

ABSTRACT
The hyperproduction of chromosomally encoded β-lactamases is a key method of acquired resistance to ceftazidime, aztreonam, and, when seen in backgrounds having reduced envelope permeability, carbapenems. Here, we show that the loss of Mpl, a UDP-muramic acid/peptide ligase, is a common and previously overlooked cause of chromosomally encoded β-lactamase hyperproduction in clinical isolates of Stenotrophomonas maltophilia and Pseudomonas aeruginosa, important pathogens notorious for their β-lactam-resistant phenotypes.
KEYWORDS: beta-lactamases, ceftazidime, regulation
TEXT
Stenotrophomonas maltophilia clinical isolates are resistant to almost all β-lactams because of the production of two β-lactamases: L1, a subclass B3 metallo-β-lactamase, and L2, a class A extended-spectrum β-lactamase (1). The production of L1 and L2 is coordinately controlled by AmpR, a LysR-type transcriptional activator, and is induced during β-lactam challenge of cells (2). Where previously characterized, AmpR regulators have been shown to bind two ligands in a competitive manner (3, 4). As summarized in Fig. 1, the AmpR activator ligand, an anhydro-muramyl-pentapeptide, is produced during β-lactam challenge via the concerted actions of lytic transglycosylases, which release N-acetylglucosamine-anhydro-muramyl-peptides from peptidoglycan (5), and AmpG, a permease that transports them into the cytoplasm (6, 7). NagZ, an enzyme that removes the N-acetylglucosamine moiety, is also necessary to release the AmpR activator ligand in some species (8), though not in S. maltophilia (9). The AmpR repressor ligand is a UDP-muramyl-pentapeptide (10). It is produced via the sequential addition of amino acids to a UDP-muramyl substrate, via four separate ligase enzymes, MurC (11), MurD (12), MurE (13), and MurF (14), with the last adding a d-alanine–d-alanine dipeptide made by a fifth ligase enzyme, Ddl (15). Mpl is an enzyme that can ligate a ready-made pentapeptide onto the UDP-muramyl substrate, skipping the MurC, MurD, MurE, Ddl, and MurF ligation reactions, each of which requires ATP hydrolysis (16). This Mpl-catalyzed reaction therefore saves considerable amounts of energy for the cell. Its pentapeptide substrate comes from the breakdown of anhydro-muramyl-pentapeptides by the peptide amidase AmpD. In this way, the breakdown of the anhydro-muramyl-pentapeptide AmpR activator ligand by AmpD is also directly linked to the production of the UDP-muramyl-pentapeptide AmpR repressor ligand by Mpl (2, 5, 17, 18) (Fig. 1).
FIG 1.
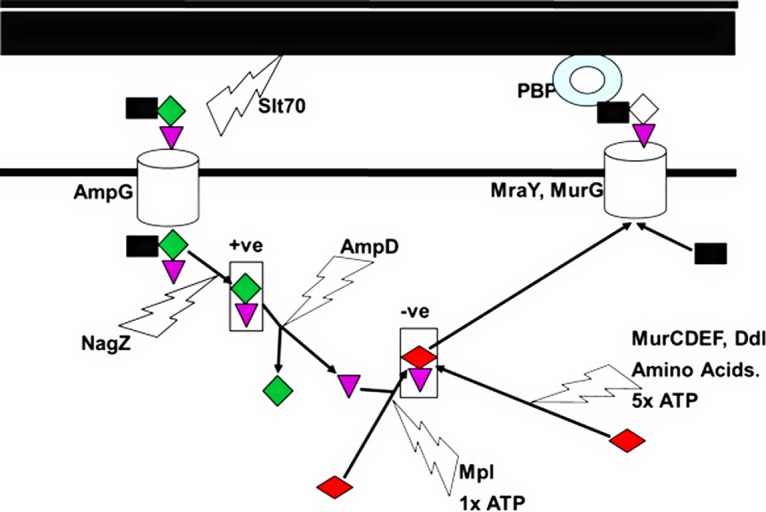
Role of Mpl in peptidoglycan recycling and AmpR activation. The schematic shows that N-acetylglucosamine (black square)-anhydro-muramyl (green diamond)-pentapeptide (purple triangle) is removed from peptidoglycan by lytic transglycosylases, such as Slt70, and enters the cytoplasm through the permease AmpG. NagZ removes the N-acetylglucosamine group to produce the anhydro-muramyl-pentapeptide AmpR activator ligand (+ve). AmpD then releases the pentapeptide ready to be linked to a UDP-muramic acid molecule (red diamond) by Mpl to produce the UDP-muramyl-pentapeptide AmpR repressor ligand (−ve). This can then be further incorporated into the biosynthetic pathway and processed by MurG and MraY, which add N-acetylglucosamine and penicillin-binding proteins, which add these high-energy N-acetylglucosamine-muramyl (white diamond)-pentapeptide substrates to the nascent peptidoglycan strand. UDP-muramyl-pentapeptide formation can also occur without peptidoglycan recycling through the sequential addition of amino acids to UDP-muramic acid. However, this requires five moles ATP per mole UDP-muramyl-pentapeptide, while the recycling pathway only requires one.
Ceftazidime is a relatively weak substrate for both L1 and L2 β-lactamases from S. maltophilia, and so many clinical isolates remain ceftazidime susceptible (1). However, mutants that have acquired ceftazidime resistance can easily be identified in the laboratory, and ceftazidime-resistant isolates are commonly encountered in the clinic. In most cases, these mutants hyperproduce L1 and L2 (19). Mutations that reduce AmpD function are known to boost L1/L2 production, because the AmpR activator ligand is broken down much less if AmpD is damaged (20). Mutations that (presumably) increase peptidoglycan turnover, releasing more muropeptides, also activate L1/L2 production, e.g., those in penicillin-binding protein 1A (PBP1A), encoded by mcrA (21), and in the lytic transglycosylase MltD, because this mutation stimulates the net production of lytic transglycosylase activity in the cell (22). Mutations in AmpR also activate L1/L2 production (4). We have previously characterized ceftazidime-resistant β-lactamase-hyperproducing laboratory-selected mutants derived from the extremely well-studied clinical isolate K279a. One of these mutants, KCAZ14, was wild type for ampR, ampD, and mcrA (19). To identify the mutation responsible, whole-genome resequencing was performed by MicrobesNG (Birmingham, UK) on a HiSeq 2500 instrument (Illumina, San Diego, CA, USA). Reads were trimmed using Trimmomatic (23) and assembled into contigs using SPAdes 3.10.1 (http://cab.spbu.ru/software/spades/). The assembled contigs were mapped to the reference genome for S. maltophilia K279a (24) obtained from GenBank (accession number NC_010943) using the progressiveMauve alignment software (25). The only mutation identified in KCAZ14 was a deletion of 18 nucleotides in the mpl gene, deleting amino acids 141 to 146 of Mpl. The level of β-lactamase production, measured as described previously (19), was similar for the mpl mutant KCAZ14, for the ampD loss-of-function mutant KCAZ10 (19), and for KM11, an ampR activatory mutant (4) (Table 1). To confirm the involvement of mpl loss in the β-lactamase-hyperproducing ceftazidime-resistant phenotype of KCAZ14, we attempted complementation in trans. K279a mpl was amplified by PCR, as previously described (19), with primers mpl_F (5′-ACCAGATCCAGGTACCGCC-3′) and mpl_R (5′-TCTCACATCCCGTGTAGGACT-3′). The product was blunt-end ligated into pBBRMCS-5 (gentamicin resistance [Gmr]) (26, 27) digested with SmaI, and the resulting recombinant plasmid was used to transform KCAZ14 to gentamicin resistance (15 μg · ml−1) via electroporation. The ceftazidime MIC against KCAZ14(pBBRMCS-5) was 64 μg · ml−1 and reduced to 4 μg · ml−1 in KCAZ14(pBBRMCS-5::mpl), the same as the MIC against wild-type K279a. The production of β-lactamase was also reduced to wild-type levels in KCAZ14(pBBRMCS-5::mpl) (Table 1), adding further confirmation of successful complementation.
TABLE 1.
β-Lactamase activity observed in S. maltophilia K279a and in ceftazidime-resistant K279a mutants and clinical isolates carrying different mutations
| Isolate | β-Lactamase activity (mean ± SEM) (nmol · min−1 · μg−1 protein nitrocefin hydrolyzed in cell extracts) | Relevant amino acid changes (relative to K279a) |
|---|---|---|
| K279a | 0.02 ± 0.004 | WTa |
| KM11 | 0.99 ± 0.03 | Asp135Asn in AmpR |
| KCAZ10 | 1.52 ± 0.04 | 159–168del in AmpD |
| KCAZ14 | 0.72 ± 0.01 | 140–146del in Mpl |
| 49-6147 | 0.45 ± 0.12 | 92–109del Mpl |
| 3800 | 0.73 ± 0.03 | Truncation at 368 in Mpl |
| 98 | 1.76 ± 0.07 | IS insertion in ampD, Ala85Glyb in Mpl |
| ula-511 | 1.19 ± 0.01 | Truncation at 360 in Mpl |
| KCAZ14(pBBRMCS-5) | 1.14 ± 0.10 | |
| KCAZ14(pBBRMCS-5::mpl) | 0.03 ± 0.003 |
WT, wild type.
Random genetic drift.
We have four ceftazidime-resistant β-lactamase-hyperproducing clinical S. maltophilia clinical isolates in our collection, isolates 49-6147, 3800, and 98 (19), and ULA-511 (28) (Table 1). Isolate 98 has an insertion sequence element disrupting ampD (19). While we also found a mutation causing an Ala85Gly change in Mpl, the same mutation is carried by ∼5% of S. maltophilia genomes in the GenBank database and therefore is probably insignificant. The other three clinical isolates have mpl mutations. In 49-6147, the mutation causes the deletion of amino acids 92 to 109, which disrupts the conserved Ser-Gly-Pro region (29). In 3800, there is a frameshift at codon 368, and in ULA-511, there is a nonsense mutation at codon 360.
The result of Mpl loss in KCAZ14 and these clinical isolates will be a build-up of pentapeptides released by AmpD (Fig. 1). Even though there are other enzymes that can break down these pentapeptides, it seems reasonable to hypothesize that this net accumulation of pentapeptide will affect AmpD activity by feedback inhibition, increasing the concentration of its substrate, the AmpR activator ligand, causing β-lactamase hyperproduction (18).
This is the first report of mpl disruption causing β-lactamase hyperproduction in S. maltophilia, and to find it in 3/4 clinical isolates was striking. It is also interesting to find that mpl loss-of-function mutations have been seen to accumulate in Pseudomonas aeruginosa populations carried by people with cystic fibrosis during long-term colonization in two separate studies (30, 31) and in 3/4 patients with P. aeruginosa-mediated ventilator-associated pneumonia (32). Indeed, mpl mutation has been identified as a cause of AmpC β-lactamase hyperproduction in one P. aeruginosa PAO1 laboratory selected transposon-insertion mutant (33). While this did not dramatically increase β-lactam MICs (33), PAO1 is relatively permeable to β-lactams, because it lacks many of the efflux pump/porin-altering mutations seen in clinical isolates (34). Therefore, it would seem reasonable to propose that these clinically acquired P. aeruginosa mpl mutations are being selected by β-lactam therapy. We have a small collection of ceftazidime-resistant P. aeruginosa clinical isolates, 2/5 of which have previously been confirmed to hyperproduce AmpC (35). Both have a mutation in mpl, according to whole-genome sequencing. The mutations in isolates 86-14571 and 73-56826 cause Met297Val and an Arg103His changes in Mpl, respectively. We conclude, therefore, that mpl loss in S. maltophilia and P. aeruginosa is a clinically important and previously underreported cause of β-lactamase hyperproduction and acquired β-lactam resistance.
ACKNOWLEDGMENTS
This work was funded, in part, by grant MR/N013646/1 to M.B.A. from the Antimicrobial Resistance Cross Council Initiative supported by the seven United Kingdom research councils. K.C. received a postgraduate scholarship from SENESCYT, Ecuador.
We declare no conflicts of interest.
REFERENCES
- 1.Calvopiña K, Hinchliffe P, Brem J, Heesom KJ, Johnson S, Cain R, Lohans CT, Fishwick CWG, Schofield CJ, Spencer J, Avison MB. 2017. Structural/mechanistic insights into the efficacy of nonclassical beta-lactamase inhibitors against extensively drug resistant Stenotrophomonas maltophilia clinical isolates. Mol Microbiol 106:492–504. doi: 10.1111/mmi.13831. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs C, Frere JM, Normark S. 1997. Cytosolic intermediates for cell wall biosynthesis and degradation control inducible beta-lactam resistance in Gram-negative bacteria. Cell 88:823–832. doi: 10.1016/S0092-8674(00)81928-5. [DOI] [PubMed] [Google Scholar]
- 3.Kraft AR, Prabhu J, Ursinus A, Holtje JV. 1999. Interference with murein turnover has no effect on growth but reduces beta-lactamase induction in Escherichia coli. J Bacteriol 181:7192–7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okazaki A, Avison MB. 2008. Induction of L1 and L2 beta-lactamase production in Stenotrophomonas maltophilia is dependent on an AmpR-type regulator. Antimicrob Agents Chemother 52:1525–1528. doi: 10.1128/AAC.01485-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vadlamani G, Thomas MD, Patel TR, Donald LJ, Reeve TM, Stetefeld J, Standing KG, Vocadlo DJ, Mark BL. 2015. The beta-lactamase gene regulator AmpR is a tetramer that recognizes and binds the d-Ala-d-Ala motif of its repressor UDP-N-acetylmuramic acid (MurNAc)-pentapeptide. J Biol Chem 290:2630–2643. doi: 10.1074/jbc.M114.618199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindquist S, Westonhafer K, Schmidt H, Pul C, Korfmann G, Erickson J, Sanders C, Martin HH, Normark S. 1993. AmpG, a signal transducer in chromosomal beta-lactamase induction. Mol Microbiol 9:703–715. doi: 10.1111/j.1365-2958.1993.tb01731.x. [DOI] [PubMed] [Google Scholar]
- 7.Huang YW, Lin CW, Hu RM, Lin YT, Chung TC, Yang TC. 2010. AmpN-AmpG Operon is essential for expression of L1 and L2 beta-lactamases in Stenotrophomonas maltophilia. Antimicrob Agents Chemother 54:2583–2589. doi: 10.1128/AAC.01283-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vötsch W, Templin MF. 2000. Characterization of a beta-N-acetylglucosaminidase of Escherichia coli and elucidation of its role in muropeptide recycling and beta-lactamase induction. J Biol Chem 275:39032–39038. doi: 10.1074/jbc.M004797200. [DOI] [PubMed] [Google Scholar]
- 9.Huang YW, Hu RM, Lin CW, Chung TC, Yang TC. 2012. NagZ-dependent and NagZ-independent mechanisms for beta-lactamase expression in Stenotrophomonas maltophilia. Antimicrob Agents Chemother 56:1936–1941. doi: 10.1128/AAC.05645-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uehara T, Park JT. 2002. Role of the murein precursor UDP-N-acetylmuramyl-l-Ala-gamma-d-Glu-meso-diaminopimelic acid-d-Ala-d-Ala in repression of beta-lactamase induction in cell division mutants. J Bacteriol 184:4233–4239. doi: 10.1128/JB.184.15.4233-4239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falk PJ, Ervin KM, Volk KS, Ho HT. 1996. Biochemical evidence for the formation of a covalent acyl-phosphate linkage between UDP-N-acetylmuramate and ATP in the Escherichia coli UDP-N-acetylmuramate:l-alanine lipase-catalyzed reaction. Biochemistry 35:1417–1422. doi: 10.1021/bi952078b. [DOI] [PubMed] [Google Scholar]
- 12.Pratviel-Sosa F, Mengin-Lecreulx D, Van Heijenoort J. 1991. Over-production, purification and properties of the uridine-diphosphate N-acetylmuramoyl-l-alanine-d-glutamate ligase from Escherichia coli. Eur J Biochem 202:1169–1176. doi: 10.1111/j.1432-1033.1991.tb16486.x. [DOI] [PubMed] [Google Scholar]
- 13.Michaud C, Mengin-Lecreulx D, Van Heijenoort J, Blanot D. 1990. Over-production, purification and properties of the uridine-diphosphate-N-acetylmuramoyl-l-alanyl-d-glutamate-meso-2,6-diaminopimelate ligase from Escherichia coli. Eur J Biochem 194:853–861. doi: 10.1111/j.1432-1033.1990.tb19479.x. [DOI] [PubMed] [Google Scholar]
- 14.Duncan K, Van Heijenoort J, Walsh CT. 1990. Purification and characterization of the d-alanyl-d-alanine adding enzyme from Escherichia coli. Biochemistry 29:2379–2386. doi: 10.1021/bi00461a023. [DOI] [PubMed] [Google Scholar]
- 15.Zawadzke LE, Bugg TDH, Walsh CT. 1991. Existence of two d-alanine:d-alanine ligases in Escherichia coli–cloning and sequencing of the ddlA gene and purification and characterization of the DdlA and DdlA enzymes. Biochemistry 30:1673–1682. doi: 10.1021/bi00220a033. [DOI] [PubMed] [Google Scholar]
- 16.Mengin-Lecreulx D, van Heijenoort J, Park JT. 1996. Identification of the mpl gene encoding UDP-N-acetylmuramate:l-alanyl-gamma-d-glutamyl-meso-diaminopimelate ligase in Escherichia coli and its role in recycling of cell wall peptidoglycan. J Bacteriol 178:5347–5352. doi: 10.1128/jb.178.18.5347-5352.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park JT, Uehara T. 2008. How bacteria consume their own exoskeletons. Microbiol Mol Biol Rev 72:211–227. doi: 10.1128/MMBR.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobs C, Joris B, Jamin M, Klarsov K, Van Beeumen J, Mengin-Lecreulx D, Van Heijenoort J, Park JT, Normark S, Frere JM. 1995. AmpD, essential for both beta-lactamase regulation and cell-wall recycling, is a novel cytosolic N-acetylmuramyl-l-alanine amidase. Mol Microbiol 15:553–559. doi: 10.1111/j.1365-2958.1995.tb02268.x. [DOI] [PubMed] [Google Scholar]
- 19.Talfan A, Mounsey O, Charman M, Townsend E, Avison MB. 2013. Involvement of mutation in ampD I, mrcA, and at least one additional gene in beta-lactamase hyperproduction in Stenotrophomonas maltophilia. Antimicrob Agents Chemother 57:5486–5491. doi: 10.1128/AAC.01446-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang TC, Huang YW, Hu RM, Huang SC, Lin YT. 2009. AmpD(I) is involved in expression of the chromosomal L1 and L2 beta-lactamases of Stenotrophomonas maltophilia. Antimicrob Agents Chemother 53:2902–2907. doi: 10.1128/AAC.01513-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin CW, Lin HC, Huang YW, Chung TC, Yang TC. 2011. Inactivation of mrcA gene derepresses the basal-level expression of L1 and L2 beta-lactamases in Stenotrophomonas maltophilia. J Antimicrob Chemother 66:2033–2037. doi: 10.1093/jac/dkr276. [DOI] [PubMed] [Google Scholar]
- 22.Huang YW, Wu CJ, Hu RM, Lin YT, Yang TC. 2015. Interplay among membrane-bound lytic transglycosylase D1, the CreBC two-component regulatory system, the AmpNG-AmpD(I)-NagZ-AmpR regulatory circuit, and L1/L2 beta-lactamase expression in Stenotrophomonas maltophilia. Antimicrob Agents Chemother 59:6866–6872. doi: 10.1128/AAC.05179-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crossman LC, Gould VC, Dow JM, Vernikos GS, Okazaki A, Sebaihia M, Saunders D, Arrowsmith C, Carver T, Peters N, Adlem E, Kerhornou A, Lord A, Murphy L, Seeger K, Squares R, Rutter S, Quail MA, Rajandream MA, Harris D, Churcher C, Bentley SD, Parkhill J, Thomson NR, Avison MB. 2008. The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome Biol 9:R74. doi: 10.1186/gb-2008-9-4-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jain A, Srivastava P. 2013. Broad host range plasmids. FEMS Microbiol Lett 348:87–96. doi: 10.1111/1574-6968.12241. [DOI] [PubMed] [Google Scholar]
- 27.Obranic S, Babic F, Maravic-Vlahovfcek G. 2013. Improvement of pBBR1MCS plasmids, a very useful series of broad-host-range cloning vectors. Plasmid 70:263–267. doi: 10.1016/j.plasmid.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Felici A, Amicosante G. 1995. Kinetic analysis of extension of substrate specificity with Xanthomonas maltophilia, Aeromonas hydrophila, and Bacillus cereus metallo-beta-lactamases. Antimicrob Agents Chemother 39:192–199. doi: 10.1128/AAC.39.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das D, Herve M, Feuerhelm J, Farr CL, Chiu HJ, Elsliger MA, Knuth MW, Klock HE, Miller MD, Godzik A, Lesley SA, Deacon AM, Mengin-Lecreulx D, Wilson IA. 2011. Structure and function of the first full-length murein peptide ligase (Mpl) cell wall recycling protein. PLoS One 6:e17624. doi: 10.1371/journal.pone.0017624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diaz Caballero J, Clark ST, Coburn B, Zhang Y, Wang PW, Donaldson SL, Tullis DE, Yau YCW, Waters VJ, Hwang DM, Guttman DS. 2015. Selective sweeps and parallel pathoadaptation drive Pseudomonas aeruginosa evolution in the cystic fibrosis lung. mBio 6:e00981-15. doi: 10.1128/mBio.00981-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams D, Evans B, Haldenby S, Walshaw MJ, Brockhurst MA, Winstanley C, Paterson S. 2015. Divergent, coexisting Pseudomonas aeruginosa lineages in chronic cystic fibrosis lung infections. Am J Respir Crit Care Med 191:775–785. doi: 10.1164/rccm.201409-1646OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K, Chen YQ, Salido MM, Kohli GS, Kong JL, Liang HJ, Yao ZT, Xie YT, Wu HY, Cai SQ, Drautz-Moses DI, Darling AE, Schuster SC, Yang L, Ding YC. 2017. The rapid in vivo evolution of Pseudomonas aeruginosa in ventilator-associated pneumonia patients leads to attenuated virulence. Open Biol 7:170029. doi: 10.1098/rsob.170029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsutsumi Y, Tomita H, Tanimoto K. 2013. Identification of novel genes responsible for overexpression of ampC in Pseudomonas aeruginosa PAO1. Antimicrob Agents Chemother 57:5987–5993. doi: 10.1128/AAC.01291-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castanheira M, Mills JC, Farrell DJ, Jones RN. 2014. Mutation-driven β-lactam resistance mechanisms among contemporary ceftazidime-nonsusceptible Pseudomonas aeruginosa isolates from U.S. hospitals. Antimicrob Agents Chemother 58:6844–6850. doi: 10.1128/AAC.03681-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takebayashi Y, Wan Nur Ismah WAK, Findlay J, Heesom KJ, Zhang J, Williams M, MacGowan AP, Avison MB. 2017. Prediction of cephalosporin and carbapenem susceptibility in multi-drug resistant Gram-negative bacteria using liquid chromatography-tandem mass spectrometry. bioRxiv doi: 10.1101/138594. [DOI]