

Abstract
Purpose
To characterize the molecular genetics of autosomal recessive Noonan syndrome.
Methods
Families underwent phenotyping for features of Noonan syndrome in children and their parents. Two multiplex families underwent linkage analysis. Exome, genome, or multigene panel sequencing was used to identify mutations. The molecular consequences of observed splice variants were evaluated by reverse-transcription PCR.
Results
Twelve families with a total of 23 affected children with features of Noonan syndrome were evaluated. The phenotypic range included mildly affected patients, but it was lethal in some, with cardiac disease and leukemia. All of the parents were unaffected. Linkage analysis using a recessive model supported a candidate region in chromosome 22q11, which includes LZTR1, previously shown to harbor mutations in patients with Noonan syndrome inherited in a dominant pattern. Sequencing analyses of 21 liveborn patients and a stillbirth identified biallelic pathogenic variants in LZTR1, including putative loss of function, missense, and canonical and non-canonical splicing variants in the affected children, with heterozygous, clinically unaffected parents and heterozygous or normal genotypes in unaffected siblings.
Conclusion
These clinical and genetic data confirm the existence of a form of Noonan syndrome that is inherited in an autosomal recessive pattern and identify biallelic mutations in LZTR1.
Keywords: Noonan syndrome, Autosomal recessive inheritance, Multiple congenital anomalies, Cardiomyopathy, Leukemia
Introduction
Noonan syndrome is part of a spectrum of disorders with overlapping phenotypes that include craniofacial features1 and cardiovascular abnormalities2,3 and overlaps with cardiofaciocutaneous and Costello syndromes4. Most of the genes mutated in patients with Noonan syndrome cause dysregulated RAS-MaPK signaling5-7. Genes shown to be mutated in these disorders include PTPN11, SOS1, SOS2, RAF1, KRAS, NRAS, BRAF, SHOC2, CBL, RIT1, and LZTR1, among others. For LZTR1, no role in RAS-MaPK signaling was known prior to its association with autosomal dominant Noonan syndrome.8 Germline and somatic biallelic loss of function in LZTR1 is involved in schwannomatosis9. Previously, all molecularly confirmed forms of Noonan syndrome were inherited in an autosomal dominant pattern. Here we describe the results of clinical and molecular evaluations of 12 families with Noonan syndrome inherited in an autosomal recessive pattern.
Materials and Methods
The studies in Bethesda, Houston, Seattle, and Oxford were approved by their respective research ethics bodies. The work done at Utrecht and Magdeburg was not considered research. All DNA analyses were performed using standard techniques. Linkage analyses of families 1 and 12 was performed by genotyping nuclear family members on an Illumina Human Omni Express Bead Chip or cytoSNP12v2 Bead Chip. Next generation sequencing analyses were performed at several centers. Families 1 and 3 were exome sequenced at the NIH Intramural Sequencing Center (NISC), as described.10 Family 2, who was in the Undiagnosed Diseases Network (UDN) program, and family 6 had exome sequencing at Baylor Genetics Laboratories, as described elsewhere.11 Families 4 and 5 were sequenced in the Utrecht clinical exome laboratory following standard procedures (see supplemental methods for details). Individuals in families 7, 8, 10, and 11 had targeted next-generation sequencing of the coding sequence of 19 candidate genes (Illumina Nextera Rapid Capture): PTN11, SOS1, SOS2, RAF1, RIT1, KRAS, NRAS, RRAS, HRAS, SHOC2, LZTR1, A2ML1, BRAF, MAP2K2, MAP2K2, NF1, SPRED1, RASA1. As described in the supplement, some probands were tested by Sanger sequencing for subsets of these 19 genes and in some cases for PPP1CB. Genome sequencing was performed on family 9 by the Mary-Claire King laboratory at the University of Washington. Family 12 was exome sequenced at the Oxford Genomics Centre (www.well.ox.ac.uk/ogc/) as described.12
RNA Analyses
The consequences of the splice variants in families 1 and 3 were analyzed using total lymphoblasts RNA (RNeasy, Qiagen, Hilden, Germany). One microliter of RNA (300 – 500 ng) was amplified using the OneStep RT-PCR kit (Qiagen). Products were separated on a 1.5% agarose gel and visualized using ethidium bromide, gel-isolated (QIAquick Gel Extraction Kit, Qiagen), and sequenced (BigDye Terminator v3.1 Cycle Sequencing Kit, Thermo Fisher Scientific, Waltham, MA).
Mutations were specified in HGVS nomenclature re NM_006767.3, (ENST00000215739.12) and GRCh37/hg19.
Results
Clinical Evaluations
Brief summaries are provided here. Table 1 lists the individual features and the online supplement includes full clinical descriptions.
Table 1. Clinical Features of Patients with Autosomal Recessive LZTR1-Associated Noonan syndrome.
| Family 1 II-1 |
Family 1 II-2 |
Family 1 II-3 |
Family 1 II-4 |
Family 2 II-4 |
Family 2 II-5 |
Family 3 II-1 |
Family 3 II-4 |
Family 4 II-1 |
Family 4 II-2 |
Family 5 II-4 |
Family 5 II-1/2 |
Family 6 II-6 |
Family 7 II-1 |
Family 8 II-11 |
Family 9 II-2 |
Family 10 II-3 |
Family 11 II-6 |
Family 12 V-1 |
Family 12 V-4 |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | F | F | F | M | M | M | F | M | M | M | F | F | F | M | M | F | F | M | F | F |
| Prenatal hydrops, nuchal transl, or cardiac findings | - | + | - | + | + | + | + | + | + | + | + | - | + | + | + | - | - | |||
| BW (kg) | 4.1 (90-95th) | 3.9 (75-90th) | 4.5 (>97th) | 4.2 (90-97th) | 3.3 (>97th) | 4.5 (>97th) | 3.32 (50-75th) | 2.3 (AGA) | 4.1 | 3.9 (90th) | 3.46 (50th) | 0.4/0.75 | 2.0 (90th) | 3.5 (AGA) | 3.4 (AGA) | 4 (AGA) | 3.74 (75th-90th) | 2.6 (60th) | 2.55 (AGA) | 3.78 |
| BL (cm) | 55.9 (>97th) | 50.8 (50th) | 52.7 (75-90th) | 52 (50-75th) | 43 (10-25th) | 52 (95th) | 50 (50-75th) | 52 (70th) | 48 (2-16th) | 41 (50th) | 50 (AGA) | 51 (AGA) | ||||||||
| BOFC (cm) | U | 35.6 (75-90th) | U | 36.5 (90th) | 34.5 (97th) | 37 (98th) | 36 (>90th) | 36 | 34.5 (50th) | 36.2 (50-98th) | 31.5 (90th) | 34 (AGA) | 34 (25th-50th) | 31 (90th) | ||||||
| Age at evaluation | 7y6m | 5y6m | 3y8m | 10m | 8y | 3y | 1y | 3y2m | 3y | 3y10m | 2y1m | 7y | 3y4m | 2y10m | 4y9m | 6m | 3y | 3y9m | ||
| W (kg) | 22 (25th) | 17.3 (10-25th) | 16.1 (75-90th) | 7.6 (<3rd) | 23.5 (30th) | 12.5 (11th) | 6.0 (<3rd) | 12 (<3rd) | 12 (10th) | 13.5 (25th) | 10.4 (<5th) | 21.4 (10-25th) | 13.8 (<10-25th) | 13 kg (50th) | 14 (3rd) | 3.1 (25th) | (25th-50th) | 12.7 (0.4th-2nd) | ||
| L/Ht (cm) | 121 (25th) | 106 (10-25th) | 98 (75-90th) | 67 (<3rd) | 113 (<3rd) | 89 (3rd) | 64 (<3rd) | 85 (5-10th) | 93 (25th) | 89 (3rd) | 78.2 (<5th) | 113 (<3rd) | 88 (<3rd) | 86 (3rd) | 96.5 (<3rd) | (9th-25th) | 93.7 (<3rd) | |||
| OFC (cm) | 52 (50-75th) | 52 (50-75th) | 49 (75-90th) | 45.5 (50th) | 53 (57th) | 50 (42nd) | 44 (3rd) | 49.5 (53rd) | 51 (50-75th) | 48.5 (25-50th) | 52 (25-50th) | 51 (50-75th) | 51 (97th) | 48 (3rd - 10th) | (50th) | 47 (<3rd) | ||||
| Ptosis | + | - | - | - | + | - | + | + | + | - | + | - | + | +/- | ||||||
| Short nose or anteverted nares | - | + | - | - | - | - | - | - | - | + | - | + | + | - | ||||||
| Depressed or wide bridge | - | - | - | + | - | + | + | + | + | + | - | - | + | + | + | + | - | |||
| Widely spaced eyes | - | - | - | - | + | - | + | - | + | + | + | + | - | + | - | |||||
| Downslanted palpebral fissures | - | - | - | - | + | - | + | + | + | + | + | + | + | - | + | + | - | |||
| Low-set ears | - | + | - | + | + | - | + | + | + | + | + | + | + | + | + | + | ||||
| Post angulated ears | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||||
| Malformed ears | - | - | + | + | + | + | + | + | - | - | - | - | - | |||||||
| Midface retrusion | + | + | - | - | + | + | - | +/- | + | +/- | - | - | + | + | ||||||
| Micrognathia | +/- | +/- | +/- | +/- | + | + | + | + | - | - | +/- | - | - | |||||||
| Broad/short neck | + | + | + | + | + | + | + | + | + | - | + | + | + | + | + | + | + | - | - | |
| Low posterior hairline | + | + | + | + | + | - | + | + | - | - | + | - | + | + | ||||||
| Wide-spaced nipples/Broad chest | - | - | + | - | + | - | + | + | + | + | + | + | + | + | + | |||||
| Pectus carinatum or excavatum | + | + | + | - | + | - | + | - | + | - | - | + | + | + | ||||||
| Curly hair | - | + | + | - | - | - | - | + | + | + | - | - | + | - | ||||||
| Cardio-myopathy | - | - | - | - | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||
| CHD or Valvular disease | + | + | - | + | + | + | - | + | + | + | + | + | + | + | + | - | + | + | + | |
| Cryptorchid-ism | NA | - | + | - | - | + | - | + | ||||||||||||
| Dev delay/ID | - | +/- | - | - | + | + | + | - | + | + | + | + | + | + | ||||||
| Paternal variant | c.628C>T p.R210* | c.2178C>A p.Y726* | c.1943-256C>T | c.1687G>C p.E563Q | c.2407-2A>G | c.27delG p.Q10fs | c.361C>G p.H121D | c.508C>T; c.614T>C p.R170W; I205T | c.650A>C, p.E217A | c.2062C>G, p.R688G | c.2325+1G>A | c.2462T>C p.I821T | c.2462T>C p.I821T | |||||||
| Maternal variant | c.2220-17C>A | c.1943-256C>T | c.1943-256C>T | c.1687G>C p.E563Q | c.2090G>A p.R697Q | c.1149+1G>A | c.2264G>A p.R755Q | c.508C>T; c.614T>C p.R170W; I205T | c.650A>C, p.E217A | c.1943-256C>T | c.1943-256C>T | c.2462T>C p.I821T | c.2462T>C p.I821T | |||||||
| Pat Var frequency in ExAC | 12/120,308 | NF | See prior | NF | NF | 1/15,734 | NF | 2/120,622; NF | NF | NF | NF | NF | See prior | |||||||
| Mat Var frequency in ExAC | NF | 1/13,270 | See prior | NF | 4/120,894 | NF | 1:120,614 | See above | See prior | See prior | See prior | See prior | See prior | |||||||
| Pat Var frequency in gnomAD | 13/220,926 | NF | See prior | 1/245,294 | NF | NF | NF | 3/245,898; 2/245,670 | NF | NF | NF | 1/30,936 | See prior | |||||||
| Mat Var frequency in gnomAD | NF | NF | See prior | See prior | 7/246,118 | 1/241,846 | 1/246,052 | See prior | See prior | See prior | See prior | See prior | See prior | |||||||
| Pat Var CADD | 40 | 40 | Spl | 24 | 24.6 | 29.8 | 29.8 | 34;23.6 | 23.2 | 34 | Spl | 27.3 | 27.3 | |||||||
| Mat Var CADD | Spl | Spl | Spl | 24 | 35 | 23.7 | 35 | 34;23.6 | 23.2 | Spl | Spl | 27.3 | 27.3 |
Note that the twins in Family 5 are here presented as a single occurrence. Insufficient clinical data for individual II-2 in Family 7 was available to populate this table. Nuchal transl indicates nuchal translucency. V1 and V2 are the two genetic variants in the affected patients. BW, BL, and BOFC are birth weight, birth length, and birth occipitofrontal circumference. In the age entries, y and m are years and months, respectively, W, L/Ht, and OFC are weight, length/height, and occipitofrontal circumference. Post angulated ears is posterior angulation of the ears. CHD is congenital heart defect. ID is intellectual disability. + indicates feature present, - indicates feature absent, +/- indicates feature ambiguous or mild, NA is not applicable. NF indicates that the variant was not present in the database. Pat Var is the variant in that sibship that was inherited from the father. Mat Var is the variant in that sibship that was inherited from the mother. CADD is the combined annotation dependent deletion Phred-like score. CADD scores are not relevant to frameshift or non-canonical splice variants, and therefore “Fs” or “Spl” are specified there instead of numbers, when applicable.
Family 1
This family comprised four siblings and parents (Figure 1A). The parents had no facial features of Noonan syndrome (Figure 2 A,B) and normal echocardiograms. All four siblings had variable features of Noonan syndrome including curly hair, depressed and wide nasal bridges, low-set ears, increased posterior angulation of the ears, broad neck with low posterior hairline (Figure 2C-F) and cardiac anomalies. Individual (II-3) developed acute lymphoblastic leukemia at 5 years old (y.o.), which progressed to acute myeloblastic leukemia at 7 y.o. and she died two years later. The maternal grandmother had onset of unilateral hearing loss in her 60's, which was associated with a tumor, assessed as a meningioma on imaging. Several individuals in this family had subtle imaging findings compatible with schwannomas.
Figure 1.

Pedigrees of the 12 affected families showing 23 affected liveborn offspring, 21 of whom underwent molecular analysis with mutational data on the affected children and carrier parents. Clinically unaffected children shown not to have two mutant alleles are indicated with an asterisk (carrier status of minors thereby not disclosed).
Figure 2.
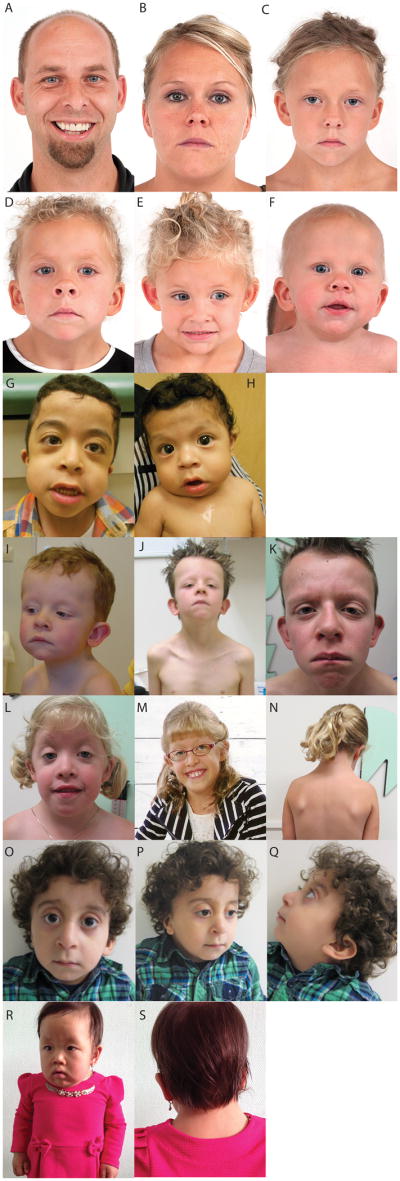
Facial features of Family 1 including unaffected parents (A, B) and four affected children (C-F). In the children note variable features of short or upturned nose, depressed bridge, low-set, posteriorly angulated, or malformed ears, midface retrusion, broad/short neck, low posterior hairline, and curly hair. Facial features of the affected brothers from Family 2 (G, H) demonstrating widely-spaced eyes, downslanted palpebral fissures, midface retrusion, full cheeks, a long philtrum, and overfolded, posteriorly angulated ears. Features of individual II-1 from Family 4 at three ages. At three years of age (I), at 7 years of age (J), and 14 years of age (K). Notice the short, broad neck, widely-spaced eyes with downslanted palpebral fissures and bilateral ptosis, low set and posteriorly angulated ears, and pectus excavatum. Affected individual from Family 5 at 4 years of age (L), 8 years of age (M) and 6 years of age (N). Her features included downslanted palpebral fissures, bilateral epicanthus and ptosis, broad neck, low-set, small, cupped ears, and a wide thorax. Affected individual from Family 8 showing widely-spaced eyes, downslanted palpebral fissures, mild ptosis, low-set and posteriorly angulated ears, a broad, webbed neck, and curly hair (O-Q). Features of individual II-2 from Family 9. Note her widely set eyes, strabismus, bilateral epicanthus, downslanted palpebral fissures, depressed nasal bridge, short and upturned nose, short and broad neck, thickened vermilion of the lips (R,S).
Family 2
This family included six boys, four full siblings and two half siblings. The affected boy (Figure 1B, individual II-4) had a prenatal course complicated by arthrogryposis and polyhydramnios and his postnatal features included widely-spaced eyes, downslanted palpebral fissures, midface retrusion, full cheeks, a long philtrum, overfolded ears, and increased posterior angulation of the ears (Figure 2G). He had an atrial septal defect (ASD), pulmonary artery stenosis, a dysplastic pulmonary valve, and mild left ventricular and moderate right ventricular hypertrophy. His younger brother (Figure 1A, individual II-5) had similar features (Figure 2H) but did not have arthrogryposis. Both boys had hypotonia, developmental delay, recurrent metabolic acidosis episodes with cyclic vomiting, and had clinical laboratory findings of mitochondrial complex I+III deficiency. The parents were non-dysmorphic.
Family 3
The proband (Figure 1C, individual II-4) had prenatal hypertrophic cardiomyopathy (HCM) and postnatally had coarse facial features, low set ears, depressed nasal bridge, gingival overgrowth and thickened vermilion of upper and lower lips, short neck with low posterior hairline, small nails, and widely-spaced nipples. He was confirmed to have HCM and a small ASD. His older sister (Figure 1C, individual II-1) had a cystic hygroma and polyhydramnios at 6 months gestation. Postnatally she had proptosis, ptosis, a wide mouth, low-set ears, a bulbous nose, and relative macrocephaly. She was found to have septal HCM with a small ASD, short stature, and growth hormone deficiency. The father had mild short stature and ptosis. Parents were consanguineous, but non-dysmorphic.
Family 4
The proband (Figure 1D, individual II-1) was found to have biventricular HCM neonatally. He had a short, broad neck, widely-spaced eyes with downslanted palpebral fissures and bilateral ptosis, low set ears, increased posterior angulation of the ears, and pectus excavatum (Figure 2I-K). He had mild short stature. At 3 y.o., he developed acute lymphoblastic leukemia and is in remission (in his teens). His younger brother (II-2) had atrioventricular septal defect (AVSD), severe biventricular HCM, and a large sacral meningomyelocele. He died at day 4 of an inoperable cardiac defect. At postmortem, several organs were enlarged, the heart showed severe hypertrophy with muscular disarray. The parents were non-consanguineous, nondysmorphic, and had no cardiac abnormalities.
Family 5
The proband (Figure 1E, individual II-4) had prenatal findings of cystic hygroma and a small VSD. Postnatally, she had muscular VSDs, valvular pulmonary stenosis, a type II ASD, and biventricular HCM. She had short stature, downslanted palpebral fissures, bilateral epicanthus and ptosis, broad neck, wide thorax, low-set, small, cupped ears, a narrow palate, and pes planus (Figure 2L-N). The twins (II-1 and II-2) had prenatal cystic hygroma; they died with hydrops at midgestation. Postmortem showed a large perimembranous VSD in one twin, and the other had a VSD and a dysplastic, not completely patent tricuspid valve, and a hypoplastic right ventricle. Dysmorphic features were not assessed. The parents were non-dysmorphic and had no cardiac anomalies.
Family 6
The proband (Figure 1F, individual II-6) had prenatal polyhydramnios. She had a depressed, broad nasal bridge, midface retrusion with marked frontal bossing, high anterior hair line, nevus flammeus on forehead, downslanted palpebral fissures, bilateral epicanthus with widely-spaced eyes, long philtrum, full, sagging cheeks, short neck, broad chest and relatively short arms and legs. The mother had one stillbirth with polyhydramnios and arthrogryposis, which was not investigated. The parents were non-dysmorphic.
Family 7
Prenatally, the proband (Figure 1G, individual II-1) had increased nuchal translucency. Postnatally, he had left ventricle and interventricular septum hypertrophy, a VSD, and a small ASD. He had short stature, developmental delay, curly hair, low-set ears, increased posterior angulation of the ears, widely-spaced eyes, down-slanting palpebral fissures, a narrow nasal bridge and tip, and a mildly pointed chin, a broad neck and chest with mild pectus excavatum, relatively short fingers, and mild hypertrichosis. A brother (II-2) of the proband had fetal hydrops and died neonatally. The parents were non-dysmorphic.
Family 8
Postnatally, the proband (Figure 1H, individual II-11) had an ASD, valvular and supravalvular pulmonary stenosis, and right ventricular HCM. His facial features included widely-spaced eyes, downslanted palpebral fissures, mild ptosis, low-set ears, increased posterior angulation of the ears, a broad, webbed neck, and curly hair (Figure 2O-Q). His psychomotor development was apparently delayed. Other family members were non-dysmorphic. The parents are first cousins once removed.
Family 9
The proband (Figure 1I, individual II-2) is a female with an unaffected 6 y.o. sister. There was no known consanguinity. The findings noted at birth included a broad neck. She had developmental delay from an early age. Her findings at a later examination included widely-spaced eyes, strabismus, bilateral epicanthus, downslanted palpebral fissures, depressed nasal bridge, short and upturned nose, short and broad neck, thickened vermilion of the lips, broad great toes and thumbs, and hypermobility (Figure 2R,S). She had a thickened ventricular septum and valvular insufficiency and left axis deviation with prolonged QT interval on ECG. A head MRI showed ventriculomegaly. Her parents were non-dysmorphic.
Family 10
This female proband (individual II-3) was born to healthy non-dysmorphic, non-consanguineous parents. Their first pregnancy (46,XX) was lost with a tetralogy of Fallot and fetal hydrops. The proband came to attention for increased nuchal translucency and polyhydramnios. Postnatally, a prominent Chiari network in the right atrium was detected by ultrasound, and at age 6 weeks, HCM was diagnosed. She had mild muscular hypotonia, a broad chest with widely-spaced nipples, and pes planus. She had telecanthus, epicanthus, long philtrum, broad neck, and low set, posteriorly angulated ears. She developed mild short stature. Formal IQ tests showed a mean IQ of 95 at age 4 years and 83 at age 6 years.
Family 11
The parents were non-consanguineous and the family history was notable for an elective termination of a fetus with cystic hygroma, hydrops, and CHD. In the proband (II-6), cystic hygroma and polyhydramnios were identified prenatally. Postnatal echocardiography showed left HCM, left ventricular outlet stenosis and moderate mitral insufficiency. He had downslanted palpebral fissures, broad nasal bridge, upturned nares, low-set ears, increased posteriorly angulated ears, broad neck with redundant nuchal tissue, wide thorax with pectus excavatum, and single left palmar crease. The parents were non-dysmorphic except for a mildly broad neck in the mother.
Family 12
The proband (V-1) was dysmorphic at birth. At initial assessment, mild developmental delay was noted. Noonan syndrome was suspected. Echocardiography showed a small perimembranous VSD and non-obstructive HCM with mild mitral valve prolapse. Her sister (individual V-4) was had a murmur neonatlly. HCM and dysplastic mitral valve was diagnosed by echocardiography. There was delay in attaining motor and speech milestones. At age 3 years 9 months features of Noonan syndrome were more apparent. At age 6 years mitral valve repair with sub-aortic membrane resection was undertaken. The parents were consanguineous.
Molecular data
Most probands underwent DNA sequencing either to search for causative variants or to exclude genes known at the time of sequencing to cause Noonan syndrome. One proband had genome sequencing, three probands had exome sequencing, and most probands had targeted sequencing for various subsets of the 20 candidate genes listed in Methods. The LZTR1 variants were detected by a number of methods amongst the various centers. These included exome and genome sequencing, NGS panel sequencing, and/or Sanger sequencing (see methods and online supplemental methods). All affected patients had biallelic LZTR1 variants, with heterozygous parents. None of the nine molecularly evaluated, clinically unaffected children had biallelic variants. The genotypes are presented in Figure 1 and Table 1, detailed data are presented in the supplemental material. Other molecular and genetic data are summarized below.
Family 1
Analysis of the SNP genotyping data under a model of autosomal recessive inheritance identified a chromosome 22 region with shared alleles in all four children. Further analyses showed that the 9 Mb shared region containing LZTR1 on chr22 was the only extended shared region that could explain recessive inheritance and the only shared region that achieved the maximum achievable multipoint LOD score of 1.8 over many consecutive markers. Surprisingly, there was only one other autosomal region that had LOD >1.3, a 3 Mb region near 8pter in which multi-marker analysis showed that any possible fully shared multi-marker region must be small (<1 Mb).
The effect of the c.2220-17C>A splice variant was tested using RTPCR from three of the siblings, both parents, and a control. In the affected children and mother a large, unspliced product was evident and showed a modest but noticeable decrease in properly spliced cDNA (Figure 3A). This splice variant retained intron 18, which predicts p.(Tyr741Hisfs*89) and that the cDNA of this intron had only the A allele of the c.2220-17C>A variant (Supplemental Figure 1A), demonstrating that retention of this intron is associated with that allele. Finally, we Sanger sequenced an exonic heterozygous SNP (rs13054014) in the gene and showed that the cDNA is skewed toward the allele with the splice variant, with relatively reduced levels of the nonsense variant, confirming a moderate level of nonsense-mediated mRNA decay. In Figure 3A, a minor aberrant 386 bp splice product is an inconsistently incorporated intron 19 (84 nucleotides), with an in-frame stop codon (Supplemental Figures 1B and 2).
Figure 3.
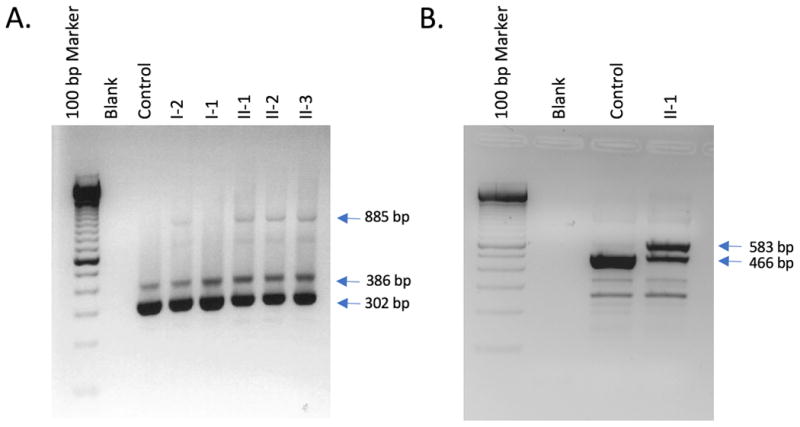
A. Image of RTPCR products associated with the splice variant in Family 1. Total lymphoblast RNA was reverse-transcribed and PCR-amplified with primers from exons 18 and 21 of LZTR1. Two products were present in all individuals: normally spliced product of 302 bp and an alternatively spliced product of 386 bp retaining intron 19. The carrier mother (lane 4) and affected children (lanes 6 – 8) had an additional RT-PCR product of 885 bp retaining intron 18. This product was not seen in either the father (lane 5) or control lymphoblast RNA (lane 3). Size markers are shown in lane 1 and a no-RNA control in lane 2. B. Image of RTPCR products associated with the splice variant in Family 3. Total lymphoblast RNA was reverse transcribed and PCR-amplified with primers from exons 15 and 18 of LZTR1. A normally spliced product of 465 bp was present in both a control (lane 3) and an affected individual (lane 4). The affected child (lane 4) had an additional RT-PCR product of 583 bp retaining a 117 bp alternate exon from within intron 16. This product was not seen at a significant level in control lymphoblast RNA (lane 3). Size markers are shown in lane 1 and a no-RNA control in lane 2.
Family 3
RTPCR data from lymphoblasts showed the c.1943-256C>T splice variant retained a 117 bp alternate exon that lies within intron 16 (Figure 3B).
Family 8
Homozygosity for the familial disease-associated allele was found in the (not phenotyped) abortus.
Family 12
Linkage analysis under a model of autosomal recessive inheritance identified candidate regions summing to 357 Mb of shared IBD in the affected siblings, not shared in the unaffecteds. Including multiple loops of consanguinity into the analysis increased the maximum multipoint LOD score from 0.82 to 2.30 and identified two regions of shared autozygosity; a 4.8 Mb region on chr22 (17,782,813 to 22,590,873) and a 3.9 Mb region on chr7 (20,461,417 to 24,349,255). No other regions achieved a LOD score >1.00.
Discussion
Here we present clinical and molecular data on 12 families with children with Noonan syndrome caused by biallelic, pathogenic LZTR1variants. In contrast to prior molecularly confirmed occurrences of Noonan syndrome that were inherited in an autosomal dominant pattern, the inheritance in these families is compatible with autosomal recessive inheritance. Like all other forms of Noonan syndrome, the clinical phenotype was variable but included bilateral epicanthus, downslanted palpebral fissures, ptosis, low-set ears, increased posterior angulation of the ears, prenatal cystic hygromas with broad neck and low posterior hairline, short stature, and cardiac anomalies including HCM and valvular abnormalities, abnormal cardiac septation, and dysrhythmias. The children's parents were not affected with Noonan syndrome or known cardiac anomalies. Two parents did have short stature and one had ptosis, but we cannot distinguish coincidence from causation for these. This disorder could have a semidominant inheritance pattern (mild phenotype in heterozgyotes), which necessitates further studies.
The existence of a recessive form of Noonan syndrome was suggested from clinical analysis (OMIM: NS2, 605275), but to date has not been molecularly confirmed. In 1969, Abdel-Salaam and Temtamy13 described a male-female sib pair, offspring of a first cousin marriage with ‘Turner syndrome’. Their features were similar to the families described here, especially the siblings in Family 3, who had linear growth delay and developmental compromise. Maximilian et al14 described three siblings with unaffected parents. The clinical features in this family were similar to the present families, with cardiac disease including pulmonic stenosis, (but not cardiomyopathy) as did several of the children described here. Van der Burgt and Brunner15 described four families with apparent recessive Noonan syndrome, but molecular characterization was not done. A potential explanation for sibling recurrence of a Noonan syndrome phenotype with unaffected parents is gonadal mosaicism for a gene known to cause autosomal dominant Noonan syndrome.16 However, in the individuals described here, extensive analyses failed to identify heterozygous pathogenic variants in genes other than LZTR1 associated with Noonan syndrome.
Autosomal dominant inheritance of Noonan syndrome has been associated with LZTR1 variants.8 Three families had vertical transmission of the phenotype. They had co-segregation of c.742G>A p.(Gly248Arg), c.850C>T p.(Arg284Cys), and c.740G>A p.(Ser247Asn) in LZTR1. Two other families had a single affected individual with a de novo heterozygous LZTR1 variant (c.859C>T p.(His287Tyr) and the other c.356A>C p.(Tyr119Cys)). We couldn't compare the dysmorphic features in the present patients with those of Yamamoto et al because they were described only as having “typical facial features”. Several did have webbed necks, short stature, and cardiac valvular disease, and two had developmental delay. Heterozygous variants (c.710G>A p.(Arg237Gln) and c.745G>C p.(Ala249Pro)) in LZTR1 were also identified in two individuals by Chen et al17, although they did not consider them to be pathogenic. We agree with Yamamoto et al8 that the two families of Chen et al17 have autosomal dominant LZTR1-associated Noonan syndrome. These data confirm that some heterozygous LZTR1 variants cause autosomal dominant Noonan syndrome.
In contrast to these papers, we present clinical and molecular data on 23 offspring from 12 families that implicate LZTR1 in an autosomal recessive form of Noonan syndrome. All 12 families have variants that are absent or rare in databases (Table 1). The most common variant (c.628C>T p.(Arg210Ter)) was found in 13/220,526 alleles with a non-Finnish European MAF of 0.000098. Other variants were less common or absent in databases, compatible with a rare recessive disorder. The variants were found in the biallelic state in affected patients and co-segregated with affection status in several multiplex sibships. Five of the implicated variants are nonsense or canonical splice variants. The U12-dependent branch site variant in family 1 increased the level of unspliced cDNA (Figure 3A). This is similar to a known LZTR1 c.2220-15_2220-14delCTT variant in the same branch site.9 That variant, from a NF2/SMARCB1-negative schwannomatosis patient, did not show altered splicing by (non-quantitative) RT-PCR, was considered likely pathogenic based on affecting evolutionary-conserved nucleotides (PhastCons 1.00 and phyloP 4.81-4.92), retention of the variant in the tumors with loss of the remaining wild-type allele, a molecular signature typical of LZTR1-associated schwannomas (L. Messiaen, personal communication). The deep intronic splice variant in families 2, 3, and 10 also affected cDNA splicing (Figure 3B).
The role of this gene in Noonan syndrome is supported by linkage in families 1 and 12, the latter of which generated a maximum LOD score of 2.3, and a combined LOD score of 4.1.
The eight missense variants were predicted damaging by CADD scores: p.(His121Asp); 29.8, p.(Arg170Trp); 34, p.(Glu217Ala); 23.2, p.(Glu563Gln); 24, p.(Arg688Gly); 34, p.(Arg697Gln); 35, p.(Arg755Gln); 35, and p.(Ile821Thr); 27.3. Family 8 had a p.(Ile205Thr) variant in cis with p.(Arg170Trp) and has a CADD score of 23.6. We suggest that p.(Arg170Trp) is more likely pathogenic, but we cannot exclude the hypothesis that the pathogenic allele/haplotype comprises both and neither would be pathogenic alone. The c.*70G>A variant was in cis with c.1943-256C>T in family 3 but was not present on the c.1943-256C>T allele in three other families. We conclude c.*70G>A variant alone is benign. Three missense variants we detected (p.(Arg170Trp), p.(Arg697Gln), and p.(Arg688Gly)) were similar to schwannomatosis variants (p.(Arg170Gln)18, p.Arg697Trp)19, and p.(Arg688Cys)9). One putative loss of function variant was identical to a schwannomatosis variant p.(Gln10fs*15).9
The existence of recessive and dominant forms of Noonan syndrome associated with LZTR1 variants has implications for the pathogenetic model for this disorder. To date, all mutations associated with the dominant form are missense mutations, although this number is small (six, including the putatively pathogenic variants of Chen et al17). In contrast, we identified missense, nonsense, and splicing variants in the recessive form (Figure 4). There are several pathogenetic models for these observations. The first specifies a reduction of LZTR1 function below 50% resulting in Noonan syndrome, either through monoallelic dominant negative LZTR1 missense variants (autosomal dominant form) or the combination of hypomorphic and loss of function variants in the recessive form. Homozygous loss of function of Lztr1 in the mouse causes preweaning lethality and is probably lethal in humans, as suggested by the report of a fetus with non-immune hydrops fetalis and a homozygous LZTR1 c.2317G>A; p.(Val773Met) variant.20 To date, no examples of biallelic germline loss of function alleles have been identified in the mouse (see web link, below). The second model specifies that the mutational position determines whether missense LZTR1 variants cause dominant or recessive Noonan syndrome. Although the numbers of missense variants in dominant LZTR1-associated Noonan syndrome are small, they all reside between codons 119 and 287, whereas the mutations we have identified in the recessive form range throughout the protein (Figure 4). A robust genotype-phenotype analysis awaits the description of additional patients with dominant and recessive LZTR1-associated Noonan syndrome.
Figure 4.

Cartoon of LZTR1 mutations showing the variants identified here in autosomal recessive Noonan syndrome above the protein and the previously reported in autosomal dominant Noonan syndrome below the protein. The ˆ symbol indicates that these two variants were in cis on this mutant allele and it is hypothesized that p.(Arg170Trp) is pathogenic; see text.
The LZTR1 pathogenicity model must also take into account data on schwannomatosis susceptibility. There are two potential models of schwannomatosis susceptibility that involve a three-step mutational process beginning with a germline LZTR1 mutation, with three or four additional hits in two or three genes.21 A germline LZTR1 variant in combination with a somatic deletion of wild-type 22q including LZTR1, NF2, and potentially SMARCB1 in combination with one or more point mutations, can lead to tumors.21 Several Family 1 members had suggestive signs of schwannomas on MRI. As well, one variant in family 5 (c.27delG, p.(Gln10Argfs*15)) was also identified as a schwannomatosis susceptibility variant.9 This model of schwannomatosis has implications for the relatively common 22q11.2 deletion. LZTR1 lies within the 3 Mb A-D proximal region, which is found in ∼85% of patients with the phenotype22. Every patient with that deletion is haploinsufficient for LZTR1 and should be susceptible to schwannomas. Additionally, to our knowledge, there are no patients reported with both Noonan syndrome and a typical 22q11.2 deletion syndrome phenotype (one deleted allele plus a point variant in trans). We predict that such patients exist. This combined phenotype may be subtle, as has been shown for Bernard-Soulier, CEDNIK, and Van den Ende-Gupta syndromes with the typical 22q11.2 3 Mb deletion and a point mutation in trans (GP1BB, SNAP29, or SCARF2, respectively)23-25. We predict that patients with mild Noonan syndrome, with or without features of 22q11.2 deletion syndrome exist and encourage physicians to be alert for this combination of phenotypic findings.
Autosomal recessive LZTR1-associated Noonan syndrome has variable expressivity and includes several mildly affected patients, but the severe end includes lethality, most commonly from cardiac disease, which can be prenatal. Several patients had significant cardiac dysfunction and while we cannot prove that hydrops and prenatal losses were attributable to LZTR1 variants, we strongly suspect that they are. Also, two of these patients developed ALL. ALL is not the most common leukemia in known forms of Noonan syndrome26. The possibility that these leukemias were coincidental is discounted by the role of LZTR1 in tumorigenesis.9,21 These data suggest, but do not prove, that autosomal recessive LZTR1-associated Noonan syndrome is a cancer susceptibility disorder.
We conclude that biallelic LZTR1 variants can cause Noonan syndrome inherited in an autosomal recessive pattern, based on 12 families with a total of 21 molecularly characterized affected offspring, unaffected parents, cosegregation in unaffected siblings, rare, biallelic LZTR1 variants. These data have important implications for the molecular diagnosis of Noonan syndrome, evaluating recurrence risks for families with LZTR1 variants, the pathogenesis of familial schwannomatosis, and the heterogeneity of phenotypes associated with 22q11.2 deletions.
Supplementary Material
Acknowledgments
The authors dedicate this paper to the children whose lives were claimed by this disease. We thank the families who supported this work and permitted us to share their information. The NIH authors thank Dr. Jean-Pierre Guadagnini for dental evaluations and Ms. Cantelmo and Ms. King for audiologic evaluations. The authors thank Christina Lissewski, for molecular evaluation of families 7 and 8, Franziska Waldmann and Melanie Graf for clinical evaluation of family 8, Rebecca Okashah Littlejohn and James Gibson for evaluation of family 2, Hugh Watkins, John Taylor, and the Oxford RGL for clinical evaluation and genetic studies in family 12. Jessica Chen provided the cDNA coordinates for the variants in the Chen et al reference17. Tabitha Banks performed RTPCR analyses on samples from Family 1. Julia Fekecs at NIH provided expert graphics support. The views expressed here are solely those of the authors and do not necessarily represent the views of the institutions the authors are funded by, or affiliated with.
Conflict of Interest: LGB, JJJ, and JCS received support from the Intramural Research Program of the National Human Genome Research Institute grants HG200328-11 and HG200388-03. AAS received support from the Intramural Research Program of the National Library of Medicine. DMM received honoraria for lectures on 22q11.2 deletion syndrome for Natera. MZ received support from German Federal Ministry of Education and Research (BMBF) NSEuroNet (FKZ 01GM1602A), GeNeRARe (FKZ 01GM1519A). JAR, WC, LE, MJ, and BL received support from the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number U01 HG007709-01. The High-Throughput Genomics Group at the Wellcome Centre for Human Genetics is funded by the Wellcome Trust (grant reference 090532/Z/09/Z). This work was also supported by the National Institute for Health Research (NIHR) Biomedical Research Centre Oxford with funding from the Department of Health's NIHR Biomedical Research Centre's funding scheme. LGB receives royalties from Genentech Corp, is an advisor to the Illumina Corp, and receives honoraria from Wiley-Blackwell. The Department of Molecular & Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered at the Baylor Genetics Laboratories.
Footnotes
Contributor Information
Jennifer J. Johnston, Medical Genomics and Metabolic Genetics Branch, National Human Genome Research Institute, NIH, Bethesda, MD, USA.
Jasper J. van der Smagt, Department of Genetics University Medical Center Utrecht, Utrecht, The Netherlands.
Jill A. Rosenfeld, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Alistair T. Pagnamenta, National Institute for Health Research Oxford Biomedical Research Centre, Wellcome Centre for Human Genetics, University of Oxford, Oxford, UK.
Abdulrahman Alswaid, King Abdulaziz Medical City, Riyadh, Saudi Arabia.
Eva H. Baker, Department of Radiology and Imaging Services; Clinical Center; NIH, Bethesda, MD, USA
Edward Blair, Oxford Centre for Genomic Medicine, Oxford University Hospitals NHS Foundation Trust, Oxford, UK.
Guntram Borck, Institute of Human Genetics, University of Ulm, Ulm, Germany.
Julia Brinkmann, Institute of Human Genetics, University Hospital, Magdeburg, Germany.
William Craigen, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Vu Chi Dung, Rare Disease and Newborn Screening Service, The National Children's Hospital, Hanoi, Vietnam, Department of Medical Genetics and Metabolism.
Lisa Emrick, Division of Neurology and Developmental Neuroscience and Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA.
David B. Everman, Greenwood Genetic Center, Greenwood, SC, USA
Koen L. van Gassen, Department of Genetics University Medical Center Utrecht, Utrecht, The Netherlands
Suleyman Gulsuner, Division of Medical Genetics, University of Washington, Seattle, WA, USA.
Margaret H. Harr, Center for Applied Genomics, Children's Hospital of Philadelphia, Philadelphia, PA, USA
Mahim Jain, Department of Molecular & Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Alma Kuechler, Institut für Humangenetik, Universitätsklinikum Essen, Universität Duisburg-Essen, Essen, Germany.
Kathleen A. Leppig, Genetic Services, Kaiser Permanente of Washington, Seattle, WA, USA
Donna M. McDonald-McGinn, Division of Human Genetics and Department of Pediatrics, Children's Hospital of Philadelphia and the Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
Can Thi Bich Ngoc, Rare Disease and Newborn Screening Service, The National Children's Hospital, Hanoi, Vietnam, Department of Medical Genetics and Metabolism.
Amir Peleg, Institute of Human Genetics, Carmel Medical Center, Haifa, Israel.
Elizabeth R. Roeder, Department of Pediatrics and Molecular and Human Genetics, Baylor College of Medicine, San Antonio, TX, USA
R. Curtis Rogers, Greenwood Genetic Center, Greenwood, SC, USA.
Lena Sagi-Dain, Institute of Human Genetics, Carmel Medical Center, Haifa, Israel.
Julie C. Sapp, Medical Genomics and Metabolic Genetics Branch, National Human Genome Research Institute, NIH, Bethesda, MD, USA
Alejandro A. Schäffer, Computational Biology Branch, National Center for Biotechnology Information, NIH, Bethesda, MD, USA
Denny Schanze, Institute of Human Genetics, University Hospital, Magdeburg, Germany.
Helen Stewart, Oxford Centre for Genomic Medicine, Oxford University Hospitals NHS Foundation Trust, Oxford, UK.
Jenny C. Taylor, National Institute for Health Research Oxford Biomedical Research Centre, Wellcome Centre for Human Genetics, University of Oxford, Oxford, UK
Nienke E. Verbeek, Department of Genetics University Medical Center Utrecht, Utrecht, The Netherlands
Magdalena A. Walkiewicz, Baylor Genetics and the Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA
Elaine H. Zackai, Division of Human Genetics and Department of Pediatrics, Children's Hospital of Philadelphia and the Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
Christiane Zweier, Institute of Human Genetics, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Erlangen, Germany.
Martin Zenker, Institute of Human Genetics, University Hospital, Magdeburg, Germany.
Brendan Lee, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Leslie G. Biesecker, Medical Genomics and Metabolic Genetics Branch, National Human Genome Research Institute, NIH, Bethesda, MD, USA
Bibliography
- 1.Allanson JE. Objective studies of the face of Noonan, Cardio-facio-cutaneous, and Costello syndromes: A comparison of three disorders of the Ras/MAPK signaling pathway. Am J Med Genet A. 2016;170:2570–2577. doi: 10.1002/ajmg.a.37736. [DOI] [PubMed] [Google Scholar]
- 2.Gelb BD, Roberts AE, Tartaglia M. Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog Pediatr Cardiol. 2015;39:13–19. doi: 10.1016/j.ppedcard.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colquitt JL, Noonan JA. Cardiac findings in Noonan syndrome on long-term follow-up. Congenit Heart Dis. 2014;9:144–150. doi: 10.1111/chd.12102. [DOI] [PubMed] [Google Scholar]
- 4.Allanson JE, Roberts AE. Noonan Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 5.Aoki Y, Niihori T, Inoue S, Matsubara Y. Recent advances in RASopathies. J Hum Genet. 2016;61:33–39. doi: 10.1038/jhg.2015.114. [DOI] [PubMed] [Google Scholar]
- 6.Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14:355–369. doi: 10.1146/annurev-genom-091212-153523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevenson DA, Schill L, Schoyer L, et al. The Fourth International Symposium on Genetic Disorders of the Ras/MAPK pathway. Am J Med Genet A. 2016;170:1959–1966. doi: 10.1002/ajmg.a.37723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto GL, Aguena M, Gos M, et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet. 2015;52:413–421. doi: 10.1136/jmedgenet-2015-103018. [DOI] [PubMed] [Google Scholar]
- 9.Piotrowski A, Xie J, Liu YF, et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet. 2014;46:182–187. doi: 10.1038/ng.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnston JJ, Sanchez-Contreras MY, Keppler-Noreuil KM, et al. A Point Mutation in PDGFRB Causes Autosomal-Dominant Penttinen Syndrome. Am J Hum Genet. 2015;97:465–474. doi: 10.1016/j.ajhg.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piret SE, Gorvin CM, Pagnamenta AT, et al. Identification of a G-Protein Subunit-alpha11 Gain-of-Function Mutation, Val340Met, in a Family With Autosomal Dominant Hypocalcemia Type 2 (ADH2) J Bone Miner Res. 2016;31:1207–1214. doi: 10.1002/jbmr.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdel-Salam E, Temtamy SA. Familial Turner phenotype. J Pediatr. 1969;74:67–72. doi: 10.1016/s0022-3476(69)80009-0. [DOI] [PubMed] [Google Scholar]
- 14.Maximilian C, Ioan DM, Fryns JP. A syndrome of mental retardation, short stature, craniofacial anomalies with palpebral ptosis and pulmonary stenosis in three siblings with normal parents. An example of autosomal recessive inheritance of the Noonan phenotype? Genet Couns. 1992;3:115–118. [PubMed] [Google Scholar]
- 15.van Der Burgt I, Brunner H. Genetic heterogeneity in Noonan syndrome: evidence for an autosomal recessive form. Am J Med Genet. 2000;94:46–51. doi: 10.1002/1096-8628(20000904)94:1<46::aid-ajmg10>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 16.Yoon SR, Choi SK, Eboreime J, Gelb BD, Calabrese P, Arnheim N. Age-dependent germline mosaicism of the most common noonan syndrome mutation shows the signature of germline selection. Am J Hum Genet. 2013;92:917–926. doi: 10.1016/j.ajhg.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen PC, Yin J, Yu HW, et al. Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc Natl Acad Sci U S A. 2014;111:11473–11478. doi: 10.1073/pnas.1324128111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith MJ, Isidor B, Beetz C, et al. Mutations in LZTR1 add to the complex heterogeneity of schwannomatosis. Neurology. 2015;84:141–147. doi: 10.1212/WNL.0000000000001129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paganini I, Chang VY, Capone GL, et al. Expanding the mutational spectrum of LZTR1 in schwannomatosis. Eur J Hum Genet. 2015;23:963–968. doi: 10.1038/ejhg.2014.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shamseldin HE, Kurdi W, Almusafri F, et al. Molecular autopsy in maternal-fetal medicine. Genet Med. 2017 doi: 10.1038/gim.2017.111. [DOI] [PubMed] [Google Scholar]
- 21.Kehrer-Sawatzki H, Farschtschi S, Mautner VF, Cooper DN. The molecular pathogenesis of schwannomatosis, a paradigm for the co-involvement of multiple tumour suppressor genes in tumorigenesis. Hum Genet. 2017;136:129–148. doi: 10.1007/s00439-016-1753-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11.2 Deletion Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [PubMed] [Google Scholar]
- 23.Kunishima S, Imai T, Kobayashi R, Kato M, Ogawa S, Saito H. Bernard-Soulier syndrome caused by a hemizygous GPIbbeta mutation and 22q11.2 deletion. Pediatr Int. 2013;55:434–437. doi: 10.1111/ped.12105. [DOI] [PubMed] [Google Scholar]
- 24.Bedeschi MF, Colombo L, Mari F, et al. Unmasking of a Recessive SCARF2 Mutation by a 22q11.12 de novo Deletion in a Patient with Van den Ende-Gupta Syndrome. Mol Syndromol. 2010;1:239–245. doi: 10.1159/000328135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDonald-McGinn DM, Fahiminiya S, Revil T, et al. Hemizygous mutations in SNAP29 unmask autosomal recessive conditions and contribute to atypical findings in patients with 22q11.2DS. J Med Genet. 2013;50:80–90. doi: 10.1136/jmedgenet-2012-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasle H. Malignant diseases in Noonan syndrome and related disorders. Horm Res. 2009;72(Suppl 2):8–14. doi: 10.1159/000243773. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.