

Abstract
Ewing sarcoma is a highly aggressive small round cell malignant neoplasm of bone and soft tissue that typically is manifested in children and young adults. It is most commonly a primary bone tumor; however, extraosseous cases have been increasingly reported. We report a case of metastatic extraosseous Ewing sarcoma with the primary lesion occurring within a limb affected by primary lymphedema. Lymphedema, in this case, played a role not only in the genesis of the tumor because of the relative local immunosuppression but also in masking the development of the lower limb mass.
Keywords: Ewing sarcoma, Lymphedema, Lymphatic disorders, Histopathology, Oncology, Radiology
Ewing sarcoma is a highly aggressive small round cell malignant neoplasm of bone and soft tissue that typically is manifested in children and young adults.1 In the majority of patients (∼90%), a translocation t(11;22) between the Ewing sarcoma breakpoint region 1 (EWSR1) and the Friend leukemic integration 1 (FLI1) genes is seen, producing the EWS-FLI1 chimeric fusion oncogene.2, 3 In addition, CD99 antigen is strongly expressed on the cell membrane in Ewing sarcoma and represents a sensitive although nonspecific immunohistochemical marker.1 Ewing sarcoma is most commonly a primary bone tumor; however, extraosseous primary tumors are documented to represent up to 31% in a large-scale analysis of the U.S. National Cancer Institute's Surveillance, Epidemiology and End Results database, with both having similar patient outcomes.4 The most common extraosseous sites include the torso, paravertebral regions, limbs, retroperitoneum, and head and neck.1, 5 Five-year overall survival for localized disease is 65% to 75%, whereas <30% is reported for extensive metastatic disease.6 This report outlines a case of extraosseous Ewing sarcoma in a patient with primary lymphedema of the left leg. Informed consent was obtained before publication.
Case report
A 34-year-old man presented to the emergency department the morning after developing sudden-onset left-sided pleuritic chest pain and shortness of breath. Six months previously, a large parapneumonic effusion had been drained, followed by a video-assisted thoracoscopic surgical pleurodesis. The pathologic examination of the pleural fluid did not show any signs of malignancy, and computed tomography (CT) of the chest did not show any pulmonary lesions. In this 6-month period, he had experienced two similar episodes that had spontaneously resolved. He had been born with primary lymphedema to the left midthigh and in the previous 6 to 12 months had experienced significant pain in this leg. He reported periodic disabling crescendo episodes of pain that would occur every few weeks, persisting for a couple of days, responding only to tramadol. During these pain episodes, he would present in crisis to varying primary care physicians as he did not have a regular physician. His past medical history was otherwise significant for recurrent left leg cellulitis, asthma, and depression. Physical examination at presentation was unremarkable with the exceptions of tachycardia (110 beats/min) and a nontender lymphedematous left leg, with pitting edema to the waist and no palpable mass (Fig 1).
Fig 1.
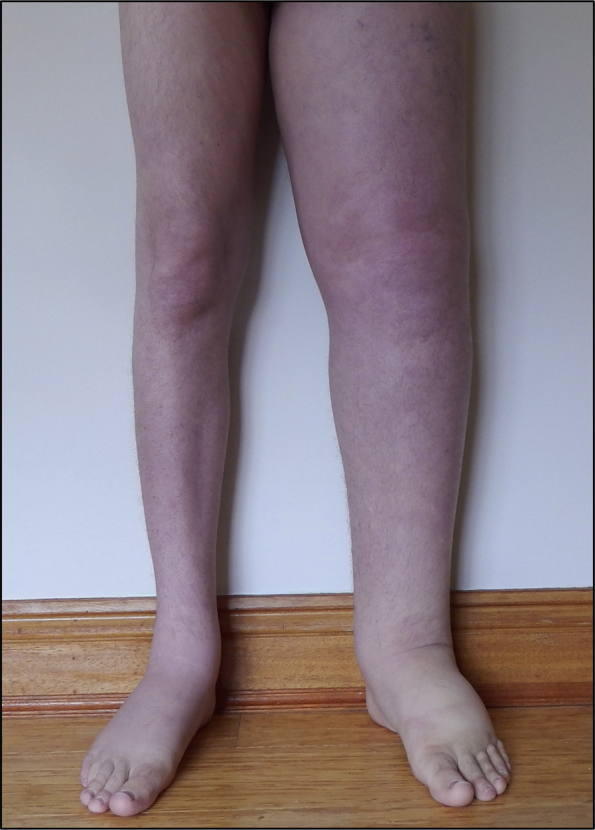
Clinical photograph demonstrating left leg lymphedema.
Initial blood tests revealed a microcytic anemia (mean corpuscular volume, 74 fL; hemoglobin value, 109 g/L), hypoalbuminemia (27 g/L), elevated lactate dehydrogenase (331 IU/L), and positive D-dimer (2.99 mg/L). CT pulmonary angiography revealed numerous abnormalities (Fig 2, A-C). These included bilateral pulmonary emboli; innumerable pulmonary and pleural nodules within both lungs; bilateral pleural effusions; hilar and mediastinal lymphadenopathy; lytic lesions in T9, T11, and T12 vertebral bodies and sternum; and a bulky mass within the tail of the pancreas. CT of the abdomen and pelvis demonstrated the pancreatic lesion to be round, 29 × 24 mm, with mild contrast enhancement and a left external iliac-femoral vein thrombus (Fig 2, D). Findings on CT of the brain and testicular ultrasound were unremarkable. Carcinoembryonic antigen, cancer antigen 19-9, α-fetoprotein, and β-human chorionic gonadotropin were all within normal limits.
Fig 2.

Computed tomography (CT) of the chest and abdomen. Coronal (A), parasagittal (B), and axial (C) maximal intensity projections of the chest highlight innumerable pulmonary nodules involving the parenchyma and pleura (example nodules are indicated with arrows). Abdominal CT revealed a round, solitary, hypodense lesion in the tail of the pancreas (D; arrow).
Fine-needle aspiration of a pleura-based lesion showed scattered tumor cells with a high nuclear to cytoplasmic ratio, primitive-looking nuclei with slightly irregular nuclear outlines, nuclear molding, and cytoplasmic vacuolation, all features reminiscent of Ewing sarcoma (Fig 3). Flow cytometry was unremarkable. Core biopsies of the same lesion showed sheets of small tumor cells, with some arranged in a circular fashion resembling rosettes (Fig 4). There was weak patchy reactivity with antibody to synaptophysin and moderate to strong membranous reactivity with antibody to CD99. Endothelial markers CD31 and CD34 stained vessels present in the core biopsy sections but not the tumor cells. Interphase fluorescent in situ hybridization showed a signal pattern consistent with FLI1-EWSR1 rearrangement. EWSR1 rearrangement was confirmed by a second (break-apart) probe set. This gene rearrangement results from the translocation t(11;22)(q24;q12) and, in the presence of typical microscopy and immunohistochemistry, confirmed the provisional diagnosis of Ewing sarcoma/primitive neuroectodermal tumor.
Fig 3.

Cytology from the initial lung fine-needle aspirate. A, A smear from the lung fine-needle aspirate shows a population of cells larger than the adjacent neutrophils (white arrows). The nuclei have an open fine chromatin with one or two tiny pale blue nucleoli. Note the vacuoles in the blue cytoplasm (gray arrows). B, Sections from the cell block show a population of poorly cohesive cells with a diffuse fine nuclear chromatin, slight variation in nuclear size and outline, and no obvious nucleoli. The cytoplasm appears delicate, vacuolated, and disrupted (arrow).
Fig 4.

Core biopsy sections. A, A population of poorly cohesive primitive cells showing no differentiating features was seen. B, A closer view shows poorly cohesive cells with a high nuclear to cytoplasmic ratio and circular pseudorosette-like (gray arrow) and rosette-like arrangements of nuclei (white arrow). C, Strong membranous reactivity was seen with antibody to CD99.
Because of the surprising histopathologic findings of Ewing sarcoma, with a definite primary site unknown, a fluorodeoxyglucose positron emission tomography (FDG-PET) scan was performed. Appearances were consistent with disseminated malignant disease, with intense uptake in the known pulmonary, nodal, and pancreatic anatomic lesions. In addition, a large hypodense soft tissue lesion was identified in the posterior compartment of the thigh, 71 × 125 mm in the axial plane, associated with markedly increased tracer activity. This lesion could be seen extending proximally into the pelvis, deep to the inguinal ligament along the course of the left iliac vein (Fig 5).
Fig 5.

Fluorodeoxyglucose positron emission tomography (FDG-PET) scan coronal (A) and parasagittal (B) images show a large soft tissue mass lesion in the posterior compartment of the left thigh with proximal extension to the pelvis, deep to the inguinal ligament, along the course of the iliac vein. Note the relative size difference of the lymphedematous left leg.
Given the burden of metastatic disease, the patient was not a candidate for surgical management. Treatment involved chemotherapy on an outpatient basis, alternating vincristine, doxorubicin, and cyclophosphamide with ifosfamide and etoposide. Repeated FDG-PET scan after eight cycles of chemotherapy showed a favorable response to therapy, with reduction in size and intensity of FDG uptake of the primary and metastatic lesions. At the time of writing, 12 months from diagnosis, the patient was doing well.
Discussion
Given the recent history of weight loss and probable recurrent pulmonary emboli, malignant neoplasm was thought the most likely underlying disease process, and the initial impression was that of a primary metastatic pancreatic tumor. The histopathologic findings from a pleura-based lesion were surprising, suggesting sarcoma and favoring Ewing sarcoma. Before the results of the fluorescent in situ hybridization, which ultimately detected the FLI1-EWSR1 rearrangement, a PET scan was performed to assist in diagnosis and to plan treatment. This revealed the occult primary within the lymphedematous limb, which had been given less attention because of its chronicity and the focus on the chest and abdominal findings. In retrospect, the thrombus visualized in the left iliac vein on CT of the abdomen was likely tumor thrombus.
Extraosseous Ewing sarcoma is consistently reported as a rare presentation; however, it does represent approximately one-third of cases. Features of extraosseous tumors in contrast to osseous include the following: patients are less likely to be male (no gender predilection); overall survival is poorer in the first 24 months; overall survival is improved beyond 24 months; and average age at onset is later (bimodal distribution, more commonly found in patients younger than 5 years and older than 35 years).4 Signs and symptoms are dependent on the site of origin7 and tend to be due to space-occupying effects, such as pain and swelling.8 Local control is best achieved with surgery, which should be performed whenever a marginal or wide resection is achievable.6 Amputations are rarely indicated (<10% of cases) but may be used in inoperable lesions if radiotherapy is predicted to lead to a poor functional outcome because of tumor location or patient factors.6 Case reports of extraosseous Ewing sarcoma of varying locations (eg, leg,9 spinal,10 intracranial,11 and intra-abdominal7) indicate positive outcomes after surgery and adjuvant chemotherapy with or without radiotherapy, although follow-up has been limited to <2 years. The thigh is a reasonably common site for extraosseous Ewing sarcoma8; however, evidence of long-term outcomes in the literature is lacking.
The fact that the primary tumor in this case arose within a lymphedematous limb is significant, as the chronic condition masked the presence of a mass lesion. Lymphedema may be primary, which may or may not be congenital, or secondary to surgery (eg, lymph node dissection or vein harvesting), chronic bacterial or parasitic infection, irradiation, trauma, or lymphoma.12 Lymphedema produces local immunosuppression as a result of lymphatic fluid stasis and the relative upregulation of growth factors within the microenvironment.13 This is thought to be associated with the potential development of malignant disease.12 The most common malignant neoplasms reported to occur in the context of chronic lymphedema are angiosarcoma and lymphangiosarcoma, with >400 documented cases.12 Other tumors arising within lymphedematous regions include Kaposi sarcoma, nonmelanoma skin cancer, melanoma, and cutaneous lymphoma.12 Single case reports have also identified porocarcinoma after vein harvesting14 and breast surgery,15 fibrous histiocytoma,16 and Merkel cell carcinoma.17
To the best of our knowledge, there have been no reported cases of Ewing sarcoma arising within a lymphedematous region. Ewing sarcoma has been sporadically reported to arise within immunocompromised patients, including after renal transplantation,18 in children with human immunodeficiency virus infection,19 and as a secondary malignant neoplasm after treatment for hematologic and solid malignant neoplasms.20
Primary lymphedema, congenital or acquired, is associated with a higher albeit rare increased risk of a number of different malignant neoplasms, including angiosarcoma, Kaposi sarcoma, B-cell lymphoma,21 and, as demonstrated in this case, Ewing sarcoma. As such, a high index of suspicion is needed because patients will not have the usual clinical findings of a palpable mass. Any significant change in symptoms, especially pain or an increase in swelling, should be thoroughly investigated, including imaging of the limb with CT or magnetic resonance imaging. Early diagnosis and early surgical intervention with or without radiotherapy for local control combined with multiagent chemotherapy are critical to improve patient outcomes, given significantly poorer overall survival associated with metastatic disease.
Footnotes
Author conflict of interest: none.
The editors and reviewers of this article have no relevant financial relationships to disclose per the Journal policy that requires reviewers to decline review of any manuscript for which they may have a conflict of interest.
References
- 1.Tural D., Molinas Mandel N., Dervisoglu S., Oner Dincbas F., Koca S., Colpan Oksuz D. Extraskeletal Ewing's sarcoma family of tumors in adults: prognostic factors and clinical outcome. Jpn J Clin Oncol. 2012;42:420–426. doi: 10.1093/jjco/hys027. [DOI] [PubMed] [Google Scholar]
- 2.Delattre O., Zucman J., Plougastel B., Desmaze C., Melot T., Peter M. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–165. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 3.Zucman J., Delattre O., Desmaze C., Plougastel B., Joubert I., Melot T. Cloning and characterization of the Ewing's sarcoma and peripheral neuroepithelioma t(11;22) translocation breakpoints. Genes Chromosomes Cancer. 1992;5:271–277. doi: 10.1002/gcc.2870050402. [DOI] [PubMed] [Google Scholar]
- 4.Applebaum M.A., Worch J., Matthay K.K., Goldsby R., Neuhaus J., West J. Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer. 2011;117:3027–3032. doi: 10.1002/cncr.25840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung C.C., Kandel R.A., Bell R.S., Mathews R.E., Ghazarian D.M.D. Extraskeletal Ewing sarcoma in a 77-year-old woman. Arch Pathol Lab Med. 2001;125:1358–1360. doi: 10.5858/2001-125-1358-EESIAY. [DOI] [PubMed] [Google Scholar]
- 6.Gaspar N., Hawkins D.S., Dirksen U., Lewis I.J., Ferrari S., Le Deley M.C. Ewing sarcoma: current management and future approaches through collaboration. J Clin Oncol. 2015;33:3036–3046. doi: 10.1200/JCO.2014.59.5256. [DOI] [PubMed] [Google Scholar]
- 7.Saif M.W., Kaley K. Extraosseous Ewing's sarcoma of the pancreas: an uncommon but treatable disease. Cureus. 2017;9:e1882. doi: 10.7759/cureus.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rud N.P., Reiman H.M., Pritchard D.J., Frassica F.J., Smithson W.A. Extraosseous Ewing's sarcoma. A study of 42 cases. Cancer. 1989;64:1548–1553. doi: 10.1002/1097-0142(19891001)64:7<1548::aid-cncr2820640733>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 9.Boufettal M., Azouz M., Rouhi A.H., Mahfoud M., El Bardouni A., Berrada M.S. Extraosseous Ewing’s sarcoma of the leg: a case report. Scholars Acad J Biosci. 2014;2:510–512. [Google Scholar]
- 10.Bostelmann R., Leimert M., Steiger H.J., Gierga K., Petridis A.K. The importance of surgery as part of multimodal therapy in rapid progressive primary extraosseous Ewing sarcoma of the cervical intra- and epidural space. Clin Pract. 2016;6:897. doi: 10.4081/cp.2016.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar V., Singh A., Sharma V., Kumar M. Primary intracranial dural-based Ewing sarcoma/peripheral primitive neuroectodermal tumor mimicking a meningioma: a rare tumor with review of literature. Asian J Neurosurg. 2017;12:351–357. doi: 10.4103/1793-5482.185060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee R., Saardi K.M., Schwartz R.A. Lymphedema-related angiogenic tumors and other malignancies. Clin Dermatol. 2014;32:616–620. doi: 10.1016/j.clindermatol.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Ruocco V., Schwartz R.A., Ruocco E. Lymphedema: an immunologically vulnerable site for development of neoplasms. J Am Acad Dermatol. 2002;47:124–127. doi: 10.1067/mjd.2002.120909. [DOI] [PubMed] [Google Scholar]
- 14.Lynch M.D., Jones A.E., Marker A., Grant J.W., Purushotham A.D. Malignant eccrine poroma in breast cancer-related lymphoedema. Ann R Coll Surg Engl. 2004;86:W32–W35. doi: 10.1308/147870804155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baroni A., Russo T., Piccolo V., Siano M., Russo D., Nacca L. Opportunistic metastatic porocarcinoma after saphenous venectomy for coronary bypass surgery. Clin Exp Dermatol. 2013;38:507–510. doi: 10.1111/ced.12032. [DOI] [PubMed] [Google Scholar]
- 16.Fergusson C.M., Copeland S.A., Horton L. Unusual sarcoma arising in lymphoedema. J R Soc Med. 1985;78:497–498. doi: 10.1177/014107688507800618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peterson C.M., Lane J.E., Guill M.A. Merkel cell carcinoma after postmastectomy lymphedema. J Am Acad Dermatol. 2003;48:983. doi: 10.1067/mjd.2003.288. [DOI] [PubMed] [Google Scholar]
- 18.Balakrishnan R., Khairullah Q.T., Giraldo A., Provenzano R. Extraskeletal Ewing's sarcoma in a kidney transplant patient. Am J Kidney Dis. 1999;33:1164–1167. doi: 10.1016/S0272-6386(99)70157-5. [DOI] [PubMed] [Google Scholar]
- 19.Lyall E.G., Langdale-Brown B., Eden O.B., Mok J.Y., Croft N.M. Ewing's sarcoma in a child with human immunodeficiency virus (type 1) infection. Med Pediatr Oncol. 1993;21:127–131. doi: 10.1002/mpo.2950210209. [DOI] [PubMed] [Google Scholar]
- 20.Spunt S.L., Rodriguez-Galindo C., Fuller C.E., Harper J., Krasin M.J., Billups C.A. Ewing sarcoma-family tumors that arise after treatment of primary childhood cancer. Cancer. 2006;107:201–206. doi: 10.1002/cncr.21962. [DOI] [PubMed] [Google Scholar]
- 21.Fan P., Nong L., Sun J., Liu X., Kadin M.E., Li T. Primary cutaneous anaplastic large cell lymphoma with intralymphatic involvement associated with chronic lymphedema. J Cutan Pathol. 2017;44:616–619. doi: 10.1111/cup.12933. [DOI] [PubMed] [Google Scholar]