

Plazomicin is a novel aminoglycoside with potent in vitro activity against multidrug- and carbapenem-resistant Enterobacteriaceae. The objective of this study was to assess the efficacy of plazomicin exposure, alone and in combination with meropenem or tigecycline, against Enterobacteriaceae in the immunocompetent murine septicemia model.
KEYWORDS: aminoglycosides, pharmacodynamics, pharmacokinetics
ABSTRACT
Plazomicin is a novel aminoglycoside with potent in vitro activity against multidrug- and carbapenem-resistant Enterobacteriaceae. The objective of this study was to assess the efficacy of plazomicin exposure, alone and in combination with meropenem or tigecycline, against Enterobacteriaceae in the immunocompetent murine septicemia model. ICR mice were inoculated intraperitoneally with bacterial suspensions. Eight Enterobacteriaceae isolates with wide ranges of plazomicin, meropenem, and tigecycline MICs were utilized. Treatment mice were administered plazomicin, meropenem, or tigecycline human-equivalent doses alone or in combinations of plazomicin-meropenem and plazomicin-tigecycline. Treatments were initiated at 1 h postinfection and continued for 24 h. Efficacy was assessed by determination of mouse survival through 96 h. Compared with the survival of the controls, plazomicin monotherapy produced a significant improvement in survival for all mice infected with the isolates (P < 0.05) and resulted in overall survival rates of 86% (n = 50) and 53.3% (n = 30) for mice infected with isolates with plazomicin MICs of ≤4 and ≥8 mg/liter, respectively (P < 0.05). The survival of the meropenem and tigecycline groups correlated well with susceptibilities of their respective isolates, with incremental increases in survival being observed at lower MIC values. For mice infected with isolate Klebsiella pneumoniae 561 (plazomicin, meropenem, and tigecycline MICs, 8, >32, and 2 mg/liter, respectively), combination therapies showed a significant reduction in mortality compared with that achieved with any monotherapy (P < 0.05). Plazomicin monotherapy resulted in improved survival in the immunocompetent murine septicemia model, notably, for mice infected with isolates with plazomicin MICs of ≤4 mg/liter. As evidenced by our current data, coadministration of meropenem or tigecycline could potentially lead to a further improvement in survival. These data support a role for plazomicin in the management of septicemia due to Enterobacteriaceae with plazomicin MICs of ≤4 mg/liter, including carbapenem-resistant isolates.
INTRODUCTION
Carbapenem-resistant Enterobacteriaceae (CRE) are a group of Gram-negative bacteria that are very challenging to treat. These isolates exhibit high levels of resistance to carbapenem antibiotics, a last-line treatment for patients with serious resistant Enterobacteriaceae infections (1), as they express a group of hydrolytic enzymes known as carbapenemases, such as Klebsiella pneumoniae carbapenemase (KPC) and New Delhi metallo-β-lactamase (NDM). Although they are branded “carbapenemases,” many of these enzymes recognize almost all β-lactam antibiotics, rendering them ineffective.
CRE infections are associated with poor outcomes because of the limited therapeutic options with activity against these isolates (2). In a report published in 2013, the Centers for Disease Control and Prevention (CDC) classified CRE as one of the top urgent drug-resistant threats to the United States, accounting for 9,000 drug-resistant infections and 600 fatalities per year (3). Septicemia due to CRE is particularly very serious, resulting in high rates of mortality. A recent study reported overall 14-day and 30-day mortality rates of 34% and 49%, respectively, among patients with CRE bacteremia (4). The high mortality rates are attributed in part to the delay in the initiation of appropriate therapy with activity against CRE and the high rate of resistance emergence to the currently available agents, such as tigecycline, polymyxins, and aminoglycosides (2). The development of new agents to treat serious bacterial infections due to CRE, inclusive of septicemia, is crucial.
Plazomicin is a novel aminoglycoside that is resilient to the activity of all clinically relevant aminoglycoside-modifying enzymes (AMEs), the primary mechanism of aminoglycoside resistance (5). Plazomicin shows activity against both Gram-positive and Gram-negative bacteria, including multidrug-resistant Enterobacteriaceae and CRE (6). Owing to its spectrum of activity, plazomicin is a promising novel agent for the treatment of infections due to these resistant isolates. It has been studied in clinical trials for the treatment of serious bacterial infections due to CRE (7).
The purpose of this study was to evaluate the efficacy of plazomicin, alone and in combination with two broad-spectrum antibiotics, meropenem or tigecycline, and compare these efficacy profiles with the efficacy of either meropenem or tigecycline alone against Enterobacteriaceae isolates exhibiting various resistance profiles, including CRE, using an immunocompetent murine septicemia model, a preclinical model commonly utilized for the assessment of antimicrobial efficacy. In an attempt to further improve the translational application of the outcomes from our animal model into clinical practice, human-simulated regimens of the antimicrobial agents were utilized in this investigation.
(This study was presented in part at IDWeek 2017, San Diego, CA, USA, 4 to 8 October 2017 [8].)
RESULTS
Bacterial isolates.
A summary of the 8 Enterobacteriaceae isolates utilized in this study and their susceptibilities to plazomicin, meropenem, and tigecycline as well as their genotypes is shown in Table 1.
TABLE 1.
Summary of the isolates selected for the in vivo efficacy studies
| Isolate | JMI Laboratories isolate identifier | MIC (mg/liter) |
Positive molecular test results | Notes |
||||
|---|---|---|---|---|---|---|---|---|
| Plazomicin | Meropenem | Tigecycline | OmpC/OmpK36 | OmpF/OmpK35 | OmpK37 | |||
| Klebsiella pneumoniae 557 | 34215 | 2 | 8 | 0.5 | aadA2, blaTEM-1, blaSHV-11, aac(6′)-Ib, blaKPC-2, aph(3′)-Ia | Alterations | Disrupted | Alterations |
| Klebsiella pneumoniae 558 | 45846 | 2 | >32 | 1 | aac(3)-IId, aadA2, blaTEM-1, aph(6)-Ia, blaSHV-11, blaKPC-3, aph(6)-Id | Alterations | Disrupted | Alterations |
| Escherichia coli 471 | 31278 | 4 | 0.03 | ≤0.06 | aac(3)-IId, blaTEM-1, EC-6 (intrinsic AmpC) | Alterations | Wild type | NAa |
| Klebsiella oxytoca 92 | 41599 | 4 | ≤0.015 | 0.12 | blaOXY-6-4 (intrinsic gene) | Alterations | Alterations | Wild type |
| Citrobacter freundii 38 | 50925 | 4 | 0.06 | 0.5 | blaCMY-48-like (intrinsic gene), aac(6′)-If | Alterations | Alterations | NA |
| Morganella morganii 65 | 39292 | 8 | 0.06 | 1 | aac(3)-IId, blaDHA-9 (intrinsic gene), aadA5, aph(3′)-Ia | Wild type | NA | NA |
| Klebsiella pneumoniae 561 | 51020 | 8 | >32 | 2 | blaTEM-1, blaSHV-11, aac(3)-IIa, blaCTX-M-15, aac(6′)-Ib-cr, blaOXA-48, blaOXA-1, blaOXA-30 | Alterations | Disrupted | Alterations |
| Klebsiella pneumoniae 559 | 51015 | 16 | >32 | 2 | blaTEM-1, blaSHV-11, aac(3)-IIa, blaCTX-M-15, aac(6′)-Ib-cr, blaOXA-48, blaOXA-1, blaOXA-30 | Alterations | Disrupted | Alterations |
NA, not applicable.
Human-simulated plazomicin exposure pharmacokinetic studies.
Plazomicin was adequately detected in mouse plasma for all examined doses in the single-dose pharmacokinetic studies following subcutaneous administration. The observations were satisfactorily described by a two-compartment linear model. The relationship between the plazomicin area under the total-drug plasma concentration-time curve (AUC) from 0 to 24 h (AUC0–24) and the range of doses examined was linear with a coefficient of determination (R2) of 0.9862.
Based on the observed AUC0–24, it was predicted that a dose of 28 mg/kg of body weight every 24 h (q24h) in mice would achieve an exposure similar to that observed in humans following administration of plazomicin at 15 mg/kg q24h. A confirmatory pharmacokinetic study of the selected regimen was conducted in the murine infection model; the plasma AUC0–24 achieved following administration of a single 28-mg/kg dose was 269 mg · h/liter, which was comparable to the human target AUC0–24 exposure (mean AUC0–24, 265 mg · h/liter).
Human-simulated exposure pharmacokinetic studies of comparator agents.
Based on the percentage of the dosing interval during which the free drug concentrations remained above the MIC (%fT>MIC), a meropenem exposure in mice similar to that expected in humans following administration of 2 g every 8 h (q8h) as a 3-h infusion was attained when mice were administered a 6-dose regimen (14 mg/kg at 0 h, 19 mg/kg at 1.25 h, 21 mg/kg at 2.5 h, 18 mg/kg at 3.75 h, 13 mg/kg at 5 h, and 10.5 mg/kg at 6.25 h), repeated every 8 h.
On the basis of the observed area under the free-drug plasma concentration-time curve (fAUC0–24), it was predicted that a tigecycline dose of 1.66 mg/kg every 12 h (q12h) in mice would achieve an exposure similar to that observed in humans following administration of tigecycline at 50 mg q12h. The observed plasma fAUC0–24 achieved following administration of 1.66 mg/kg q12h was 0.83 mg · h/liter, which was comparable to the human target AUC0–24 exposure (fAUC0–24 = 0.94 mg · h/liter).
Human-simulated exposure pharmacokinetic studies of combination treatment.
After the human-simulated regimens of plazomicin and the comparators were selected, confirmatory combination pharmacokinetic studies were conducted; animals were dosed with plazomicin at 28 mg/kg q24h and coadministered either meropenem (14 mg/kg at 0 h, 19 mg/kg at 1.25 h, 21 mg/kg at 2.5 h, 18 mg/kg at 3.75 h, 13 mg/kg at 5 h, and 10.5 mg/kg at 6.25 h), repeated every 8 h, or tigecycline at 1.66 mg/kg q12h. The concomitant administration of plazomicin and meropenem did not alter the plazomicin or meropenem profiles. However, the tigecycline fAUC0–24 was slightly reduced in the presence of plazomicin (observed fAUC0–24 = 0.55 mg · h/liter). This was associated with a shortening of the elimination half-life, and, thus, an increase in the tigecycline dose to 2 mg/kg q12h was warranted to attain the target human exposure. The tigecycline fAUC0–24 achieved when tigecycline was dosed at 2 mg/kg q12h concomitantly with plazomicin at 28 mg/kg q24h was 0.96 mg · h/liter. The mechanism of interaction between tigecycline and plazomicin was not examined, as it was beyond the scope of this investigation. The concomitant administration of plazomicin and tigecycline did not alter the plazomicin profile. Table 2 shows the observed plazomicin plasma AUC0–24 achieved following administration of the human-simulated regimen alone or in the presence of comparators as well as the observed tigecycline plasma fAUC0–24 achieved following administration of the human-simulated regimen alone or in combination with plazomicin.
TABLE 2.
Plazomicin AUC0–24 and tigecycline fAUC0–24 values in mice and humans
| Agent | Species | Daily dose | Regimen | AUC0–24 or fAUC0–24 (mg · h/liter) |
|---|---|---|---|---|
| Plazomicin | Human | 15 mg/kg q24h | Plazomicin | 265.00a |
| Mouse | 28 mg/kg q24h | Plazomicin alone | 269.06 | |
| Plazomicin + meropenem | 284.98 | |||
| Plazomicin + tigecycline | 259.96 | |||
| Tigecycline | Human | 50 mg q12h | Tigecycline | 0.94b |
| Mouse | 1.66 mg/kg q12h | Tigecycline alone | 0.83 | |
| 1.66 mg/kg q12h | Tigecycline + plazomicin | 0.55 | ||
| 2 mg/kg q12h | Tigecycline + plazomicin | 0.96 | ||
| 2 mg/kg q12h | Tigecycline alone | 1.34 |
Corresponding to an AUC0–24/MIC of 132.5 to 16.6 for isolates with plazomicin MICs of 2 to 16 mg/liter.
Corresponding to an fAUC0–24/MIC of 15.7 to 0.47 for isolates with tigecycline MICs of 0.06 to 2 mg/liter.
Survival studies in immunocompetent septicemia model.
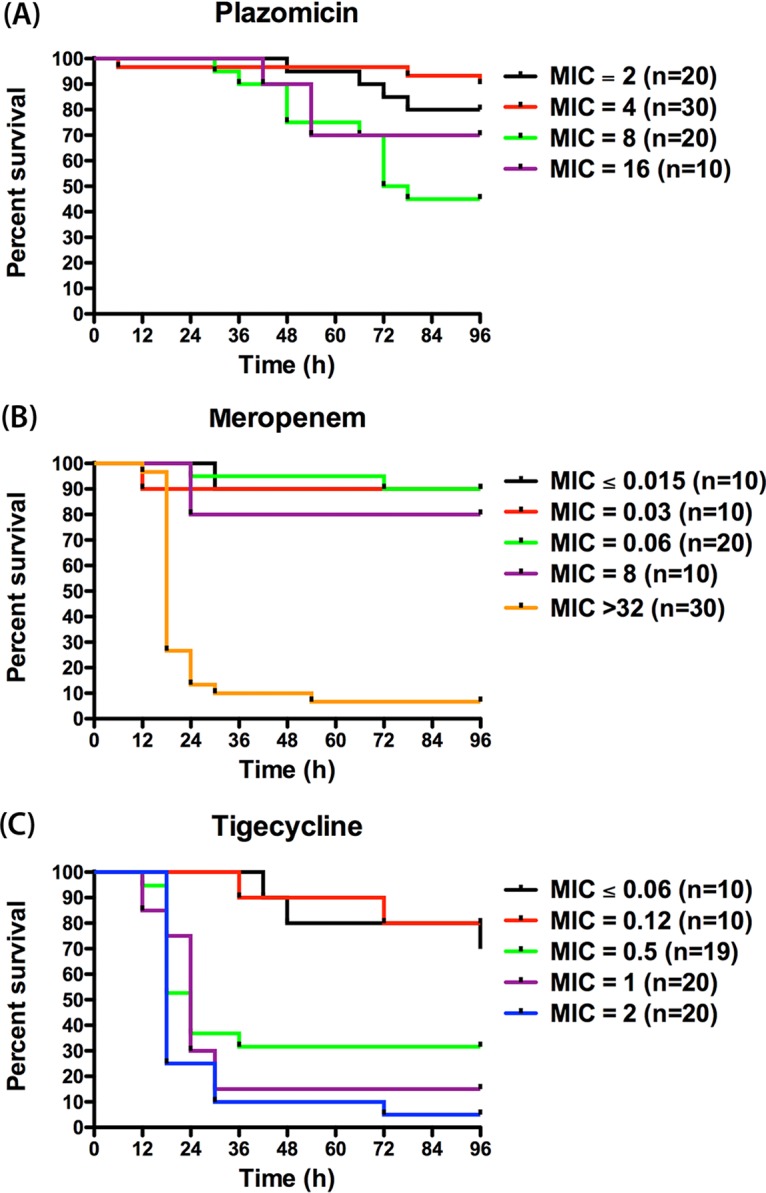
The blood cultures of the 0-h groups (1 h postinfection) showed bacterial growth for all 8 isolates. The average bacterial burden observed at 0 h was 5.07 ± 0.23 log10 CFU/ml. This confirmed the capability of the tested isolates to induce septicemia in the murine model utilized. Figure 1A to H shows the Kaplan-Meier survival curves for the 8 isolates, and Table 3 summarizes the survival percentages at 96 h with each therapy. The vehicle-dosed controls exhibited a high degree of mortality for all tested isolates. The survival percentages among the control groups at 96 h ranged from 0% to 20%, with the majority of the mortalities occurring within the first 24 h of the study period.
FIG 1.

Survival curves for isolates K. pneumoniae (KP) 557 (MICs, plazomicin [PLZ], 2 mg/liter; meropenem [MEM], 8 mg/liter; tigecycline [TGC], 0.5 mg/liter) (A), K. pneumoniae 558 (MICs, PLZ, 2 mg/liter; MEM, >32 mg/liter; TGC, 1 mg/liter) (B), Escherichia coli (EC) 471 (MICs, PLZ, 4 mg/liter; MEM, 0.03 mg/liter; TGC, ≤0.06 mg/liter) (C), Klebsiella oxytoca (KO) 92 (MICs, PLZ, 4 mg/liter; MEM, ≤0.015 mg/liter; TGC, 0.12 mg/liter) (D), Citrobacter freundii (CF) 38 (MICs, PLZ, 4 mg/liter; MEM, 0.06 mg/liter; TGC, 0.5 mg/liter) (E), Morganella morganii (MM) 65 (MICs, PLZ, 8 mg/liter; MEM, 0.06 mg/liter; TGC, 1 mg/liter) (F), K. pneumoniae 561 (MICs, PLZ, 8 mg/liter; MEM, >32 mg/liter; TGC, 2 mg/liter) (G), and K. pneumoniae 559 (MICs, PLZ, 16 mg/liter; MEM, >32 mg/liter; TGC, 2 mg/liter) (H).
TABLE 3.
Mouse survival at 96 h with different treatments in the in vivo efficacy studies
| Isolate | MIC (mg/liter) |
Survival (%) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Plazomicin | Meropenem | Tigecycline | Controls | Plazomicin | Meropenem | Tigecycline | Plazomicin + meropenem | Plazomicin + tigecycline | |
| K. pneumoniae 557 | 2 | 8 | 0.5 | 0 | 60 | 80 | 0 | 60 | 80 |
| K. pneumoniae 558 | 2 | >32 | 1 | 10 | 100 | 10 | 30 | 80 | 100 |
| Escherichia coli 471 | 4 | 0.03 | ≤0.06 | 0 | 100 | 90 | 70 | 70 | 80 |
| Klebsiella oxytoca 92 | 4 | ≤0.015 | 0.12 | 10 | 80 | 90 | 80 | 90 | 90 |
| Citrobacter freundii 38 | 4 | 0.06 | 0.5 | 20 | 90 | 100 | 60 | 100 | 100 |
| Morganella morganii 65 | 8 | 0.06 | 1 | 0 | 70 | 80 | 0 | 100 | 80 |
| K. pneumoniae 561 | 8 | >32 | 2 | 0 | 20 | 0 | 10 | 100 | 90 |
| K. pneumoniae 559 | 16 | >32 | 2 | 0 | 70 | 10 | 0 | 70 | 80 |
Compared with the survival of the vehicle-dosed controls, human-simulated exposure of plazomicin monotherapy produced significant improvement in survival for mice infected with all isolates (P < 0.05), with the survival percentages ranging from 20% to 100% by the end of the study period (Fig. 1A to H). When the survival outcome was grouped by the plazomicin MIC of the causative pathogen, the survival percentages with plazomicin monotherapy were 80% (n = 20), 90% (n = 30), 45% (n = 20), and 70% (n = 10) for mice infected with isolates with MICs of 2, 4, 8, and 16 mg/liter, respectively, as shown in Fig. 2A. Thus, plazomicin monotherapy resulted in overall survival percentages of 86% (n = 50) and 53.3% (n = 30) for mice infected with isolates with plazomicin MICs of ≤4 and ≥8 mg/liter, respectively (P < 0.05).
FIG 2.
Overall survival curves for mice treated with plazomicin (A), meropenem (B), or tigecycline (C) monotherapy.
The administration of the human-simulated exposure of meropenem monotherapy was associated with survival percentages ranging from 0% to 100% among the mice infected with the 8 isolates by the end of the study period. Likewise, the human-simulated exposure of tigecycline monotherapy was associated with survival percentages ranging from 0% to 80% (Fig. 1A to H). Figure 2B and C shows the overall survival proportions of the mice receiving meropenem or tigecycline monotherapy grouped by the MIC of the examined isolates. The survival percentages with meropenem monotherapy were 90% (n = 10), 90% (n = 10), 90% (n = 20), 80% (n = 10), and 6.667% (n = 30) for mice infected with isolates with MICs of ≤0.015, 0.03, 0.06, 8, and >32 mg/liter, respectively, while those with tigecycline monotherapy were 70% (n = 10), 80% (n = 10), 31.579% (n = 19), 15% (n = 20), and 5% (n = 20) for mice infected with isolates with MICs of ≤0.06, 0.12, 0.5, 1, and 2 mg/liter, respectively.
Overall, both combination treatments (plazomicin-meropenem and plazomicin-tigecycline) were associated with good survival for all the test agents, with survival percentages ranging from 60 to 100%. For isolate K. pneumoniae 561 (plazomicin, meropenem, and tigecycline MICs, 8, >32, and 2 mg/liter, respectively), the plazomicin-meropenem and plazomicin-tigecycline combinations showed significant improvement in survival (100% and 90%, respectively) compared with that achieved with plazomicin, meropenem, or tigecycline monotherapy (20%, 0%, and 10%, respectively; P < 0.05) as shown in Fig. 1G.
Detection of plazomicin-resistant mutants.
Less than 5% of the samples collected from the surviving mice treated with plazomicin showed positive bacterial growth on the drug-free plates at either 24 h or 48 h. For all the tested isolates, no growth was detected on the plazomicin-supplemented plates at either drug concentration (4-fold and 8-fold the plazomicin MIC). These results suggest the absence of growth of a resistant subpopulation during treatment.
DISCUSSION
Infections due to CRE are on the rise, particularly among health care settings. Recent FDA approval of new agents with activity against CRE, such as ceftazidime-avibactam, has provided important therapeutic alternatives for clinicians to address this threat (9). However, shortly after introduction to the market, the first case of a K. pneumoniae isolate expressing the KPC-3 carbapenemase and resistance to ceftazidime-avibactam was reported (10). Given the rapid evolution and spread of carbapenemases among Gram-negative bacteria, continuous development of new agents with activity against these isolates is warranted.
In this study, we examined the efficacy of plazomicin, meropenem, and tigecycline monotherapies as well as that of the combinations of plazomicin-meropenem and plazomicin-tigecycline using clinically relevant exposures of the test agents in the immunocompetent murine septicemia model. The human-equivalent doses in mice were selected to mimic the mean exposure achieved in humans on the basis of the parameter that correlated well with the efficacy of each agent: fAUC0–24 for plazomicin and tigecycline and %fT>MIC for meropenem. Eight Enterobacteriaceae isolates were utilized in this investigation. The selection of the isolates was based on the ability of the isolates to establish viable infection in the murine model, as evident by a high degree of mouse mortality in the absence of antimicrobial therapy, as well as the phenotypic and the genotypic profiles of these isolates. In order to account for the worst possible outcome and increase the robustness of the study results, Enterobacteriaceae isolates with MICs at the upper end of the plazomicin MIC distribution were selected, with the MICs being 2 to 16 mg/liter (MIC90 ≤ 1 mg/liter) (6). All isolates were positive for genes encoding AMEs and/or had alterations in outer membrane porins. Four of the isolates were CRE expressing various carbapenemases, including KPC-2, KPC-3, and OXA-48, as well as extended spectrum β-lactamases (ESBLs), such as CTX-M-15.
For meropenem monotherapy, the human-simulated regimen was associated with high survival rates for mice infected with isolates with MICs of ≤8 mg/liter, which was the expected outcome of the exposure attained with the prolonged infusion of 2 g q8h, as the fT>MIC was ≥75% (the threshold for carbapenem efficacy is a 40% fT>MIC [11]). On the other hand, the survival for mice infected with isolates with MICs of >32 mg/liter was low, with the predicted fT>MIC being 0% (11). The survival with tigecycline monotherapy correlated well with the isolates' susceptibilities, with an incremental increase in survival being observed at lower MIC values. Data from the tigecycline and meropenem monotherapy groups provided validation to the discriminatory capability of the model utilized, as the efficacy profiles achieved in the mice with the human-simulated exposures were in accordance with the expected clinical outcomes in humans receiving these exposures (11, 12).
Plazomicin as monotherapy or in combination with tigecycline or meropenem was associated with a high probability of survival for mice infected with isolates with plazomicin MICs of ≤4 mg/liter, inclusive of those expressing various AMEs. Plazomicin monotherapy resulted in erratic survival rates for mice infected with isolates with plazomicin MICs of ≥8 mg/liter, with mice infected with isolate K. pneumoniae 561 (MIC, 8 mg/liter) showing reduced survival compared with mice infected with isolate K. pneumoniae 559 (MIC, 16 mg/liter), which could allude to a difference in the fitness and/or in vivo expression of the resistance genes between the two bacterial strains. Furthermore, plazomicin combinations were associated with a high probability of survival for mice infected with isolates with plazomicin MICs up to 16 mg/liter, which is much higher than the reported plazomicin MIC90 among Enterobacteriaceae isolates (≤1 mg/liter) (6). These results were in general agreement with those from the phase 3 Combating Antibiotic Resistant Enterobacteriaceae (CARE) trial; plazomicin therapy in combination with meropenem or tigecycline was associated with a significantly lower rate of mortality or serious disease-related complications than colistin-based combination therapy among patients with serious infections, such as bloodstream infections as well as hospital-acquired and ventilator-associated bacterial pneumonia due to CRE (7). The results from isolate K. pneumoniae 561 in the murine septicemia model suggest that a potential in vivo synergistic effect may have contributed to the outcomes observed in the clinical trial. However, further data on more isolates are needed to draw definite conclusions. Unfortunately, more isolates with phenotypic profiles comparable to those of K. pneumoniae 561 (with reduced susceptibilities to the three agents) were not available at the time of conduct of this study.
We further examined the blood of the mice receiving plazomicin for the presence of viable bacterial cells 24 h or 48 h from the initiation of therapy. The limited growth on the drug-free plates indicated that plazomicin had the capability to rapidly eradicate the bacteria from the blood. Given that the plasma concentrations of all test agents were negligible at the time of the assessment, we ruled out the possibility that the lack of growth on the drug-free plates was attributed to drug carryover effects. Thus, the lack of growth on the plazomicin-supplemented plates provided some evidence for the absence of resistance development in vivo during treatment, albeit only a few samples showed bacterial growth on the drug-free plates due to the effectiveness of the compound. Additional in vitro and in vivo studies are warranted to better evaluate the resistance emergence during plazomicin therapy.
In summary, our data showed that plazomicin monotherapy resulted in improved survival in the immunocompetent murine septicemia model, particularly for mice infected with isolates with plazomicin MICs of ≤4 mg/liter. Moreover, data from one isolate suggested that coadministration of meropenem or tigecycline could have potentially led to a further improvement in survival. These preclinical data utilizing clinically relevant exposures corroborate the finding from the phase 3 CARE trial and support a potential role for plazomicin combinations with meropenem or tigecycline in the management of septicemia due to Enterobacteriaceae, including carbapenem-resistant isolates.
MATERIALS AND METHODS
Antimicrobial agents.
Plazomicin sulfate for injection (50 mg/ml; batch number JGC1-ENG13003) was supplied by Achaogen, Inc., for in vivo testing. Plazomicin solution was diluted in sterile 0.9% sodium chloride solution (B. Braun Medical Inc., Irvine, CA, USA) to attain final concentrations that would deliver the required doses based on the mean weight of the study mouse population. Plazomicin was administered via subcutaneous (s.c.) injections of 0.1 ml.
Commercially available meropenem 500-mg vials (lot 39D55; WG Fresenius Kabi USA, LLC, Lake Zurich, IL, USA) and tigecycline 50-mg vials (lot AJP312; Wyeth Pharmaceuticals Inc., Philadelphia, PA, USA) were reconstituted and diluted with 0.9% normal saline solution and administered as 0.1-ml s.c. injections.
Bacterial isolates.
A total of 8 Enterobacteriaceae isolates with various phenotypic and genotypic profiles, including CRE isolates, were provided by JMI Laboratories (North Liberty, IA, USA). The MICs of plazomicin, meropenem, and tigecycline were derived by JMI Laboratories using the broth microdilution methodology, as outlined by the Clinical and Laboratory Standards Institute (CLSI) criteria (13). The isolates were characterized through whole-genome sequencing by JMI Laboratories using methods previously reported (14, 15). Furthermore, these isolates were screened for their ability to induce septicemia in the immunocompetent infection model utilized.
Immunocompetent murine septicemia model.
Specific-pathogen-free, female ICR mice weighing 20 to 22 g were obtained from Envigo RMS, Inc. (Indianapolis, IN, USA). The animals were allowed to acclimate for a minimum of 48 h before commencement of experimentation and were provided food and water ad libitum. The protocol was reviewed and approved by the Institutional Animal Care and Use Committee at Hartford Hospital. Mice were administered uranyl nitrate at 5 mg/kg intraperitoneally (i.p.) 3 days prior to inoculation to produce a controlled degree of renal impairment to assist with humanizing the target exposures of the test agents.
All bacterial isolates had previously been frozen at −80°C in skim milk (BD BioSciences, Sparks, MD, USA). Prior to mouse inoculation, two transfers of the organisms were performed onto Trypticase soy agar plates with 5% sheep blood (TSA II; Becton, Dickinson & Co., Sparks, MD, USA) and incubated at 37°C. After 18 to 24 hours of incubation of the second transfer, a bacterial suspension of approximately 106.5 CFU/ml in 5% hog gastric mucin was made for inoculation. This inoculum was selected following a series of pilot studies to produce mortality within a reasonable window of 18 to 48 hours postinfection in the absence of appropriate antimicrobial therapy. Final inoculum concentrations were confirmed by serial dilution and plating techniques. Septicemia was produced by i.p. injection of 0.5 ml of the inoculum 1 h prior to the initiation of antimicrobial therapy (16, 17).
Human-simulated plazomicin exposure pharmacokinetic studies.
Pharmacokinetic studies of plazomicin were carried out to identify a regimen that provided an exposure similar to that achieved in infected patients following the administration of 15 mg/kg as a 0.5-h infusion q24h, based on the AUC0–24 (18). Since the percentage of plazomicin protein binding is low and comparable between humans and mice (19.6% ± 8.8% and 19.9% ± 10.5%, respectively) (19), no correction for the free fraction was applied.
Initially, four single-dose pharmacokinetic studies were undertaken to identify the plazomicin concentration-time profile and exposures in the infection model utilized. The doses examined were 5, 20, 40, and 80 mg/kg. The AUC0–24 values achieved with these doses were used to predict a dose in mice that would simulate that achieved in infected patients (265 mg · h/liter) (20). Infected animals were administered plazomicin, and then groups of 6 mice were euthanized at 8 predefined time points. Terminal blood samples from CO2-asphyxiated mice were collected via cardiac puncture and placed in K2EDTA BD Microtainer tubes (BD, Franklin Lakes, NJ, USA). Plasma was separated by centrifugation for 10 min at 4°C at 10,000 × g and then stored at −80°C until analyzed for the plazomicin concentrations using a validated liquid chromatography-tandem mass spectrometry method by Alturas Analytics, Inc. (Moscow, ID, USA). The pharmacokinetic parameters of plazomicin were estimated (WinNonlin, version 5.0.1; Pharsight Corp., Mountain View, CA, USA). Additionally, the AUC0–24 was estimated using the trapezoidal rule and used to predict the human-simulated regimen. After the human-simulated regimen of plazomicin was identified mathematically, a confirmatory pharmacokinetic study was undertaken using the same methodology to confirm that the target AUC0–24 exposure was achieved.
Human-simulated exposure pharmacokinetic studies of comparator agents.
Pharmacokinetic studies of meropenem were carried out to identify a regimen that provided an exposure similar to that achieved in humans following the administration of 2 g as a 3-h infusion q8h on the basis of the %fT>MIC across a range of MICs (4 to 64 mg/liter) using the meropenem protein binding data (8% in both humans and mice) (21). Similarly, pharmacokinetic studies of tigecycline were undertaken to identify a regimen that provided an exposure similar to that achieved in humans following the administration of 50 mg q12h on the basis of the fAUC0–24 using the average tigecycline protein binding data (80% and 87.86% in humans and mice, respectively) (22, 23). Infected animals were administered either the selected meropenem or tigecycline regimen, and then groups of 6 mice each were euthanized at 6 to 8 predefined time points. Terminal blood samples from CO2-asphyxiated mice were collected via cardiac puncture and placed in K2EDTA BD Microtainer tubes (BD, Franklin Lakes, NJ, USA). Plasma was separated by centrifugation for 10 min at 4°C at 10,000 × g and then transferred into polypropylene tubes. These tubes were stored at −80°C until analyzed. Plasma samples were assayed for meropenem and tigecycline concentrations at the Center for Anti-Infective Research and Development laboratory at Hartford Hospital using previously established validated high-performance liquid chromatography (HPLC) assays (22, 24). The pharmacokinetic parameters of meropenem and tigecycline were estimated on the basis of the total drug concentrations (WinNonlin, version 5.0.1; Pharsight Corp., Mountain View, CA, USA) and thereafter used to confirm that the target human exposures were achieved.
Human-simulated exposure pharmacokinetic studies of combination treatment.
Once the human-simulated regimens of all three agents were identified, confirmatory combination pharmacokinetic studies were undertaken to ascertain that coadministration of the human-simulated regimens of the comparator agents (meropenem or tigecycline) did not considerably alter the concentration-time profile or the exposure of plazomicin or that of the comparators. Infected animals were administered the plazomicin human-simulated regimen in combination with the meropenem or tigecycline human-simulated regimens, and then groups of 6 mice each were euthanized at 6 to 8 predefined time points. Terminal blood samples were collected and processed as described above. The plasma samples from each mouse were divided into two aliquots; one aliquot was utilized to determine the comparator agent concentrations (meropenem or tigecycline), while the other aliquot was utilized for plazomicin concentration determination. Once the concentration data were compiled, the exposure of each agent was compared with that previously achieved when the agent was administered alone.
Survival studies in immunocompetent septicemia model.
The purpose of this section of the study was to assess the in vivo activity of the human-simulated plazomicin exposure alone and compare it with that of the human-simulated meropenem and tigecycline exposures alone as well as the plazomicin-meropenem and plazomicin-tigecycline human-simulated combination exposures against the selected 8 Enterobacteriaceae isolates.
Mice were prepared and inoculated as described above. For each isolate tested, mice were randomly assigned to treatment and control groups of 10 mice each. Treatments were initiated with one of the five predetermined human-simulated plazomicin, meropenem, or tigecycline monotherapies or plazomicin-meropenem or plazomicin-tigecycline combination regimens at 1 h postinfection. For each isolate tested, one group of control animals received sterile normal saline in the same volume and by the same route and schedule as the most frequent treatment regimen (vehicle-dosed control). Additionally, another group of controls was sacrificed at 1 h postinfection and served as 0-h controls to test for the presence of viable bacterial cells in blood and confirm the establishment of septicemia. Briefly, mice were euthanized by CO2 exposure followed by cardiac puncture. Blood samples (10 to 100 μl) were plated onto Trypticase soy agar plates with 5% sheep blood (TSA II; Becton, Dickinson & Co., Sparks, MD, USA). The plates were incubated overnight at 37°C and then assessed for bacterial growth.
Treatments were administered for 24 h. Treated and vehicle-dosed control mice were followed up for survival every 6 h for up to 96 h. The rate and proportion of mortality were recorded and assessed relative to those for the treatment regimen received over the 4-day study period using Kaplan-Meier analysis and compared statistically between the different regimens using the log-rank test.
Detection of plazomicin-resistant mutants.
In addition, we assessed whether the administration of plazomicin was associated with the selection of resistant mutants in vivo using the infection model utilized. This part of the study was run in conjunction with the efficacy studies described above for 6 out of 8 isolates. For this purpose, blood samples with volumes of 10 to 100 μl were collected though the tail vein from all the surviving mice receiving plazomicin as monotherapy or in combination with meropenem or tigecycline at 24 h (K. pneumoniae isolates 557, 559, 558, and 561) or 48 h (Klebsiella oxytoca 92 and Citrobacter freundii 38). Prior to the tail bleed, animals were warmed under a heating lamp (up to 35°C, or 95°F) for 10 min to dilate the tail blood vessels. The collected blood samples were plated on Mueller-Hinton agar plates (BBL; Becton, Dickinson & Co., Sparks, MD, USA) supplemented with plazomicin at final concentrations equivalent to 4-fold and 8-fold the plazomicin MIC of the test isolate. Blood samples of equal volumes were simultaneously plated on Trypticase soy agar plates with 5% sheep blood (TSA II; Becton, Dickinson & Co., Sparks, MD, USA). The plates were incubated for 2 days at 37°C and checked daily for the growth of bacteria. The growth observed on the plazomicin-supplemented plates was compared with that observed on the drug-free plates at the same time point.
ACKNOWLEDGMENTS
This study was sponsored by Achaogen, Inc., South San Francisco, CA, USA (sponsor study no. A16040-1490-EF-M).
We acknowledge the superior assistance of Christina Sutherland, Sara Giovagnoli, Deborah Santini, Jennifer Tabor-Rennie, Elizabeth Cyr, Kimelyn Greenwood, Mordechai Grupper, Marguerite Monogue, Alissa Padgett, Safa Abuhussain, and Sean Stainton in the performance of this study. We acknowledge Raymond Diokno and Julie Seroogy from Achaogen, Inc., for their assistance with the bioanalytical assessment of plazomicin. We also acknowledge Mariana Castanheira from JMI Laboratories for her kind assistance with the genotypic characterization of the bacterial isolates.
A.K. and K.M.K. are employers and shareholders of Achaogen, Inc. K.A. and D.P.N. have no conflicts of interest to declare.
REFERENCES
- 1.Gupta N, Limbago BM, Patel JB, Kallen AJ. 2011. Carbapenem-resistant Enterobacteriaceae: epidemiology and prevention. Clin Infect Dis 53:60–67. doi: 10.1093/cid/cir202. [DOI] [PubMed] [Google Scholar]
- 2.Morrill HJ, Pogue JM, Kaye KS, LaPlante KL. 2015. Treatment options for carbapenem-resistant Enterobacteriaceae infections. Open Forum Infect Dis 2:ofv050. doi: 10.1093/ofid/ofv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States. Centers for Disease Control and Prevention, U.S. Department of Health and Human Services, Atlanta, GA. [Google Scholar]
- 4.Satlin MJ, Chen L, Patel G, Gomez-Simmonds A, Weston G, Kim AC, Seo SK, Rosenthal ME, Sperber SJ, Jenkins SG, Hamula CL, Uhlemann AC, Levi MH, Fries BC, Tang YW, Juretschko S, Rojtman AD, Hong T, Mathema B, Jacobs MR, Walsh TJ, Bonomo RA, Kreiswirth BN. 2017. Multicenter clinical and molecular epidemiological analysis of bacteremia due to carbapenem-resistant Enterobacteriaceae (CRE) in the CRE epicenter of the United States. Antimicrob Agents Chemother 61:e02349-16. doi: 10.1128/AAC.02349-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhanel GG, Lawson CD, Zelenitsky S, Findlay B, Schweizer F, Adam H, Walkty A, Rubinstein E, Gin AS, Hoban DJ, Lynch JP, Karlowsky JA. 2012. Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin. Expert Rev Anti Infect Ther 10:459–473. doi: 10.1586/eri.12.25. [DOI] [PubMed] [Google Scholar]
- 6.Walkty A, Adam H, Baxter M, Denisuik A, Lagace-Wiens P, Karlowsky JA, Hoban DJ, Zhanel GG. 2014. In vitro activity of plazomicin against 5,015 Gram-negative and Gram-positive clinical isolates obtained from patients in Canadian hospitals as part of the CANWARD study, 2011-2012. Antimicrob Agents Chemother 58:2554–2563. doi: 10.1128/AAC.02744-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKinnell JA, Connolly LE, Pushkin R, Jubb AM, O'Keeffe B, Serio AW, Smith A, Gall J, Riddle V, Krause KM, Pogue JM. 2017. Improved outcomes with plazomicin (PLZ) compared with colistin (CST) in patients with bloodstream infections (BSI) caused by carbapenem-resistant enterobacteriaceae (CRE): results from the CARE study, abstr 1853. Abstr IDWeek 2017, San Diego, CA, USA. Infectious Diseases Society of America, Arlington, VA. [Google Scholar]
- 8.Abdelraouf K, Kim A, Krause KM, Nicolau DP. 2017. Assessment of the in vivo efficacy of plazomicin (PLZ) alone or in combination with meropenem (MEM) or tigecycline (TGC) against Enterobacteriaceae (EB) isolates exhibiting various resistance mechanisms in an immunocompetent (I+) murine septicemia model, abstr 1506. IDWeek 2017, San Diego, CA, USA. Infectious Diseases Society of America, Arlington, VA. [Google Scholar]
- 9.Sader HS, Castanheira M, Flamm RK, Farrell DJ, Jones RN. 2014. Antimicrobial activity of ceftazidime-avibactam against Gram-negative organisms collected from U.S. medical centers in 2012. Antimicrob Agents Chemother 58:1684–1692. doi: 10.1128/AAC.02429-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humphries RM, Yang S, Hemarajata P, Ward KW, Hindler JA, Miller SA, Gregson A. 2015. First report of ceftazidime-avibactam resistance in a KPC-3-expressing Klebsiella pneumoniae isolate. Antimicrob Agents Chemother 59:6605–6607. doi: 10.1128/AAC.01165-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crandon JL, Ariano RE, Zelenitsky SA, Nicasio AM, Kuti JL, Nicolau DP. 2011. Optimization of meropenem dosage in the critically ill population based on renal function. Intensive Care Med 37:632–638. doi: 10.1007/s00134-010-2105-0. [DOI] [PubMed] [Google Scholar]
- 12.Kontopidou F, Giamarellou H, Katerelos P, Maragos A, Kioumis I, Trikka-Graphakos E, Valakis C, Maltezou HC. 2014. Infections caused by carbapenem-resistant Klebsiella pneumoniae among patients in intensive care units in Greece: a multi-centre study on clinical outcome and therapeutic options. Clin Microbiol Infect 20:O117–O123. doi: 10.1111/1469-0691.12341. [DOI] [PubMed] [Google Scholar]
- 13.Clinical and Laboratory Standards Institute. 2017. Performance standards for antimicrobial susceptibility testing, 27th ed CLSI supplement M100. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 14.Mendes RE, Castanheira M, Gasink L, Stone GG, Nichols WW, Flamm RK, Jones RN. 2015. β-Lactamase characterization of Gram-negative pathogens recovered from patients enrolled in the phase 2 trials for ceftazidime-avibactam: clinical efficacies analyzed against subsets of molecularly characterized isolates. Antimicrob Agents Chemother 60:1328–1335. doi: 10.1128/AAC.01173-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castanheira M, Deshpande L, Hubler C, Mendes R, Serio A, Krause K, Flamm R. 2017. Activity of plazomicin against Enterobacteriaceae isolates collected in the United States including isolates carrying aminoglycoside-modifying enzymes detected by whole genome sequencing, abstr 1235. Abstr IDWeek 2017, San Diego, CA, USA. Infectious Diseases Society of America, Arlington, VA. [Google Scholar]
- 16.Docobo-Perez F, Lopez-Cerero L, Lopez-Rojas R, Egea P, Dominguez-Herrera J, Rodriguez-Bano J, Pascual A, Pachon J. 2013. Inoculum effect on the efficacies of amoxicillin-clavulanate, piperacillin-tazobactam, and imipenem against extended-spectrum beta-lactamase (ESBL)-producing and non-ESBL-producing Escherichia coli in an experimental murine sepsis model. Antimicrob Agents Chemother 57:2109–2113. doi: 10.1128/AAC.02190-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ambrose PG, Drusano GL, Craig WA. 2012. In vivo activity of oritavancin in animal infection models and rationale for a new dosing regimen in humans. Clin Infect Dis 54(Suppl 3):S220–S228. doi: 10.1093/cid/cis001. [DOI] [PubMed] [Google Scholar]
- 18.Wart SV, Rubino CM, Reynolds DK. 2012. Non-compartmental pharmacokinetic analysis for study ACHN-490-006, a randomized, double-blind, placebo and positive-controlled, crossover study to evaluate the effect of intravenous ACHN-490 injection on the QT/QTc interval in healthy volunteers. Institute for Clinical Pharmacodynamics, Inc, Latham, NY. [Google Scholar]
- 19.Wu S. 2017. Study report. Assessment of protein binding of ACHN-490 in plasma. Quintara Discovery, Hayward, CA. [Google Scholar]
- 20.Seroogy J, Choi T, Gall J, Van Wart S. 2017. Pharmacokinetics (PK) of plazomicin in healthy adults, abstr 206. Abstr ASM Microbe 2017, New Orleans, LA, USA. American Society for Microbiology, Washington, DC. [Google Scholar]
- 21.Ghazi IM, Crandon JL, Lesho EP, McGann P, Nicolau DP. 2015. Efficacy of humanized high-dose meropenem, cefepime, and levofloxacin against Enterobacteriaceae isolates producing Verona integron-encoded metallo-beta-lactamase (VIM) in a murine thigh infection model. Antimicrob Agents Chemother 59:7145–7147. doi: 10.1128/AAC.00794-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crandon JL, Banevicius MA, Nicolau DP. 2009. Pharmacodynamics of tigecycline against phenotypically diverse Staphylococcus aureus isolates in a murine thigh model. Antimicrob Agents Chemother 53:1165–1169. doi: 10.1128/AAC.00647-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wyeth Pharmaceuticals Inc. 2005. Tygacil® (tigecycline) for injection for intravenous use. Wyeth Pharmaceuticals Inc, Philadelphia, PA: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021821s021lbl.pdf. [Google Scholar]
- 24.Elkhaili H, Niedergang S, Pompei D, Linger L, Leveque D, Jehl F. 1996. High-performance liquid chromatographic assay for meropenem in serum. J Chromatogr B Biomed Appl 686:19–26. doi: 10.1016/S0378-4347(96)00205-8. [DOI] [PubMed] [Google Scholar]