermediate is apparent (lower left). (b) Predicted KIE as a function of geometry for reaction A. The lowest-energy stepwise mechanism follows the white path, avoiding the region of large KIEs. (c) Potential energy surface for reaction B. The “Meisenheimer region” is very high in energy (white). (d) Predicted KIE as a function of geometry for reaction B. The lowest-energy concerted mechanism follows the black path, resulting in a KIE that approaches the maximum possible value. (e) Potential energy (top) and charge distribution (bottom) along the intrinsic reaction coordinate for reaction B. Negative charge is distributed between the nucleophile, the leaving group, and the ring in the non-aromatic TS. Breaks in the curves indicate the position of transition state B. (f) Potential energy surface for reaction C. A typical trajectory (dotted path) reflects a fleeting intermediate or long-lived TS in the Meisenheimer region (lower left).
In contrast, computational analysis of reaction B reveals no evidence of an intermediate, either on its energy surface (Figure 3c) or intrinsic reaction coordinate (Figure 3e). Additionally, quasiclassical61 dynamics initialized from transition state B proceed quickly to product (mean time, 91 fs) and nearly all trajectories are productive (94%), indicating that the reaction is truly concerted. The predicted KIE of 1.039 is in reasonable agreement with the experimental value of 1.035(3).
Discussion
Although the experimentally determined KIEs for stepwise reaction A and concerted reaction B are the same (1.035), the calculations reveal that those values are in fact of very different magnitudes relative to the maximum values possible for the two reactions. For a given reaction, a ‘KIE surface’ (e.g., Figure 3b) can be generated by predicting KIEs for each point on the energy surface (e.g., Figure 3a). The largest value on this KIE surface may then be defined as the maximum KIE. When the bonds in the ground state are strong, more vibrational energy can be lost in the TS, resulting in a larger maximum KIE. In reaction A, a strong C–F bond is broken, and the maximum KIE is 1.070 (Figure 3b, red). By contrast, in reaction B, a weak C–Br bond is broken, and the maximum KIE is 1.045 (Figure 3d, green).
The maximum KIE in any particular reaction is obtained when the bonds to both the nucleophile and electrophile are weakest in the transition state. Thus, the size of the actual KIE relative to its theoretical maximum is diagnostic of a stepwise or concerted mechanism. Stepwise mechanisms alter one bond at a time, leading to small KIEs (47% of the maximum value for reaction A). Conversely, concerted mechanisms alter both bonds simultaneously, passing through a nearly symmetric region in which both bonds are relatively weak, leading to large KIEs (87% of the maximum value for reaction B). This correspondence between small relative KIEs for stepwise mechanisms and large relative KIEs for concerted mechanisms parallels the behavior of SN2 reactions.62
Reaction C is a borderline case. Once again, the experimentally determined 12C/13C KIE of 1.045(3) is in good agreement with the predicted KIE of 1.040. This prediction is 73% of the maximum calculated value of 1.055, and lies between the values for the stepwise (47%) and concerted (87%) pathways. Borderline behavior is observed because the putative intermediate would be stabilized by two electron-withdrawing groups (NO2), but destabilized by a good leaving group (Cl). Accordingly, the minimum energy path does not pass through an intermediate, even though structures in the vicinity of the Meisenheimer complex are relatively stable (Figure 3f). As a result, trajectories exiting the formal TS encounter this shallow region and linger for multiple vibrations before passing to product (mean time: 233 fs). The reaction mechanism can therefore be viewed either as concerted, with a long-lived TS, or stepwise, with a short-lived intermediate.
The transition from stepwise to concerted behavior can be rationalized using qualitative Marcus theory.63–65 When the Meisenheimer complex is lower in energy than the intersection of the potential energy surfaces of the starting materials and products, the reaction is stepwise (Figure 4a). When the intermediate is higher in energy than the intersection, the reaction mechanism is concerted with the TS resembling the minimum of the Meisenheimer curve (Figure 4b). In borderline cases, such as when a stabilized anion is adjacent to a good leaving group, the Meisenheimer complex may intersect the reaction coordinate as a shallow minimum or shoulder (Figure 4c).
Figure 4.

Simplified Marcus analysis of stepwise vs. concerted SNAr reactions. (a) In stepwise reactions, the Meisenheimer structure is highly stabilized, leading to a minimum along the reaction coordinate (reaction A). (b) Concerted reactions result if the Meisenheimer structure is less stable. This is the typical situation (reaction B). (c) When the Meisenheimer structure is highly stabilized, but leaving group elimination is facile, a borderline situation (reaction C) results. (d) Meisenheimer transition states are stabilized by concurrent donor-acceptor interactions (transition state B is shown).
At first glance, concerted SNAr mechanisms may appear difficult to rationalize on stereoelectronic grounds given that backside attack by the nucleophile on the C–X σ* orbital of an aryl halide is precluded.66 However, this argument assumes that the bonding in the transition state resembles that of the starting material. In fact, concerted transition structures actually possess characteristics of a Meisenheimer intermediate. For example, charge,67 NICS,68 and NBO69 analyses all confirm that TS B is essentially a delocalized, but non-aromatic, anion (Figure 3e). This anion is generated by two concurrent, but asynchronous processes: C–F bond formation by donation of the fluoride lone pair into the C=N π* orbital, and C–Br bond cleavage by donation of the incipient nitrogen lone pair into the C–Br σ* orbital (Figure 4d). The result can be viewed as a ‘Meisenheimer transition state’.
The qualitative picture of a continuum between stepwise and concerted SNAr mechanisms depicted in Figure 4 is supported quantitatively by the close agreement between the theoretical and experimental KIEs. To evaluate the overall prevalence of concerted SNAr pathways, we carried out a computational survey of 120 SNAr reactions spanning a variety of typical ring types, nucleophiles, and leaving groups using B3LYP/6–31+G*/PCM(DMSO). This more economical level of theory also corresponds closely to the coupled cluster energy surface (see Supplementary Information Section 4g), and similar outcomes would be expected from other DFT methods. Remarkably, 99 of these reactions (83%) are predicted to proceed via concerted mechanisms.
Where an SNAr reaction lies on the stepwise/concerted continuum is determined primarily by the structural features of the reactants, rather than by the rate of their reaction. For nucleophilic substitutions on aryl rings, stepwise mechanisms are only predicted to occur when both a strongly electron-withdrawing substituent (e.g., nitro) is present and fluoride is the nucleophile or leaving group. For substitutions on pyridine, pyrazine, and pyrimidine, no stepwise mechanisms are predicted. Given that many SNAr reactions of interest are performed on heterocycles with good leaving groups (e.g. Cl, Br),1,2 it is likely that concerted mechanisms are actually very common.
The mechanistic analysis described herein was enabled by the sensitivity and practicality of the MQF method for determining 12C/13C KIEs. Our observations confirm that the nucleophilic adducts identified by Jackson70 and Meisenheimer71 over a century ago are indeed involved in SNAr reactions, but often as transition states rather than intermediates. We anticipate that the MQF method will enable the study of other fluorination and defluorination pathways of synthetic interest.
Methods
Data Availability
User-friendly software pipelines that can be used to measure and predict KIEs are freely available at www.github.com/ekwan/PyKIE and www.github.com/ekwan/PyQuiver. Raw NMR spectra and computed quasiclassical trajectories are available from the corresponding author upon reasonable request. All other data supporting the findings of this study are available within the paper and its supplementary information files.
Synthesis
Reactions were carried out in round-bottomed flasks under nitrogen. Commercially available reagents were purchased and used as received unless otherwise noted. The conversion of reactions was determined by quantitative 19F NMR spectroscopy using p-difluorobenzene as an internal standard, with the exception of reaction A, which was analyzed by HPLC using naphthalene as an internal standard. Products and recovered starting materials were purified by column chromatography, except for ‘impure’ samples, which were only subjected to an aqueous extraction. For detailed procedures and spectroscopic characterization data, please see Supplementary Information Section 1.
Isotope effect methodology
NMR samples greater than 10 mg in mass were prepared in Wilmad 528-PP-9 tubes and flame-sealed under air at room temperature. 10 mg samples were prepared in Shigemi tubes and sealed with parafilm. All spectroscopic measurements were performed at 25 °C on a Varian Inova 500 MHz machine fitted with an indirect detection probe (HFC) and a gradient driver. Specific details of the MQF pulse sequence can be found in Supplementary Information Section 3 and the PyKIE repository. For 12C-referenced experiments, standard 19F and MQF experiments were interwoven (eg. MQF then 19F then MQF). This pattern was repeated throughout the duration of the measurement.
NMR data were processed using Python and the nmrglue package32 using the PyKIE pipeline. Each FID was zero-filled (4×), apodized (exponential), Fourier transformed, and baseline corrected. Peaks were picked manually and centered on regions of interest. Widths of the peak-containing regions of interest were held constant as follows: MQF 0.045 ppm, 19F 0.15 ppm, 1H 0.08 ppm, 13C 0.12 ppm and 0.14 ppm. S/N ratios were calculated for each peak by dividing the signal intensity at half height by the root-mean-square deviation of the signal in a noisy region.
Each KIE measurement was made by comparing the fractionation between a partial conversion sample and a full conversion sample (for product-based analyses) or between recovered and unreacted starting material. When multiple samples were available, data for the full conversion (or unreacted starting material) sample was pooled to give two independent estimates of the kinetic isotope effect. The values reported in the text correspond to the average of these estimates. Error bars for individual measurements correspond to standard errors and were propagated from known standard deviations in conversion and integral area. Detailed procedures and Excel spreadsheets for reproducing these calculations can be found in Supplementary Information Section 3 and the PyKIE repository.
Calculations
DFT calculations were performed with Gaussian 09 or Gaussian 16.73 Bigeleisen–Mayer KIE predictions were made using PyQuiver.74 Tunnelling corrections were calculated using the one-dimensional Bell method. For the M06-2X prediction of the SN2 isotope effect, a multidimensional CVT/SCT correction was also calculated using GAUSSRATE75/POLYRATE76 and found to agree closely with the Bell correction. Coupled cluster calculations were carried out using ORCA 4.0.0 using TightPNO cutoffs.77
Direct quasiclassical trajectory studies were carried out on the B3LYP-D3(BJ)/6–31+G*/PCM surface as previously described. Trajectories were initialized from a thermal distribution (298 K) of vibrational states using the position eigenstates of a quantum harmonic oscillator. No displacements were made in any modes with a frequency of less than 50 cm−1, including the transition vector. The sign of the velocity in all modes was randomized except for the transition vector, which was directed towards product. 1.0 fs timesteps were calculated by the velocity Verlet method and each simulation was run 500 fs in both the forward and reverse directions.
The predicted KIEs for reactions A, B, and C were calculated with B3LYP-D3BJ/may-cc-pVQZ at 298 K (jun-cc-pVTZ is extremely similar) using PCM (i.e., IEFPCM) implicit solvation and a scaling factor78–80 of 1.2 for the sphere radii. The addition of explicit methanol molecules to reaction A or explicit water molecules to reaction B had a negligible effect. The inclusion of at least one explicit water molecule was crucial for reaction C. The effect of solvation parameters is examined thoroughly in Supplementary Information Section 4f.
The energy and KIE surfaces shown in Figure 3a–d and 3f were calculated at B3LYP-D3BJ/jun-cc-pVTZ/PCM with no explicit solvent molecules. The intrinsic reaction coordinate energetics and charges shown in Figure 3e were calculated at B3LYP-D3BJ/6–31+G*/PCM. Each colored square in Figure 3 corresponds to a structure in which the carbon–nucleophile and carbon–electrophile bond distances have been held frozen, but all other geometric parameters have been allowed to relax. Bigeleisen–Mayer KIEs were then calculated for each grid point using the harmonic frequencies of the isolated substrate and the grid point geometry at 298 K. The maximum KIE was then taken as the maximum predicted KIE over the entire grid. The trajectory overlaid on Figure 3f was calculated with an explicit water molecule at B3LYP-D3BJ/6–31+G*. All calculations in Figure 3 used default PCM settings.
120 SNAr reactions spanning a range of ring types (benzene, (NO2)n=1,2,3-benzene, H3C-C=O-benzene, pyridine, pyrazine, pyrimidine), nucleophiles (F−, MeO−, N3−, Me2N−, formate−), and leaving groups (F−, Cl−, Br−) were thoroughly examined at B3LYP-D3(BJ)/6–31+G*/PCM(DMSO). Consistent with previous studies,39–50 in all but 21 cases, it was not possible to locate a Meisenheimer intermediate by beginning optimizations in the expected neighborhood of the intermediate. Intrinsic reaction coordinate (IRC) searches were attempted for each reaction. In no case did any converged IRC calculation show the unexpected existence of an intermediate. For the significant number of remaining unconverged cases, growing string81 calculations were performed. Likewise, no string calculations revealed the unexpected existence of an intermediate. Transition states and intermediates were located for nearly all of the reactions and each transition state was confirmed to connect to the expected starting materials and products by the means above. In a small number of cases involving highly exothermic reactions it was not possible to locate saddle points on the potential energy surface (details are given in Supplementary Information Section 4g).
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM-43214). We thank Professors William F. Reynolds and Daniel A. Singleton for helpful discussions. We thank Dr. Shaw G. Huang and Mr. Bill Collins for assistance with NMR spectroscopy.
Footnotes
Supplementary Information
Supplementary Information is linked to the online version of the paper at www.nature.com/nchem.
Author Contributions
E.E.K., Y.Z., and H.A.B. developed the isotope effect methodology; Y.Z. synthesized the materials; E.E.K. and H.A.B. carried out the calculations; E.E.K and E.N.J. wrote the manuscript; E.N.J. guided the research.
Competing Interests
The authors declare no competing financial and non-financial interest.
References
- 1.Williams A. Concerted Organic and Bio-Organic Mechanisms. CRC; Boca Raton: 1999. [Google Scholar]
- 2.Terrier F. Modern Nucleophilic Aromatic Substitution. Wiley-VCH; Weinheim: 2013. [Google Scholar]
- 3.Terrier F. Rate and equilibrium studies in Jackson–Meisenheimer complexes. Chem Rev. 1982;82:77–152. [Google Scholar]
- 4.Brown DG, Boström J. Analysis of past and present synthetic methodologies on medicinal chemistry: where have all the new reactions gone? J Med Chem. 2016;59:4443–4458. doi: 10.1021/acs.jmedchem.5b01409. [DOI] [PubMed] [Google Scholar]
- 5.Neumann CN, Hooker JM, Ritter T. Concerted nucleophilic aromatic substitution with 19F− and 18F−. Nature. 2016;534:369–373. doi: 10.1038/nature19311. [DOI] [PubMed] [Google Scholar]
- 6.Neumann CN, Ritter T. Facile C–F bond formation through a concerted nucleophilic aromatic substitution mediated by the PhenoFluor reagent. Acc Chem Res. 2017;50:2822–2833. doi: 10.1021/acs.accounts.7b00413. [DOI] [PubMed] [Google Scholar]
- 7.Lucchini V, Modena G, Pasquato L. An authentic case of in-plane nucleophilic vinylic substitution: the anionotropic rearrangement of di-tert-butyl thiirenium ions into thietium ions. J Am Chem Soc. 1993;115:4527–4531. [Google Scholar]
- 8.Glukhovtsev MN, Pross A, Radom L. Is SN2 substitution with inversion of configuration at vinylic carbon feasible? J Am Chem Soc. 1994;116:5961–5962. [Google Scholar]
- 9.Lucchini V, Modena G, Pasquato L. SN2 and AdN-E mechanisms in bimolecular nucleophilic substitutions at vinyl carbon: the relevance of the LUMO Symmetry of the electrophile. J Am Chem Soc. 1995;117:2297–2300. [Google Scholar]
- 10.Okayama T, Takino T, Sato K, Ochiai M. In-plane vinylic SN2 substitution and intramolecular β-elimination of β-alkylvinyl(chloro)-λ3-iodanes. J Am Chem Soc. 1998;120:2275–2282. [Google Scholar]
- 11.Bach RD, Baboul AG, Schlegel HB. Inversion vs. retention of configuration for nucleophilic substitution at vinylic carbon. J Am Chem Soc. 2001;123:5787–5793. doi: 10.1021/ja010234y. [DOI] [PubMed] [Google Scholar]
- 12.Williams A. Concerted mechanisms of acyl group transfer reactions in solution. Acc Chem Res. 1989;22:387–392. [Google Scholar]
- 13.Curran TP, Farrar CR, Niazy O, Williams A. Structure activity studies on the equilibrium reaction between phenolate ions and 2-aryoxazolin-5-one – data consistent with a concerted acyl group transfer mechanism. J Am Chem Soc. 1980;102:6828–6837. [Google Scholar]
- 14.Chrystiuk E, Williams A. A single transition state in the transfer of methoxycarbonyl group between isoquinoline and substituted pyridines. J Am Chem Soc. 1987;109:3040–3046. [Google Scholar]
- 15.Ba-Saif SA, Luthra AK, Williams A. Concertedness in acyl group transfer: a single transition state in acetyl transfer between phenolate ion nucleophiles. J Am Chem Soc. 1987;109:6362–6368. [Google Scholar]
- 16.Ba-Saif SA, Luthra AK, Williams A. Concerted acetyl group transfer between substituted phenolate ion nucleophiles: variation of transition state structure as a function of substituent. J Am Chem Soc. 1989;111:2647–2652. [Google Scholar]
- 17.Hunter A, Renfrew M, Taylor JA, Whitmore JMJ, Williams A. A single transition state in nucleophilic aromatic substitution - reaction of phenolate ions with 2-(4-nitrophenoxy)-4,6-dimethoxy-1,3,5-triazine in aqueous solution. J Chem Soc Perkin Trans 2. 1993;10:1703–1704. [Google Scholar]
- 18.Han C, Braumann JI. Gas phase nucleophilic displacement reactions of negative ions with carbonyl compounds. J Am Chem Soc. 1979;101:3715–3724. [Google Scholar]
- 19.Kim JK, Caserio MC. Acyl-transfer reactions in the gas phase: the question of tetrahedral intermediates. J Am Chem Soc. 1981;103:2124–2127. [Google Scholar]
- 20.Guthrie JP. Concerted mechanism for alcoholysis of esters: an examination of the requirements. J Am Chem Soc. 1991;113:3941–3949. [Google Scholar]
- 21.Guthrie JP, Pike DC. Hydration of acylimidazoles: tetrahedral intermediates in acylimidazole hydrolysis and nucleophilic attack by imidazoles on esters: the question of concerted mechanisms for acyl transfer. Can J Chem. 1987;65:1951–1969. [Google Scholar]
- 22.Hengge AC, Hess R. A concerted or stepwise mechanisms for acyl transfer reactions of p-nitrophenyl acetate? transition state structures from isotope effects. J Am Chem Soc. 1994;116:11256–11263. [Google Scholar]
- 23.Blake JF, Jorgensen WL. Ab initio Study of the displacement reactions of chloride ion with formyl and acetyl chloride. J Am Chem Soc. 1987;109:3856–3861. [Google Scholar]
- 24.Fox JM, Dmitrenko O, Liao L, Bach RD. Computational studies of nucleophilic substitution at carbonyl carbon: the Sn2 mechanism versus the tetrahedral intermediate in organic synthesis. J Org Chem. 2004;69:7317–7328. doi: 10.1021/jo049494z. [DOI] [PubMed] [Google Scholar]
- 25.Xu S, Held I, Kempf B, Mayr H, Steglich W, Zipse H. The DMAP-catalyzed acetylation of alcohols–a mechanistic study. Chem Eur J. 2005;11:4751–4757. doi: 10.1002/chem.200500398. [DOI] [PubMed] [Google Scholar]
- 26.Skoog MT, Jencks WP. Reactions of pyridines and primary amines with N-phosphorylated pyridines. J Am Chem Soc. 1984;106:7597–7606. [Google Scholar]
- 27.Bourne N, Chrystiuk E, Davis AM, Williams A. A single transition state in the reaction of aryl diphenylphosphinate esters with phenolate ions in aqueous solution. J Am Chem Soc. 1988;110:1890–1895. [Google Scholar]
- 28.Bourne N, Williams A. Evidence for a single transition state in the transfer of the phosphoryl group to nitrogen nucleophiles from pyridino-N-phosphonates. J Am Chem Soc. 1984;106:7591–7596. [Google Scholar]
- 29.Buchwald SL, Friedman JM, Knowles JR. Stereochemistry of nucleophilic displacement on two phosphoric monoesters and a phosphoguanidine: the role of metaphosphate. J Am Chem Soc. 1984;106:4911–4916. [Google Scholar]
- 30.Andersen KK, Caret RI, Karup–Nielsen I. Nucleophilic substitution at tricoordinate sulfur(IV): stereochemistry of dialkylarylsulfonium salt formation from alkyl aryl sulfoxides. J Am Chem Soc. 1974;96:8026–8032. [Google Scholar]
- 31.Bourne N, Hopkins A, Williams A. Single transition state for sulfuryl group transfer between pyridine nucleophiles. J Am Chem Soc. 1985;107:4327–4331. [Google Scholar]
- 32.D’Rozario P, Smyth RL, Williams A. Evidence for a single transition state in the intramolecular transfer of a sulfonyl group between oxyanion donor and acceptors. J Am Chem Soc. 1984;106:5027–5028. [Google Scholar]
- 33.Deacon T, Farrar CR, Sikkel BJ, Williams A. Reactions of nucleophiles with strained cyclic sulfonate esters: Bronsted relationships for rate and equilibrium constants for variation of phenolate anion nucleophile and leaving group. J Am Chem Soc. 1978;100:2525–2534. [Google Scholar]
- 34.Koh HJ, Um IH. Kinetic study on quinuclidinolysis of o-phenyl o-y-substituted-phenyl thionocarbonates: effects of changing nonleaving group from thionobenzoyl to phenyloxythionocarbonyl on reactivity and transition-state structure. Bull Korean Chem Soc. 2017;38:1091–1096. [Google Scholar]
- 35.Renfrew AHM, Rettura D, Taylor JA, Whitmore JMJ, Williams A. Stepwise versus concerted mechanisms at trigonal carbon: transfer of the 1,3,5-triazinyl group between aryl oxide ions in aqueous solution. J Am Chem Soc. 1995;117:5484–5491. [Google Scholar]
- 36.Renfrew AHM, Taylor JA, Whitmore JMJ, Williams A. A single transition state in nucleophilic aromatic substitution: reaction of phenolate ions with 2-(4-nitrophenoxy)-4,6-dimethoxy-1,3,5-triazine in aqueous solution. J Chem Soc Perkin Trans 2. 1993:1703–1704. [Google Scholar]
- 37.Cullum NR, Renfrew AHM, Rettura D, Taylor JA, Whitemore JMJ, Williams A. Effective charge on the nucleophile and leaving group during the stepwise transfer of the triazinyl group between pyridines in aqueous solution. J Am Chem Soc. 1995;117:9200–9205. [Google Scholar]
- 38.Renfrew AHM, Taylor JA, Whitmore JMJ, Williams A. Timing of bonding changes in fundamental reactions in solution: pyridinolysis of a triazinylpyridinium salt. J Chem Soc Perkin Trans 2. 1994:2383–2384. [Google Scholar]
- 39.Kikushima K, Grellier M, Ohashi M, Ogoshi S. Transition-metal-free hydrodefluorination of polyfluoroarenes by a concerted nucleophilic aromatic substitution with a hydrosilicate. Angew Chem Int Ed. 2017;56:16191–16196. doi: 10.1002/anie.201708003. [DOI] [PubMed] [Google Scholar]
- 40.Ong DY, Tejo C, Xu K, Hirao H, Chiba S. Hydrodehalogenation of haloarenes by a sodium hydride–iodide composite. Angew Chem Int Ed. 2017;56:1840–1844. doi: 10.1002/anie.201611495. [DOI] [PubMed] [Google Scholar]
- 41.Sun H, DiMagno S. Room-temperature nucleophilic aromatic fluorination: experimental and theoretical studies. Angew Chem Int Ed. 2006;45:2720–2725. doi: 10.1002/anie.200504555. [DOI] [PubMed] [Google Scholar]
- 42.Zheng YJ, Bruice TC. On the dehalogenation mechanism of 4-chlorobenzoyl CoA by4-chlorobenzoyl CoA dehalogenase: insights from study on the nonenzymatic reaction. J Am Chem Soc. 1997;119:3868–3877. [Google Scholar]
- 43.Baker J, Muir M. The Meisenheimer model for predicting the principal site of for nucleophilic substitution in aromatic perfluorocarbons – Generalization to include ring-nitrogen atoms and non-fluorine ring substituents. Can J Chem. 2010;88:588–597. [Google Scholar]
- 44.Goryunov LI, Grobe J, Le Van D, Shteingarts VD, Mews R, Lork E, Würthwein E-U. Eur J Org Chem. 2010:1111–1123. [Google Scholar]
- 45.Cairns AG, Senn HM, Murphy MP, Hartley RC. Expanding the palette of phenanthridinium cations. Chem – Eur J. 2014;20:3742–3751. doi: 10.1002/chem.201304241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glukhovtsev MN, Bach RD, Laiter S. Single-step and multistep mechanisms of aromatic nucleophilic substitution of halobenzenes and halonitrobenzenes with halide anions: ab initio computational study. J Org Chem. 1997;62:4036–4046. [Google Scholar]
- 47.Giroldo T, Xavier LA, Riveros JM. An unusually fast nucleophilic aromatic displacement reaction: the gas-phase reaction of fluoride ions with nitrobenzene. Angew Chem Int Ed. 2004;43:3588–3590. doi: 10.1002/anie.200454230. [DOI] [PubMed] [Google Scholar]
- 48.Fernández I, Frenking G, Uggerud E. Rate-determining factors in nucleophilic aromatic substitution reactions. J Org Chem. 2010;75:2971–2980. doi: 10.1021/jo100195w. [DOI] [PubMed] [Google Scholar]
- 49.Liljenberg M, Brinck T, Rein T, Svensson M. Predicting regioselectivity in nucleophilic aromatic substitution. J Org Chem. 2012;77:3262–3269. doi: 10.1021/jo202569n. [DOI] [PubMed] [Google Scholar]
- 50.Liljenberg M, Brinck T, Rein T, Svensson M. Utilizing the σ-complex stability for quantifying reactivity in nucleophilic substitutions of aromatic fluorides. Beil J Org Chem. 2013;9:791–799. doi: 10.3762/bjoc.9.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clayden J, Greeves N, Warren S. Organic Chemistry. 2. Oxford University Press; 2012. p. 518. [Google Scholar]
- 52.Persson J, Axelsson S, Matsson O. Solvent dependent leaving group fluorine kinetic isotope effect in a nucleophilic aromatic substitution reaction. J Am Chem Soc. 1996;118:20–23. [Google Scholar]
- 53.Singleton DA, Thomas AA. High-precision simultaneous determination of multiple small kinetic isotope effects at natural abundance. J Am Chem Soc. 1995;117:9357–9358. [Google Scholar]
- 54.Claridge TD. High-Resolution NMR Techniques in Organic Chemistry. Elsevier; 2016. [Google Scholar]
- 55.Chan J, Tang A, Bennett AJ. A stepwise solvent-promoted SNi reaction of α-D-glucopyranosyl fluoride: mechanistic implications for retaining glycosyltransferases. J Am Chem Soc. 2012;134:1212–1220. doi: 10.1021/ja209339j. [DOI] [PubMed] [Google Scholar]
- 56.Westaway KC. Determining transition state structure using kinetic isotope effects. J Label Compd Radiopharm. 2007;50:989–1005. [Google Scholar]
- 57.Matsson O, Dybala-Defratyka A, Rostkowski M, Paneth P, Westaway KC. A theoretical investigation of α-carbon kinetic isotope effects and their relationship to the transition-state structure of SN2 reactions. J Org Chem. 2005;10:4022–4027. doi: 10.1021/jo047987y. [DOI] [PubMed] [Google Scholar]
- 58.Kwan EE, Park Y, Besser HA, Anderson TL, Jacobsen EN. Sensitive and accurate 13C kinetic isotope effect measurements enabled by polarization transfer. J Am Chem Soc. 2017;139:43–46. doi: 10.1021/jacs.6b10621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Papajak E, Zheng J, Xu X, Leverentz HR, Truhlar DG. Perspectives on basis sets beautiful: seasonal plantings of diffuse basis functions. J Chem Theory Comput. 2011;7:3027–3034. doi: 10.1021/ct200106a. [DOI] [PubMed] [Google Scholar]
- 60.Riplinger C, Neese F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J Chem Phys. 2013;138:034106. doi: 10.1063/1.4773581. [DOI] [PubMed] [Google Scholar]
- 61.Karplus M, Porter R, Sharma R. Exchange reactions with activation energy. I Simple barrier potential for (H, H2) J Chem Phys. 1965;43:3259–3287. [Google Scholar]
- 62.Melander LC, Saunders WH. Reaction Rates of Isotopic Molecules. Wiley; New York: 1980. [Google Scholar]
- 63.Marcus RA, Sutin N. Electron transfers in chemistry and biology. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 64.Shaik SS, Hiberty PC. A Chemist’s Guide to Valence Bond Theory. Wiley; 2007. [Google Scholar]
- 65.Silverstein TP. Marcus theory: thermodynamics can control the kinetics of electron transfer reactions. J Chem Educ. 2012;89:1159–1167. [Google Scholar]
- 66.Bunnett JF, Zahler RE. Aromatic nucleophilic substitution reactions. Chem Rev. 1951;49:273–412. [Google Scholar]
- 67.Marenich AV, Jerome SV, Cramer CJ, Truhlar DG. Charge model 5: an extension of Hirshfeld population analysis for the accurate description of molecular interactions in gaseous and condensed phases. J Chem Theory Comput. 2012;8:527–541. doi: 10.1021/ct200866d. [DOI] [PubMed] [Google Scholar]
- 68.Chen Z, Wannere CS, Corminboeuf C, Puchta R, Schleyer PvR. Nucleus-independent chemical shifts (NICS) as an aromaticity criterion. Chem Rev. 2005;105:3842–3888. doi: 10.1021/cr030088+. [DOI] [PubMed] [Google Scholar]
- 69.Glendening ED, et al. NBO 6.0. Theoretical Chemistry Institute; 2013. [Google Scholar]
- 70.Jackson CJ, Gazzolo FH. Am Chem J. 1900;23:376. [Google Scholar]
- 71.Meisenheimer J. Ueber reactionen aromatischer nitrokörper. Justus Liebigs Ann Chem. 1902;323:205–246. [Google Scholar]
- 72.Helmus JJ. nmrglue. ( www.nmrglue.com)
- 73.Frisch MJ, et al. Gaussian 09 and 16. Gaussian Inc; [Google Scholar]
- 74.Kwan EE, Anderson TL. PyQuiver. ( www.github.com/ekwan/pyquiver)
- 75.Zheng J, et al. GAUSSRATE 2016. University of Minnesota; [Google Scholar]
- 76.Zheng J, et al. POLYRATE 2016. University of Minnesota; [Google Scholar]
- 77.Neese F. The ORCA program system. Wiley Interdis Rev Comp Mol Sci. 2012;2:73–78. [Google Scholar]
- 78.Takano Y, Houk KN. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J Chem Theory Comput. 2005;1:70–77. doi: 10.1021/ct049977a. [DOI] [PubMed] [Google Scholar]
- 79.Kongsted J, Mennucci B. How to model solvent effects on molecular properties using quantum chemistry? Insights from polarizable discrete or continuum solvation models. J Phys Chem A. 2007;111:9890–9900. doi: 10.1021/jp074343w. [DOI] [PubMed] [Google Scholar]
- 80.Cappelli C, Monti S, Scalmani G, Barone V. On the calculation of vibrational frequencies for molecules in solution beyond the harmonic approximation. J Chem Theory Comput. 2010;6:1660–1669. doi: 10.1021/ct100048g. [DOI] [PubMed] [Google Scholar]
- 81.Zimmerman P. Reliable transition state searches integrated with the growing string method. J Chem Theory Comput. 2013;9:3043–3050. doi: 10.1021/ct400319w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
User-friendly software pipelines that can be used to measure and predict KIEs are freely available at www.github.com/ekwan/PyKIE and www.github.com/ekwan/PyQuiver. Raw NMR spectra and computed quasiclassical trajectories are available from the corresponding author upon reasonable request. All other data supporting the findings of this study are available within the paper and its supplementary information files.