

The emergence of resistance toward artemisinin combination therapies (ACTs) by the malaria parasite Plasmodium falciparum has the potential to severely compromise malaria control. Therefore, the development of new artemisinins in combination with new drugs that impart activities toward both intraerythrocytic proliferative asexual and transmissible gametocyte stages, in particular, those of resistant parasites, is urgently required.
KEYWORDS: Plasmodium falciparum, artemisinins, gametocytes, oxidative stress, reactive oxygen species, synergism
ABSTRACT
The emergence of resistance toward artemisinin combination therapies (ACTs) by the malaria parasite Plasmodium falciparum has the potential to severely compromise malaria control. Therefore, the development of new artemisinins in combination with new drugs that impart activities toward both intraerythrocytic proliferative asexual and transmissible gametocyte stages, in particular, those of resistant parasites, is urgently required. We define artemisinins as oxidant drugs through their ability to oxidize reduced flavin cofactors of flavin disulfide reductases critical for maintaining redox homeostasis in the malaria parasite. Here we compare the activities of 10-amino artemisinin derivatives toward the asexual and gametocyte stages of P. falciparum parasites. Of these, artemisone and artemiside inhibited asexual and gametocyte stages, particularly stage V gametocytes, in the low-nanomolar range. Further, treatment of both early and late gametocyte stages with artemisone or artemiside combined with the pro-oxidant redox partner methylene blue displayed notable synergism. These data suggest that modulation of redox homeostasis is likely an important druggable process, particularly in gametocytes, and this finding thereby enhances the prospect of using combinations of oxidant and redox drugs for malaria control.
INTRODUCTION
Chemotherapy, coupled with vector control, has reduced malaria disease mortality by over 66% since 2000 (1). However, the emergence of drug resistance by Plasmodium falciparum parasites to the artemisinin derivatives dihydroartemisinin (DHA), artesunate, and artemether, currently used clinically in artemisinin combination therapies (ACTs), represents a crushing setback to global malaria control (2–5). Therefore, the need to develop new drugs for use in ACTs that counter the resistance and thereby supplant the combinations of current artemisinin derivatives with other antimalarial drugs becomes a particularly urgent task (6). In addition, new partner drugs other than those normally used in ACTs—amodiaquine, lumefantrine, mefloquine, piperaquine, and pyronaridine—are also mandated, given that reduced susceptibility of the parasite to the current partner drugs is now established (7–11). The seriousness of the problem is starkly illustrated by the rapid spread of a single mutant associated with artemisinin resistance that has also acquired resistance to piperaquine (12).
In addition to activity against asexual intraerythrocytic stages of resistant parasites, any new drug used either alone or in combination with another drug must also be highly active against the sexual gametocyte stages so that by blocking transmission it may inhibit the spread of parasites with the resistant phenotypes (13). This property is not manifest in current drug combinations. In the absence of resistance, ACTs such as artemether-lumefantrine, DHA-piperaquine, and sulfadoxine-pyrimethamine-artesunate are curative of intraerythrocytic asexual-stage parasite infections and decrease gametocyte densities and clearance times but are only moderately effective in clearing mature-stage gametocytes (14, 15). As part of a large-scale international interdisciplinary research program involving the design and development of new triple-drug combinations for the treatment of malaria, tuberculosis, and toxoplasmosis, we are evaluating the utility of combinations of oxidant (16–18) and redox (or pro-oxidant) drugs (19–21) coupled with a third partner drug with a different mechanism of action. The third partner drug being considered in the first instance incorporates a 4(1H)-quinolone scaffold (22). Quinolones are used clinically for the treatment of tuberculosis (23); selected examples have acquired lead status as antimalarial drugs (24–26) and display potent dual target effects when combined with the appropriate drug partner (25). Thus, the ultimate aim is to develop compounds that, when used in combination, not only are effective against drug-resistant intraerythrocytic asexual-stage parasites but also are highly active against the sexually differentiated transmissible gametocyte stages (13).
It is widely accepted that artemisinins undergo reductive bioactivation either by heme-iron(II) or by labile nonheme iron(II) to produce toxic C radicals that alkylate vital intraparasitic proteins (27–29) through chemoproteomic analyses (30–32). However, the complicated polypharmacology of these compounds may additionally be explained by defining artemisinins and other antimalarial peroxides as oxidant drugs. This is based on the potential inability of such radicals to act as alkylating agents in the oxygen-rich essentially oxidizing environment of the intraerythrocytic parasite (18, 33, 34). Further, artemisinins are recalcitrant reaction partners for iron under biologically realistic conditions. What is clear is that intraerythrocytic parasites are oxidatively stressed (19–21, 33, 34), as reflected in the greatly increased turnover of glucose-6-phosphate dehydrogenase (G6PD), required to generate NADPH (35, 36) for reduction of the flavin cofactors flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), etc., by glutathione reductase (GR), thioredoxin reductase (TrxR), and other flavin disulfide reductases (37). The reduced flavin cofactors provide electrons for reduction of endogenous disulfides, e.g., oxidized glutathione (GSSG), to the corresponding thiol required for interception of reactive oxygen species (ROS), thereby maintaining redox homeostasis (33, 38, 39). However, artemisinins rapidly oxidize such reduced flavin cofactors (Fig. 1Ai) (16, 17, 39). Thus, the electron supply from the reduced flavin cofactor is diverted to the artemisinin, and the buildup of ROS with the abrogation of redox homeostasis in P. falciparum thereby takes place (Fig. 1B). This is compatible with the observations that the activities of artemisinins against P. falciparum are enhanced in an oxygen-enriched atmosphere (40) and that the formation of ROS occurs upon treatment of malaria parasites with artemisinin (41–43). Further, it is strikingly apparent that artemisinin resistance, as associated with mutations in the P. falciparum Kelch13 (PfK13) propeller domain (99) and induction of dormancy of ring-stage parasites as part of a complex signaling cascade, reflects an enhanced inertness of the parasite to oxidative stress (3, 44). It is notable that DHA is labile under physiological conditions, yet it is modeled intact into the P. falciparum phosphatidylinositol-3-kinase (PfPI3K) target (45). Given the structural diversity of artemisinins and synthetic peroxides that elicit potent antimalarial activities (4), the presentation of a model invoking classical inhibition by binding into an endogenous receptor site is not convincing. According to precedent for this type of target involving redox-sensitive signal transduction pathways, the nature of the inhibition may equate with the generation of ROS by the artemisinin and, evidently, inhibition of phosphatidylinositol-3-phosphate (PI3P) generation by ROS (46–48).
FIG 1.

(Ai) Formalism indicating the irreversible reduction of the oxidant drug artemisinin by reduced conjugates of flavin cofactors flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and other reduced flavins, e.g., riboflavin (RF), to the inert deoxyartemisinin. (Aii) Redox cycling of methylene blue (MB) via its reduced conjugate, leucomethylene blue (LMB), initiated through reduction of MB by FADH2 to LMB and reoxidation of the latter by oxygen to generate MB and reactive oxygen species (ROS). (B) Perturbation of redox homeostasis by artemisinin through oxidation of reduced flavin cofactors, e.g., reduced flavin adenine dinucleotide (FADH2) of thioredoxin reductase (TrxR), glutathione reductase (GR), and other flavin disulfide reductases, resulting in the buildup of ROS; the artemisinin is thereby irreversibly reduced. Addition of MB maintains ROS generation via redox cycling through its reduced form, LMB, which is oxidized to MB with the concomitant generation of ROS; the reduction of MB to LMB by FADH2 is complementary to the reduction of artemisinin to deoxyartemisinin by the same reduced cofactor, yet by reoxidation of the LMB to MB by oxygen, redox cycling now ensues with the sustained attrition of FADH2.
Hence, by combining an artemisinin with a redox drug, the action of each should be promoted. Although several structurally disparate redox drugs are under active examination in this project, we illustrate the principle here with methylene blue (MB) (Fig. 1Aii) (38, 49). MB undergoes redox cycling via oxidation of its reduced conjugate, leucomethylene blue (LMB), to MB by oxygen, a process that also generates ROS (50, 51). As must be closely related to the case with artemisinins, intraparasitic GR, TrxR, lipoamide dehydrogenase, and others are directly affected by MB (51, 52). MB rapidly oxidizes the reduced cofactor FADH2 (53, 54) and thereby enhances the consumption of NADPH associated with these reductases (38, 39, 54). Despite its relatively poor physicochemical properties (51, 54), MB has potent antimalarial activity, with a mean 50% inhibitory concentration (IC50) of 3.62 nM in vitro against asexual intraerythrocytic drug-sensitive and -resistant P. falciparum parasites (55). Additionally, the activity of MB against early asexual ring-stage parasites (56) and especially gametocytes (57) greatly increases the potential of this drug for malaria treatment in combination with artemisinins. The overall benefit of combining an oxidant, artemisinin, with a redox drug is clear: the artemisinin abruptly induces oxidative stress, which is then maintained or enhanced by redox cycling of the redox drug partner (Fig. 1B).
Although current clinical artemisinins are the most rapidly acting of the antimalarial drugs, these are unsuitable for the artemisinin component of the planned triple-drug combinations. The resistance issue aside, the drugs show variable pharmacokinetic profiles and low bioavailability (58) and, especially for DHA, elicit concerns of neurotoxicity based on established data from in vitro and in vivo studies (59–61). All are thermally and chemically fragile, and for DHA, the thermal instability engenders problems during formulation and storage (62). Therefore, emphasis has to be placed on the development of derivatives incapable of providing the active metabolite, DHA, common to the current clinical artemisinins. Thus, the focus here is on newer derivatives bearing amino groups at C-10, referred to as 10-amino artemisinins, that are readily obtained from DHA and that are optimally active against P. falciparum in vitro (63–66). For artemisone at least, a lack of neurotoxicity has been unequivocally established (64, 67).
As the individual compounds were screened in the past against P. falciparum in different laboratories at different times using different methods, it is important to conduct at the same time comparative efficacy studies in order to gain a true appreciation of the optimally active compounds. We therefore describe here the results of such screens involving artemisone, artemiside, the 10-sulfamide (64) and 10-arylamine (63) derivatives, and the new 10-piperazine and 10-phenylurea derivatives (Fig. 2). The 10-piperazine derivative attracts because it completes assessment of a series of compounds bearing a six-membered amino-heterocyclic ring attached via the nitrogen atom to C-10, as exemplified by artemisone with its thiomorpholine-S,S-dioxide ring, artemiside with its thiomorpholine ring, and the corresponding highly active morpholino derivative (68), the last whose further examination is precluded on toxicity grounds (64). Importantly, in the case of artemisone, DHA is not a product of metabolism. Metabolic profiles for the other compounds wherein DHA is not produced have also been established; the results will be published elsewhere.
FIG 2.

Amino artemisinins bearing alkyl- and arylamino groups attached to C-10. The oxygen atom attached to C-10 in the current clinical artemisinins is replaced by a nitrogen atom.
We describe the assessment of the activities of the amino artemisinins against intraerythrocytic asexual-stage parasites and early- and late-stage gametocytes of P. falciparum in accord with prerequisites for the development of new antimalarial drugs. We thereupon select the best of the derivatives and examine their combination with MB as a proof of concept for enhancing oxidative stress in the parasites, in particular as this bears on the sensitivity of gametocytes to perturbation of redox homeostasis.
RESULTS
Chemistry.
Artemisone, artemiside, and the 10-sulfamide were originally obtained by application of N-glycosylation technology to activate the hydroxyl group in DHA by conversion into the trimethylsilyl ether and thence into the β-bromide by treatment with trimethylsilyl bromide in dichloromethane (64, 68). All compounds were more economically obtained via conversion of DHA into the β-chloride via direct treatment with anhydrous hydrogen chloride in the presence of lithium chloride in dichloromethane (65, 69). Addition of the amine nucleophile to the halide generated in situ provided the 10-amino artemisinin. The 10-arylamine derivative was prepared directly from DHA through activation of the hydroxyl group by a facile phase transfer process in the presence of p-fluoroaniline (63). The 10-piperazine derivative was obtained by addition of the β-bromide of DHA, prepared as described above, to an excess of anhydrous piperazine in dichloromethane. The most economical and experimentally facile method, especially on a larger scale, was to treat a relatively concentrated solution of DHA (0.7 M) in dichloromethane with oxalyl chloride (1.1 equivalents) to convert it into the β-chloride in situ and to add the resulting solution to an excess of piperazine (Fig. 3). The 10-piperazine derivative, formed in 80% yield by the latter method, is a basic highly polar compound that could not be readily purified except by flash chromatography over silica gel and elution with a solvent combination containing triethylamine followed by crystallization from ethyl acetate. The 10-phenylurea derivative was obtained in 60% isolated yield by treatment of the 10-piperazine derivative with phenyl isocyanate in dichloromethane (Fig. 3). This nicely crystalline compound (melting point [mp], 162 to 163°C) was readily purified, is thermally relatively stable, and represents a distinct artemisinin subclass. Spectroscopic and other information on the amino artemisinins used here is given in the supplemental material.
FIG 3.
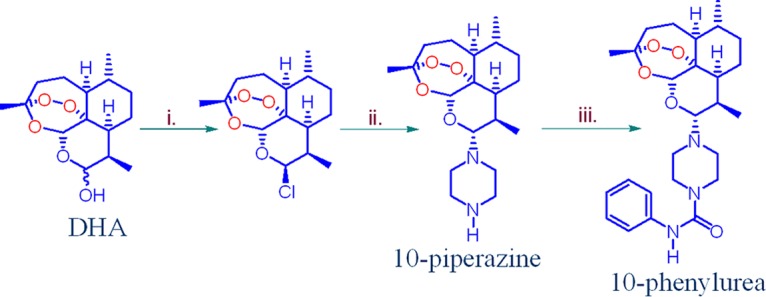
Preparation of the 10-piperazine and 10-phenylurea derivatives by conversion of DHA. (i) DHA, 17.6 mmol, 0.7 M in CH2Cl2, (COCl)2 (1.1 equivalent), N2, room temperature, 30 min; (ii) solution added to piperazine (52.8 mmol, 3.0 equivalent) in CH2Cl2, room temperature, 3 h, 80%; (iii) compound 3 (1 mmol), phenyl-N=C=O (1 mmol), CH2Cl2, 24 h, 60%.
Efficacy studies. (i) Amino artemisinins inhibit asexual parasite proliferation and metabolic activity.
The in vitro activities of the 10-amino artemisinins along with those of the reference drugs, artemisinin, DHA, artesunate, artemether, chloroquine (CQ), and MB, against drug-sensitive (NF54) and drug-resistant (Dd2, K1 and W2) P. falciparum intraerythrocytic asexual parasites were determined using both metabolic (parasite lactate dehydrogenase [pLDH]) and proliferative (SYBR green I fluorescence) viability readouts. These two different assay platforms allowed for comparative analysis of the overall activities of the compounds toward the asexual stages. The IC50s obtained for the NF54 strain with these two assay platforms were closely associated (r2 = 0.63), showing good intra-assay variability with average Z-factors of 0.70 for the metabolic pLDH assay and 0.69 for the proliferative SYBR green I assay. Acceptable interassay reproducibility with an average percent coefficient of variance (CV) of 4.1% for the metabolic assay and 5.2% for the proliferative assay was obtained.
The 10-amino artemisinins were potently active against drug-sensitive NF54 asexual parasites with IC50s in the low-nanomolar range (∼10 nM) for both assay platforms, with the reference compounds showing similar activities (Table 1). Artemisone and artemiside were confirmed to be the most active compounds with IC50s of ∼2 nM (Table 1) (69). Moreover, the 10-amino artemisinins were equally active against the drug-resistant strains (Table 1) with resistance indices (IC50 ratio between drug-resistant and -sensitive strains) of <3.5 for all compounds; in comparison, the resistance index typically observed for CQ is >10 (Table 1) (70). Therefore, the 10-amino artemisinins showed negligible cross-resistance potential against the Dd2, K1, and W2 genetic backgrounds (Table 1). The majority of compounds did not show overt cytotoxicity against HepG2 or CHO mammalian cell lines, with only ∼10% inhibition of cell viability being observed even at 1,000× the IC50 against asexual NF54 parasites for HepG2 cells. The 50% effective concentrations (EC50s) for CHO cells were, on average, higher than the EC50 of the reference drug, emetine (IC50, 340 ± 90 nM). However, the 10-arylamine and 10-phenylurea derivatives may have potential toxicity issues, as indicated by their EC50s against CHO cells (2.9 ± 1.4 and 2.4 ± 1.0 μM, respectively; Table 1).
TABLE 1.
In vitro activity of amino artemisinins against asexual P. falciparum parasites
| Compound | Metabolic readout |
Proliferation readout |
Cytotoxicity |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50a (nM) |
RIb | IC50 (nM) |
RIc | IC50 (nM) W2 | RId | HepG2 cell viability (%)e | EC50 (μM) for CHO cells | |||
| NF54 | Dd2 | NF54 | K1 | |||||||
| CQ | 31.8 ± 9.9 | 119.0 ± 20.9 | 3.7 | 10 ± 3 | 154 ± 14 | 15.4 | 233 ± 49 | 23.3 | >55f (ref. 93) | NDg |
| MB | 0.3 ± 0.8 | 12.6 ± 4.0 | 43.3 | 5.0 ± 0.8 | 6.45 ± 0.30 | 1.29 | 5.13 ± 0.31 | 1.03 | ND | 52.6 ± 4.5 |
| Artemisinin | 10.8 ± 1.6 | 17.5 ± 2.5 | 1.6 | 0.57 ± 0.01 | 0.8 ± 0.5 | 1.40 | 0.37 ± 0.11 | 0.65 | ∼90 | ND |
| DHA | 0.8 ± 0.1 | 5.7 ± 2.0 | 7.1 | 2.51 ± 0.19 | 1.51 ± 0.33 | 0.60 | 1.74 ± 0.22 | 0.69 | ND | 25.2 ± 3.7 |
| Artesunate | 10.3 ± 0.8 | 40.5 ± 5.0 | 4.0 | 3.4 ± 0.7 | 4 ± 1 | 1.24 | 2.4 ± 0.4 | 0.72 | ∼90 | >354 |
| Artemether | 2.0 ± 0.1 | 17.3 ± 2.2 | 8.7 | 8 ± 1 | 9 ± 2 | 1.13 | 7 ± 1 | 0.86 | ∼90 | >335 |
| Artemisone | 3.0 ± 0.8 | 2.7 ± 0.3 | 0.9 | 1.2 ± 0.4 | 1.01 ± 0.19 | 0.85 | 1.6 ± 0.4 | 1.36 | ∼75 | >249 |
| Artemiside | 6.0 ± 1.8 | 8.2 ± 1.4 | 1.4 | 1.11 ± 0.17 | 1.6 ± 0.4 | 1.47 | 1.75 ± 0.27 | 1.58 | ∼80 | >271 |
| 10-Sulfamide | 10.9 ± 3.4 | 16.9 ± 2.6 | 1.55 | 3 ± 1 | 1.79 ± 0.26 | 0.56 | 2.04 ± 0.11 | 0.64 | ∼90 | 56.0 ± 4.6 |
| 10-Arylamine | 7.8 ± 1.9 | 11.1 ± 1.0 | 1.41 | 1.3 ± 0.6 | 0.64 ± 0.10 | 0.48 | 3 ± 1 | 2.55 | ∼90 | 2.9 ± 1.4 |
| 10-Piperazine | 3.2 ± 1.4 | 1.7 ± 0.2 | 0.5 | 3.1 ± 0.4 | 1.9 ± 0.5 | 0.61 | 1.4 ± 0.7 | 0.45 | ∼77 | ND |
| 10-Phenylurea | 1.3 ± 0.8 | 7.5 ± 0.4 | 5.8 | 4.7 ± 1.5 | 2.9 ± 0.6 | 0.61 | 1.7 ± 0.5 | 0.36 | ∼85 | 2.4 ± 1.0 |
The IC50s for asexual parasites were determined against drug-sensitive NF54 parasites or drug-resistant Dd2, K1, and W2 parasites using either a metabolic pLDH assay or a proliferative SYBR green I-based fluorescence assay. Data are means from at least three independent biological repeats performed in triplicate for both the metabolic and proliferative assays ± SEM and the mean from a single experiment performed in duplicate for cytotoxicity assays on the HepG2 and CHO cell lines ± SD.
Resistance index (RI) = IC50 for Dd2/IC50 for NF54.
Resistance index = IC50 for K1/IC50 for NF54.
Resistance index = IC50 for W2/IC50 for NF54.
Percent viability at 1,000× the IC50 against P. falciparum parasites.
The EC50 was 22 ± 0.4 μM (93).
ND, not determined.
(ii) Amino artemisinins display gametocytocidal activity.
The inhibition of both early-stage (>95% stages I to III) and late-stage (∼10% stage III and ∼90% stages IV and V) gametocyte viability was evaluated with P. falciparum NF54 luciferase reporter lines (71). Good intra-assay variability (Z-factors, 0.8) and an acceptable interassay reproducibility (CV, 14.5%) were observed. In vitro dose-response analysis showed, on average, a 2-fold improved activity for the 10-amino artemisinins against early-stage gametocytes, with IC50s ranging from 1 to 83 nM; in comparison, the IC50s of the reference compounds ranged from 37 to 95 nM (Table 2). Artemisone was the most potent 10-amino artemisinin, displaying activities against early-stage gametocytes (IC50 = 1.94 ± 0.11 nM) similar to those observed against asexual parasites (IC50 = 1.2 ± 0.4 nM) (Table 2).
TABLE 2.
Activity of amino artemisinins against early- and late-stage gametocytesa
| Compound | IC50 (nM) |
|
|---|---|---|
| Early-stage gametocytes | Late-stage gametocytes | |
| MB | 95.0 ± 11.3 | 143.0 ± 16.7 |
| Artemisinin | 37.0 ± 4.0 | 77.6 ± 50.0 |
| DHA | 43.0 ± 3.9 | 33.66 ± 1.98 |
| Artesunate | 37.7 ± 2.0 | 136.2 ± 85.9 |
| Artemether | 62.8 ± 3.0 | 259.4 ± 80.0 |
| Artemisone | 1.94 ± 0.11 | 42.4 ± 3.3b |
| Artemiside | 16.4 ± 1.0 | 1.5 ± 0.5b |
| 10-Sulfamide | 15.0 ± 2.0 | 419.4 ± 59.5 |
| 10-Arylamine | 38.2 ± 9.0 | 16.42 ± 6.38 |
| 10-Piperazine | 54.91 ± 5.11 | 25.7 ± 17.5b |
| 10-Phenylurea | 83 ± 2 | 1.70 ± 0.99b |
The IC50s of MB and the 10-amino artemisinins against early-stage (stage I to III) and late-stage (stage IV and V) gametocytes were determined using the luciferase assay. Data are means from at least two independent biological replicates (n = 2 to 4) performed in technical triplicate ± SEM.
Data are from a 72-h incubation period.
In vitro dose-response analyses showed IC50s for DHA against late-stage gametocytes similar to those against early-stage gametocytes (Table 2). However, 2- to 4-fold increases in the IC50s for artemisinin, artesunate, and artemether against late-stage gametocytes compared to those against the early stages were observed (71). Decreased potency was also observed for the 10-sulfamide derivative that showed a preference for specificity toward early-stage gametocytes.
Artemiside and the 10-arylamine and 10-phenylurea derivatives displayed incomplete kill curves after 48 h of drug pressure against late-stage gametocytes (see Fig. S1 in the supplemental material), as previously observed for current clinical artemisinins (72). However, complete dose-response kill curves were obtained after 72 h (73–75) of drug pressure (Table 2), indicating a slow speed of action against these metabolically more latent stages (76). Under these conditions, artemiside and 10-phenylurea were the most potent compounds against late-stage gametocytes (IC50s, 1.5 nM and 1.7 nM, respectively; Table 2). The 10-arylamine derivative was slightly less potent (IC50 = 16 nM). However, four of the 10-amino artemisinin derivatives (artemiside and the 10-sulfamide, 10-piperazine, and 10-phenylurea derivatives) preferentially targeted late-stage gametocytes, ranging from 2-fold late-stage specificity for the 10-piperazine derivative to 49-fold late-stage specificity for the 10-phenylurea derivative.
Previous reports indicated that artemisinins do not target mature stage V gametocytes (72). We therefore interrogated the effect of the 10-amino artemisinins showing late-stage gametocyte specificity on late-stage (stage IV and V) gametocytes (10% stage III, 50% stage IV, and 40% stage V populations) compared to their activity against enriched, mature stage V gametocytes (>95% stage V) with DHA as a reference (Fig. 4 and S2). As with DHA, artemisone, artemiside, and the 10-arylamine, 10-piperazine, and 10-phenylurea derivatives lost activity between stage IV and V gametocyte populations after 72 h of drug exposure. However, the activities for artemisone, artemiside, and the 10-phenylurea derivative against stage V gametocytes persisted at <250 nM (IC50s, 126 nM, 209 nM, and 100 nM, respectively; Fig. S2), whereas a >10-fold loss in activity for DHA and the compounds 10-arylamine and 10-piperazine was observed (IC50s, 1,287 nM, 1,158 nM, and 800 nM, respectively; Fig. S2). We further evaluated the efficacy of these drugs for an additional 24 h after drug washout. Under these conditions, the five 10-amino artemisinins tested were potently active against stage V gametocytes at concentrations below 100 nM, even though this activity was still significantly (P < 0.05) higher than that observed against stage IV and V gametocytes (Fig. 4). In all cases, the observed efficacies were due to the ability of the compounds to induce a death phenotype in the gametocytes, irrespective of the stage of gametocyte maturation. Dead/nonviable gametocytes were morphologically detected through Giemsa-stained smears by a rounded appearance showing a punctate crystal formation indicative of unhealthy or dead gametocytes (Fig. 4) (73). Moreover, these stage V gametocytes were functionally compromised due to the inability of microgametocytes to exflagellate after 48 or 72 h of treatment with the 10-amino artemisinins (results not shown). Therefore, despite the preference toward stage IV gametocytes and apparent slow kill kinetics, the five 10-amino artemisinins tested show low-nanomolar activities against stage V gametocytes, making them at least twice as effective against stage V gametocytes as DHA.
FIG 4.

Gametocytocidal stage-specific and kill kinetic evaluation of compounds against late-stage gametocytes. Late-stage gametocytes in stages IV and V (consisting of a 10% stage III, 50% stage IV, and 40% stage V population) or mature stage V (a >95% stage V population) were used to determine the differential stage specificity and kill kinetics (speed of action) of DHA, artemisone, artemiside, and the 10-arylamine, 10-piperazine, and 10-phenylurea derivatives between stage IV and V gametocytes. Dose-responses were determined using the luciferase reporter lines exposed to 10-fold serial drug dilutions for 72 h and 72 plus 24 h at 37°C under 90% N2, 5% CO2, and 5% O2 atmospheric conditions. Population compositions were determined microscopically using Giemsa-stained smears before (0 h) and after (72 h or 72 plus 24 h) incubation for both treated and untreated populations at 2× the IC50. Data are the means from a single independent biological experiment with technical triplicates; error bars indicate ±SD. P values were determined by an unpaired t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(iii) Amino artemisinins show panreactivity toward asexual and sexual P. falciparum life cycle stages.
Evaluation of the panreactivity—the ability to target asexual parasites as well as early- and late-stage gametocytes—of the 10-amino artemisinins indicated that, on average, a 13-fold loss of activity was seen between asexual and early-stage gametocyte activities (P = 0.0002; Fig. 5). Conversely, a 34-fold loss of activity against late-stage gametocytes (P = 0.03) (77, 78) compared to that against asexual parasites was seen, with only a 3-fold loss of associated activity between early- and late-stage gametocytes being seen. However, even though a preference toward asexual parasites was observed, the compounds were still potent against both early- and late-stage gametocytes, with IC50s being <500 nM (Table 2), indicative of their ability to target multiple stages of intraerythrocytic P. falciparum parasites. The reference compounds all had a continuous decline in activity from the asexual parasites to early- and late-stage gametocytes, a trend generally observed for most antimalarial drugs (15, 78, 79). The 10-sulfamide derivative was the only 10-amino artemisinin showing a similar trend (Fig. 5). Although the 10-phenylurea derivative was equipotent with artemiside, there is a potential toxicity issue with the compound, as noted above (Table 1). No other physicochemical properties, including solubility and lipophilicity, could be correlated to a stage-specific preference (data not shown).
FIG 5.
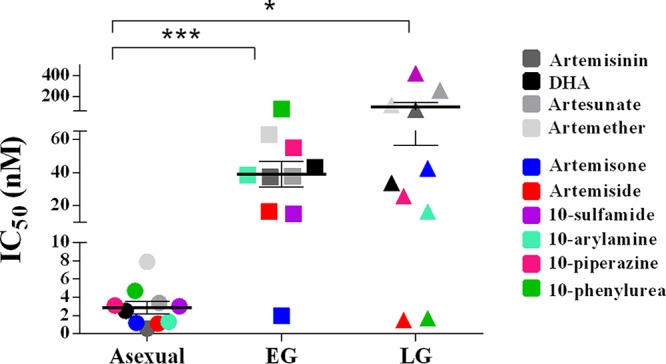
Antiplasmodial activity of 10-amino artemisinins compared to their cross-stage activity and gametocyte stage specificity. A scatter plot of the IC50 data for the compounds against asexual, early gametocyte (EG), and late gametocyte (LG) stages is shown. The results for the reference compounds (artemisinin, DHA, artesunate, and artemether) are indicated in gray scale, and those for the 10-amino artemisinins are indicated in color. The 10-sulfamide showed a consistent loss of activity from the asexual to the late gametocyte stages, like that observed for the reference compounds, whereas artemisone showed early gametocyte stage specificity with a loss of activity toward late gametocyte stages. Both artemiside and 10-phenylurea showed late gametocyte stage-specific 10-amino groups. Data are means of the IC50s for the oxidant compounds for each life cycle stage. P values were determined by an unpaired t test. *, P = 0.03; ***, P = 0.0002.
(iv) Each of artemisone and artemiside with MB synergistically inhibits P. falciparum gametocytes.
Fixed-ratio isobole analysis (80) was employed to evaluate the effect of combinations of the most potent oxidant drugs, artemisone and artemiside, with the redox drug MB (Fig. 6). Against asexual parasites, the combination of either artemisone or artemiside with MB resulted only in additive interactions [mean sums of the fractional inhibitory concentration (ΣFIC) of 1.14 and 1.02 against artemisone and artemiside, respectively]. In contrast, the interactions of either artemisone or artemiside with MB were synergistic against both early- and late-stage gametocytes, as evident from concave isobolograms (Fig. 6) with mean ΣFIC values for artemisone and artemiside of 0.75 and 0.56, respectively, against early-stage gametocytes and 0.61 and 0.73, respectively, against late-stage gametocytes. This synergism was confirmed with an independent isobole using the pLDH assay (Fig. 6), confirming the combined activity observed with the luciferase assay. The strongly synergistic interactions observed with the oxidant and redox drugs against both early- and late-stage gametocytes strongly support the idea that redox homeostasis regulated through several metabolic pathways is a mechanism essential for maintaining parasite viability in these stages in the parasite life cycle.
FIG 6.

Isobologram analysis of artemisone and artemiside in combination with MB. Isobolograms were determined using the fixed-ratio isobole analysis method by treating intraerythrocytic asexual-stage parasites and early- and late-stage gametocytes with fixed drug combination ratios (artemisone or artemiside/MB) of 5:0, 4:1, 3:2, 2:3, 1:4, and 0:5. Independent dose-response curves of the fixed drug combinations were determined using the SYBR green I-based assay against intraerythrocytic asexual parasites for 96 h and the luciferase assay for early- and late-stage gametocytes at 48-h and 72-h incubations, respectively. The dashed lines correspond to an indifferent interaction. The solid line indicates the respective isobole curve of the drug combination. Black lines with circles represent the results of the SYBR green assay against asexual parasites and the luciferase assay against early- and late-stage gametocytes. The gray line with blocks represents the results of the pLDH assay against late-stage gametocytes for artemisone and MB only. Data are the means from three or four independent biological repeats (n = 3 or 4) performed in technical triplicate; where shown, error bars indicate ±SEM. FIC50, 50% FIC.
DISCUSSION
The development of a triple drug combination using an oxidant 10-amino artemisinin derivative with a redox drug and a third partner drug, such as a 4(1H)-quinolone, may provide a novel strategy in targeting not only erythrocytic stages of drug-resistant malaria parasites but also the transmissible stages, effectively retarding the rate of resistance development and disease transmission. Here, we take the first step toward this aim and have established unambiguously the efficacy of lead 10-amino artemisinins in combination with a redox drug in effectively targeting various forms of malaria parasites in the erythrocytic stages.
Conversion of DHA into 10-amino artemisinins presents us with derivatives showing enhanced potency against drug-sensitive asexual parasites, with preliminary data indicating that these compounds also maintain activity against C580Y mutant Cambodian field isolates (81, 82; data to be presented elsewhere; Dennis E. Kyle, University of Georgia, USA, personal communication). This provides at least a preliminary indication that these derivatives may not be as prone to the resistance exerted against the current clinical artemisinins, although the possibility of the eventual development of resistance mechanisms, such as the K13-independent resistance phenotype, cannot be excluded at this stage (4, 5, 99). However, the fact that the 10-amino artemisinins inhibit not only asexual parasites but also early- and late-sexual-stage gametocytes within such a low-nanomolar range is most promising. In particular, the potencies of artemisone and artemiside against both the asexual and sexual stages support the identification of these compounds as the lead panreactive oxidant compounds in this study.
A wide selection of assay platforms has shown that the peroxidic antimalarial drugs, including artemisone, have nanomolar activities against late-stage gametocytes (83–85). However, some recent reports seemingly contradict these observations (15, 72, 86). The detailed kinetic evaluation of the 10-amino artemisinins in the current study indicates that artemisone and artemiside are indeed potently active (IC50s, 10 to 100 nM) against mature stage V gametocytes but notably have slower kill kinetics against these forms of gametocytes. These activities correlate with the general ∼10-fold decrease in activity between early- and late-stage gametocytes typically seen with other compounds as well (72, 78). Moreover, the potent activities of artemisone and artemiside against stage V gametocytes should translate into a substantial reduction of oocyst formation in a standard membrane feed assay, the results of which will be reported elsewhere. Moreover, future studies are required to show that these 10-amino artemisinins indeed translate to improved in vivo transmission blocking compared to the rather poor activity of DHA (72, 87).
The activities of the oxidant drugs artemisone and artemiside against gametocytes, including mature stage V gametocytes, support recent reports wherein targeting of redox metabolism significantly affects these forms of the parasite (76). Mature stage V gametocytes are regarded as metabolically hypoactive, due to their perceived inability to digest hemoglobin (88) and inability to respond to drug pressure on particular metabolic pathways (72). However, our data and those from Siciliano et al. (76) rather suggest that selective metabolic processes do remain active and are targetable in stage V gametocytes. In particular, the importance of antioxidant defense that is reliant on NADPH in transmissible gametocytes (76), coupled with our observation of the synergism between oxidant and redox-reactive drugs, strongly supports the need for a systematic investigation of redox maintenance and drug susceptibility in mature gametocytes. The maintenance of such potent activities of these artemisinins against late-stage gametocytes tends to suggest that heme after all is not a prerequisite for activation of artemisinins.
It is of some note that artemisone and artemiside are active in rather different parasite environments. In asexual parasites and early-stage gametocytes of P. falciparum, these artemisinins retain their ability to enhance oxidative stress. Such parasites, as noted above, have an enhanced turnover of enzymes important for regulating redox homeostasis in P. falciparum. Hemoglobin digestion and proper regulation of intracellular oxidative stress are essential in the development of asexual and metabolically active early gametocyte stages (89). The additive effect observed with each of artemisone and artemiside with MB against asexual parasites correlates with the findings of a previous study performed with artemiside and MB (69). This can be attributed to an upregulation in redox homeostasis in the asexual stages due to hemoglobin digestion, resulting in improved recovery from increased ROS production. Notably, such additive effects are maintained in vivo and are reflected in faster parasite clearance times for malaria patients treated with an oral artesunate-MB combination (90) and in two animal models infected with different Plasmodium species treated with intravenous MB and artesunate (91). However, asexual parasites conferring CQ resistance have increased glutathione (GSH) production due to a fitness cost in hemoglobin digestion and are therefore inherently more oxidatively stressed (92). This could explain why isobole analyses on asexual K1 parasites with artemether or artesunate and MB show synergism (56).
For late-stage gametocytes, the slower kill kinetics still reflect the same mode of action. The important issue is that in the absence of hemoglobin digestion for late-stage gametocytes (37, 52), redox homeostasis is downregulated, making the parasite more sensitive to increased ROS production. Therefore, the combinations of each of artemisone and artemiside with MB result in synergism, where the induction of oxidative stress by artemisone and artemiside is sustained or even enhanced by the redox cycling action of MB in gametocytes. These results are similar to those observed for plasmodione, a P. falciparum glucose-6-phosphate dehydrogenase-6-phosphogluconolactonase (PfGluPho) inhibitor, and MB against stage V gametocytes (76). Overall, redox homeostasis is an exploitable target, especially for the intraerythrocytic gametocyte stages in the parasite life cycle.
In conclusion, this study demonstrates that the 10-amino artemisinin derivatives artemisone and artemiside have potent panreactive and potential transmission-blocking activity against the intraerythrocytic stages of P. falciparum parasites. Although the 10-sulfamide, 10-arylamine, and 10-piperazine derivatives are the most accessible of the amino artemisinins, with the toxicity issue with the 10-arylamine derivative aside, their further development is not warranted on the basis of the >10-fold loss in activity against late-stage gametocytes. Although the 10-phenylurea derivative is equipotent with artemiside, there is a potential toxicity issue with the compound, as pointed out above (Table 1), and accordingly, newer derivatives of the 10-phenylurea derivative are being prepared and examined in all screens.
The low-nanomolar activities of artemisone and artemiside against asexual, early- and late-stage gametocytes, their favorable resistance and selectivity indices, and their synergism with MB against gametocytes make these ideal candidate compounds for further development within combination therapies. Monitoring of the evolution of oxidant and redox drug combinations as described here and the further examination of efficacies with selected lead 4(1H)-quinolones or other drugs with different mechanisms of action are now warranted.
MATERIALS AND METHODS
Chemistry.
The reference compounds used in this study as well as all tested compounds were ≥95% pure, unless otherwise indicated. Purity was determined using reverse-phase high-performance liquid chromatography (HPLC). Purities, instrumental conditions, and additional data are presented in the supplemental material. Artemisone was from an original sample batch prepared on a pilot scale from DHA by activation with HCl-LiCl, treatment of the intermediate β-chloride with thiomorpholine, and oxidation of the crude artemiside so obtained by hydrogen peroxide-acetonitrile, the product of which was recrystallized from ethyl acetate-hexane to provide needles (mp, 152 to 153°C) (64, 65). Artemiside (needles; mp, 152.5 to 153°C) (64, 69), the 10-sulfamide derivative (needles; mp, 168 to 169°C) (64), and the 10-arylamino derivative (fine hair-like crystals; mp, 170 to 171°C) (63) were original samples prepared and purified by recrystallization. Analysis by HPLC of the 10-sulfamide derivative under reverse-phase conditions indicated a purity of 93%.
10α-(Piperazin-1′-yl)-10-deoxo-10-dihydroartemisinin was prepared as follows. A solution of dihydroartemisinin (5.05 g, 17.6 mmol) in dichloromethane (25 ml) at room temperature was treated with oxalyl chloride (1.7 ml, 19.4 mmol, 1.1 equivalents) and stirred for 30 min under nitrogen. After 30 min, the reaction mixture was transferred via cannula into a flask containing a solution of anhydrous piperazine (4.55 g, 52.8 mmol, 3 equivalents) in dichloromethane (50 ml) and stirred for 3 h at room temperature under nitrogen. The reaction mixture was then diluted with dichloromethane (100 ml). It was washed with deionized water (3 times with 30 ml each time) followed by brine (2 times with 30 ml each time), dried over MgSO4, and then filtered. The filtrate was concentrated by evaporation under reduced pressure. The residue was purified by flash column chromatography over silica gel with dichloromethane-methanol-triethylamine (10:1:0.1) to obtain the highly polar product as a white solid (4.96 g, 80%). While recrystallization from diethyl ether or diethyl ether-ethyl acetate gave the 10-piperazine compound as fine needles (mp, 163 to 164°C), the compound was best used as the fraction isolated directly by chromatography followed by drying under high vacuum. 1H nuclear magnetic resonance (NMR) (600 MHz, CDCl3) δ (ppm) 0.74 (d, J = 7.3 Hz, 3H, H-14), 0.91 (d, J = 6.3 Hz, 3H, H-15), 1.36 (s, 3H, H-13), 1.62 to 1.69 (m, 2H, H-7), 2.29 (td, J = 14.0, 3.9 Hz, 1H, H-4), 2.48 to 2.53 (m, 1H, H-8a), 2.93 to 3.01 (m, 2H, H-2′, H-6′), 3.13 to 3.23 (m, 6H, H-2′, H-3′, H-4′, H-5′), 4.0 (d, J = 10.2 Hz, 1H, H-10), 5.24 (s, 1H, H-12); 13C NMR (151 MHz, CDCl3) δ (ppm) 103.96 (C-3), 91.64 (C-10), 91.06 (C-12), 80.34 (C-12a), 51.74 (C-5a), 50.70 (C-2′, C-6′), 45.84 to 45.69 (C-3′, C-5′), 26.02 (C-13), 24.78 (C-5), 21.67 (C-7), 20.31 (C-15), 13.45 (C-14); infrared (IR) νmax = 3,261, 2,926, 2,869, 2,839, 1,738, 1,453, 1,408, 1,375, 1,349 cm−1 (reciprocal wavelength). Mass spectrometry (MS) (electrospray ionization [ESI]) m/z calculated for C19H32N2O4 + H, 353.2440; found, 353.2469 [M+H]+. It was not possible to assess the purity of the compound under the reverse-phase conditions used for the other amino artemisinins, evidently due to decomposition. However, as gauged by its crystallinity and melting point and its clean conversion into the 10-phenylurea, the compound was suitable for assessment of its biological activities.
10α-[4′-(Phenylaminocarbonyl)piperazin-1′-yl]-10-deoxo-10-dihydroartemisinin was prepared as follows. A solution of phenyl isocyanate (0.11 ml, 1 mmol, 1 equivalent) and the 10-piperazine derivative (352 mg) in dichloromethane (5 ml) was stirred for 24 h. The mixture was then concentrated directly by evaporation under reduced pressure. Analysis of the residue by 1H NMR spectroscopy indicated essentially quantitative conversion of the 10-piperazine derivative into 10-phenylurea. The latter was isolated by chromatography of the residue over silica gel with ethyl acetate-hexane (40:60), evaporation of the eluate, and crystallization from ethyl acetate-hexane to give the 10-phenylurea as white plates (287 mg, 60%; mp, 162 to 163°C; [α]D20 +12.41 [c = 0.58, CHCl3]). 1H NMR spectrum (400 MHz) δ (ppm) 0.83 (d, J = 7.2Hz, 3 H, 6-methyl [6-Me]), 0.95 (d, J = 6 Hz, 3 H, 9-Me), 0.96 to 1.06 (m, 1 H), 1.38 (s, 3 H, 3-Me), 1.20 to 1.34 (m, 3 H), 1.45 to 1.49 (m, 1 H), 1.53 to 1.58 (m, 1 H), 1.69 to 1.74 (m, 2 H), 1.84 to 1.88 (m, 1 H), 1.97 to 2.03 (m, 1 H), 2.31 to 2.39 (m, 1 H), 2.57 to 2.63 (m, 1 H), 2.69 to 3.06 (m, 4 H, piperazine), 3.47 to 3.48 (m, 4 H, piperazine), 4.06 (d, J = 10 Hz, 1 H, H-10), 5.28 (s, 1 H, H-12), 6.40 (s, 1 H, NH), 7.02 (t, J = 7.2 Hz, 1 H, ArH), 7.25 to 7.29 (m, 2 H, ArH), 7.34 to 7.36 (m, 2 H, ArH); 13C NMR δ (ppm) 13.48, 20.78, 21.63, 24.74, 25.99, 28.48, 34.26, 36.29, 37.39, 44.44, 45.79, 47.14, 51.69, 80.30, 90.65, 91.61, 103.97, 119.91, 122.95, 128.83, 139.08, 154.99; IR (KBr) νmax = 500, 552, 694, 757, 880, 925, 984, 1,022, 1,041, 1,061, 1,113, 1,134, 1,160, 1,186, 1,207, 1,240, 1,316, 1,351, 1,380, 1,399, 1,445, 1,501, 1,527, 1,599, 1,661, 2,849, 2,868, 2,922, 2,954, 3,415 cm−1; MS (ESI): m/z = calculated for C26H37N3O5 + H, 472.2811; found, 472.2806 [M++H].
Efficacy studies. (i) In vitro maintenance of asexual parasites and gametocyte production.
In vitro P. falciparum parasites (NF54, K1, and W2) were cultured in human type O-positive erythrocytes under 90% N2, 5% CO2, and 5% O2 atmospheric conditions with supplemented RPMI 1640 medium containing AlbuMAX I lipid-rich bovine serum albumin, as previously described (93). Synchronized, ring-stage parasites (>95%) were obtained using a 5% d-sorbitol (Sigma-Aldrich) treatment (93). Parasite proliferation was monitored microscopically using Giemsa-stained smears. Gametocytogenesis was induced and maintained as previously described (71) through a combination of glucose depletion and a decrease in hematocrit from a >95% synchronized, ring-stage asexual population (∼10% parasitemia). Gametocyte cultures were kept stationary under 90% N2, 5% CO2, and 5% O2 atmospheric conditions at 37°C and treated with 50 mM N-acetylglucosamine (NAG; Sigma-Aldrich) to eliminate the residual invasion of asexual parasites.
(ii) In vitro antimalarial assays against asexual P. falciparum parasites.
Artemisinin, DHA, artesunate, artemether, the 4-aminoquinoline chloroquine (CQ), and MB were used as reference drugs for asexual-stage parasite assays. Compound working solutions were prepared from a 10 mM stock solution in 100% (vol/vol) dimethyl sulfoxide (DMSO; Sigma-Aldrich) in supplemented RPMI 1640 medium containing AlbuMAX I with a final DMSO concentration of <0.1% (vol/vol), previously determined to be nontoxic to intraerythrocytic asexual parasites and gametocytes. The dose-responses of the 10-amino artemisinins were assayed using a 2-fold serial drug dilution on in vitro >95% ring-stage intraerythrocytic P. falciparum parasites at 37°C under 90% N2, 5% CO2, and 5% O2 atmospheric conditions, detecting either parasite lactate dehydrogenase (pLDH) absorbance (94) as the metabolic indicator following a 48-h drug treatment (1.5 to 2% parasitemia and 2% hematocrit) or SYBR green I fluorescence as the proliferative marker following a 96-h drug treatment (1% parasitemia and 1% hematocrit) (93, 95). No drug washout steps were performed during drug incubation periods prior to the assays. Activity against the P. falciparum drug-sensitive NF54 strain and the drug-resistant Dd2 (resistant to CQ, pyrimethamine, mefloquine, and cycloguanil), K1 (resistant to CQ, quinine, pyrimethamine, and cycloguanil), and W2 (resistant to CQ, quinine, pyrimethamine, and cycloguanil) strains was evaluated. Data analysis was performed using GraphPad Prism (version 6) software, intra-assay variability was monitored with Z-factors, and acceptable interassay reproducibility was determined from the percent CV (71). The data for each compound are from at least three independent biological replicates, each performed in technical triplicate, and results are expressed as the compound concentration at which 50% parasite viability/proliferation is affected (IC50).
(iii) In vitro cytotoxicity determination against mammalian cells.
Caucasian hepatocellular carcinoma (HepG2) cells were maintained as previously described (93), and the cytotoxicity (at 1,000× the IC50 against asexual P. falciparum parasites) was determined using a lactate dehydrogenase assay (LDH) viability assay (BioVision Inc.). Cytotoxicity (EC50) was also determined against the Chinese hamster ovary (CHO) mammalian cells using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (96, 97) with emetine as the reference drug. Assays were performed for a single biological repeat in technical triplicate.
(iv) In vitro P. falciparum gametocytocidal assays.
Excluding CQ, the same reference compounds used against asexual P. falciparum parasites were used for intraerythrocytic early- and late-stage gametocyte assays. Gametocytocidal activity was determined using luciferase reporter lines (57, 71) to derive dose-responses with 2-fold serial drug dilutions for 48 h against early-stage gametocytes (day 5 postinduction population, ∼95% stages I to III) and 10-fold serial drug dilutions for 48 h and 72 h against late-stage gametocytes (day 10 postinduction population, ∼90% late stage [stages IV and V]) (2 to 3% gametocytemia, 2% hematocrit) at 37°C under 90% N2, 5% CO2, and 5% O2 atmospheric conditions. No drug washout steps were performed during the drug incubation periods prior to the assays. In all cases, an interference assay was run in parallel to eliminate false positives if possible compound interference with the luciferase readout existed. The data for each compound are from at least three independent biological replicates, unless otherwise indicated, each performed in technical triplicate, and results are expressed as the compound concentration at which 50% parasite viability was affected (IC50).
(v) Gametocytocidal stage-specific and kill kinetic evaluation of compounds against late-stage gametocytes.
Late-stage gametocytes, stages IV and V (a 10% stage III, 50% stage IV, and 40% stage V population on day 10 postinduction) or mature stage V (a >95% stage V population on day 13 postinduction), were used to determine the differential stage specificity and kill kinetics (speed of action) of DHA, artemisone, artemiside, and the 10-arylamine, 10-piperazine, and 10-phenylurea derivatives between stage IV and V gametocytes. Dose-responses were determined using a luciferase reporter line exposed to 10-fold serial drug dilutions for 72 h at 37°C under 90% N2, 5% CO2, and 5% O2 atmospheric conditions. Treatment for shorter periods (e.g., 24 h) did not result in an accurate dose-response determination for any compound, irrespective of the gametocyte population used, confirming the insensitivity of gametocytes to short periods of perturbation (57). Additionally, a drug washout step was included by replacing the drug-containing spent medium with fresh medium (without drug), followed by a further 24 h of incubation prior to measuring luciferase activity. In addition, gametocyte viability was monitored morphologically and by detection of exflagellation events at the initiation and completion of the experiment with a 20-min exposure to 1 mM xanthurenic acid (XA; Sigma-Aldrich) at room temperature. Population compositions were determined microscopically, using Giemsa-stained smears, before (0 h) and after (72 h or 72 plus 24 h) incubation for both treated and untreated populations at 2× the IC50. Data are the means from a single independent biological experiment with technical triplicates, and the results are expressed as the compound concentration at which 50% parasite viability was affected (IC50).
(vi) Combination of artemisone or artemiside with MB.
The in vitro interactions of artemisone and artemiside with MB were determined using a fixed-ratio isobole analysis (80) for NF54 asexual parasites (SYBR green I-based fluorescence) and early- and late-stage gametocytes (luciferase reporter lines). Shortly, the drugs were applied alone and in fixed drug combination ratios (artemisone or artemiside/MB) of 5:0, 4:1, 3:2, 2:3, 1:4, and 0:5 and serially diluted (2-fold for asexual parasites with 96-h incubations and early-stage gametocytes with 48-h incubations and 10-fold for late-stage gametocytes with 72-h incubations) to obtain the IC50 dose-response curves for each drug alone and in the fixed drug ratio. To confirm the drug interactions observed on late-stage gametocytes, isobole analysis for artemisone and MB only was also performed using the pLDH assay (73). Four fixed doses of MB (25.00, 6.25, 1.56, 0.39, 0 nM) were added to the dose-response curves of artemisone (from doses of 100 nM to 0.8 nM).
In addition, an independent dose-response curve of MB was performed to obtain the IC50 of MB. The fractional inhibitory concentration (FIC) for each drug in the combination was calculated as follows: FIC for artemisone or artemiside = IC50 of artemisone or artemiside in combination with MB/IC50 of artemisone or artemiside.
The paired FIC values for the drugs in each combination were linearly plotted, producing an isobologram. The ΣFIC of the FIC of artemisone or artemiside in combination with the FIC of MB was determined by calculating the mean FIC value to obtain the representative FIC value for the drug combination. A mean ΣFIC of <1.0 represents a synergistic interaction, a mean ΣFIC of >1.3 represents an antagonistic interaction, and a mean ΣFIC of 1 represents an indifferent/additive interaction (98). These assay platforms correlate well for the gametocyte stages, as they both measure viability. Data for each isobologram obtained using the SYBR green I fluorescence and the luciferase reporter line and data for the artemisone and MB isobologram obtained using the pLDH assay are from at least three independent biological replicates, each performed in technical triplicate.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the assistance of Carmen de Kock and Daniel J. Watson from the Division of Pharmacology, Department of Medicine, Groote Schuur Hospital, University of Cape Town, and Lung Chung Man, Yuet Wu, and Kwan Wing Cheu from the Department of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong.
This work was funded by the South African Medical Research Council (MRC) Flagship Project MALTB-Redox with funds from the National Treasury under its Economic Competitiveness and Support Package to Richard K. Haynes; a South African MRC Strategic Health Innovation Partnership (SHIP) grant, a South African MRC Collaborative Centre for Malaria Research grant, and a South African National Research Foundation grant (UID 84627) to Lyn-Marie Birkholtz; and South African National Research Foundation grants to Richard K. Haynes (UIDs 90682 and 98934). Donatella Taramelli and Sarah D'Alessandro acknowledge the support from the Global Health Program of the Bill & Melinda Gates Foundation (grant OPP1040394 to Donatella Taramelli, Pietro Alano coordinator, and COST Action CM1307).
Any opinions, findings and conclusions, or recommendations expressed in this material are those of the authors, and therefore, the NRF does not accept any liability in regard thereto.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02214-17.
REFERENCES
- 1.World Health Organization. 2016. WHO world malaria report 2016. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Lubell Y, Dondorp A, Guérin PJ, Drake T, Meek S, Ashley E, Day NP, White NJ, White LJ. 2014. Artemisinin resistance—modelling the potential human and economic costs. Malar J 13:452. doi: 10.1186/1475-2875-13-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paloque L, Ramadani AP, Mercerea-Puijalon O, Augereau JM, Benoit-Vical F. 2016. Plasmodium falciparum: multifaceted resistance to artemisinins. Malar J 15:149. doi: 10.1186/s12936-016-1206-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sutherland CJ, Lansdell P, Sanders M, Muwanguzi J, van Schalkwyk DA, Kaur H, Nolder D, Tucker J, Bennett HM, Otto TD, Berriman M, Patel TA, Lynn R, Gkrania-Klotsas E, Chiodini PL. 2017. Pfk13-independent treatment failure in four imported cases of Plasmodium falciparum malaria treated with artemether-lumefantrine in the United Kingdom. Antimicrob Agents Chemother 61:e02382-. doi: 10.1128/AAC.02382-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu F, Culleton R, Zhang M, Ramaprasad A, von Seidlein L, Zhou H, Zhu G, Tang J, Liu Y, Wang W, Cao Y, Xu S, Gu Y, Li J, Zhang C, Gao Q, Menard D, Pain A, Yang H, Zhang Q, Cao J. 2017. Emergence of indigenous artemisinin-resistant Plasmodium falciparum in Africa. N Engl J Med 376:991–993. doi: 10.1056/NEJMc1612765. [DOI] [PubMed] [Google Scholar]
- 6.Price RN. 2013. Potential of artemisinin-based combination therapies to block malaria transmission. J Infect Dis 207:1627–1629. doi: 10.1093/infdis/jit079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dama S, Niangaly H, Ouattara A, Sagara I, Sissoko S, Traore OB, Bamadio A, Dara N, Djimde M, Alhousseini ML, Goita S, Maiga H, Dara A, Doumbo OK, Djimde AA. 2017. Reduced ex vivo susceptibility of Plasmodium falciparum after oral artemether-lumefantrine treatment in Mali. Malar J 15:59. doi: 10.1186/s12936-017-1700-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amato R, Lim P, Miotto O, Amaratunga C, Dek D, Pearson RD, Almargo-Garcia J, Neal AT, Sreng S, Suon S, Drury E, Jyothi D, Stalker J, Kwiatkowski DP, Fairhurst RM.. 2017. Genetic markers associated with dihydroartemisinin-piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype-phenotype association study. Lancet Infect Dis 17:164–173. doi: 10.1016/S1473-3099(16)30409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leang R, Taylor WRJ, Bouth DM, Song L, Tarning J, Char MC, Kim S, Witkowski B, Duru V, Domergue A, Khim N, Ringwald P, Menard D. 2015. Evidence of Plasmodium falciparum malaria multidrug resistance to artemisinin and piperaquine in western Cambodia: dihydroartemisinin-piperaquine open-label multicenter clinical assessment. Antimicrob Agents Chemother 59:4719–4726. doi: 10.1128/AAC.00835-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pascual A, Madamet M, Briolant S, Gaillard T, Amalvict R, Benoit N, Travers D, Pradines B. 2015. French National Reference Centre for Imported Malaria Study Group. Multinormal in vitro distribution of Plasmodium falciparum susceptibility to piperaquine and pyronaridine. Malar J 14:49. doi: 10.1186/s12936-015-0586-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conrad MD, LeClair N, Arinaitwe E, Wanzira H, Kakuru A, Bigira V, Muhindo M, Kamya MR, Tappero JW, Greenhouse B, Dorsey G, Rosenthal PJ. 2014. Comparative impacts over 5 years of artemisinin-based combination therapies on Plasmodium falciparum polymorphisms that modulate drug sensitivity in Ugandan children. J Infect Dis 210:344–353. doi: 10.1093/infdis/jiu141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thanh NV, Thuy-Nhien N, Tuyen NT, Tong NT, Nha-Ca NT, Dong LT, Quang HH, Farrar J, Thwaites G, White NJ, Wolbers M, Hien TT. 2017. Rapid decline in the susceptibility of Plasmodium falciparum to dihydroartemisinin-piperaquine in the south of Vietnam. Malar J 16:27. doi: 10.1186/s12936-017-1680-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burrows JN, Duparc S, Gutteridge WE, Hooft van Huijsduijnen R, Kaszubska W, Macintyre F, Mazzuri S, Möhrle JJ, Wells TNC. 2017. New developments in anti-malarial target candidate and product profiles. Malar J 16:26. doi: 10.1186/s12936-016-1675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.WWARN Gametocyte Study Group. 2016. Gametocyte carriage in uncomplicated Plasmodium falciparum malaria following treatment with artemisinin combination therapy: a systematic review and meta-analysis of individual patient data. BMC Med 14:79. doi: 10.1186/s12916-016-0621-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peatey CL, Leroy D, Gardiner DL, Trenholme KR. 2012. Anti-malarial drugs: how effective are they against Plasmodium falciparum gametocytes? Malar J 11:34. doi: 10.1186/1475-2875-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haynes RK, Chan WC, Wong HN, Li KY, Wu W, Fan K, Sung HY, Williams ID, Prosperi D, Melato S, Coghi P, Monti D. 2010. Facile oxidation of leucomethylene blue and dihydroflavins by artemisinins: relationship with flavoenzyme function and antimalarial mechanism of action. ChemMedChem 5:1282–1299. doi: 10.1002/cmdc.201000225. [DOI] [PubMed] [Google Scholar]
- 17.Haynes RK, Cheu KW, Chan HW, Wong HN, Li KY, Tang MM, Chen MJ, Guo ZF, Guo ZH, Sinniah K, Witte AB, Coghi P, Monti D. 2012. Interactions between artemisinins and other antimalarial drugs in relation to the cofactor mode—a unifying proposal for drug action. ChemMedChem 7:2204–2226. doi: 10.1002/cmdc.201200383. [DOI] [PubMed] [Google Scholar]
- 18.Haynes RK, Cheu K-W, N′Da DD, Coghi P, Monti D. 2013. Considerations on the mechanism of action of artemisinin antimalarials: part 1—the ‘carbon radical’ and ‘heme’ hypotheses. Infect Disord Drug Targets 13:217–277. doi: 10.2174/1871526513666131129155708. [DOI] [PubMed] [Google Scholar]
- 19.Müller T, Johann L, Jannack B, Brückner M, Lanfranchi DA, Bauer H, Sanchez C, Yardley V, Deregnaucourt D, Schrével J, Lanzer M, Schirmer RH, Davioud-Charvet E. 2011. Glutathione reductase-catalyzed cascade of redox reactions to bioactivate potent antimalarial 1,4-naphthoquinones—a new strategy to combat malarial parasites. J Am Chem Soc 133:11557–11571. doi: 10.1021/ja201729z. [DOI] [PubMed] [Google Scholar]
- 20.Mohring F, Pretzel J, Jortzik E, Becker K. 2014. The redox systems of Plasmodium falciparum and Plasmodium vivax: comparison, in silico analyses and inhibitor studies. Curr Med Chem 21:1728–1756. doi: 10.2174/0929867321666131201144612. [DOI] [PubMed] [Google Scholar]
- 21.Rathore S, Datta G, Kaur I, Malhotra P, Mohmmed A. 2015. Disruption of cellular homeostasis induces organelle stress and triggers apoptosis like cell-death pathways in malaria parasite. Cell Death Dis 6:e1803. doi: 10.1038/cddis.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beteck RM, Coertzen D, Smit FJ, Birkholtz L-M, Haynes RK, N′Da DD. 2016. Straightforward conversion of decoquinate into inexpensive tractable new derivatives with significant antimalarial activities. Bioorg Med Chem Lett 26:3006–3009. doi: 10.1016/j.bmcl.2016.05.024. [DOI] [PubMed] [Google Scholar]
- 23.Andriole VT. 2005. The quinolones—past, present and future. Clin Infect Dis 41:S113. doi: 10.1086/428051. [DOI] [PubMed] [Google Scholar]
- 24.Beteck RM, Smit FJ, Haynes RK, N′Da DD. 2014. Recent progress in the development of anti-malarial quinolones. Malar J 13:339. doi: 10.1186/1475-2875-13-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monastyrskyi A, Kyle DE, Manetsch R. 2014. 4(1H)-Pyridone and 4(1H)-quinolone derivatives as antimalarials with erythrocytic, exoerythrocytic, and transmission blocking activities. Curr Top Med Chem 14:1693–1705. doi: 10.2174/1568026614666140808124638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stickles AM, Smilkstein MJ, Morrisey JM, Li Y, Forquer IP, Kelly JX, Pou S, Winter RW, Nilsen A, Vaidya AB, Riscoe MK. 2016. Atovaquone and ELQ-300 combination therapy as a novel dual-site cytochrome bc1 inhibition strategy for malaria. Antimicrob Agents Chemother 60:4853–4859. doi: 10.1128/AAC.00791-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Neill PM, Posner GH. 2004. A medicinal chemistry perspective on artemisinin and related endoperoxides. J Med Chem 47:2945–2964. doi: 10.1021/jm030571c. [DOI] [PubMed] [Google Scholar]
- 28.Klonis N, Creek DJ, Tilley L. 2013. Iron and heme metabolism in Plasmodium falciparum and the mechanism of action of artemisinins. Curr Opin Microbiol 16:722–727. doi: 10.1016/j.mib.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Xu C, Lun ZR, Meshnick SR. 2017. Unpacking ‘artemisinin resistance.’ Trends Pharmacol Sci 38:506–511. doi: 10.1016/j.tips.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Zhang CJ, Chia WN, Loh CC, Li Z, Lee YM, He Y, Yuan LX, Lim TK, Liu M, Liew CX, Lee YQ, Zhang J, Lu N, Lim CT, Hua ZC, Liu B, Shen HM, Tan KS, Lin Q. 2015. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun 6:10111. doi: 10.1038/ncomms10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ismail HM, Barton V, Phanchana M, Charoensutthivarakul S, Wong MH, Hemingway J, Biagini GA, O'Neill PM, Ward SA. 2016. Artemisinin activity-based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proc Natl Acad Sci U S A 113:2080–2085. doi: 10.1073/pnas.1600459113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ismail HM, Barton VE, Panchana M, Charoensutthivarakul S, Biagini GA, Ward SA, O'Neill PM. 2016. A click chemistry-based proteomic approach reveals that 1,2,4-trioxolane and artemisinin antimalarials share a common protein alkylation profile. Angew Chem Int Ed Engl 55:6401–6405. doi: 10.1002/anie.201512062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krauth-Siegel RL, Bauer H, Schirmer RH. 2005. Dithiol proteins as guardians of the intracellular redox milieu in parasites: old and new drug targets in trypanosomes and malaria-causing plasmodia. Angew Chem Int Ed Engl 44:690–715. doi: 10.1002/anie.200300639. [DOI] [PubMed] [Google Scholar]
- 34.Goyal M, Alam A, Bandyopadhyay U. 2012. Redox regulation in malaria: current concepts and pharmacotherapeutic implications. Curr Med Chem 19:1475–1503. doi: 10.2174/092986712799828328. [DOI] [PubMed] [Google Scholar]
- 35.Atamna H, Pascarmona G, Ginsburg H. 1994. Hexose monophosphate shunt activity in intact Plasmodium falciparum-infected erythrocytes and in free parasites. Mol Biochem Parasitol 67:79–89. doi: 10.1016/0166-6851(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 36.Becker K, Rahlfs S, Nickel C, Schirmer RH. 2003. Glutathione—functions and metabolism in the malarial parasite Plasmodium falciparum. Biol Chem 384:551–566. [DOI] [PubMed] [Google Scholar]
- 37.Müller S. 2004. Redox and antioxidant systems of the malaria parasite Plasmodium falciparum. Mol Microbiol 53:1291–1305. doi: 10.1111/j.1365-2958.2004.04257.x. [DOI] [PubMed] [Google Scholar]
- 38.Kinoshita A, Nakayama Y, Kitayama T, Tomita M. 2007. Simulation study of methemoglobin reduction in erythrocytes. Differential contribution of two pathways to tolerance to oxidative stress. FEBS J 274:1449–1458. [DOI] [PubMed] [Google Scholar]
- 39.Schirmer RH, Müller JG, Krauth-Siegel RL. 1995. Disulfide-reductase inhibitors as chemotherapeutic agents: the design of drugs for trypanosomiasis and malaria. Angew Chem Int Ed Engl 34:141–154. doi: 10.1002/anie.199501411. [DOI] [Google Scholar]
- 40.Parapini S, Basilico N, Mondani M, Olliaro P, Taramelli D, Monti D. 2004. Evidence that haem iron in the malaria parasite is not needed for the antimalarial effects of artemisinin. FEBS Lett 575:91–94. doi: 10.1016/j.febslet.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 41.Klonis N, Crespo-Ortiz MP, Bottova I, Abu-Bakar N, Kenny S, Rosenthal PJ, Tilley L. 2011. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci U S A 108:11405–11410. doi: 10.1073/pnas.1104063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Antoine T, Fisher N, Amewu R, O'Neill PM, Ward SA, Biagini GA. 2014. Rapid kill of malaria parasites by artemisinin and semi-synthetic endoperoxides involves ROS-dependent depolarization of the membrane potential. J Antimicrob Chemother 69:1005–1016. doi: 10.1093/jac/dkt486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gopalakrishnan AM, Kumar N. 2015. Antimalarial action of artesunate involves DNA damage mediated by reactive oxygen species. Antimicrob Agents Chemother 59:317–325. doi: 10.1128/AAC.03663-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Hook AM. 2015. Antimalarial drugs inhibit PI3P production. Sci Signal 8:ec118. doi: 10.1126/scisignal.aac4781. [DOI] [Google Scholar]
- 45.Mbengue A, Bhattacharjee S, Pandharkar T, Liu H, Estiu G, Stahelin RV, Rizk SS, Njimoh DL, Ryan Y, Chotivanich K, Nguon C, Ghorbal M, Lopez-Rubio J, Pfrender M, Emrich S, Mohandas N, Dondorp AM, Wiest O, Haldar K. 2015. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 520:683–687. doi: 10.1038/nature14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okoh VO, Felty Q, Parkash J, Poppiti R, Roy D. 2013. Reactive oxygen species via redox signaling to PI3K/AKT pathway contribute to the malignant growth of 4-hydroxy estradiol-transformed mammary epithelial cells. PLoS One 8:e54206. doi: 10.1371/journal.pone.0054206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu J, Zhou J, Xing D. 2012. Phosphatidylinositol 3-kinase plays a vital role in regulation of rice seed vigor via altering NADPH oxidase activity. PLoS One 7:e33817. doi: 10.1371/journal.pone.0033817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim JH, Chu SC, Gramlich JL, Pride YB, Babendreier E, Chauhan D, Salgia R, Podar K, Griffin JD, Sattler M. 2005. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood 105:1717–1723. doi: 10.1182/blood-2004-03-0849. [DOI] [PubMed] [Google Scholar]
- 49.Schirmer RH, Coulibaly B, Stich A, Scheiwein M, Merkle H, Eubel J, Becker K, Becher H, Müller O, Zich T, Schiek W, Kouyate B. 2003. Methylene blue as an antimalarial agent. Redox Rep 8:272–276. doi: 10.1179/135100003225002899. [DOI] [PubMed] [Google Scholar]
- 50.Buchholz K, Schirmer RH, Eubel JK, Akoachere MB, Dandekar T, Becker K, Gromer S. 2008. Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum. Antimicrob Agents Chemother 52:183–191. doi: 10.1128/AAC.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vennerstrom JL, Makler MT, Angerhofer CK, Williams JA. 1995. Antimalarial dyes revisited: xanthenes, azines, oxazines, and thiazines. Antimicrob Agents Chemother 39:2671–2677. doi: 10.1128/AAC.39.12.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sarma GN, Savvides SN, Becker K, Schirmer M, Schirmer RH, Karplus PA. 2003. Glutathione reductase of the malarial parasite Plasmodium falciparum: crystal structure and inhibitor development. J Mol Biol 328:893–907. doi: 10.1016/S0022-2836(03)00347-4. [DOI] [PubMed] [Google Scholar]
- 53.Haynes RK, Cheu KW, Tang MM, Chen MJ, Guo ZF, Guo ZH, Coghi P, Monti D. 2011. Reactions of antimalarial peroxides with each of leucomethylene blue and dihydroflavins: flavin reductase and the cofactor model exemplified. ChemMedChem 6:279–291. doi: 10.1002/cmdc.201000508. [DOI] [PubMed] [Google Scholar]
- 54.Rengelshausen J, Burhenne J, Fröhlich M, Tayrouz Y, Singh SK, Riedel K-D, Müller O, Hoppe-Tichy T, Haefeli WE, Mikus G, Walter-Sack I. 2004. Pharmacokinetic interaction of chloroquine and methylene blue combination against malaria. Eur J Clin Pharmacol 60:709–715. doi: 10.1007/s00228-004-0818-0. [DOI] [PubMed] [Google Scholar]
- 55.Pascual A, Henry M, Briolant S, Charras S, Baret E, Amalvict R, des Etages EH, Feraud M, Rogier C, Pradines B. 2011. In vitro activity of Proveblue (methylene blue) on Plasmodium falciparum strains resistant to standard antimalarial drugs. Antimicrob Agents Chemother 55:2472–2474. doi: 10.1128/AAC.01466-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akoachere M, Buchholz K, Fischer E, Burhenne J, Haefeli WE, Schirmer RH, Becker K. 2005. In vitro assessment of methylene blue on chloroquine-sensitive and -resistant Plasmodium falciparum strains reveals synergistic action with artemisinins. Antimicrob Agents Chemother 49:4592–4597. doi: 10.1128/AAC.49.11.4592-4597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adjalley SH, Johnston GL, Li T, Eastman RT, Ekland EH, Eappen AG, Richman A, Sim BK, Lee MC, Hoffman SL, Fidock DA. 2011. Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc Natl Acad Sci U S A 108:1214–1223. doi: 10.1073/pnas.1112037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Navaratnam V, Mansor SM, Sit NW, Grace J, Li Q, Olliaro P. 2000. Pharmacokinetics of artemisinin-type compounds. Clin Pharmacokinet 39:255–270. doi: 10.2165/00003088-200039040-00002. [DOI] [PubMed] [Google Scholar]
- 59.Wesche DL, DeCoster MA, Tortella FC, Brewer TG. 1994. Neurotoxicity of artemisinin analogs in vitro. Antimicrob Agents Chemother 38:1813–1819. doi: 10.1128/AAC.38.8.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Genovese RF, Newman DB. 2008. Understanding artemisinin-induced brainstem neurotoxicity. Arch Toxicol 82:379–385. doi: 10.1007/s00204-007-0252-z. [DOI] [PubMed] [Google Scholar]
- 61.Ramos-Martin V, Gonzalez-Martinez C, Mackenzie I, Schmutzhard J, Pace C, Lalloo DG, Terlouw DJ. 2014. Neuroauditory toxicity of artemisinin combination therapies—have safety concerns been addressed? Am J Trop Med Hyg 91:62–73. doi: 10.4269/ajtmh.13-0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haynes RK, Chan HW, Lung CM, Ng NC, Wong HN, Shek LY, Williams ID, Gomes MF, Cartwright A. 2007. Artesunate and dihydroartemisinin (DHA): unusual decomposition products formed under mild conditions and comments on the fitness of DHA as an antimalarial drug. ChemMedChem 2:1448–1463. doi: 10.1002/cmdc.200700064. [DOI] [PubMed] [Google Scholar]
- 63.Haynes RK, Chan HW, Ho WY, Ko CK, Gerena L, Kyle DE, Peters W, Robinson BL. 2005. Convenient access both to highly antimalaria-active 10-arylaminoartemisinins, and to 10-alkyl ethers including artemether, arteether, and artelinate. Chembiochem 6:659–667. doi: 10.1002/cbic.200400366. [DOI] [PubMed] [Google Scholar]
- 64.Haynes RK, Fugmann B, Stetter J, Rieckmann K, Heilmann HD, Chan HW, Cheung MK, Lam WL, Wong HN, Croft SL, Vivas L, Rattray L, Stewart L, Peters W, Robinson BL, Edstein MD, Kotecka B, Kyle DE, Beckermann B, Gerisch M, Radtke M, Schmuck G, Steinke W, Wollborn U, Schmeer K, Romer A. 2006. Artemisone—a highly active antimalarial drug of the artemisinin class. Angew Chem Int Ed Engl 45:2082–2088. doi: 10.1002/anie.200503071. [DOI] [PubMed] [Google Scholar]
- 65.Haynes RK. 2008. Strategies in the development and chemical modification of the new artemisinin antimalarial artemisone. Synform 3:34–36. [Google Scholar]
- 66.Wu Y, Wu RW, Cheu KW, Williams ID, Krishna S, Slavic K, Gravett AM, Liu WM, Wong HN, Haynes RK.. 2016. Methylene homologues of artemisone: an unexpected structure-activity relationship and a possible implication for the design of C10-substituted artemisinins. ChemMedChem 11:1469–1479. doi: 10.1002/cmdc.201600011. [DOI] [PubMed] [Google Scholar]
- 67.Schmuck G, Temerowski M, Haynes RK, Fugmann B. 2003. Identification of non-neurotoxic artemisinin derivatives in vivo and in vitro. Res Adv Antimicrob Agents Chemother 3:35–47. [Google Scholar]
- 68.Haynes RK, Ho W-Y, Chan H-W, Fugmann B, Stetter J, Croft SL, Vivas L, Peters W, Robinson BL. 2004. Highly antimalaria-active artemisinin derivatives: biological activity does not correlate with chemical reactivity. Angew Chem Int Ed Engl 43:1381–1385. doi: 10.1002/anie.200352343. [DOI] [PubMed] [Google Scholar]
- 69.Guo J, Guiguemde AW, Bentura-Marciano A, Clark J, Haynes RK, Chan WC, Wong HN, Hunt NH, Guy RK, Golenser J. 2012. Synthesis of artemiside and its effects in combination with conventional drugs against severe murine malaria. Antimicrob Agents Chemother 56:163–173. doi: 10.1128/AAC.05006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nzila A, Mwai L. 2010. In vitro selection of Plasmodium falciparum drug-resistant parasite lines. J Antimicrob Chemother 65:390–398. doi: 10.1093/jac/dkp449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reader J, Botha M, Theron A, Lauterbach SB, Rossouw C, Engelbrecht D, Wepener M, Smit A, Leroy D, Mancama D, Coetzer LC, Birkholtz L. 2015. Nowhere to hide: interrogating different metabolic parameters of Plasmodium falciparum gametocytes in a transmission blocking drug discovery pipeline towards malaria elimination. Malar J 14:213. doi: 10.1186/s12936-015-0718-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Plouffe DM, Wree M, Du AY, Meister S, Li F, Patra K, Lubar A, Okitsu SL, Flannery EL, Kato N, Tanaseichuk O, Comer E, Zhou B, Kuhen K, Zhou Y, Leroy D, Schreiber SL, Scherer CA, Vinetz J, Winzeler EA. 2016. High-throughput assay and discovery of small molecules that interrupt malaria transmission. Cell Host Microbe 19:114–126. doi: 10.1016/j.chom.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.D'Alessandro S, Silvestrini F, Dechering K, Corbett Y, Parapini S, Timmerman M, Galastri L, Basilico N, Sauerwein R, Alano P, Taramelli D. 2013. A Plasmodium falciparum screening assay for anti-gametocyte drugs based on parasite lactate dehydrogenase detection. J Antimicrob Chemother 68:2048–2058. doi: 10.1093/jac/dkt165. [DOI] [PubMed] [Google Scholar]
- 74.Bolscher JM, Koolen KM, van Gemert GJ, van de Vegte-Bolmer MG, Bousema T, Leroy D, Sauerwein RW, Dechering KJ. 2015. A combination of new screening assays for prioritization of transmission-blocking antimalarials reveals distinct dynamics of marketed and experimental drugs. J Antimicrob Chemother 70:1357–1366. doi: 10.1093/jac/dkv003. [DOI] [PubMed] [Google Scholar]
- 75.Sun W, Tanaka TQ, Magle CT, Huang W, Southall N, Huang R, Dehdashti SJ, McKew JC, Williamson KC, Zheng W. 2014. Chemical signatures and new drug targets for gametocytocidal drug development. Sci Rep 4:3743. doi: 10.1038/srep03743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Siciliano G, Santha Kumar TR, Bona R, Camarda G, Calabretta MM, Cevenini L, Davioud-Charvet E, Becker K, Cara A, Fidock DA, Alano P. 2017. A high susceptibility to redox imbalance of the transmissible stages of Plasmodium falciparum revealed with a luciferase-based mature gametocyte assay. Mol Microbiol 104:306–331. doi: 10.1111/mmi.13626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Almela MJ, Lozano S, Lelievre J, Colmenarejo G, Coteron JM, Rodrigues J, Gonzalez C, Herreros E. 2015. A new set of chemical starting points with Plasmodium falciparum transmission-blocking potential for antimalarial drug discovery. PLoS One 10:e0135139. doi: 10.1371/journal.pone.0135139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Duffy S, Avery VM. 2013. Identification of inhibitors of Plasmodium falciparum gametocyte development. Malar J 12:408. doi: 10.1186/1475-2875-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lucantoni L, Avery V. 2012. Whole-cell in vitro screening for gametocytocidal compounds. Future Med Chem 4:2337–2360. doi: 10.4155/fmc.12.188. [DOI] [PubMed] [Google Scholar]
- 80.Fivelman QL, Adagu IS, Warhurst DC. 2004. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob Agents Chemother 48:4097–4102. doi: 10.1128/AAC.48.11.4097-4102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hott A, Casandra D, Sparks KN, Morton LC, Castanares GG, Rutter A, Kyle DE. 2015. Artemisinin-resistant Plasmodium falciparum parasites exhibit altered patterns of development in infected erythrocytes. Antimicrob Agents Chemother 59:3156–3167. doi: 10.1128/AAC.00197-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lanteri CA, Chaorattanakawee S, Lon C, Saunders DL, Rutvisuttinunt W, Yingyuen K, Bathurst I, Ding XC, Tyner SD. 2014. Ex vivo activity of endoperoxide antimalarials, including artemisone and arterolane, against multidrug-resistant Plasmodium falciparum isolates from Cambodia. Antimicrob Agents Chemother 58:5831–5840. doi: 10.1128/AAC.02462-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.D'Alessandro S, Camarda G, Corbett Y, Siciliano G, Parapini S, Cevenini L, Michelini E, Roda A, Leroy D, Taramelli D, Alano P. 2016. A chemical susceptibility profile of the Plasmodium falciparum transmission stages by complementary cell-based gametocyte assays. J Antimicrob Chemother 71:1148–1158. doi: 10.1093/jac/dkv493. [DOI] [PubMed] [Google Scholar]
- 84.Lucantoni L, Duffy S, Adjalley SH, Fidock DA, Avery VM. 2013. Identification of MMV malaria box inhibitors of Plasmodium falciparum early-stage gametocytes using a luciferase-based high-throughput assay. Antimicrob Agents Chemother 57:6050–6062. doi: 10.1128/AAC.00870-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lucantoni L, Fidock DA, Avery VM. 2016. Luciferase-based, high-throughput assay for screening and profiling transmission-blocking compounds against Plasmodium falciparum gametocytes. Antimicrob Agents Chemother 60:2097–2107. doi: 10.1128/AAC.01949-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lelievre J, Almela MJ, Lozano S, Miguel C, Franco V, Leroy D, Herreros E. 2012. Activity of clinically relevant antimalarial drugs on Plasmodium falciparum mature gametocytes in an ATP bioluminescence “transmission blocking” assay. PLoS One 7:e35019. doi: 10.1371/journal.pone.0035019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vos MW, Stone WJR, Koolen KM, van Gemert G-J, van Schaijk B, Leroy D, Sauerwein RW, Bousema T, Dechering KJ. 2015. A semi-automated luminescence based standard membrane feeding assay identifies novel small molecules that inhibit transmission of malaria parasites by mosquitoes. Sci Rep 5:18704. doi: 10.1038/srep18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hanssen E, Knoechel C, Dearnley M, Dixon MW, Le Gros M, Larabell C, Tilley L. 2012. Soft X-ray microscopy analysis of cell volume and hemoglobin content in erythrocytes infected with asexual and sexual stages of Plasmodium falciparum. J Struct Biol 177:224–232. doi: 10.1016/j.jsb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Becker K, Tilley L, Vennerstrom JL, Roberts D, Rogerson S, Ginsburg H. 2004. Oxidative stress in malaria parasite-infected erythrocytes: host-parasite interactions. Int J Parasitol 34:163–189. doi: 10.1016/j.ijpara.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 90.Zoungrana A, Coulibaly B, Sié A, Walter-Sack I, Mockenhaupt FP, Kouyaté B, Schirmer RH, Klose C, Mansmann U, Meissner P, Müller O. 2008. Safety and efficacy of methylene blue combined with artesunate or amodiaquine for uncomplicated falciparum malaria: a randomized controlled trial from Burkina Faso. PLoS One 3:e1630. doi: 10.1371/journal.pone.0001630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ohrt C, Li Q, Obaldia N, Im-erbsin R, Xie L, Berman J. 2014. Efficacy of intravenous methylene blue, intravenous artesunate, and their combination in preclinical models of malaria. Malar J 13:415. doi: 10.1186/1475-2875-13-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ginsburg H, Golenser J. 2003. Glutathione is involved in the antimalarial action of chloroquine and its modulation affects drug sensitivity of human and murine species of Plasmodium. Redox Rep 8:276–279. doi: 10.1179/135100003225002907. [DOI] [PubMed] [Google Scholar]
- 93.Verlinden BK, Niemand J, Snyman J, Sharma SK, Beattie RJ, Woster PM, Birkholtz L. 2011. Discovery of novel alkylated (bis)urea and (bis)thiourea polyamine analogues with potent antimalarial activities. J Med Chem 54:6624–6633. doi: 10.1021/jm200463z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Makler MT, Hinrichs DJ. 1993. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am J Trop Med Hyg 48:205–210. doi: 10.4269/ajtmh.1993.48.205. [DOI] [PubMed] [Google Scholar]
- 95.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. 2004. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother 48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 97.Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S, Skehan P, Scudiero DA, Monks A, Boyd MR. 1990. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst 82:1113–1118. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- 98.Ohrt C, Willingmyre GD, Lee P, Knirsch C, Milhous W. 2002. Assessment of azithromycin in combination with other antimalarial drugs against Plasmodium falciparum in vitro. Antimicrob Agents Chemother 46:2518–2524. doi: 10.1128/AAC.46.8.2518-2524.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ariey F, Witkowski B, Amarantunga C, Beghain J, Langlois KKA, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chour CM, Bout DM, Menard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale JC, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Menard D. 2014. A molecular marker of artemisinin resistant Plasmodium falciparum malaria. Nature 51:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.