

Tuberculosis is the ninth-leading cause of death worldwide. Treatment success is approximately 80% for susceptible strains and decreases to 30% for extensively resistant strains.
KEYWORDS: Mycobacterium tuberculosis, acid-phase-growth bacteria, bedaquiline, combination therapy, linezolid, log-phase-growth bacteria, nonreplicating-persister-phase-growth bacteria
ABSTRACT
Tuberculosis is the ninth-leading cause of death worldwide. Treatment success is approximately 80% for susceptible strains and decreases to 30% for extensively resistant strains. Shortening the therapy duration for Mycobacterium tuberculosis is a major goal, which can be attained with the use of combination therapy. However, the identification of the most promising combination is a challenge given the quantity of older and newer agents available. Our objective was to identify promising 2-drug combinations using an in vitro strategy to ultimately be tested in an in vitro hollow fiber infection model (HFIM) and in animal models. We studied the effect of the combination of linezolid (LZD) and bedaquiline (BDQ) on M. tuberculosis strain H37Rv in log- and acid-phase growth and M. tuberculosis strain 18b in log- and nonreplicating-persister-phase growth in a plate system containing a 9-by-8 matrix of concentrations of both drugs alone and in combinations. A characterization of the interaction as antagonistic, additive, or synergistic was performed using the Greco universal response surface approach (URSA) model. Our results indicate that the interaction between LZD and BDQ is additive for bacterial killing in both strains for both of the metabolic states tested. This prescreen strategy was suitable to identify LZD and BDQ as a promising combination to be further tested in the HFIM. The presence of nonoverlapping mechanisms of drug action suggests each drug in the combination will likely be effective in suppressing the emergence of resistance by M. tuberculosis to the companion drug, which holds promise in improving treatment outcomes for tuberculosis.
INTRODUCTION
According to the World Health Organization, tuberculosis is the ninth-leading cause of death worldwide. The estimates from 2016 indicate that the overall incidence was at 10.3 million cases; deaths were estimated at 2 million for the same year (1). Treatment success is around 80% for drug-susceptible Mycobacterium tuberculosis and decreases to 50% in multidrug-resistant (MDR) and 30% in extensively resistant (XDR) isolates (2).
The treatment for drug-susceptible M. tuberculosis consists of 2 months of rifampin, isoniazid, pyrazinamide, and ethambutol in the intensive phase followed by 4 months of rifampin and isoniazid in the continuation phase (2). In humans, M. tuberculosis exists in several metabolic phases. The most commonly mentioned metabolic states are logarithmic-growth-phase M. tuberculosis, slowly replicating acid-phase-growth M. tuberculosis, and nonreplicating-persister-phase M. tuberculosis. Six months of treatment are needed to cure pan-susceptible strains, because the standard regimen has not be optimized to maximize the killing of M. tuberculosis in all metabolic phases.
The conversion from drug-susceptible to drug-resistant M. tuberculosis is related to the duration of treatment and patient adherence to the regimen. To optimize drug therapy, it is important to use drug regimens which maximize the killing of M. tuberculosis. This may lead to shorter treatment durations which, in turn, may improve patient adherence. At the same time, optimally designed regimens should suppress resistance amplification (3).
These goals cannot be achieved by single-agent therapy. They require the use of combination therapy, which has been proven to be effective to suppress resistance in patients with tuberculosis (4). However, new combinations should be developed and optimized for the treatment of tuberculosis, as drug-resistant strains are on the rise. Combination therapy aims to promote bacterial kill and the suppression of resistance. A decreased treatment duration may also be observed if drug combinations promote the death of slowly or nongrowing organisms (e.g., M. tuberculosis in nonreplicating-persister phase or acid phase).
Selected regimens should ideally have synergistic or at least additive treatment effects on bacterial killing and resistance suppression. Antagonistic interactions may also be effective in suppressing resistance development; however, this type of interaction leads to a slow bacterial killing, as was shown for the combination of rifampin (RIF) and moxifloxacin (MOX) in an in vitro hollow fiber infection model (HFIM) for M. tuberculosis (5). This combination suppressed the amplification of less-susceptible subpopulations of M. tuberculosis but was antagonistic for the nonreplicative-persister (NRP)-phenotype organisms. These in vitro results were confirmed in the REMOX trial, where the objective of shortening the duration of therapy to 4 months for fully susceptible M. tuberculosis by substituting MOX for isoniazid in both the intensive and continuation phases of therapy was not achieved (6). Given that the combination of MOX and RIF cleared the sputum significantly more rapidly in the intensive phase, it is likely that the failure to achieve the therapy-shortening endpoint was related to the antagonistic interaction of these 2 drugs for killing NRP organisms. As a result, we hypothesized that the shortening of therapy can be achieved by selecting nonantagonistic drug combinations with increased activities against NRP-phase and acid-phase organisms.
Bedaquiline (BDQ) and linezolid (LZD) were chosen for evaluation in this study because both drugs have been shown in vitro to be active against replicating and nonreplicating M. tuberculosis (3, 7–9). BDQ is approved by the U.S. Food and Drug Administration and the European Medicines Agency for the treatment of multidrug-resistant TB. LZD is licensed for pneumonia due to Staphylococcus aureus and Streptococcus pneumoniae and uncomplicated and complicated skin and skin structure infections due to staphylococci and streptococci (10–12). LZD is currently being evaluated in phase III clinical trials as a repurposed drug for the treatment of tuberculosis (13, 14). Since these drugs have different mechanisms of action (15, 16), it is possible that these drugs, in combination, will interact additively or synergistically to kill M. tuberculosis and may suppress the emergence of resistance to each other.
The selection of the optimal treatment regimen can be supported by the use of mathematical and statistical models in conjunction with experimental data. Greco and colleagues (17) proposed a universal response surface approach (URSA) method to characterize drug interactions based on Loewe additivity. The method was initially used for anticancer agents but was also successfully applied to anti-infective agents (18–20). The type of interaction is determined in this approach by the interaction term “α.” If α is positive and the lower 95% confidence bound does not overlap zero, the interaction is significantly synergistic. If the value is negative and the upper 95% confidence bound does not overlap zero, the interaction is significantly antagonistic. If the confidence bounds overlap zero for any value of α, the interaction is deemed additive. The Greco model can be informed by experimental data evaluating the effect of different drug combinations against different metabolic states of M. tuberculosis. This evaluation can also be made in the in vitro HFIM, where organisms are exposed to changing drug concentrations over time (4). However, given the quantity of older and newer agents, as well as the number of possible drug-drug combinations, we decided to prescreen these treatment options for synergy, additivity, or antagonism under static conditions using a plate system in order to identify the most promising combinations to be taken forward into the HFIM.
RESULTS
MIC determination.
The LZD MIC was 1 mg/liter for M. tuberculosis H37Rv in both log and acid phases and for strain 18b in log phase. The BDQ MIC was 0.25 mg/liter for the H37Rv strain in log phase, 0.125 mg/liter for the H37Rv strain in acid phase, and 0.5 mg/liter for the 18b strain in log phase. Since M. tuberculosis in NRP phase does not replicate, the MICs of LZD and BDQ could not be performed for bacteria in this metabolic state.
In vitro drug interaction studies in the plate system.
Figure 1 displays the estimated values and confidence intervals (CIs) for the interaction parameter α for two replicates of M. tuberculosis strain H37Rv in log and acid phases and strain 18b in log and NRP phases (the latter tested in acidic and neutral pH environments). The estimates for α (9 of 10) were positive. The tenth determination had a very small negative α value. All CIs overlapped zero. These results indicate that the interaction between LZD and BDQ was additive for all the tested M. tuberculosis strains and in all metabolic states and pH values evaluated.
FIG 1.
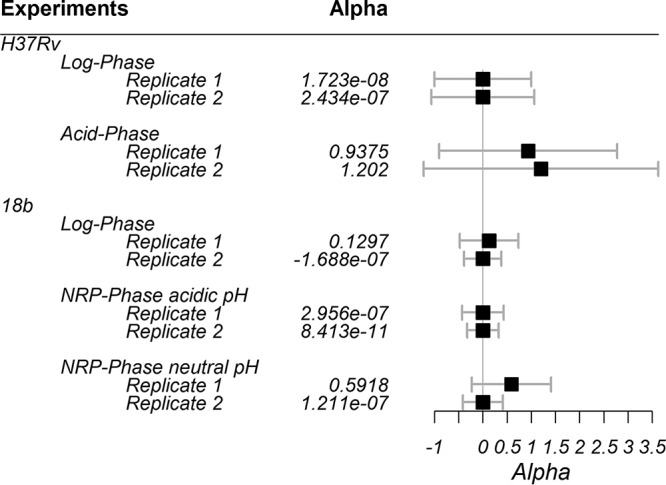
Estimated values for the interaction parameter α for each population. Black boxes represent the mean estimates; error bars represent the 95% CIs.
The point estimates and 95% CIs for the remaining model parameters for each studied strain and phenotype are displayed in Table 1. The estimated IC50s show there are significant differences between strains and metabolic states for both LZD and BDQ. Those IC50 estimates suggest lower LZD potency (i.e., higher IC50) against the H37Rv strain in log phase than against the studied 18b metabolic states. Among the metabolic states for 18b, the results indicate lower LZD potency against this isolate when it is in NRP phase in a neutral pH environment. Regarding the BDQ IC50 estimates, our results show that BDQ is more potent against H37Rv strain in log phase (i.e., lower IC50) than against the 18b strains in the metabolic states studied. In addition, the comparison between the two study drugs showed BDQ is more potent than LZD against H37Rv in log phase and against all tested 18b phenotypes.
TABLE 1.
Estimated parameter values from URSA Greco model in ADAPT 5 for each strain/phenotype
| Parametera (drug) | Value (95% CI) for: |
||||
|---|---|---|---|---|---|
| H37Rv |
18b |
||||
| Log phase | Acid phase | Log phase | NRP in acidic pH | NRP in neutral pH | |
| ECON (log CFU/ml) | 7.79 (7.07–8.51) | 9.13 (6.19–12.1) | 7.68 (7.31–8.06) | 5.57 (5.12–6.02) | 6.28 (5.95–6.61) |
| IC50,1 (LZD) (mg/liter) | 4.72b (2.85–6.59) | 2.0 (−1.31–5.31) | 1.89b,c (1.60–2.18) | 1.27b,c (0.994–1.54) | 2.76c (2.36–3.16) |
| IC50,2 (BDQ) (mg/liter) | 0.265d,e (0.230–0.301) | 0.15 (−0.145–0.444) | 0.855e (0.735–0.975) | 0.760e (0.563–0.956) | 0.701e (0.562–0.839) |
| m1 (LZD) | 0.845 (0.489–1.20) | 0.428 (0.179–0.676) | 1.34 (1.08–1.61) | 1.33 (1.04–1.61) | 2.07 (1.59–2.56) |
| m2 (BDQ) | 2.49 (1.24–3.74) | 0.326 (0.224–0.428) | 1.72 (1.23–2.21) | 1.16 (0.87–1.45) | 1.20 (0.974–1.42) |
IC50,1 and IC50,2, concentration for half maximal effect; m1 and m2, Hill constant; ECON, effect for the control.
P < 0.05 between H37Rv strain at log phase, 18b strain at log phase, and 18b strain at NRP phase in acidic pH environment for LZD.
P < 0.05 between 18b strain at log phase and 18b strain at NRP phase in acidic and neutral pH environments for LZD.
P < 0.05 between H37Rv strain at log phase, 18b strain at log phase, and 18b strain at NRP phase in acidic and neutral pH environments for BDQ.
P < 0.05 between IC50,1 (LZD) and IC50,2 (BDQ).
Figure 2 shows the predicted effect of the LZD and BDQ combination on the total colony counts of M. tuberculosis strains H37Rv at log and acid phases and 18b at log and NRP phases as a response surface. It also shows that the model fits to the data were acceptable. Figure 2B (strain H37Rv, acid phase) shows that there is some model bias, because colony counts associated with lower BDQ concentrations and higher LZD concentrations are located below the fitted response surface. This indicates that the antimicrobial activity is underpredicted for these concentrations.
FIG 2.

Effect of LZD and BDQ on total colony counts of M. tuberculosis metabolic states and pH environments according to the URSA model by Greco et al. (17). (A) H37Rv strain in log-phase growth; (B) H37Rv strain in acid phase; (C) 18b strain in log-phase growth; (D) 18b strain in NRP-phase phenotype in acidic pH environment; (E) 18b strain in NRP-phase phenotype in neutral pH environment. Blue surfaces represent model predictions, green spheres represent observations above the fitted surface, and red cubes represent observations below the fitted surface. Experiments were performed in duplicates. Plots show the results from a representative replicate.
Mathematical model.
Modeling results from ADAPT 5 (21) are displayed in Fig. 3. Figure 3 shows the observed versus predicted colony counts for each replicate of strain H37Rv at log and acid phases and strain 18b at log and NRP phases at acidic and neutral pH environments. These results show the observed versus predicted pairs are evenly distributed around the unity line. Figure 3 also shows that the observed versus predicted regression line superimposes the unity line. All regression slopes are below 1.000 and above 0.941; coefficients of determination (r2) are greater than 0.771. Figure 3 shows a trend toward lower colony counts being below the regression line (Fig. 3D1, D2, E1, and E2), which is due to the presence of data below the lower limit of quantitation (LLOQ). Overall, these results indicate the model was adequate to describe the data.
FIG 3.

Observed versus predicted colony counts for H37Rv strain at log phase (A1 and A2) and acid phase (B1 and B2), for 18b strain at log phase (C1 and C2), NRP phase in sn acidic pH environment (D1 and D2), and NRP phase in a neutral pH environment (E1 and E2). 1 and 2 are replicates for the same experiment. Red lines represent the regression lines over the observed versus model predicted colony counts, the black lines represent the unity lines; ●, observation/prediction pairs.
DISCUSSION
The manuscript describes a novel in vitro screening procedure to identify optimal antituberculosis drug combinations for the main metabolic states of M. tuberculosis. A drug combination was evaluated in a static in vitro plate system where constant fractions and multiples of MICs for LZD and BDQ were incubated with H37Rv and 18b M. tuberculosis strains in log phase, H37Rv in acid phase, and streptomycin (STR)-starved 18b in NRP phase in acidic (pH 6) and neutral (pH 7) pH environments. The activities of and interaction between LZD and BDQ were tested against NRP in media adjusted to two pHs, since it is possible this metabolic state may exist in humans in microniches with neutral or acidic pH environments and the activities of the drugs in the two pHs may differ. M. tuberculosis quantitative culture data were analyzed with the Greco URSA model (17). Our results indicate that the interaction between LZD and BDQ is additive in all metabolic states tested. This in vitro screening is the first step in the process to identify an optimal drug combination to improve the treatment of tuberculosis through decreasing the treatment duration.
Once promising drug combinations are identified, the next step will be their evaluation in an in vitro HFIM. In this model, drug exposures and human pharmacokinetic profiles of LZD and BDQ in the target site will be simulated to evaluate the impact of this combination for both cell killing and the suppression of resistance. Obtained data will be analyzed by the Drusano-Greco model, which expands the original model to analyze both susceptible and resistant populations of bacteria (3).
Should additivity or synergy be observed between LZD and BDQ in the HFIM, the evaluation of this drug combination in preclinical animal models of tuberculosis will be performed. Drug combinations will be evaluated in mice and nonhuman primates (NHP), where the impact of disease on drug pharmacokinetics, the activity of the immune system, and drug toxicity increase the complexity of this scenario (22, 23).
Model-estimated IC50 values (IC50,1) (Table 1) suggest LZD is more potent in killing the 18b strain at NRP phase in an acidic pH environment than in killing the 18b strain at log phase, 18b strain at NRP phase in a neutral pH environment, and H37Rv strain at log phase. It is noteworthy to mention the distinct potency of LZD according to the medium pH in NRP phase, which might be attributed to changes in bacterial turnover. In NRP phase, the rates of growth are reduced to near zero, but there are some ongoing metabolic functions (e.g., protein synthesis) (24). Differences in LZD potency between the 18b strain at log phase and NRP phase in an acidic environment and the H37Rv strain at log phase could be explained by the mechanism of action of the drug. LZD inhibits protein synthesis by binding to the bacterial ribosomal 30S subunit, which inhibits the formation of the preinitiation complex between mRNA, tRNA, and the ribosomal unit and halts the transcription process (15). It is quite possible that more LZD is needed to fully inhibit protein synthesis of bacteria at a faster metabolic rate (i.e., log-phase bacteria), as it presents a higher ribosomal content. Regarding BDQ, our model estimates indicate that this drug has a higher potency against the H37Rv strain in log phase than against the 18b strain at all tested metabolic states. This difference can also be explained by the mechanism of action of BDQ. This drug acts by binding to subunit c of mycobacterial ATP synthase, leading to a disruption of bacterial ATP production and cell death (16). In this scenario, it is a reasonable hypothesis that the effect on energy production would present a greater impact on bacterial cells at log phase (i.e., a high-energy consumption state). It is important to note that the relative potencies were determined in vitro with static concentrations. The true potencies of these agents will be made manifest when the pharmacokinetic profiles and the impact of protein binding are taken into account.
Our model approach was applied to characterize the interaction of LZD and BDQ on H37Rv in log and acid phases and 18b strains at log phase and NRP phase. The activities of the drugs were evaluated against NRP M. tuberculosis incubated at two different pHs to determine whether an acidic environment altered the killing effect of either drug alone and/or in combination. The characterization of combination therapy is traditionally performed by the calculation of the fractional inhibitory concentration (FIC) index (25). Although it also enables characterization of the combination as synergistic, additive, and antagonistic, it is a descriptive index, and we did not use it because it has limited applicability to a clinical scenario. The Greco URSA model, on the other hand, is a regression-based approach: parameters and their associated precision (i.e., confidence intervals) are estimated on the basis of statistical criteria and can be further associated with pharmacokinetic data to simulate the bacterial kill promoted by drug combinations in a therapeutic regimen. Previous studies from our group showed the use of the Drusano-Greco URSA model as an approach to characterize the combination effect of LZD and RIF on M. tuberculosis in an HFIM. Additivity was observed on H37Rv strain at a log-phase phenotype; however, the failure to suppress resistance indicates that this drug combination would not lead to a shortening of therapy, at least with standard doses of RIF (3).
One drawback of the current plate system containing a 9-by-8 matrix of concentrations of both drugs alone and in combination is its inability to evaluate the effect of the drugs on the suppression of resistance, as the probability of a resistant colony to grow and amplify is very low due to the small burden of M. tuberculosis in each well (5). However, it is highly likely that this combination of drugs with nonoverlapping mechanisms of action (i.e., inhibition of protein synthesis for LZD and inhibition of ATP production by BDQ) would suppress resistance amplification. Previous studies from our group support that hypothesis: the combination of MOX and RIF suppressed resistance (inhibition of DNA replication plus inhibition of protein synthesis) while the combination of RIF with LZD did not (inhibition of protein synthesis plus inhibition of protein synthesis). MOX kills bacterial cells by blocking DNA replication and repair through the inhibition of DNA gyrase (topoisomerase II) and DNA topoisomerase IV enzymes, which does not overlap the rifampin inhibition of protein synthesis through the inhibition of RNA polymerase (26–28). In conclusion, the proposed screening strategy enabled the study of drug interaction on the bacterial killing of M. tuberculosis in log, acid, and NRP phases and whether pH alters the killing effect of the drugs against NRP bacteria. The LZD-BDQ combination was identified as a promising option to promote M. tuberculosis bacterial killing in log, acid, and NRP phases and will be evaluated for resistance suppression in the HFIM.
MATERIALS AND METHODS
Bacterium and generation of metabolic phases.
M. tuberculosis strains H37Rv and 18b were used. M. tuberculosis 18b is a clinical isolate that is an STR-resistant STR auxotroph. It requires at least 10 mg/liter of STR to be added to agar and broth medium for it to propagate in log phase. In STR-free medium, it converts to the NRP phase and reverts back to log phase when STR is added back to the medium. Stocks of the bacterium were stored at −80°C. For experiments using log-phase H37Rv, an aliquot of the stock culture was thawed and incubated at 37°C at 5% CO2 with shaking in Middlebrook 7H9 broth supplemented with 10% albumin, dextrose, and catalase (ADC), 0.05% Tween 80, and 0.3% dimethyl sulfoxide (DMSO) (TB broth) for 7 to 10 days to achieve log-phase growth. To generate log phase for M. tuberculosis strain 18b, the same procedure was followed, except STR 50 mg/liter was added to the medium. To transition M. tuberculosis 18b to an NRP state, log-phase 18b grown in STR-containing medium was washed thrice by centrifugation with PBS containing 0.05% Tween 80 and resuspended in STR-free TB broth at pH 7 (neutral pH environment) or adjusted to pH 6 (acidic pH environment) with citric acid. These cultures were incubated at 37°C at 5% CO2 with shaking for 7 to 10 days to allow the microbe to transition to the NRP phase. Acid-phase bacteria were generated by transferring 100 μl of log-phase H37Rv to 40 ml of TB broth at pH 6. The culture was incubated at 37°C at 5% CO2 for 7 to 10 days before they were used in susceptibility testing or in the plate system (9-by-8 matrix of concentrations of both drugs alone and in combinations) studies (3, 24, 29, 30).
Drugs.
LZD solution (600 mg/300 ml) was purchased from TEVA Pharmaceuticals (North Wales, PA), while pharmaceutical BDQ was purchased from BOC Sciences (Shirley, NY); both compounds were stored according to the manufacturers' instructions. BDQ was dissolved in dimethyl sulfoxide (DMSO). LZD was dissolved with sterile water. STR was purchased from Sigma-Aldrich (St. Louis, MO) and was dissolved in sterile water.
MIC determination.
Broth microdilution MIC values of LZD and BDQ were determined for H37Rv strain at log phase and acid phase and for 18b strain at log phase. The final bacterial inoculum added to round-bottom 96-well dilution plates was 1 × 105 CFU/well. The bacteria in the different metabolic phases were prepared as described earlier in TB broth. The bacterial suspensions were added to wells containing geometric 2-fold dilutions of LZD or BDQ. For studies with acid-phase M. tuberculosis, the medium was adjusted to pH 6. After 21 days of incubation at 37°C at 5% CO2, the MICs were read. The MIC was defined as the lowest concentration that resulted in no visible growth. The susceptibility studies for both drugs were performed using polystyrene 96-well plates and dilution tubes to minimize nonspecific drug binding by BDQ.
In vitro drug interaction studies in the plate system.
Each bacterial state (H37Rv in log phase and acid phase, 18b in log phase and NRP phase at acidic [pH 6] and neutral [pH 7] environments) was prepared in TB broth, with adjustment of the medium pH to 6, when needed. The bacterial suspensions were inoculated at 105 CFU/well in 96-well round-bottom microdilution plates (Falcon, Corning, NY) containing a 9-by-8 matrix consisting of no drug or serial 2-fold-increments of BDQ (0, 0.03, 0.06, 0.125, 0.25, 0.5, 1, 2, and 4× MIC) and LZD (0, 0.06, 0.125, 0.25, 0.5, 1, 2, and 4× MIC) alone and in all possible two-drug combinations. The checkerboard studies were performed using polystyrene 96-well plates and dilution tubes to minimize the nonspecific binding reported for BDQ. Since antibiotic MICs cannot be determined for M. tuberculosis strain 18b in NRP phase, the middle concentration of LZD and BDQ in the range of concentrations evaluated singly and in combination in the checkerboard experiments was the MIC of the respective drug identified in the susceptibility studies for log-phase strain 18b. Log-phase and acid-phase phenotypes were incubated for 21 days, while NRP phenotypes (in acidic and neutral pH environments) were incubated for 14 days. The M. tuberculosis suspensions were washed twice with normal saline to remove drug carryover and then quantitatively plated on 7H10 agar supplemented with 10% oleic acid-ADC (OADC) and 0.05% Tween. The cultures were incubated at 37°C at 5% CO2 for 4 weeks before the colonies were enumerated. For studies using 18b in log-phase growth, the TB broth was supplemented with 75 μg/ml STR, while the studies using 18b in NRP states used STR-free TB broth. For all studies with 18b, the agar used for the quantitative cultures was also supplemented with 75 μg/ml of STR (24).
Mathematical model.
The quantitative culture counts obtained from the combination regimens for each metabolic state (log, acid, and NRP phases) were analyzed in ADAPT 5 (21) using the maximum likelihood estimation method. Data were described by the universal response surface approach (URSA) equation of Greco and colleagues (17)
where drug1 (LZD) and drug2 (BDQ) are the drug concentrations, IC50,1 and IC50,2 are the concentrations of the drugs for which the effect is half maximal, m1 and m2 are Hill's constants, ECON is the effect for the control, α is the interaction parameter, and E is the fractional effect. Figure 2 depicts the three-dimensional view of the Greco equation for different parameter values given in Table 1. The plots were generated using Mathematica (v. 11.1; Wolfram Research Inc., Champaign, IL). The use of the Greco model enabled us to distinguish between the presence of additivity, synergy, and antagonism in a quantitative manner by evaluating the α value and its associated confidence interval. Additivity is declared if α and its attendant 95% confidence interval include zero. Synergy is declared if α and its attendant 95% confidence interval are positive and do not include zero. Antagonism is declared if α and its attendant 95% confidence interval are negative and do not include zero.
ACKNOWLEDGMENTS
These studies were supported by P01 AI123036-01 from NIAID.
The authors declare no conflict of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.World Health Organization. 2017. Chapter 3, TB disease burden, p 21–62. Global tuberculosis report 2017. World Health Organization, Geneva, Switzerland: http://www.who.int/tb/publications/global_report/en/, Accessed 23 March 2018. [Google Scholar]
- 2.World Health Organization. 2017. Chapter 4, Diagnosis and treatment: TB, HIV-associated TB and drug-resistant TB, p 63–96. Global tuberculosis report 2017. World Health Organization, Geneva, Switzerland. http://www.who.int/tb/publications/global_report/en/ Accessed 23 March 2018.
- 3.Drusano GL, Neely M, Van Guilder M, Schumitzky A, Brown D, Fikes S, Peloquin C, Louie A. 2014. Analysis of combination drug therapy to develop regimens with shortened duration of treatment for tuberculosis. PLoS One 9:e101311. doi: 10.1371/journal.pone.0101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selkon JB, Devadatta S, Kullarna KG, Mitchinson DA, Narayana AS, Nair CN, Ramachandran K. 1964. The emergence of isoniazid-resistant cultures in patients with pulmonary tuberculosis during treatment with isoniazid alone or isoniazid plus PAS. Bull World Health Organ 31:273–294. [PMC free article] [PubMed] [Google Scholar]
- 5.Drusano GL, Sgambati N, Eichas A, Brown DL, Kulawy R, Louie A. 2010. The combination of rifampin plus moxifloxacin is synergistic for suppression of resistance but antagonistic for cell kill of Mycobacterium tuberculosis as determined in a hollow-fiber infection model. mBio 1:e00139-. doi: 10.1128/mBio.00139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, Pappas F, Phillips PPJ, Nunn AJ. 2014. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med 371:1577–1587. doi: 10.1056/NEJMoa1407426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang M, Sala C, Dhar N, Vocat A, Sambandamurthy VK, Sharma S, Marriner G, Balasubramanian V, Cole ST. 2014. In vitro and in vivo activities of three oxazolidinones against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:3217–3223. doi: 10.1128/AAC.02410-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koul A, Vranckx L, Dendouga N, Balemans W, Van den Wyngaert I, Vergauwen K, Göhlmann HW, Willebrords R, Poncelet A, Guillemont J, Bald D, Andries K. 2008. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J Biol Chem 283:25273–25280. doi: 10.1074/jbc.M803899200. [DOI] [PubMed] [Google Scholar]
- 9.Andries K, Verhasselt P, Guillemont J, Göhlmann HWH, Neefs J-M, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 10.Food and Drug Administration, Center for Drug Evaluation and Research. 2012. NDA 204384. Food and Drug Administration, Silver Spring, MD: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/204384Orig1s000ltr.pdf Accessed 17 April 2018. [Google Scholar]
- 11.European Medicines Agency Committee for Medicinal Products for Human Use. 2013. Assessment report 329898/2013. European Medicines Agency, London, UK: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002614/WC500163215.pdf Accessed 17 April 2018. [Google Scholar]
- 12.Food and Drug Administration, Center for Drug Evaluation and Research. 2000. NDA 21-130, 21-131, 21-132. Food and Drug Administration, Silver Spring, MD: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021130s022lbl.pdf Accessed 17 April 2018. [Google Scholar]
- 13.National Institutes of Health, National Library of Medicine. 2015. Clinical trial identifier NCT02333799. A phase 3 study assessing the safety and efficacy of bedaquiline plus PA-824 plus linezolid in subjects with drug resistant pulmonary tuberculosis. National Library of Medicine, Bethesda, MD: https://clinicaltrials.gov/ct2/show/NCT02333799 Accessed April 17 2018. [Google Scholar]
- 14.National Institutes of Health, National Library of Medicine. 2015. Clinical trial identifier NCT02454205. An open-label RCT to evaluate a new treatment regimen for patients with multi-drug resistant tuberculosis (NEXT). National Library of Medicine, Bethesda, MD: https://clinicaltrials.gov/ct2/show/study/NCT02454205 Accessed April 17 2018. [Google Scholar]
- 15.Swaney SM, Aoki H, Ganoza MC, Shinabarger DL. 1998. The oxazolidinone linezolid inhibits initiation of protein synthesis in bacteria. Antimicrob Agents Chemother 42:3251–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koul A, Dendouga N, Vergauwen K, Molenberghs B, Vranckx L, Willebrords R, Ristic Z, Lill H, Dorange I, Guillemont J, Bald D, Andries K. 2007. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol 3:323–324. doi: 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- 17.Greco WR, Bravo G, Parsons JC. 1995. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev 47:331–385. [PubMed] [Google Scholar]
- 18.Drusano GL, D'Argenio DZ, Symonds W, Bilello PA, McDowell J, Sadler B, Bye A, Bilello JA. 1998. Nucleoside analog 1592U89 and human immunodeficiency virus protease inhibitor 141W94 are synergistic in vitro. Antimicrob Agents Chemother 42:2153–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drusano GL, D'Argenio DZ, Preston SL, Barone C, Symonds W, LaFon S, Rogers M, Prince W, Bye A, Bilello JA. 2000. Use of drug effect interaction modeling with Monte Carlo simulation to examine the impact of dosing interval on the projected antiviral activity of the combination of abacavir and amprenavir. Antimicrob Agents Chemother 44:1655–1659. doi: 10.1128/AAC.44.6.1655-1659.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snyder S, D'Argenio DZ, Weislow O, Bilello JA, Drusano GL. 2000. The triple combination of indinavir, zidovudine plus lamivudine is highly synergistic. Antimicrob Agents Chemother 44:1051–1058. doi: 10.1128/AAC.44.4.1051-1058.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D'Argenio DZ, Schumitzky A, Wang X. 2009. ADAPT 5 user's guide: pharmacokinetic/pharmacodynamic systems analysis software. Biomedical Simulations Resource, Los Angeles, CA. [Google Scholar]
- 22.Balasubramanian V, Solapure S, Gaonkar S, Mahesh Kumar KN, Shandil RK, Deshpande A, Kumar N, Vishwas KG, Panduga V, Reddy J, Ganguly S, Louie A, Drusano GL. 2012. Effect of coadministration of moxifloxacin and rifampin on Mycobacterium tuberculosis in a murine aerosol infection model. Antimicrob Agents Chemother 56:3054–3057. doi: 10.1128/AAC.06383-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.White AG, Maiello P, Coleman MT, Tomko JA, Frye LJ, Scanga CA, Lin PL, Flynn JL. 2017. Analysis of 18FDG PET/CT imaging as a tool for studying Mycobacterium tuberculosis infection and treatment in non-human primates. J Vis Exp 127:e56375. doi: 10.3791/56375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sala C, Dhar N, Harkoorn RC, Zhang M, Ha YH, Schneider P, Cole ST. 2010. Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 54:4150–4158. doi: 10.1128/AAC.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.European Committee for Antimicrobial Susceptibility Testing of the European Society of Clinical Microbiology and Infectious Diseases. 2000. EUCAST definitive document E. Def 1.2, May 2000: terminology relating to methods for the determination of susceptibility of bacteria to antimicrobial agents. Clin Microbiol Infect 6:503–508. [DOI] [PubMed] [Google Scholar]
- 26.Wehrli W. 1983. Rifampin: mechanisms of action and resistance. Rev Infect Dis 5(Suppl 3):S407–S411. doi: 10.1093/clinids/5.Supplement_3.S407. [DOI] [PubMed] [Google Scholar]
- 27.Willmott CJ, Critchlow SE, Eperon IC, Maxwell A. 1994. The complex of DNA gyrase and quinolone drugs with DNA forms a barrier to transcription by RNA polymerase. J Mol Biol 242:351–363. doi: 10.1006/jmbi.1994.1586. [DOI] [PubMed] [Google Scholar]
- 28.Shea ME, Hiasa H. 1999. Interactions between DNA helicases and frozen topoisomerase IV-quinolone-DNA ternary complexes. J Biol Chem 274:22747–22754. doi: 10.1074/jbc.274.32.22747. [DOI] [PubMed] [Google Scholar]
- 29.Zhang M, Sala C, Hartkoorn RC, Dhar N, Mendoza-Losana A, Cole ST. 2012. Streptomycin-starved Mycobacterium tuberculosis 18b, a drug discovery tool for latent tuberculosis. Antimicrob Agents Chemother 56:5782–5789. doi: 10.1128/AAC.01125-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gumbo T, Dona CS, Meek C, Leff R. 2009. Pharmacokinetics-pharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: a paradigm for faster assessment of new antituberculosis drugs. Antimicrob Agents Chemother 53:3197–3204. doi: 10.1128/AAC.01681-08. [DOI] [PMC free article] [PubMed] [Google Scholar]