

Abstract
The autonomic nervous system is a powerful regulator of circulatory adjustments to acute hemodynamic stresses. Here we focus on new concepts that emphasize the chronic influence of the sympathetic and parasympathetic systems on cardiovascular pathology. The autonomic neurohumoral system can dramatically influence morbidity and mortality from cardiovascular disease through newly discovered influences on the innate and adaptive immune systems. Specifically, the end-organ damage in heart failure or hypertension may be worsened or alleviated by pro- or anti-inflammatory pathways of the immune system, respectively, that are activated through neurohumoral transmitters. These concepts provide a major new perspective on potentially life-saving therapeutic interventions in the deadliest of diseases.
Keywords: sympathetic, parasympathetic, hypertension, immune system, cytokines
the autonomic nervous system (ANS), through an enormous array of reflexes, can regulate almost all physiological functions in the body. This brief review focuses on the role of the ANS as a key chronic regulator of the pathological states of the cardiovascular system rather than on its role in homeostasis of the normal physiological state. The main message is that not only does the ANS act directly on the cardiovascular hemodynamic state, but it also is a powerful contributor to the development of cardiovascular pathology and disease through its regulation of the immune system.
Autonomic Dysregulation in Cardiovascular Disease
The beginnings of our understanding of the ANS in cardiovascular control can be traced back to the work of the Nobel Prize winner Corneille Heymans in 1938 for identifying the carotid sinus nerves (33) (Fig. 1). These are tiny baroreceptor and chemoreceptor nerves that sense minute changes in hemodynamic pressure and humoral factors and respond by activating autonomic reflexes transmitted by the sympathetic and parasympathetic neural outputs. The barosensory nerves in the carotid sinus and aortic arch exert powerful bidirectional regulation of hemodynamic states. On one hand, their activation can inhibit the sympathetic drive to prevent a surge in the blood pressure from causing a stroke, while, on the other hand, they can increase sympathetic activity if the arterial pressure falls.
Fig. 1.

Discovery of sensory receptors and autonomic neurotransmitters. Sensory afferents from the carotid body, carotid sinus, and aortic arch relay their signals through the petrosal and nodose ganglia to the nucleus tractus solitarius. The sympathetic ganglia neurons are the soma of the sympathetic efferent innervation releasing norepinephrine (NE). The parasympathetic cholinergic (ACh) innervation of the heart and gut originates in the dorsal motor nucleus of the vagus in the medulla (red lines).
The carotid body chemosensory nerves are activated by glomus cells that sense hypoxia and acidosis, release of ATP, acetylcholine (ACh), and other transmitters, and initiate reflex increases in ventilation as well as in sympathetic nerve activity.
Since the discovery of carotid and aortic baro- and chemoreceptors, it took 30 yr to define the efferent part, the neurotransmitters of the reflex responses, when Axelrod (6), Del Castillo and Katz (16), and Von Euler (80) identified ACh as a transmitter for the parasympathetic nerves and norepinephrine (NE) as a transmitter for the sympathetic nerves. Their studies elucidated how these neurotransmitters are stored, released, and degraded.
More recent work has focused on the molecular identity of the mechanosensitive ion channels in baroreceptor endings (2, 17, 18, 51) and the chemosensing channels in glomus cells of the carotid body (62, 64, 77). The clinical relevance of identifying these channels is in their contribution to the morbid increase in sympathetic nerve activity in heart failure and hypertension. In both of these fatal cardiovascular states, the reduced baroreceptor sensitivity and enhanced chemoreceptor sensitivity (1, 78) sustain the high sympathetic drive. Ongoing clinical trials attempt to activate the carotid sinus baroreceptors electrically or ablate the carotid body in patients (32, 61, 82) to decrease sympathetic nerve activity.
The powerful influence of the sympathetic nerves can be illustrated by the following examples, where the sympathetic nerve activity either is suppressed suddenly as in syncope, or is enhanced chronically in pathological states, as in heart failure and hypertension.
Sympathoinhibition and vagal stimulation during syncope.
The first example is that of the sudden suppression of the sympathetic activity. Prolonged standing under stressful situations often causes collapse and fainting. Intense anxiety can also induce fainting. It is estimated that most people have experienced similar episodes at some time in their lifetime. The sudden loss of motor tone is transient, as these individuals quickly recover their normal postures. Measurements of the sympathetic nerve activity in the peroneal nerve suggest that, immediately before the syncopal episode, the sympathetic activity is dramatically increased, and then it abruptly stops (Fig. 2). This sympathetic withdrawal is accompanied by bradycardia, vasodilatation, and a dramatic drop in the arterial pressure. Although one would expect that the fall in the arterial pressure should trigger the baroreceptor reflex to increase sympathetic activity, quite oppositely the sympathetic activity is inhibited (81).
Fig. 2.

Hemodynamics of syncope: sympathoinhibition, hypotension, bradycardia, and vasodilatation (finger plethysmography). The asterisk marks the sudden loss of sympathetic activity. [From Wallin and Sundlof (81) with permission from Elsevier.]
The reason is that, in addition to the baroreceptor regulation of the sympathetic and parasympathetic outflows, there are cardiac sensory afferents and other splanchnic vagal afferents that are sympathoinhibitory as well (1). Thus the vagus nerve does not simply carry an efferent parasympathetic cholinergic innervation; it also includes sensory fibers with nerve endings in the heart and other viscera that can be inhibitory. In addition to the mechanical activation of baroreceptors that reflexly decrease sympathetic drive, the signals from cardiac and other vagal afferent sensory receptors may also inhibit the sympathetic drive and accentuate the parasympathetic drive, causing vasodilatation, bradycardia, and hypotension. Hence these episodes of loss of consciousness are referred to as “vasovagal” syncope (2). Some of the episodes are referred to as “neurocardiogenic syncope” when the sympathoinhibitory sensory signal is originating in the heart, in cardiac afferents that may be the cause of syncope in patients with aortic stenosis (2, 56).
Sympathoexcitation in heart failure.
The second example of autonomic dysregulation illustrates a heightened sympathetic activity. It has been reported that patients with severe heart failure may have circulating blood levels of NE well over 800 pg/ml (13). These patients had a <10% chance of survival in 2 yr. However, patients with <400 pg/ml circulating levels of NE had a 50% survival chance in 5 yr. The levels of NE are powerful biomarkers of the excessive sympathetic activation and the mortality of patients in heart failure. A significant positive correlation between left ventricular filling pressures, plasma NE levels, and muscle sympathetic nerve activity was observed in patients in heart failure (46) (Fig. 3). Clearly, the baroreflexes are not effective in suppressing the fatal increase in sympathetic activity in congestive heart failure. This raises a question of how can the excessive sympathetic activity be inhibited to reverse heart failure. For a long time it was thought that it is not possible to induce and sustain inhibition of sympathetic activity by electrical nerve stimulation of the baroreceptor nerves (21). However, in 2004, Lohmeier et al. (50) demonstrated that stimulating the carotid sinus nerves in conscious dogs can cause bradycardia and hypotension that last for 1 wk. This study raised the possibility that excessive sympathetic drive may indeed be inhibited in patients over a prolonged period of time.
Fig. 3.

Muscle sympathetic nerve activity (SNA) in heart failure. Left: direct recordings of muscle SNA from the peroneal nerve in patients with heart failure are shown. SNA is highest in the patient with the most severe heart failure. MAP, mean arterial pressure; HR, heart rate; bpm, beats/min; CI, cardiac index. Right: bar graphs show the marked increase in muscle SNA in congestive heart failure (CHF) compared with age-matched controls. The SNA increases progressively with the left ventricular (LV) filling pressure. Values are means ± SE; n, no. of subjects. [Left panel from Ferguson et al. (21) with permission from Elsevier; right panel from Leimbach et al. (46) with permission from Wolters Kluwer Health.]
The following two examples in animal models of heart failure demonstrate that the autonomic system can be modulated to improve survival.
First is a landmark study of carotid sinus nerve stimulation in dogs with heart failure induced with chronic cardiac pacing (88). The plasma levels of NE in heart failure were markedly increased in the unstimulated group (Fig. 4), but remained low in dogs that also underwent carotid sinus nerve stimulation. Without stimulation of the carotid sinus nerves, the mortality was very high. In just ~40 days, heart failure was uniformly fatal, while the NE levels were very high. On the other hand, in the group that underwent carotid stimulation, the survival was impressively prolonged, and NE levels were reduced (Fig. 4).
Fig. 4.

Prolonged survival with sympathoinhibition and vagal stimulation (VS). Improved survival in heart failure in dogs with carotid sinus baroreceptor nerve stimulation (top left) and in rats with vagus nerve stimulation (top right) is shown. Bottom: the survival was dramatically prolonged and associated with significant reductions in norepinephrine (NE) levels (shaded line and bar). Values are means ± SE; nos. in parentheses, no. of animals. SS, sham stimulation. *P < 0.01. [Right panels from Li et al. (49) and left panels from Zucker et al. (88) with permission from Wolters Kluwer Health.]
In a second study using a rat model of heart failure after myocardial infarction (49), vagal nerve stimulation dramatically improved survival. Despite the relatively short period of vagal stimulation (between 0 and 40 days post-myocardial infarction), the protective effect was sustained over 4 mo. Improved survival was associated with reduced NE level during stimulation of the vagus nerve (Fig. 4).
Thus sympathoinhibition and parasympathetic activation improve survival in these two models of heart failure. In one model, this was achieved with baroreceptor stimulation, and in the other with vagus nerve stimulation. The question, then, is whether similar benefits can be attained in humans. As mentioned above, there are currently clinical trials that test the effectiveness of carotid sinus nerve stimulation and vagal nerve stimulation in patients with drug-resistant hypertension, and in heart failure.
Autonomic Regulation of the Immune System
How does the autonomic system influence cardiovascular pathology to affect survival?
A “conspiracy” triangle between the ANS, the immune system, and cardiovascular disease is proposed (Fig. 5) (3). Vertex A of the triangle represents increased mortality from autonomic dysregulation. Vertex B represents the inflammatory response that is integral to almost all cardiovascular manifestations of disease. Finally, and most relevant to this paper, vertex C defines how the ANS can modulate the immune system to either increase or reduce mortality. It is known that the immune system contributes significantly to atherosclerosis through deposition of foam cells, T cells, and dendritic cells in the atherosclerotic plaques. In heart failure, the cytokine storm causes significant centrally induced sympathetic drive. In atrial fibrillation, invasion of the atria by macrophages and monocytes can induce fibrosis and remodeling. In stroke, peroxiredoxins that are cytosolic antioxidants are released from ischemic cells and act as initiators of cytokine release from macrophages, causing postischemic inflammation and brain damage (23). Here the focus will be on hypertension and heart failure.
Fig. 5.
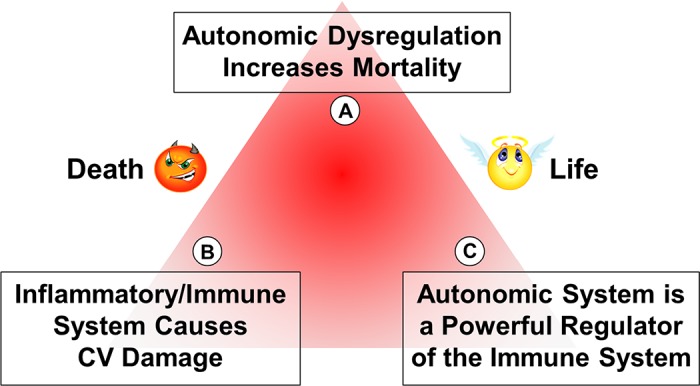
The neuroimmune triangle. The schematic portrays the fatal influences of autonomic dysregulation in A, the pathological damage of the immune system in B, and the powerful modulation of the immune response by the autonomic system in C. CV, cardiovascular.
Innate and adaptive immune modulation in hypertension.
The immune system comprises two broad components. The innate immune component, which includes monocytes, macrophages, and dendritic cells, “presents” the antigens to the second component, the T lymphocytes of the adaptive immune system. Both of these systems contribute to hypertension. The role of the immune system in hypertension was shown in a series of papers published by Svendsen’s laboratory as early as the 1970s. Those papers founded the concept of the immunogenetic basis of hypertension (74–76). The absence of the thymus (in athymic nude mice) prevents the onset of DOCA-salt hypertension, which suggests that the thymus is required for DOCA-salt hypertension (74, 75). The importance of the thymus in genetic hypertension was impressively demonstrated by Ba et al. (7). Their early work in the spontaneously hypertensive rat (SHR) demonstrated the delayed onset of hypertension associated with arterial lesions and a selective depression of T-cell function, including suppressor T cells, along with the appearance of thymocytotoxic auto-antibodies. They found that histo-compatible thymus grafts or injections of thymus extracts into neonatal SHR resulted in long-lasting recovery of suppressor T-cell immune functions, decrease in the titer of the auto-antibodies, and prevention of both the arterial lesions and the hypertension. Twenty-five years later, the importance of T cells was demonstrated in angiotensin II (ANG II)-induced hypertension (27). Adoptive transfer of wild-type (WT) T lymphocytes into RAG1−/− mice that lack both B and T cells and do not develop hypertension with ANG II infusion induces hypertension and causes significant infiltration of the vascular system with T lymphocytes (Fig. 6).
Fig. 6.

T-cell dependence of ANG II hypertension. Left: shows that vascular infiltration with CD3+ lymphocytes (purple-stained cells) and expression of T-cell receptors (TcR; red cells) are increased with ANG II infusion. Right: the pressor response to ANG II is restored in RAG1−/− mice with T-cell but not B-cell infusions. Values are means ± SE. *P < 0.01 vs. C57BL/6. [Modified from Guzik et al. (27) with permission.]
Role of IL-6 in ANG II hypertension.
T lymphocytes are activated by the innate immune system, which releases interleukin-6 (IL-6). IL-6 is a very potent inflammatory cytokine, and IL-6 knockout mice have a significantly suppressed hypertensive response during ANG II infusion (9, 45). The prevention of ANG II hypertension in IL-6−/− mice was not caused by a loss of the renal vasoconstrictor effect of ANG II, as ANG II caused significant vasoconstriction of the afferent renal arterioles of these mice as in WT mice (Fig. 7).
Fig. 7.

IL-6 dependence of ANG II hypertension. IL-6 knockout (KO) prevents ANG II-hypertension seen in wild type (WT; left), but constriction of afferent renal arterioles is preserved in the KO (right). [From Brand et al. (9) with permission from Wolters Kluwer Health.]
Yet another renal mechanism of IL-6-dependent ANG II hypertension is the phosphorylation of JAK2/STAT3 in the kidney (9). In the IL-6 knockout, the phosphorylation of JAK/STAT by ANG II is prevented and so is sodium reabsorption in the kidney, as well as the aldosterone-dependent ANG II hypertension (73).
A different IL-6-dependent component involved in ANG II hypertension is likely a central neural activation of JAK2/STAT3 pathways that was demonstrated in cultured rat brain astrocytes (40). If so, there is likely an IL-6 cytokine release in the hypothalamic area by the central microglial immune cells that drives central sympathetic activity and hypertension.
An intriguing interaction between IL-6 and IL-17 has been described in the ANG II-mediated aortic dissections (39). The transduction and activation of STAT3 signaling by IL-6 occurs via the T-helper lymphocyte 17-IL-17 axis in C57BL/6 mice (39).
Microglial activation in autonomic brain region.
The work of Santisteban et al. in Circ Res (68) has provided compelling evidence that microglial activation in autonomic brain regions represents a neuroinflammatory nidus in neurogenic hypertension. The concept is that proinflammatory bone marrow cells migrate to the hypothalamic paraventricular nucleus and activate and become part of the microglial/macrophage inflammatory central system (87). The release of cytokines, chemokines, and reactive oxygen species would lead to sustained and excessive sympathetic activity and hypertension.
The successful reconstitution of male Wistar-Kyoto (WKY) rats with bone marrow of male SHR results in a significant elevation of mean arterial pressure in the WKY (147 ± 16 mmHg) compared with 114 ± 2 mmHg in the WKY-WKY reconstitution (68, 87).
The effectiveness of minocycline, an oral antibiotic that inhibits microglial activation and attenuates hypertension in both SHR and ANG II-infused rats, in reducing central migration of bone marrow-derived cells supports the concept of central neurogenic inflammation in hypertension. The fact that minocycline has also been shown to be clinically effective in depression, in reducing gut mucosal damage in experimental colitis, and in enhancing morphine’s effectiveness in diabetic neuropathy has justified some of the ongoing clinical trials in drug-resistant hypertension (24, 25, 34).
Inflammatory immune suppression in heart failure.
The link between the immune system and hypertension and heart failure in humans has been addressed. With the availability of anti-tumor necrosis factor (TNF-α) agents, several trials have been carried out in patients with inflammatory autoimmune diseases, such as rheumatoid arthritis (86). Many such patients have associated hypertension. Although the systemic rheumatological manifestations of the disease may improve, the associated hypertension has worsened with prolonged administration of Etanercept (the recombinant TNF receptor that binds to TNF-α and neutralizes it) and Infliximab (the monoclonal antibody that binds to both soluble and transmembrane TNF-α) (60).
In heart failure, the inflammatory immune response is damaging, and TNF receptor-1 knockdown in subfornical organs, as well as inhibition of brain proinflammatory cytokines, were effectively protective in animal models (41, 84). In humans, however, the use of Etanercept was fraught with worsening of heart failure (12, 38, 55). Since TNF-α initiates its biological effects by binding to two distinct receptors, receptor 1 (R1; p55), which initiates a toxic response, and receptor 2 (R2; p75), which mediates a protective effect (57), the worsening of the patients in the anti-TNF-α clinical trials may reflect the suppression of the beneficial R2. This was shown in a murine model of myocardial infarction (57) and raises the possibility that anti-TNF-α R1 selective blockers might be more consistently effective in future clinical trials.
We believe that more promising results of clinical trials with monoclonal antibodies will be forthcoming, and the transfer of immunological information from rat to mouse to human will be much more predictable with technological advances. A compelling case in point is the most recent article from the New England Journal of Medicine (August 27, 2017) by Ridker et al. (65) showing that Canakinumab (monoclonal antibody targeting IL-1β) in 10,061 patients reduced cardiovascular mortality post-myocardial infarction, despite elevated cholesterol. Currently, the drug is approved for juvenile idiopathic arthritis and periodic fever syndrome. The authors also noted a decrease in lung cancer progression.
Sympathetic innervation of the immune system.
A direct link between the sympathetic nerves and the immune system exists. Direct ANG II administration in the central nervous system increases the splenic as well as renal sympathetic nerve activities, which are accompanied with expression of IL-1β, IL-2, IL-6, IL-16, and TGF-β (Fig. 8). However, denervation of the spleen abrogated the cytokine responses to central administration of ANG II but did not affect the renal nerve activity. This suggests a direct link between sympathetic nerve activity and the activation of the immune system in the spleen (22).
Fig. 8.

ANG II-induced splenic sympathetic overactivity enhances gene expression of cytokines. Central ANG II infusion increases splenic and renal sympathetic nerve discharge (SND; left, tracings) and activates immune system (right; bar graphs). Increased cytokine gene expression in spleen with ANG II (light shaded bars) vs. artificial cerebrospinal fluid (aCSF; solid bars) is reduced significantly with splenic denervation (dark shaded bars). Values are means ± SE. *Significantly different from aCSF-treated splenic-intact and ANG II-treated splenic-denervated, P < 0.05. †Significantly different from ANG II-treated splenic-denervated, P < 0.05. [From Ganta et al. (22).]
The lymphoid organs are innervated by sympathetic nerves that release NE at the nerve terminals and may induce immune activation (20). Immune cells, including T and B lymphocytes, predominantly express β2-adrenergic receptors (66, 67). Activation of T and B cells in the presence of NE enhances cytokine and IgG secretion, respectively (59). Thus increased sympathetic nerve activity has an immune-enhancing proinflammatory effect. Another possible mechanism of sympatho-mediated immune regulation is the NE-induced increased production of reactive oxygen species in immune cells. A redox-regulated suppressor of splenic T-lymphocyte activation has been reported in a model of sympathoexcitation (11).
In another model, myocardial infarction increases sympathetic nerve activity to the bone marrow, causing release of hematopoietic stem cells. These bone marrow-derived hematopoietic cells migrate to the spleen, where monocyte production is accelerated (Fig. 9). The monocytes/macrophages then migrate to the periphery and to the endothelium of the coronary vessels and magnify the thrombotic effects and the risk of further myocardial infarction (19). There is clear indication in patients that activation of β3-adrenergic receptor in bone marrow increases the intravascular migration of monocyte and hematopoietic stem cells.
Fig. 9.

Myocardial infarction (MI) increases sympathetic activity to bone marrow. Schematic shows that MI accelerates atherosclerosis by an immunological process involving activation of the sympathetic nervous system (SNS) to the bone marrow (BM), releasing hematopoietic, stem, and progenitor cells, and activation of the immune system of the spleen and migration of macrophages and monocytes to the coronaries. SCF, stem cell factor. [From Dutta et al. (19) with permission from Macmillan Publishers Ltd.]
The circadian oscillations of the sympathetic nerve activity can also regulate the transendothelial movements of monocytes and macrophages and govern the recruitment of hematopoietic, stem, and progenitor cells to the bone marrow and leukocytes to peripheral tissues or organs. These effects are exaggerated during increased sympathetic activity as a result of the activation of endothelial adhesion molecules. Increased transendothelial migration may increase inflammatory responses in skeletal muscle, whereas, in the bone marrow, the migration may improve bone marrow engraftment. Scheiermann et al. (69) have also shown a diurnal difference in the engraftment of bone marrow. On adoptive transfer, bone marrow engraftment in lethally irradiated rodents is more effective if the bone marrow transfer is done at night during the nocturnal increase in sympathetic activity or during the infusion of isoproterenol. It will be interesting to see whether this enhanced engraftment occurs in humans.
The recent attempts to treat drug-resistant hypertension with renal denervation in patients raises the question of whether the sympathetic innervation of the kidney contributes directly or indirectly to immune activation and hypertension. Such an effect might contribute to the drop in arterial pressure in the respondents to denervation. Xiao et al. (83) had reported an anti-inflammatory response to renal denervation. We also observed that renal denervation abrogates nicotine-induced premature hypertension in SHR and reduces renal macrophage migration (30). In confirmation, Zaldivia et al. (85) recently reported, in Hypertension, compelling evidence that renal denervation may indeed be associated with reduced monocyte activation.
Parasympathetic modulation of the immune system.
The parasympathetic nervous system has considerable effects on immunity that are even more intriguing. When the subdiaphragmatic vagi of NF-κB reporter mice were severed, the in vivo luminescence from NF-κB promoter-driven luciferase expression was markedly increased around the gut and the colon 4 wk after the denervation (58). Moreover, lymphocytes obtained from the spleens after vagotomy show proinflammatory increases in expression of interferon-γ, TNF-α, and IL-6 on CD4-T-cell activation (Fig. 10). Thus the parasympathetic nervous system must exert a constitutive tonic inhibition of inflammatory responses and NF-κB activation in the gut that are unmasked on vagotomy (42). This is a remarkable demonstration of an intrinsic anti-inflammatory influence of the vagi.
Fig. 10.

Tonic suppressive vagal influence on the immune system. Vagotomy increases NF-κB expression at 2 and 4 wk (W) after surgery (A) and enhances cytokine release from CD4+ T-cell activation (solid bars vs. open bars; B). Values are means ± SE. *P < 0.05 and **P < 0.005 (A). *P < 0.01 (B). [panel B was modified from Karimi et al. (42) with permission from Elsevier.]
In another study, Tracey et al. (8, 79) stimulated the vagi in mice that were given lipopolysaccharides (LPS). LPS, the cell wall component of gram-negative bacteria, causes septic shock from excessive inflammatory response through activation of toll-like receptor (TLR)-4 to produce cytokines such as TNF-α. Administration of LPS to mice results in a marked drop in the arterial blood pressure (Fig. 11). Moreover, administration of LPS to vagotomized mice causes an even more dramatic drop in blood pressure as seen in septic shock. In contrast, there is significant abrogation of LPS-induced hypotension in mice that are undergoing electrical stimulation of the vagus nerve.
Fig. 11.

Vagal nerve stimulation attenuates the LPS-induced TNF response and endotoxin shock. Left: schematic shows vagal efferent activation of the reticuloendothelial system and cholinergic (ACh) receptors on macrophage. Right, A–C: show that vagal denervation (VGX) worsens the fall in mean arterial blood pressure (MABP) and enhances the serum and liver TNF responses to LPS (hatched bars). Vagal nerve stimulation (STIM) attenuates the LPS-induced TNF responses and prevents hypotension, endotoxin, and shock (shaded bars and line). Values are means ± SE. *P < 0.05 and **P < 0.005 vs. sham + LPS. #P < 0.05 vs. VGX + LPS. [Right panel was modified from Borovikova et al. (8) with permission from Macmillan Publishers Ltd.]
It is noteworthy that, despite the immunoregulatory effects of the nervous system on cytokine production by the innate immune cells, the details of the autonomic circuitry innervating the spleen are disputed (5). In particular, Bratton et al. (10) tested the vagal innervation of the spleen in a series of studies and concluded that the vagus nerve does not directly innervate the spleen. The mechanism by which sympathetic nerves may induce cholinergic activation in the spleen deserves further consideration.
However, vagi innervate the splanchnic bed and most of the splanchnic organs and must modulate and suppress the inflammatory response of the extensive gut immune system. The vagal influence is sufficiently powerful to suppress the response to activation of the TLRs by LPS. This modulation of the inflammatory response occurs at the level of macrophages (8, 79). Macrophages express cholinergic receptors on their membranes (Fig. 11). It appears that the α7-nicotinic acetylcholinergic receptors that are activated during the stimulation of the vagus nerve can be protective against septic shock by suppressing the activation of macrophages. But how is that suppression of macrophage inflammatory response to LPS relevant to hypertension?
Relevance of the cholinergic anti-inflammatory response to hypertension.
In a two-kidney, one-clip model of hypertension, it was observed that, in α7-nicotinic receptor knockout mice, there were greater increases in IL-6, IL-1β, and TNF-α compared with the WT mice (Fig. 12) (48). End-organ damage was also more pronounced with increased cardiac fibrosis and renal glomerulosclerosis. Thus the damage in two-kidney, one-clip hypertensive mice is accentuated in the absence of α7-nicotinic cholinergic receptor (Fig. 12). The ANS modulates the expression of proinflammatory cytokines and immune cell proliferation (43). In addition to the macrophages, α7-nicotinic cholinergic receptors can also modulate lymphocyte activation (15). These studies further underline the pathological significance of the autonomic regulation of the immune system.
Fig. 12.

α7-Nicotinic receptors are protective in two-kidney-one-clip (2K1C) hypertension. Proinflammatory state (IL-6, IL-Iβ, and TNF-α), as well as cardiac hypertrophy [left ventricular weight/body weight (LVW/BW)] and glomerulosclerosis in 2K1C hypertension (WT; solid bars), are enhanced in α7-nicotinic receptors of KO mice (shaded bars). nAChR, nicotinic ACh receptor. Values are means ± SE. *P < 0.05 and **P < 0.01 compared with sham (unpaired t-test). #P < 0.05 compared with WT (unpaired t-test). [From Li et al. (48) with permission from Wolters Kluwer Health.]
An important question is whether an abnormality of the immune system may precede hypertension and be causative in the development of hypertension. More specifically, does the SHR that develops hypertension several weeks after birth have an intrinsic abnormality of the innate immune system that initiates the hypertension?
Abnormal Immune System in Hypertension
As mentioned above, blood pressure in SHR is dependent on the thymus. Thymus grafts or injection of thymus extracts into neonatal SHR restored suppressor T-cell immune function and delayed or prevented hypertension (7). Although thymus is required for normal establishment of the T cells that form the adaptive immune system, these cells are dependent on the innate immune system for their activation. The recent work from our laboratories has addressed the following question.
Do the innate immune cells of SHR display an inflammatory response on activation of their TLRs?
The innate immune cells express TLRs through which they can be induced to produce proinflammatory cytokines. They also express the receptors that enable the ANS to influence the inflammatory response. This regulation is an important mechanism, because the production of cytokines by the innate system initiates the process of activation of the adaptive immune cells, such as T-helper (Th) and T-regulatory cells, which migrate to peripheral organs, such as the brain, the vascular system, the heart, and the kidney, and induce end-organ damage (Fig. 13). Thus the inflammatory signal pathway can be modulated in a very effective way by the nicotinic cholinergic receptor or the ANG receptor to modify the amount and type of cytokines that are released and, in turn, induce T-cell activation, invasion of the tissues, and the development of hypertension (Fig. 14). ANG II-induced sympathetic nerve activity is proinflammatory, and, by contrast, the parasympathetic cholinergic nerve activity is anti-inflammatory. Together, these two autonomic outputs regulate the immune system through their effects on the innate immune cells.
Fig. 13.
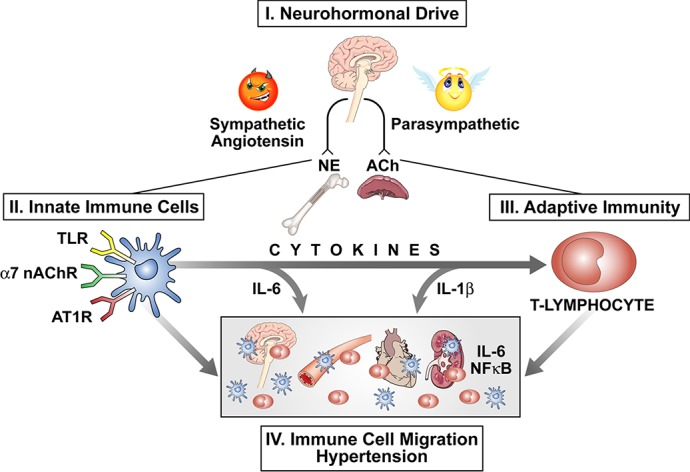
Pathways of autonomic modulation of inflammation cells and their migration to end organs. Schematic portrays the proinflammatory influence of angiotensin II and increased sympathetic neurohumoral drive (NE) balanced off by an anti-inflammatory influence of parasympathetic activity (ACh). Receptors on the innate immune cells modulate cytokine release that activates the adaptive immune system and the migration of immune cells to end organs, causing hypertension. nAChR, nicotinic ACh receptor; AT1R, angiotensin II type 1 receptor.
Fig. 14.
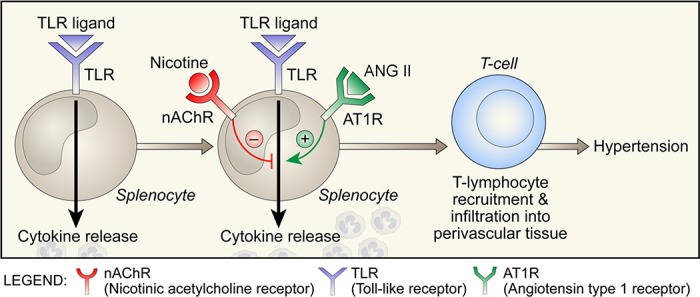
Regulation of cytokine release from immune cells during toll-like receptor (TLR) activation. Schematic shows opposing influences of activation of α7-nAChR by nicotine and AT1R by ANG II on cytokine release.
Does ANG II or the cholinergic transmitter nicotine modulate the response of innate immune cells in culture?
There are more than a dozen TLRs that are activated on innate immune cells by multiple specific exogenous ligands that represent the molecular patterns of a class of pathogens, such as bacteria, fungi, or viruses. TLRs can also recognize endogenous ligands that are relevant to autoimmunity. The endogenous ligands are generated by injury or cell damage; for example, in myocardial infarction, the cellular debris and released cellular DNA may function as ligands for the TLRs. On activation by their respective ligands, the TLR signaling pathways lead to activation of the NF-κB transcription factor and expression of a battery of proinflammatory genes, including cytokines. The interactions of the inflammatory pathways with pathways of activation of ANG receptors or ACh receptors regulate the intensity of cytokine production.
To test this conceptual model, splenocytes from WKY and SHR were isolated and exposed to various ligands of the TLRs in the presence or absence of either ANG II or nicotine. More specifically, splenocytes were stimulated with three doses of several TLR ligands, and the release of TNF-α, IL-10, and IL-6 was measured (Fig. 15) (31).
Fig. 15.

Cytokine responses of splenocytes to activation of TLR effects of specific TLR ligands on TNF-α (A), IL-10 (B), and IL-6 (C) are dose related and equivalent in WKY (open bars) and SHR (shaded bars) splenocytes. Direct effects of nicotine (Nic) and ANG II are negligible. Values are means ± SE. LTA, lipoteichoic acid; PIC, poly I:C; Flag, flagellin; CpG, CpG2395. [From Harwani et al. (31) with permission from Wolters Kluwer Health.]
TLR activation is enhanced by nicotine and ANG II in SHR.
Two important observations were immediately obvious from the results. First, there was a dose-dependent response of the cytokine production to the ligands, and, second, there was significant variability in the amount of cytokine release, depending on which TLR was activated (Fig. 15). Neither ANG II nor nicotine alone activated the cytokine production (Fig. 15). However, the release of cytokines by activation of TLR-7/8 with Cl097 and TLR-9 with CpG was significantly reduced by nicotine in WKY splenocytes (31) (Fig. 16). Thus nicotine has an ex vivo anti-inflammatory effect that reduces the release of cytokines by immune cells of the normotensive WKY. In contrast, SHR splenocytes showed significant enhancement of the cytokines by TLR activation in the presence of nicotine. It is notable that these splenocytes were from prehypertensive SHR. These results show that nicotine has an anti-inflammatory effect on normotensive WKY, whereas it has a proinflammatory effect in SHR.
Fig. 16.

Markedly enhanced IL-6 responses to TLR by nicotine (A and B) and ANG II (C and D) in SHR splenocytes (B and D) and not in WKY (A and C). IL-6 release in response to TLR 7 (Cl097) and TLR9 (CpG) ligands ex vivo is inhibited by nicotine in WKY (solid bars) and markedly enhanced in SHR (shaded bars). ANG II also enhances the response in SHR but not in WKY. Values are means ± SE. ***P < 0.001 and **P < 0.01 indicate significantly different responses to TLR alone (open bars) vs. TLR with exposure to either nicotine or ANG II (solid and shaded bars). [From Harwani et al. (31) with permission from Wolters Kluwer Health.]
ANG II had no effect on TLR-induced cytokine production in the WKY splenocytes. Yet again, in SHR, there was an exaggerated inflammatory response to TLR activation. These results show that both nicotine and ANG have a proinflammatory effect in SHR immune cells even before the onset of hypertension. The mechanisms of these differences between the normotensive WKY and the SHR are complex and will require further study.
In vivo inflammatory responses to nicotine and ANG II in SHR.
The in vitro effect of nicotine on the isolated splenocytes was reproduced in vivo. Nicotine was infused in young prehypertensive SHR for 24 h, and 4 h before the end of the 24-h period the TLR-7/8 ligand Cl097 was injected. The sera and tissues from these rats were collected. In WKY, infusion of nicotine alone did not increase IL-6 or IL-1β. However, injection of TLR-7/8 ligand Cl097 increased the cytokine release, but the infusion of nicotine resulted in a reduced cytokine response to injection of Cl097 in WKY (31). In contrast, in the SHR, infusion of nicotine enhanced the proinflammatory cytokine release on Cl097 injection (Fig. 17). Clearly the immune cells of SHR lack the anti-inflammatory suppressive effect of nicotine that is seen in the WKY. Not only was the responsiveness of the SHR immune cells to TLR-7/8 stimulation increased in the presence of nicotine (31), but their number was also increased (Fig. 18). This proliferative response of CD161+CD8+ cells in SHR was not seen in WKY splenocytes.
Fig. 17.

In vivo cytokine responses to Cl097. In vivo responses of serum levels of IL-6 and IL-1β to TLR7 ligand (Cl097) are increased in SHR (solid bars) and suppressed in WKY (shaded bars) by nicotine. Nos. above each bar graph, no. of animals in the particular group. Values are means ± SE. *P < 0.05. **P < 0.01. [From Harwani et al. (31) with permission from Wolters Kluwer Health.]
Fig. 18.

Cell sorting of activated macrophages in SHR vs. WKY. Activated macrophages (CD161+, CD8+) are increased in SHR vs. WKY and increase further with nicotine in SHR. [From Harwani et al. (31) with permission from Wolters Kluwer Health.]
Abnormal immune cell phenotype in SHR.
The large population of CD161+ cells in SHR was further investigated to determine a connection between the development of hypertension and the number of CD161+ cells (Fig. 19, A and B). First, the density of CD161+ cells in the spleens of WKY and SHR from birth (1 day old) to full adulthood (38 wk old) was compared (71).
Fig. 19.

Pronounced increases of CD161+ splenocytes in SHR over WKY is age related and antecedes hypertension. A: age-related increases in CD161+ cells in splenocyte population are associated with increases in arterial pressure in SHR (shaded bars) vs. WKY (black bars). B: the increases of CD161+ cells in SHR over WKY are not caused by an increase in the splenocyte population and spleen size (C) nor on CD4+ (D) or CD8+ cells (E). Values are means ± SE. [From Singh et al. (71) with permission from Elsevier.]
In SHR spleens, the CD161+ cell number was significantly greater at birth than in WKY and continued to increase for 5 wk before hypertension developed. Moreover, the number of CD161+ cells dramatically increased with aging, reaching to one-third of the splenocyte population, and was associated with the progressive increase in arterial pressure (71). Thus the CD161 cell surface marker is expressed in an abnormally high number of immune cells in SHR, and its expression precedes the development of hypertension. This increase in CD161+ cells is not from increased spleen size and is also independent of CD4 or CD8 cell surface markers, as the number of CD4+ or CD8+ cells was comparable in WKY and SHR (Fig. 19, C–E). The increased number of CD161+ cells was also evident in the increased infiltration of other organs, such as kidney and aorta (Fig. 20). When SHRs were infused with nicotine for 24 h, the subset of CD4+CD161+ cells increased in spleens. The identity and role of the CD161+ cells in SHR hypertension was further investigated (71).
Fig. 20.

End-organ overexpression of CD161+ cells in SHR. Overexpression of CD161+ cell population in kidney and aorta of SHRs vs. WKYs is shown. FSC, forward scatter. [From Singh et al. (71) with permission from Elsevier.]
CD161, RORγt, and Th17 molecular markers of inflammatory cells in SHR.
The CD161 marker was originally identified as the human homolog of the NKRP1 glycoproteins expressed on rodent natural killer cells. Later, it was found to be expressed on several different cell types. This marker is significantly upregulated in CD4+ Th17 cells that express the master regulator transcription factor retinoic acid receptor (RAR)-related orphan receptor γ t (RORγt) and secrete cytokines IL-17A and IL-17F (37, 71). In fact, CD161 is considered as one of the defining markers of CD4+ Th17 cells (14, 54).
Th17 cells and IL-17 cytokines are involved in several autoimmune diseases, including Crohn’s disease, psoriasis, and lupus, as well as in ANG II hypertension and end-organ damage (52, 53). Since hypertension entails several features of an autoimmune disorder (29, 63), the CD161+ Th17 cells are likely components of the inflammation response in SHR hypertension. Moreover, SHR splenocytes constitutively express high levels of RORγt transcription factor, predominantly in the CD4+ cells (71) (Figs. 21 and 22). When splenocytes are programmed into Th17 lineage by in vitro T-cell receptor activation with an agonistic anti-CD3 antibody in the presence of TGF-β and IL-6, both WKY and SHR cells induced similar levels of Il-17a RNA, but SHR spleen cells had significantly greater expression of Il-17f RNA (Fig. 22). Thus, although the genes for Il-17a and Il-17f are transcribed by RORγt, they must be regulated differently in SHR vs. WKY (4, 26).
Fig. 21.

Importance of RORγt in Th17 programming. Activated innate immune cells induce Th17 programming of naive CD4+ T cells that includes overexpression of RORγt. MHC, myosin heavy chain; TCR, T-cell receptor. [From Singh et al. (71) with permission from Elsevier.]
Fig. 22.

Pronounced gene expression of RORγt and Il-17f in SHR vs. WKY. Proinflammatory cytokine gene induction of splenocytes into Th17 lineage by T-cell receptor activation includes a marked overexpression of RORγt and an enhanced response of Il-17f, but not Il-17a, in SHR compared with WKY. Values are means ± SE. Untr, untreated. *Statistically significant difference (P < 0.05). [From Singh et al. (71) with permission from Elsevier.]
In vivo, the Th17 polarization is prompted by activation of T-cell receptors with dendritic cells. A distinctive feature of dendritic cells is high expression of TLR3. So, when rats were treated with an agonist of TLR3 (poly-IC), the expression of Il-17a was modestly increased in both WKY and SHR. However, the expression of Il-17f was considerably more enhanced in SHR than in WKY. This increase in Il-17f was significantly reduced by digoxin, a blocker of RORγt (35), which also reduced the hypertension when given over several weeks to SHRs.
These results indicate that RORγt in SHR has a greater potential to produce proinflammatory IL-17F and to a much greater extent than IL-17A cytokines, which must be contributing to the hypertension.
Selective endothelial dysfunction by IL-17F.
Immune mechanisms, including IL-17 cytokines, may cause vascular damage (53, 70). RORγt is also known to promote glomerulonephritis through increased immune cell infiltration and cytokine production in renal tissue (72). IL-17A and IL-17F are the closest members of the proinflammatory IL-17 cytokine family; they share significant sequence homology. Both are secreted as homodimers and also form IL-17A/IL-17F heterodimers. However, these cytokines have distinct as well as overlapping roles in highly pathogenic inflammatory diseases, including hypertension and end-organ damage (28).
We tested endothelium-dependent vascular relaxation in WKY rat aortic rings following treatment with IL-17A or IL-17F. IL-17F treatment impaired ACh-induced, endothelium-dependent vasorelaxation (71) (Fig. 23A). However, sodium nitroprusside-induced nonendothelial vasorelaxation was not affected in IL-17F (Fig. 23B). In contrast, IL-17A treatment did not affect endothelium-dependent vasorelaxation, even at a higher concentration (Fig. 23C), which actually enhanced sodium nitroprusside vasorelaxation (Fig. 23D). Despite their structural similarity and sharing of receptors (44), IL-17A and IL-17F display significant differences in their activities (36, 47) (Fig. 23).
Fig. 23.

Effects of IL-17A (C and D) and IL-17F (A and B) on endothelial dysfunction. Selective loss of cholinergic (ACh) endothelial-dependent vasorelaxation is seen with IL-17F (A) but not IL-17A (C). Neither 17F (B) nor 17A (D) reduced nonendothelial-mediated relaxation with sodium nitroprusside (SNP). Values are means ± SE. *Statistically significant difference (P < 0.05). [From Singh et al. (71) with permission from Elsevier.]
Summary
-
1.
The ANS exerts a powerful modulatory influence on the immune system with proinflammatory morbid cardiovascular consequences.
-
2.
Vagus nerve activity provides a protective anti-inflammatory nicotinic cholinergic effect mediated by α7-nicotinic cholinergic receptors.
-
3.
In a genetic model of hypertension (SHR), the anti-inflammatory effect of nicotine on innate immune cells is reversed to a proinflammatory response before the onset of hypertension.
-
4.
An excessively large population of CD161+ splenocytes is present in SHR in the neonatal state and increases with age as hypertension develops and progresses.
-
5.
RORγt, the master regulator transcription factor of Th17 cells, is overexpressed in CD161+CD4+ in SHR, which, when induced by a TLR-3 agonist, express high levels of the proinflammatory IL-17F cytokine.
-
6.
ACh-induced endothelium-dependent vasorelaxation is significantly impaired by IL-17F.
Conclusion
The ANS is a powerful regulator of the immune system. The innate immune system in genetic hypertension is abnormally induced by the ANS to trigger proinflammatory responses to endogenous antigens. The findings implicate a dysregulated immune system with increased IL-17F production by the overexpressed RORγt transcription factor in the genetic hypertension seen in SHR. These provoke pathological renal and vascular changes that initiate and sustain the hypertensive state.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-14388 and HL-07121.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F.M.A. and M.V.S. conceived and designed research; F.M.A. and M.V.S. analyzed data; F.M.A. and M.V.S. interpreted results of experiments; F.M.A. drafted manuscript; F.M.A. and M.V.S. edited and revised manuscript; F.M.A. and M.V.S. approved final version of manuscript; F.M.A. and M.V.S. performed experiments; F.M.A. and M.V.S. prepared figures.
ACKNOWLEDGMENTS
This review article is partly a transcript of a lecture presented by F. M. Abboud at the Neuroscience Refresher Course of the Experimental Biology Meeting 2015 entitled, “It’s All in Your Head–Brain and Systems Control.” Dr. Abboud’s presentation was entitled, “The Brain and the Immune System.” Coauthors on the work at the University of Iowa (3, 31, 71) included Z. K. Ballas, M. W. Chapleau, M. Z. Cicha, S. C. Harwani, K. J. Irani, S. Kumar, and D. K. Meyerholz. We thank Angela Hester for help in preparation of the manuscript, and Shawn Roach for artistic contributions to the figures.
REFERENCES
- 1.Abboud F, Thames MD, Mark AL. Role of cardiac afferent nerves in regulation of circulation during coronary occlusion and heart failure. In: Disturbances in Neurogenic Control of the Circulation, edited by Abboud FM, Fozzard HA, Gilmore JP, Reid DJ. Bethesda, MD: American Physiological Society, 1981, p. 65–86. [Google Scholar]
- 2.Abboud FM. Ventricular syncope: is the heart a sensory organ? N Engl J Med 320: 390–392, 1989. doi: 10.1056/NEJM198902093200609. [DOI] [PubMed] [Google Scholar]
- 3.Abboud FM, Harwani SC, Chapleau MW. Autonomic neural regulation of the immune system: implications for hypertension and cardiovascular disease. Hypertension 59: 755–762, 2012. doi: 10.1161/HYPERTENSIONAHA.111.186833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adamik J, Henkel M, Ray A, Auron PE, Duerr R, Barrie A. The IL17A and IL17F loci have divergent histone modifications and are differentially regulated by prostaglandin E2 in Th17 cells. Cytokine 64: 404–412, 2013. doi: 10.1016/j.cyto.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson C, McKinley M, Martelli D, and McAllen R.. Letter to the editor: Parasympathetic innervation of the rodent spleen? Am J Physiol Heart Circ Physiol 309: H2158, 2015. doi: 10.1152/ajpheart.00766.2015. [DOI] [PubMed] [Google Scholar]
- 6.Axelrod J. Studies on sympathomimetic amines. II. The biotransformation and physiological disposition of d-amphetamine, d-p-hydroxyamphetamine and d-methamphetamine. J Pharmacol Exp Ther 110: 315–326, 1954. [PubMed] [Google Scholar]
- 7.Ba D, Takeichi N, Kodama T, Kobayashi H. Restoration of T cell depression and suppression of blood pressure in spontaneously hypertensive rats (SHR) by thymus grafts or thymus extracts. J Immunol 128: 1211–1216, 1982. [PubMed] [Google Scholar]
- 8.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405: 458–462, 2000. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 9.Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 56: 879–884, 2010. doi: 10.1161/HYPERTENSIONAHA.110.158071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bratton BO, Martelli D, McKinley MJ, Trevaks D, Anderson CR, McAllen RM. Neural regulation of inflammation: no neural connection from the vagus to splenic sympathetic neurons. Exp Physiol 97: 1180–1185, 2012. doi: 10.1113/expphysiol.2011.061531. [DOI] [PubMed] [Google Scholar]
- 11.Case AJ, Zimmerman MC. Redox-regulated suppression of splenic T-lymphocyte activation in a model of sympathoexcitation. Hypertension 65: 916–923, 2015. doi: 10.1161/HYPERTENSIONAHA.114.05075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT; Anti-TNF Therapy Against Congestive Heart Failure Investigators . Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 107: 3133–3140, 2003. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 13.Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB, Rector T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 311: 819–823, 1984. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 14.Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F, Rodolico G, Querci V, Abbate G, Angeli R, Berrino L, Fambrini M, Caproni M, Tonelli F, Lazzeri E, Parronchi P, Liotta F, Maggi E, Romagnani S, Annunziato F. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med 205: 1903–1916, 2008. doi: 10.1084/jem.20080397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Rosa MJ, Dionisio L, Agriello E, Bouzat C, Esandi MC. Alpha 7 nicotinic acetylcholine receptor modulates lymphocyte activation. Life Sci 85: 444–449, 2009. doi: 10.1016/j.lfs.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 16.Del Castillo J, Katz B. Quantal components of the end-plate potential. J Physiol 124: 560–573, 1954. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Driscoll M, Chalfie M. The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 349: 588–593, 1991. doi: 10.1038/349588a0. [DOI] [PubMed] [Google Scholar]
- 18.Drummond HA, Price MP, Welsh MJ, Abboud FM. A molecular component of the arterial baroreceptor mechanotransducer. Neuron 21: 1435–1441, 1998. doi: 10.1016/S0896-6273(00)80661-3. [DOI] [PubMed] [Google Scholar]
- 19.Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, Iwamoto Y, Thompson B, Carlson AL, Heidt T, Majmudar MD, Lasitschka F, Etzrodt M, Waterman P, Waring MT, Chicoine AT, van der Laan AM, Niessen HW, Piek JJ, Rubin BB, Butany J, Stone JR, Katus HA, Murphy SA, Morrow DA, Sabatine MS, Vinegoni C, Moskowitz MA, Pittet MJ, Libby P, Lin CP, Swirski FK, Weissleder R, Nahrendorf M. Myocardial infarction accelerates atherosclerosis. Nature 487: 325–329, 2012. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Felten DL, Felten SY, Carlson SL, Olschowka JA, Livnat S. Noradrenergic and peptidergic innervation of lymphoid tissue. J Immunol 135, Suppl: 755s–765s, 1985. [PubMed] [Google Scholar]
- 21.Ferguson DW, Berg WJ, Sanders JS, Kempf JS. Clinical and hemodynamic correlates of sympathetic nerve activity in normal humans and patients with heart failure: evidence from direct microneurographic recordings. J Am Coll Cardiol 16: 1125–1134, 1990. doi: 10.1016/0735-1097(90)90544-Y. [DOI] [PubMed] [Google Scholar]
- 22.Ganta CK, Lu N, Helwig BG, Blecha F, Ganta RR, Zheng L, Ross CR, Musch TI, Fels RJ, Kenney MJ. Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am J Physiol Heart Circ Physiol 289: H1683–H1691, 2005. doi: 10.1152/ajpheart.00125.2005. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Bonilla L, Iadecola C. Peroxiredoxin sets the brain on fire after stroke. Nat Med 18: 858–859, 2012. doi: 10.1038/nm.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garrido-Mesa N, Camuesco D, Arribas B, Comalada M, Bailón E, Cueto-Sola M, Utrilla P, Nieto A, Zarzuelo A, Rodríguez-Cabezas ME, Gálvez J. The intestinal anti-inflammatory effect of minocycline in experimental colitis involves both its immunomodulatory and antimicrobial properties. Pharmacol Res 63: 308–319, 2011. doi: 10.1016/j.phrs.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 25.Garrido-Mesa N, Utrilla P, Comalada M, Zorrilla P, Garrido-Mesa J, Zarzuelo A, Rodríguez-Cabezas ME, Gálvez J. The association of minocycline and the probiotic Escherichia coli Nissle 1917 results in an additive beneficial effect in a DSS model of reactivated colitis in mice. Biochem Pharmacol 82: 1891–1900, 2011. doi: 10.1016/j.bcp.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Gomez-Rodriguez J, Sahu N, Handon R, Davidson TS, Anderson SM, Kirby MR, August A, Schwartzberg PL. Differential expression of interleukin-17A and -17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity 31: 587–597, 2009. doi: 10.1016/j.immuni.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guzik TJ, Mikolajczyk T. In search of the T cell involved in hypertension and target organ damage. Hypertension 64: 224–226, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03340. [DOI] [PubMed] [Google Scholar]
- 29.Harrison DG, Guzik TJ, Goronzy J, Weyand C. Is hypertension an immunologic disease? Curr Cardiol Rep 10: 464–469, 2008. doi: 10.1007/s11886-008-0073-6. [DOI] [PubMed] [Google Scholar]
- 30.Harwani S, Raikwar N, Ratcliff J, Allamargot C, Chapleau M, Abboud F. Renal denervation prevents cholinergic mediated hypertension and renal macrophage infiltration. In: Proceedings of the AHA Council on Hypertension, AHA Council on Kidney in Cardiovascular Disease, American Society of Hypertension, Joint Scientific Sessions 2017. Dallas, TX: American Heart Assoc., 2017. [Google Scholar]
- 31.Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res 111: 1190–1197, 2012. doi: 10.1161/CIRCRESAHA.112.277475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heusser K, Tank J, Engeli S, Diedrich A, Menne J, Eckert S, Peters T, Sweep FC, Haller H, Pichlmaier AM, Luft FC, Jordan J. Carotid baroreceptor stimulation, sympathetic activity, baroreflex function, and blood pressure in hypertensive patients. Hypertension 55: 619–626, 2010. doi: 10.1161/HYPERTENSIONAHA.109.140665. [DOI] [PubMed] [Google Scholar]
- 33.Heymans C, Bouckaert JJ. Sinus caroticus and respiratory reflexes. I. Cerebral blood flow and respiration. Adrenaline apnoea. J Physiol 69: 254–266, 1930. doi: 10.1113/jphysiol.1930.sp002648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu P, Thinschmidt JS, Yan Y, Hazra S, Bhatwadekar A, Caballero S, Salazar T, Miyan JA, Li W, Derbenev A, Zsombok A, Tikhonenko M, Dominguez JM II, McGorray SP, Saban DR, Boulton ME, Busik JV, Raizada MK, Chan-Ling T, Grant MB. CNS inflammation and bone marrow neuropathy in type 1 diabetes. Am J Pathol 183: 1608–1620, 2013. doi: 10.1016/j.ajpath.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huh JR, Leung MWL, Huang P, Ryan DA, Krout MR, Malapaka RRV, Chow J, Manel N, Ciofani M, Kim SV, Cuesta A, Santori FR, Lafaille JJ, Xu HE, Gin DY, Rastinejad F, Littman DR. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature 472: 486–490, 2011. doi: 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30: 108–119, 2009. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 37.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126: 1121–1133, 2006. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 38.Javed Q, Murtaza I. Therapeutic potential of tumour necrosis factor-alpha antagonists in patients with chronic heart failure. Heart Lung Circ 22: 323–327, 2013. doi: 10.1016/j.hlc.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 39.Ju X, Ijaz T, Sun H, Ray S, Lejeune W, Lee C, Recinos A III, Guo DC, Milewicz DM, Tilton RG, Brasier AR. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arterioscler Thromb Vasc Biol 33: 1612–1621, 2013. doi: 10.1161/ATVBAHA.112.301049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kandalam U, Clark MA. Angiotensin II activates JAK2/STAT3 pathway and induces interleukin-6 production in cultured rat brainstem astrocytes. Regul Pept 159: 110–116, 2010. doi: 10.1016/j.regpep.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 41.Kang YM, Zhang ZH, Xue B, Weiss RM, Felder RB. Inhibition of brain proinflammatory cytokine synthesis reduces hypothalamic excitation in rats with ischemia-induced heart failure. Am J Physiol Heart Circ Physiol 295: H227–H236, 2008. doi: 10.1152/ajpheart.01157.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karimi K, Bienenstock J, Wang L, Forsythe P. The vagus nerve modulates CD4+ T cell activity. Brain Behav Immun 24: 316–323, 2010. doi: 10.1016/j.bbi.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 43.Kawashima K, Fujii T. The lymphocytic cholinergic system and its contribution to the regulation of immune activity. Life Sci 74: 675–696, 2003. doi: 10.1016/j.lfs.2003.09.037. [DOI] [PubMed] [Google Scholar]
- 44.Kuestner RE, Taft DW, Haran A, Brandt CS, Brender T, Lum K, Harder B, Okada S, Ostrander CD, Kreindler JL, Aujla SJ, Reardon B, Moore M, Shea P, Schreckhise R, Bukowski TR, Presnell S, Guerra-Lewis P, Parrish-Novak J, Ellsworth JL, Jaspers S, Lewis KE, Appleby M, Kolls JK, Rixon M, West JW, Gao Z, Levin SD. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. J Immunol 179: 5462–5473, 2007. doi: 10.4049/jimmunol.179.8.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee DL, Sturgis LC, Labazi H, Osborne JB Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940, 2006. doi: 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
- 46.Leimbach WN Jr, Wallin BG, Victor RG, Aylward PE, Sundlöf G, Mark AL. Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation 73: 913–919, 1986. doi: 10.1161/01.CIR.73.5.913. [DOI] [PubMed] [Google Scholar]
- 47.Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, Pouly S, Murphy AJ, Valenzuela DM, Yancopoulos GD, Becher B, Littman DR, Neurath MF. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 136: 257–267, 2009. doi: 10.1053/j.gastro.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 48.Li DJ, Evans RG, Yang ZW, Song SW, Wang P, Ma XJ, Liu C, Xi T, Su DF, Shen FM. Dysfunction of the cholinergic anti-inflammatory pathway mediates organ damage in hypertension. Hypertension 57: 298–307, 2011. doi: 10.1161/HYPERTENSIONAHA.110.160077. [DOI] [PubMed] [Google Scholar]
- 49.Li M, Zheng C, Sato T, Kawada T, Sugimachi M, Sunagawa K. Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation 109: 120–124, 2004. doi: 10.1161/01.CIR.0000105721.71640.DA. [DOI] [PubMed] [Google Scholar]
- 50.Lohmeier TE, Irwin ED, Rossing MA, Serdar DJ, Kieval RS. Prolonged activation of the baroreflex produces sustained hypotension. Hypertension 43: 306–311, 2004. [Erratum. Hypertension 44: e9, 2004.] doi: 10.1161/01.HYP.0000111837.73693.9b. [DOI] [PubMed] [Google Scholar]
- 51.Lu Y, Ma X, Sabharwal R, Snitsarev V, Morgan D, Rahmouni K, Drummond HA, Whiteis CA, Costa V, Price M, Benson C, Welsh MJ, Chapleau MW, Abboud FM. The ion channel ASIC2 is required for baroreceptor and autonomic control of the circulation. Neuron 64: 885–897, 2009. doi: 10.1016/j.neuron.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madhur MS, Funt SA, Li L, Vinh A, Chen W, Lob HE, Iwakura Y, Blinder Y, Rahman A, Quyyumi AA, Harrison DG. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol 31: 1565–1572, 2011. doi: 10.1161/ATVBAHA.111.227629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55: 500–507, 2010. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, Querci V, Fambrini M, Liotta F, Levings MK, Maggi E, Cosmi L, Romagnani S, Annunziato F. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur J Immunol 40: 2174–2181, 2010. doi: 10.1002/eji.200940257. [DOI] [PubMed] [Google Scholar]
- 55.Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L, Krum H, Liu P, Nieminen M, Tavazzi L, van Veldhuisen DJ, Waldenstrom A, Warren M, Westheim A, Zannad F, Fleming T. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 109: 1594–1602, 2004. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 56.Mark AL, Kioschos JM, Abboud FM, Heistad DD, Schmid PG. Abnormal vascular responses to exercise in patients with aortic stenosis. J Clin Invest 52: 1138–1146, 1973. doi: 10.1172/JCI107280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monden Y, Kubota T, Inoue T, Tsutsumi T, Kawano S, Ide T, Tsutsui H, Sunagawa K. Tumor necrosis factor-α is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction. Am J Physiol Heart Circ Physiol 293: H743–H753, 2007. doi: 10.1152/ajpheart.00166.2007. [DOI] [PubMed] [Google Scholar]
- 58.O’Mahony C, van der Kleij H, Bienenstock J, Shanahan F, O’Mahony L. Loss of vagal anti-inflammatory effect: in vivo visualization and adoptive transfer. Am J Physiol Regul Integr Comp Physiol 297: R1118–R1126, 2009. doi: 10.1152/ajpregu.90904.2008. [DOI] [PubMed] [Google Scholar]
- 59.Padro CJ, Sanders VM. Neuroendocrine regulation of inflammation. Semin Immunol 26: 357–368, 2014. doi: 10.1016/j.smim.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Panoulas VF, Metsios GS, Pace AV, John H, Treharne GJ, Banks MJ, Kitas GD. Hypertension in rheumatoid arthritis. Rheumatology (Oxford) 47: 1286–1298, 2008. doi: 10.1093/rheumatology/ken159. [DOI] [PubMed] [Google Scholar]
- 61.Paton JF, Sobotka PA, Fudim M, Engelman ZJ, Hart EC, McBryde FD, Abdala AP, Marina N, Gourine AV, Lobo M, Patel N, Burchell A, Ratcliffe L, Nightingale A. The carotid body as a therapeutic target for the treatment of sympathetically mediated diseases. Hypertension 61: 5–13, 2013. [Erratum. Hypertension 61: e26, 2013.] doi: 10.1161/HYPERTENSIONAHA.111.00064. [DOI] [PubMed] [Google Scholar]
- 62.Peers C, Green FK. Inhibition of Ca2+-activated K+ currents by intracellular acidosis in isolated type I cells of the neonatal rat carotid body. J Physiol 437: 589–602, 1991. doi: 10.1113/jphysiol.1991.sp018613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pober JS. Is hypertension an autoimmune disease? J Clin Invest 124: 4234–4236, 2014. doi: 10.1172/JCI77766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prabhakar NR, Peng YJ. Peripheral chemoreceptors in health and disease. J Appl Physiol (1985) 96: 359–366, 2004. doi: 10.1152/japplphysiol.00809.2003. [DOI] [PubMed] [Google Scholar]
- 65.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ; CANTOS Trial Group . Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N Engl J Med 377: 1119–1131, 2017. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 66.Sanders VM. The beta2-adrenergic receptor on T and B lymphocytes: do we understand it yet? Brain Behav Immun 26: 195–200, 2012. doi: 10.1016/j.bbi.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE. Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. J Immunol 158: 4200–4210, 1997. [PubMed] [Google Scholar]
- 68.Santisteban MM, Ahmari N, Carvajal JM, Zingler MB, Qi Y, Kim S, Joseph J, Garcia-Pereira F, Johnson RD, Shenoy V, Raizada MK, Zubcevic J. Involvement of bone marrow cells and neuroinflammation in hypertension. Circ Res 117: 178–191, 2015. doi: 10.1161/CIRCRESAHA.117.305853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scheiermann C, Kunisaki Y, Lucas D, Chow A, Jang JE, Zhang D, Hashimoto D, Merad M, Frenette PS. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity 37: 290–301, 2012. doi: 10.1016/j.immuni.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schiffrin EL. Immune mechanisms in hypertension and vascular injury. Clin Sci (Lond) 126: 267–274, 2014. doi: 10.1042/CS20130407. [DOI] [PubMed] [Google Scholar]
- 71.Singh MV, Cicha MZ, Kumar S, Meyerholz DK, Irani K, Chapleau MW, Abboud FM. Abnormal CD161(+) immune cells and retinoic acid receptor-related orphan receptor γt-mediate enhanced IL-17F expression in the setting of genetic hypertension. J Allergy Clin Immunol 140: 809–821.e3, 2017. doi: 10.1016/j.jaci.2016.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Steinmetz OM, Summers SA, Gan P-Y, Semple T, Holdsworth SR, Kitching AR. The Th17-defining transcription factor RORγt promotes glomerulonephritis. J Am Soc Nephrol 22: 472–483, 2011. doi: 10.1681/ASN.2010040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sturgis LC, Cannon JG, Schreihofer DA, Brands MW. The role of aldosterone in mediating the dependence of angiotensin hypertension on IL-6. Am J Physiol Regul Integr Comp Physiol 297: R1742–R1748, 2009. doi: 10.1152/ajpregu.90995.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand A 84: 523–528, 1976. [DOI] [PubMed] [Google Scholar]
- 75.Svendsen UG. The importance of thymus on the degree of increased blood pressure and vascular disease in mice with DOCA and salt hypertension. Acta Med Scand Suppl 602: 19–21, 1976. [DOI] [PubMed] [Google Scholar]
- 76.Svendsen UG, Koch CM, Rubin B. Genetic control of the spontaneous hypertension in the NZB/Cr strain of mice. Immunogenetic considerations. Acta Pathol Microbiol Scand C 87C: 269–273, 1979. [PubMed] [Google Scholar]
- 77.Tan ZY, Lu Y, Whiteis CA, Benson CJ, Chapleau MW, Abboud FM. Acid-sensing ion channels contribute to transduction of extracellular acidosis in rat carotid body glomus cells. Circ Res 101: 1009–1019, 2007. doi: 10.1161/CIRCRESAHA.107.154377. [DOI] [PubMed] [Google Scholar]
- 78.Tan ZY, Lu Y, Whiteis CA, Simms AE, Paton JF, Chapleau MW, Abboud FM. Chemoreceptor hypersensitivity, sympathetic excitation, and overexpression of ASIC and TASK channels before the onset of hypertension in SHR. Circ Res 106: 536–545, 2010. doi: 10.1161/CIRCRESAHA.109.206946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tracey KJ. The inflammatory reflex. Nature 420: 853–859, 2002. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 80.Von Euler US. Sympathin in adrenergic nerve fibres. J Physiol 105: 26, 1946. [PubMed] [Google Scholar]
- 81.Wallin BG, Sundlöf G. Sympathetic outflow to muscles during vasovagal syncope. J Auton Nerv Syst 6: 287–291, 1982. doi: 10.1016/0165-1838(82)90001-7. [DOI] [PubMed] [Google Scholar]
- 82.Whiteis CA, Post CE, Morgan DA, Rahmouni K, Chapleau MW, Abboud FM. Peripheral chemoreceptors contribute significantly to hypertension in spontaneously hypertensive rats (SHR) (Abstract). FASEB J 26: 703.15, 2012. [Google Scholar]
- 83.Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, Chen W, Roberts J II, Osborn JW, Itani HA, Harrison DG. Renal denervation prevents immune cell activation and renal inflammation in angiotensin ii-induced hypertension. Circ Res 117: 547–557, 2015. doi: 10.1161/CIRCRESAHA.115.306010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu Y, Wei SG, Weiss RM, Felder RB. TNF-α receptor 1 knockdown in subfornical organ ameliorates sympathetic excitation and cardiac hemodynamics in heart failure rats. Am J Physiol Heart Circ Physiol 313: H744–H756, 2017. doi: 10.1152/ajpheart.00280.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zaldivia MT, Rivera J, Hering D, Marusic P, Sata Y, Lim B, Eikelis N, Lee R, Lambert GW, Esler MD, Htun NM, Duval J, Hammond L, Eisenhardt SU, Flierl U, Schlaich MP, Peter K. Renal denervation reduces monocyte activation and monocyte-platelet aggregate formation: an anti-inflammatory effect relevant for cardiovascular risk. Hypertension 69: 323–331, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08373. [DOI] [PubMed] [Google Scholar]
- 86.Zhao Q, Hong D, Zhang Y, Sang Y, Yang Z, Zhang X. Association between anti-TNF therapy for rheumatoid arthritis and hypertension: a meta-analysis of randomized controlled trials. Medicine (Baltimore) 94: e731, 2015. doi: 10.1097/MD.0000000000000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zubcevic J, Waki H, Raizada MK, Paton JF. Autonomic-immune-vascular interaction: an emerging concept for neurogenic hypertension. Hypertension 57: 1026–1033, 2011. doi: 10.1161/HYPERTENSIONAHA.111.169748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zucker IH, Hackley JF, Cornish KG, Hiser BA, Anderson NR, Kieval R, Irwin ED, Serdar DJ, Peuler JD, Rossing MA. Chronic baroreceptor activation enhances survival in dogs with pacing-induced heart failure. Hypertension 50: 904–910, 2007. doi: 10.1161/HYPERTENSIONAHA.107.095216. [DOI] [PubMed] [Google Scholar]