

EmrE is the archetypical member of the small multidrug resistance transporter family and confers resistance to a wide range of disinfectants and dyes known as quaternary cation compounds (QCCs). The aim of this study was to examine which conserved amino acids play an important role in substrate selectivity.
KEYWORDS: EmrE, multidrug resistance, quaternary cation compound, quaternary ammonium compound, polyspecificity, multidrug transporter, antimicrobial resistance, efflux pump, polyselectivity
ABSTRACT
EmrE is the archetypical member of the small multidrug resistance transporter family and confers resistance to a wide range of disinfectants and dyes known as quaternary cation compounds (QCCs). The aim of this study was to examine which conserved amino acids play an important role in substrate selectivity. On the basis of a previous analysis of EmrE homologues, a total of 33 conserved residues were targeted for cysteine or alanine replacement within E. coli EmrE. The antimicrobial resistance of each EmrE variant expressed in Escherichia coli strain JW0451 (lacking dominant pump acrB) to a collection of 16 different QCCs was tested using agar spot dilution plating to determine MIC values. The results determined that only a few conserved residues were drug polyselective, based on ≥4-fold decreases in MIC values: the active-site residue E14 (E14D and E14A) and 4 additional conserved residues (A10C, F44C, L47C, W63A). EmrE variants I11C, V15C, P32C, I62C, L93C, and S105C enhanced resistance to polyaromatic QCCs, while the remaining EmrE variants reduced resistance to one or more QCCs with shared chemical features: acylation, tri- and tetraphenylation, aromaticity, and dicationic charge. Mapping of EmrE variants onto transmembrane helical wheel projections using the highest resolved EmrE structure suggests that polyselective EmrE variants were located closest to the helical faces surrounding the predicted drug binding pocket, while EmrE variants with greater drug specificity mapped onto distal helical faces. This study reveals that few conserved residues are essential for drug polyselectivity and indicates that aromatic QCC selection involves a greater portion of conserved residues than that in other QCCs.
INTRODUCTION
The Escherichia coli EmrE protein is the archetypical member of the small multidrug resistance (SMR) transporter family identified in bacteria and archaea. This tiny yet potent transporter has been the focus of many biochemical and biophysical studies seeking to understand how such a small proton antiport-driven protein can broadly select and transport a diverse array of drugs (reviewed in reference 1). EmrE is renowned for its ability to confer resistance to a wide range of quaternary cation compounds (QCCs). QCC are chemicals that possess one or more permanently charged cations (typically, nitrogen or phosphorus) bound to 3 or 4 R groups consisting of acyl and/or aryl moieties (Table 1). QCCs are often toxic and lipophilic and commonly used as antiseptics, biocides, industrial surfactants, phase catalysts, and dyes (2). Studies of EmrE and its homologues are important due to their frequent detection on multidrug resistance plasmids and in conserved 3′ regions of mobile genetic elements known as integrons that are shared between bacteria (3). By comparison to other larger multidrug transporters (12 to 14 α-helical transmembrane [TM] strands), EmrE is distinct due to its short length (110 amino acids), consisting of only 4 TM strands connected by 3 short loops, and its extreme hydrophobicity (reviewed in reference 1). The functional arrangement and structure of EmrE have been the subjects of considerable debate (4, 5), but consensus supports an antiparallel topology of dimeric EmrE as the minimal functional unit (6–8).
TABLE 1.
Summary of the chemical properties and structural features of the 18 QCCs examined in this study
| QCCa | Known mechanism of action | Concn range tested (μg/ml) | Acyl chain length (no. of amino acids) | No. of aromatic rings | Solubility in water (mg/ml)b |
|---|---|---|---|---|---|
| Acriflavine chloride (ACR) | Antiseptic intercalating dye | 0.5–128 | 3 | 1–330 | |
| Benzalkonium chloride (BZ) | Antiseptic surfactant phase transfer agent | 0.25–64 | 8–18 | 1 | 100 |
| Cetalkonium chloride (CAC) | Antiseptic surfactant | 0.5–128 | 16 | 1 | 2 |
| Cetylpyridinium bromide (CPB) | Antiseptic surfactant | 2–512 | 16 | 1 | 0.04 |
| Cetylpyridinium chloride (CPC) | Antiseptic surfactant | 2–512 | 16 | 1 | 0.05 |
| Cetrimide bromide (CET) | Antiseptic surfactant | 0.5–128 | 8–18 | 0.56 | |
| Crystal violet chloride (CV) | Antiseptic dye | 0.25–64 | 3 | 0.02–50 | |
| Dequalinium dichloride (DQ) | Antiseptic surfactant | 2–500 | 10 | 2 | 4.7 × 10−7–10 |
| Ethidium bromide (ET) | Intercalating dye | 1–256 | 4 | 10–20 | |
| Methyl viologen dichloride (MV) | Herbicide reactive oxygen species initiator | 2–512 | 2 | 100 | |
| Methyltriphenylphosphonium bromide (MTP) | Antiseptic surfactant | 1–240 | 3 | 100 | |
| Myristalkonium chloride (MAC) | Antiseptic surfactant | 0.25–64 | 14 | 1 | 1–10 |
| Proflavin dihydrochloride (PRO) | Antiseptic intercalating dye | 0.5–128 | 3 | 22–500 | |
| Pyronin Y chloride (PY) | Antiseptic intercalating dye | 0.25–64 | 3 | 30 | |
| Rhodamine 6G chloride (R6G) | Antiseptic dye | 0.25–64 | 4 | 0.02 | |
| Stearyltrimethylammonium chloride (STAC) | Antiseptic surfactant | 7.8–2000 | 18 | 5–50 | |
| Tetraphenylarsonium chloride (TPA) | Phase transfer catalyst | 0.25–64 | 4 | 0.05 | |
| Tetraphenylphosphonium chloride (TPP) | Phase transfer catalyst | 0.125–32 | 4 | 0.005–50 |
Figure S1 in the supplemental material provides the chemical structures for all 18 QCCs tested.
Ranges indicate differences in solubility according to the product information of the different chemical suppliers.
Our understanding of how EmrE can recognize such diverse substrates remains poorly understood, and most site-directed mutagenesis studies of EmrE have focused on residue replacements within particular TM strands (e.g., TM2 and TM4), functional properties, and structural investigations (dimerization domain TM4). The most well characterized and highly conserved (98% identity [9]) E. coli EmrE residue is a negatively charged glutamate (E14), located in the first TM1 strand (Table 2), that acts as the transporter active site due to its inability to bind drugs and protons when it is replaced with aspartate or other residues in site-directed mutagenesis studies (10, 11). The replacement of other residues in EmrE and its homologues, such as Y3 and G17 (12), S43 (12, 13), and L47 (14), generated variants with resistance toward specific QCCs, suggesting that some residues are substrate selective. Replacement of conserved residue W63 in EmrE (75% identity; Table 2) (14) and moderately conserved residues H24, M39, I43, and A44 in SMR family member SugE (40 to 60% identity [9]) (15) has also shown that some residue replacements can convert these transporters from an exporter to an importer, indicating remarkable functional plasticity. Previous biochemical and biophysical studies exploring EmrE variant substrate selectivity have involved relatively few drugs; the most commonly tested have been ethidium (ET), methyl viologen (MV), acriflavine (ACR), and tetraphenylphosphonium (TPP), all of which are aromatic compounds (Table 2).
TABLE 2.
Summary of the 33 EmrE variants examined in this study, their previously known functional roles, and their conferred susceptibility/tolerance to each of the 18 QCCs, as determined in this study
| EmrE variant | Protein domain | % identitya | Previously determined functional role | Reference(s) | QCC specificityb in this study |
|---|---|---|---|---|---|
| L7A | TM1 | 98 | Possible substrate binding | 50 | ACR, BZ, CAC, CV, DQ, MV, PY, TPP |
| A10C | TM1 | 94 | Substrate/H+ binding | 50 | ACR, BZ, CAC, CET, DQ, ET, PRO, PY, MV, MTP, TPP |
| I11C | TM1 | 90 | Protein folding/stability | 50 | ACR, CET, PRO |
| E14D | TM1 | 98 | In vivo active site substrate/H+ binding | 10, 11, 19 | ACR, BZ, CAC, CET, CPB, CPC, CV, DQ, ET, PRO, PY, MAC, MTP, TPP, TPA |
| V15C | TM1 | 85 | No effect in vivo/in vitro | 50 | ACR, BZ, CAC, CV, ET, PY, MV, TPP |
| G17A | TM1 | 70 | Protein folding/stability | 36, 50 | ACR, CAC, CV, PRO, PY, MV, TPP |
| T18C | TM1 | 88 | Protein folding/stability | 50 | CAC, CPC, CV, ET, MV, PRO, PY, R6G |
| M21C | TM1 | 87 | No effect in vivo/in vitro | 50 | CET, CV, DQ, ET, PY, R6G |
| G26C | Loop 1 | 75 | In vivo substrate recognition | 36 | CET, CPB, CV, DQ, ET, PY, R6G |
| F27C | Loop 1 | 90 | Not previously determined | CET, CPB, CV, DQ, ET, PY, R6G | |
| W31A | Loop 1 | 60 | No effect in vivo/in vitro | 29, 33 | CAC, CET, CV, DQ, ET, PRO, PY, R6G, TPA, TPP |
| P32C | Loop 1 | 88 | No effect in vivo/in vitro | 29 | ACR, PRO, PY |
| Y40A | TM2 | 94 | Substrate binding | 29, 34, 51, 52 | ACR, CV, DQ, ET, MAC, PRO, TPP |
| F44C | TM2 | 94 | Possible substrate binding | 29, 34 | BZ, CET, CPB, CV, DQ, MTP, MV, PY, R6G |
| W45A | TM2 | 70 | No effect in vivo/in vitro | 29, 33, 34 | BZ, CAC, CET, DQ, ET, PY, TPP |
| L47C | TM2 | 90 | Possible substrate binding | 29 | ACR, BZ, CET, CPB, CV, DQ, ET, MV, PRO, PY, R6G |
| P55C | Loop 2 | 94 | In vivo/in vitro protein folding/stability | 29 | CET, CPC, CPB, CV, MTP, PY, R6G, TPA, TPP |
| G57C | Loop 2 | 90 | Protein folding/stability | 29, 36 | CAC, CET, CV, MV, PY, R6G |
| A59C | TM3 | 98 | Not previously determined | ACR, BZ, CAC, CET, DQ, ET, CV, MTP, PY, TPP | |
| Y60A | TM3 | 88 | In vitro substrate binding | 51 | ACR, BZ, CPC, CET, CAC, DQ, MV, PRO, TPP |
| I62C | TM3 | 98 | Not previously determined | CET, CAC, CV, DQ, ET, MV, PY, TPP | |
| W63A | TM3 | 75 | Substrate binding | 33, 52 | ACR, CPC, CPB, ET, MTP, MV, PRO, PY, R6G, TPA, TPP |
| S64C | TM3 | 87 | Not previously determined | BZ, CET, CAC, DQ, ET, MTP, MV, TPA, TPP | |
| V66C | TM3 | 98 | Not previously determined | ACR, CET, DQ, ET, MV, PY, TPP | |
| V69C | TM3 | 75 | Not previously determined | ACR, BZ, CET, DQ, ET, MTP, PY, TPA | |
| S72C | TM3 | 75 | Substrate binding | 29 | BZ, CAC, DQ, ET, PY |
| W76A | TM3/Loop 3 | 50 | Protein folding/insertion | 33 | BZ, CV, DQ, MTP, PRO, R6G, TPA, TPP |
| G90C | TM4 | 92 | In vitro protein multimer stability | 29, 36 | CET, CV, DQ, MAC, MV, PRO, PY, R6G |
| L93C | TM4 | 76 | Substrate binding | 29, 32 | ACR, BZ, CET, DQ, ET, PY, TPP |
| I94C | TM4 | 80 | No effect in vivo/in vitro | 29, 32 | ACR, BZ, CAC, ET, MV, PRO, PY, R6G, TPP |
| G97C | TM4 | 97 | In vivo protein multimer stability | 29, 36 | ACR, BZ, CET, CAC, DQ, ET, MAC, MTP, MV, PRO, PY |
| L103C | TM4 | 75 | Not previously determined | BZ, CET, CAC, DQ, ET, MAC, TPP | |
| S105C | TM4 | 80 | Not previously determined | BZ, CET, CAC, DQ, ET, MTP, TPP, PY |
Percent identity values were determined from an alignment of 92 taxonomically diverse EmrE homolog sequences (37).
QCCs in plain font indicate those for which the residue replacements led to a ≤2-fold reduction in MIC from that for the WT. QCCs highlighted in bold identify compounds for which the residue replacements led to a sensitivity similar to that in previous studies. Underlined QCCs indicate drugs for which residue replacements led to a 2-fold increase in the MIC compared to that for the WT.
The current hypothesis suggests that EmrE substrate specificity is largely determined by poor to moderately conserved residues, where conserved residues play an important role in QCC polyselectivity (12–14, 16). The aim of this study was to validate the role and importance of conserved EmrE residues toward QCC polyselectivity using a collection of 16 chemically diverse QCCs. Site-directed mutagenesis was performed on plasmid-encoded E. coli emrE to generate 33 single-residue replacements at 33 conserved EmrE residues (≥70% identity; Table 2). EmrE variants were transformed into E. coli JW0451 lacking the dominant QCC-selective efflux pump acrB, and each EmrE variant was screened against the wild type (WT) for its QCC resistance using agar spot dilution testing. To avoid drug selection biases caused by the addition of affinity tags previously used to study EmrE, such as hexahistidine (10, 17) or hemagglutinin epitopes (18), modifications to untagged wild-type EmrE sequences were made. The MIC values for each variant and drug were statistically assessed, sorted, and visualized using hierarchical agglomerative clustering, which was used to compare fold change differences in the MIC values of different QCCs for the EmrE variants and the strain with wild-type EmrE. This analysis revealed that only a few conserved residues could be considered polyselective: active-site residue E14 (E14D and E14A) and 4 additionally conserved residues (A10C, F44C, L47C, W63A). However, less than half of all EmrE variants with conserved residues conferred decreased resistance (a ≥−2-fold change in the MIC) to one or more QCCs. EmrE variants I11C, V15C, P32C, I62C, L93C, and S105C significantly enhanced resistance to one or more polyaromatic QCCs, suggesting a role in aromatic QCC selection. Variant residues were mapped onto transmembrane helical wheel projections from the highest resolved X-ray diffraction EmrE structure (3.8 Å [6]), and the results support the location of highly polyselective residues closer to the predicted substrate binding pocket than their location in variants that were more QCC specific.
RESULTS
Few EmrE variants with conserved residues contribute to QCC polyselection.
To identify conserved EmrE residues with the greatest QCC polyselectivity, agar spot dilution antimicrobial susceptibility testing of EmrE variants was performed in E. coli strain JW0451 ΔacrB to measure the MIC values of 17 QCCs for each of the 33 variants (see Tables S2 to S3 in the supplemental material). This analysis revealed unique profiles of the MICs of the 17 QCCs tested for each of the 33 EmrE variants (Table S2; Fig. S2), but only 11 QCCs showed significant ≥4-fold differences in MICs and were included in the final analysis (Fig. 1). Data for QCCs methyltriphenylphosphonium (MTP), cetylpyridinium chloride (CPC), cetylpyridinium bromide (CPB), myristalkonium chloride (MAC), and tetraphenylarsonium (TPA) were omitted from the final analysis due to their MIC values for any variant failing to exceed −2- to +2-fold changes. Hierarchical agglomerative clustering analysis was used to group variants and drugs to visualize patterns between different EmrE variants and particular QCCs (Fig. 1 and S2). EmrE E14D variants were included as a polyselective control, and as expected, fold change reductions in the MICs of the most QCCs tested (15 of 17) were demonstrated for EmrE E14D variants, reconfirming the importance of E14 as an active-site residue. Since the E14D variant showed no differences in the MICs of rhodamine 6G (R6G) and MAC from those of the WT, EmrE E14A replacements were generated and the variants were retested against the WT with the QCCs ET, dequalinium (DQ), R6G, and MAC (Table S2). The MICs of these QCCs for these E14A variants were reduced −4- to −8-fold (Table S2), confirming that more severe amino acid replacements were necessary to completely eliminate QCC resistance, similar to the findings of a previous study (19).
FIG 1.

A hierarchical agglomerative cluster heatmap of the MIC values of 11 different QCCs determined for EmrE variants. The fold change in the MIC for EmrE variants (y axis) exposed to each QCC tested (x axis) is shown on the heatmap (refer to the key). EmrE variants (V) were grouped into 2 main clusters, V1 and V2, and the letters above the indicated nodes (a to d) are discussed in the text. Each EmrE variant is listed on the right-hand y axis and colored according to its TM position. All QCCs are listed on the bottom x axis and the results presented in the heatmaps above and below the QCCS correspond to those QCCs. The purple heatmaps below the QCCs on the bottom x axis indicate specific chemical structural features for each drug. Dendrograms on the left y axis and top x axis show cladistic associations between EmrE variant MICs and QCCs, and the clusters are discussed in text according to the labels for the EmrE variant clusters (V1-V2a-V2d) and QCC clusters (Q1 to Q4).
The results of the clustering analysis in Fig. 1 highlight three main findings from the MIC data set. The first finding indicates that only 14 of the 31 EmrE variants with conserved residues showed consistent reductions in MICs similar to those for the polyselective control, the E14D variant; the V1 cluster demonstrated a ≤−4-fold change in MIC values for two or more QCCs (Fig. 1, V2b-c). Besides the E14D variant, the most significant polyselective variants were the A10C (5 QCCs), F44C (5 QCCs), L47C (6 QCCs), and W63A (5 QCCs) variants, where pyronin Y (PY) was the only QCC in common for all variants. Plotting of these polyselective variant residue locations onto TM helical wheel maps of the X-ray diffraction EmrE dimer suggested that all 4 of the most polyselective residues were located close to the drug-binding pocket, as we would expect for polyselective residues (Fig. 2). The remaining 10 polyselective residues identified in cluster V2b-c (Fig. 1) also appeared to be located on TM sides and regions of loops (Fig. 2) shown to be important for QCC binding in past experiments (Table 2). Hence, less than half of the EmrE variants with conserved residues generated in this study were significantly polyselective for more than 2 QCCs, which is in contrast to the proposed hypothesis suggesting that even conserved residues may be important for drug specificity.
FIG 2.

Locations of EmrE variants with conserved residues on TM helical wheel projections derived from the highest resolved structure of dimeric EmrE. (A and B) The 3.8-Å X-ray diffraction crystal structure of dimeric antiparallel EmrE bound to TPP (PDB accession number 3B5D) (6). Dimers are colored to identify each monomer (green, monomer A; magenta, monomer B). Front views of the dimer within the membrane bilayer plane (represented as black bars) and top-down views of the EmrE dimer relative to the membrane plane are shown. A red ball-and-stick structure of TPP is included to indicate the position of TPP within the dimer. Panel A is color coded to highlight residue positions that resulted in the enhanced polycyclic resistance shown in panel C. Panel B is color coded to highlight the increasingly polyspecific residues shown in panel D. Each TM strand in the monomer is numbered 1 to 4 from the amino to the carboxyl terminus in each monomer and lettered A or B in all panels. (C and D) Top-down helical wheel projections generated from the EmrE dimer structure shown in panel B. The residues highlighted on the structures shown in panels A and B correspond to EmrE variants with enhanced polycyclic resistance at 4-fold MIC values (solid blue symbols), and variants that resulted in a 2-fold MIC for increased polycyclic QCC resistance are colored in cyan in panel C. EmrE variant positions that conferred significant resistance (≤−4-fold changes in MIC values) to more than 5 QCCs that differed by 2 or more chemical features are colored in red in panel D, and moderately polyspecific EmrE variants that conferred significant resistance (≤−4-fold changes in MIC values) to 3 or 4 QCCs that differed by 2 or more chemical features are colored in orange in panel D. TM residues are presented as circles and dashed lines, and diamonds indicate residues located in turns or termini on the helical wheel projection diagrams. The large gray spheres in the center of each dimer labeled TPP indicate the relative position of TPP/ligand. Outlined residues highlight the locations of residues involved in QCC/substrate binding, participate in protein multimerization, influence protein folding/complex stability, or have an unknown or no effect on the basis of the findings of the previous studies listed in Table 2.
Some conserved-residue replacements enhanced aromatic QCC resistance.
The second finding was that more than half of the EmrE variants with conserved residues (in which the residue was replaced with either cysteine or alanine) showed increased resistance (+2- to +4-fold changes in MIC values) to two or more polyaromatic QCCs: ET, ACR, PY, proflavin (PRO), crystal violet (CV), and R6G (Fig. 1, V1, V2a, and V2d). The most significant increases in MIC values (+4-fold) were observed for PY- and ET-exposed EmrE variants I11C, V15C, P32C, I62C, L93C, and S105C (Fig. 1). After plotting the locations of these 6 variants on TM helical wheel projections, all 6 residues (where the I11C and V15C variants plotted to TM1, the P32C variant to loop 1, the I62C variant to TM3, and the L93C and S105C variants to TM4) were found to be located on TM sides or loops that were farther away from the proposed dimeric drug-binding pocket (Fig. 2). This may suggest that aromatic QCC interactions may require greater protein domain involvement. When variants were plotted onto Venn diagrams according to aromatic QCC MIC values (Fig. 3), the number of conserved EmrE positions involved in aromatic QCC activity enhancement may be influenced by the degree of ring planarity (ACR, PRO, PY > ET, CV, R6G) and polyaromaticity (2 rings, MV; 3 rings, ACR PRO, PY, and CV; 4 rings, ET and CV).
FIG 3.
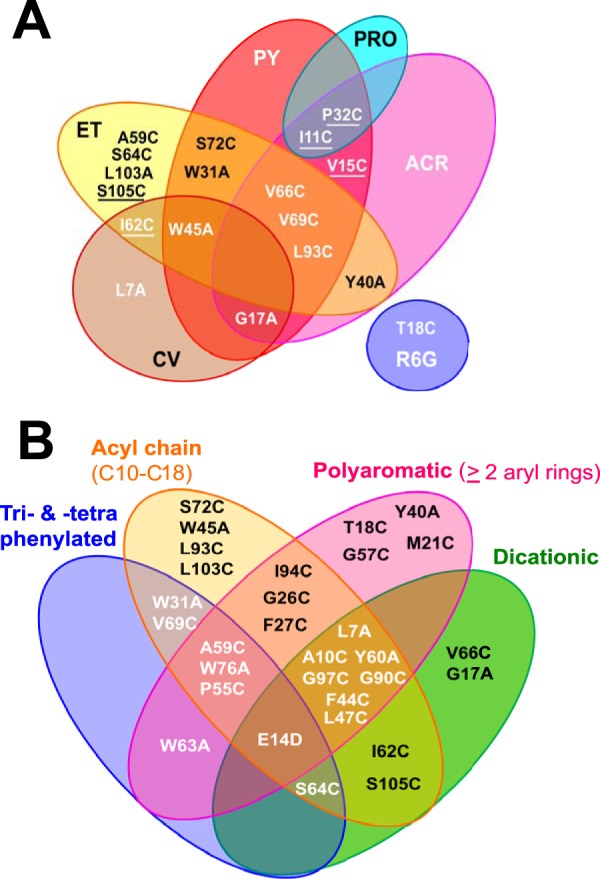
Venn diagrams summarizing MIC differences between EmrE variants and QCCs with particular chemical features. (A) EmrE variants that conferred enhanced resistance to specific polycyclic aromatic compounds. Underlined EmrE variants demonstrated significant 4-fold changes in MIC values. (B) A summary of EmrE variants that increased susceptibility (≤−4-fold change in MIC values) to ≥1 QCC separated according to 4 major structural features: dicationic (MV and DQ), acyl chained (CPC, CPB, CET, MAC, CAC, BZ, DQ), polycyclic (ET, ACR, PRO, PY, CV, R6G, DQ, MV), and tri- and tetraphenylated (MTP, TPP, TPA).
Greater QCC chemical complexity identified more EmrE variants with conserved residues.
The third finding was the relationship between QCC chemical properties and the MIC values for the EmrE variants. Dequalinium (DQ) produced the greatest MIC reductions (−4- to −64-fold changes in the MIC) for all but 7 EmrE variants (the T18C, G57C, I94C, W63C, P32C, I11C, V15C variants; Fig. 1). DQ possesses 3 of the 4 major distinguishing chemical features used for QCC selection in this study; DQ is a dicationic, acyl C10 chain-linked polyaromatic compound (Table 1; Fig. S1). All other QCCs included in the study, including MV, the only other dication in this study, possessed one or two of these features at most. MV possesses 2 aryl rings separated by a single carbon bond and is typically used as an EmrE substrate for polyvalent cation testing (Table 2). Our results suggest that the presence of 3 chemical properties rather than 1 or 2 may require the involvement of more conserved residues, as we observed for most EmrE variants (Fig. 1). Further testing of EmrE variants for susceptibility to DQ and other QCCs may be useful in future EmrE studies. Cetrimide (CET) resulted in the second most severe reduction in the MICs (<−4-fold change in the MIC) for the EmrE variants tested herein (Fig. 1). Unlike DQ, CET is an amphipathic detergent consisting of a trimethylated ammonium cation linked to a single acyl chain of various lengths (C8 to C18) (Table 1; Fig. S1). Our analysis of QCCs with fixed acyl chain lengths (cetalkonium chloride [CAC], MAC, cetylpyridinium chloride [CPC], or cetylpyridinium bromide [CPB]) or a cation located in aryl rings (benzalkonium [BZ]) often identified fewer EmrE variants with significant fold change reductions in MICs compared to the MICs for the WT. This may suggest that less chemical complexity requires fewer conserved EmrE residues (Fig. 1 and S2). Reduced MIC values were also observed for larger, tetra- and triphenylated polycyclic aromatic QCCs (TPP, tetraphenylarsonium chloride [TPA], and MTP) by specific EmrE variants that exhibited enhanced fold changes in MICs (Fig. 1). Some variations in these MIC patterns may be attributed to changes in the central cation atom (P+ in TPP versus As+ in TPA) and the replacement of a methyl group by a phenyl group (MTP versus TPP). Therefore, QCCs that possess greater structural variation, a larger size, and greater polycyclic aromaticity appear to require the involvement of more conserved residues, based on the pattern of the MIC resistance profiles of EmrE variants with conserved residues.
DISCUSSION
This study has demonstrated that only 1/4 of all conserved residues in EmrE are involved in drug binding and QCC polyselectivity, in contrast to the proposed hypothesis (12–14, 16). To emphasize this point, even the E14D variant with the replacement of the active-site E14 residue showed WT MIC values for 2 QCCs (R6G and MAC), which indicates that even the active-site residue can withstand some alteration and which highlights the dynamic plasticity of this protein. Our findings are in agreement with those of recently published studies suggesting that EmrE is highly flexible with regard to drug binding and adopts a free-exchange model for drug and proton transport (20, 21). Hence, having fewer conserved polyspecific residues in a flexible protein complex may be helpful for greater substrate recognition.
Many of the conserved EmrE residue replacements examined herein, W63, Y40, Y60, G90, and G97, were previously characterized (Table 2), and our MIC data are in agreement with those for previously tested QCC substrates, MV, TPP, ACR, and ET (see Tables S2 and S3 in the supplemental material). New insights into the QCC resistance attributed to 8 uncharacterized conserved EmrE residues (F27C, A59C, I62C, S64C, V66C, V69C, L103C, and S105C) have also been identified, some of which are important for aromatic QCC selection (I62C, S105C) (Table 2; Fig. 1). Our study also provides new MIC data for the QCCs MAC, TPA, and stearylalkonium chloride (STAC), which have been examined only as in vitro ligands in biophysical EmrE protein experiments (with DQ [22, 23] and MTP [24]). Altogether, this analysis identifies key conserved EmrE residues involved in QCC polyselectivity but also highlights residues and protein domains that may play a role in specific drug selection or protein stability.
In an effort to examine the resistance attributed to conserved residues of unmodified EmrE, all variants were expressed in E. coli ΔacrB strains without an affinity tag. Our goal was to examine the resistance conferred by EmrE which had the least amount of modifications to avoid drug selection biases stemming from the affinity tag itself, as noted in a recent study (17). The vast majority of studies involving EmrE have involved the addition of an affinity tag (as reviewed in reference 1), specifically, a C-terminal myc-His6 tag, which extends the protein by 30 amino acids and reduces its solubility but not membrane targeting (17). Furthermore, each EmrE variant that we examined conferred resistance to at least 2 or more QCCs similar to that for the WT (Fig. 1 and S2), suggesting that all variants actively accumulated in the membrane to enhance or reduce drug resistance (Fig. S2). In an effort to verify if the EmrE variant protein was indeed accumulated and targeted the membrane, we used an organic extraction technique (25) developed to isolate the accumulated EmrE protein from E. coli JW0531 membranes in strains lacking emrE. Although this technique is ideal to purify untagged protein, it was unable to accurately quantify exact amounts of the EmrE variant protein within membranes to concentrations below 10 μg/ml, since a portion of the protein (an estimated 20 to 60%) is lost as a highly insoluble lipid-bound fraction (25) (Fig. S5). This at least confirms that untagged EmrE variants accumulate in E. coli membranes, but it cannot distinguish if reduced MIC values were due to significantly less protein accumulation, protein misfolding/mistargeting, or the loss of substrate binding. Despite this factor, all EmrE variants showed WT levels of resistance to one or more QCCs, highlighting the resilience of the protein to sequence variation. It should be noted that differences in ligand binding, protein folding, and MIC values for affinity-tagged EmrE and untagged EmrE were identified and suggested that untagged EmrE binds ligands (TPP, MV, ET, CPC) more tightly and conferred higher levels of drug resistance in a recent study (17). This highlights the importance of studying unmodified versions of a protein along with tagged constructs and now provides greater insight into the resistance conferred by native EmrE protein.
Despite their low conservation in EmrE homologues, transient disulfide bonding between the three preexisting Cys residues (C39, C41, and C95) may potentially be responsible for enhancements and reductions in the QCC MIC by EmrE Cys replacement variants in this study. Stable disulfide bond formation in previous EmrE protein studies has reportedly required the exogenous addition of a strong oxidizing agent to catalyze its formation (26–29). If stable disulfide bonding was a significant factor in drug binding and function, we would expect that these residues would be well conserved, and stable disulfide bond formation would restrict drug access or multimerization for all substrates tested, which was not observed, as reported by cross-linking studies. We would also expect to observe consistent reductions in the MICs of all QCCs tested for the same EmrE variant, which was also not observed in our study. Additionally, these Cys residues have poor positional conservation within 290 taxonomically diverse EmrE homologues, indicating that Cys cross-linking is not an essential element in a QCC or H+ transport mechanism model of EmrE, as highlighted in recent biophysical studies of purified EmrE (8, 30).
Another important note is that due to the use of MIC values to interpret EmrE variants and the lack of side chain resolution in the highest resolved structure of EmrE (3.8 Å) (6), this study cannot distinguish whether EmrE variants alter the MIC due to folding or drug binding. However, it did identify distinct QCC resistance patterns for variants with conserved-residue replacements, reaffirming the importance of residues now studied to a much larger drug collection. Our study supports previous findings suggesting that the replacement of residues L7, A10, and M21 forms a polyselective substrate region on the same face of TM1 with active-site residue E14 (Table 1). We also identified that the V15, T18, and G17 residues, located on adjacent sides of the TM1 binding face, participate in polycyclic aromatic selectivity (Fig. 2 and S3). SMR protein loops are known to participate in multimerization and influence the insertion topology (31, 32), and as such, they are expected to influence QCC resistance (MICs) (29, 33). Our findings suggest that the replacement of conserved residues found in loops 1 and 2 influences QCC resistance selection. The P32C replacement in loop 1 enhanced resistance only to the polycyclic QCCs PY (4-fold), ACR (2-fold), and PRO (Fig. 3A), suggesting that the loss of this bend-inducing residue improved tricyclic aryl ring selection. In loop 2, the P55C and G57C variants had a notable loss of resistance to structurally unrelated QCCs (Fig. 3B), suggesting that the loss of either a helix-breaking proline or a turn-favoring glycine altered EmrE polyspecificity in this short loop (Fig. 2, S1, and S2).
The findings obtained with variants in which four highly conserved residues located on TM2 (Y40, F44, W45, L47) were replaced (Table 2; Fig. 1) were in good agreement with those of previous analyses suggesting that TM2 conserved residues tolerate greater residue diversity (34) and that the majority of residues in TM2 are moderately to poorly conserved (9). Our analysis suggests that the different faces of TM2 may be important for QCC polycyclic selectivity (the W45A and Y40A variants) and drug polyspecificity (the L47C and F44C variants), based on EmrE variant MIC data (Fig. 1, 2, S2, and S3). The replacement of each of the 4 tryptophans reaffirmed the importance of W63 as the most polyspecific (14, 33, 35) and also indicates that 3 of the 4 Trp replacements (W31A, W45A, and W76A) contribute toward greater QCC polyselectivity (Fig. 3B).
Previous structural studies of EmrE TM3 suggests that TM3 is flexible and plays a pivotal role in the movement of substrates across the membrane (16). Based on this role, it is not surprising that the highest number of conserved residues were identified on TM3 (Table 2) (9). In our study, the amount of different QCCs and variant combinations which were located in TM3 that we identified (Fig. 1) suggests that TM3 is the main contributor to QCC polyselectivity.
Finally, conserved residues G90C and G97C, located in TM4, were shown in a previous study to form a EmrE multimerization domain with a G90X6G97 motif (36). In our study, the G90C and G97C variants demonstrated moderate polyspecificity for 3 or 4 of the 11 QCCs, where the G97C variant differed from the G90C variant by significantly (a ≤−4-fold change in the MIC) reducing tolerance to CET (Fig. 1). It was expected that either variant with a glycine replacement in TM4 would reduce MIC values for all QCCs on the basis of previous findings (36), since stable multimerization should be essential for drug transport, as demonstrated in vitro (16). Based on our findings, the loss of individual glycines resulted in reductions in the MICs of most QCCs of ≤−2-fold, indicating that these variants were somewhat influential on polyselectivity (Fig. S2).
Conclusions.
This study emphasizes the importance of examining more substrates when determining EmrE residue involvement in QCC specificity and polyselectivity (Fig. 3A and B). Our findings confirm that few conserved residues are polyselective and suggest that drugs with greater chemical complexity require a greater involvement of more conserved residues located around the drug-binding pocket of EmrE.
MATERIALS AND METHODS
E. coli strains, plasmids, growth conditions, and QCCs used.
Keio Collection E. coli K-12 strain JW0451 with a deletion of acrB in the AcrAB-TolC efflux pump system was used for MIC determination, on the basis of the findings of previous studies demonstrating that AcrAB obscures the QCC resistance phenotypes of EmrE variants under the experimental conditions used in this study (37). E. coli DH5α was used for cloning and plasmid isolations. A previously constructed vector, pMS119EH, containing a cloned wild-type copy of the E. coli K-12 emrE gene (pEMR-11) (25) was used to generate all single-codon EmrE variants. All experiments included the parental vector pMS119EH as a negative control for antimicrobial susceptibility experiments. All plasmid-transformed strains were grown at 37°C in shaking incubators (200 rpm) in Luria-Bertani (LB) broth or agar medium with 100 μg/ml ampicillin (AMP) to maintain plasmids.
Construction of EmrE variants.
PCR-based site-directed mutagenesis of the emrE gene in pEMR-11 was performed according to a previously described method (38) to generate each of the 33 EmrE variants using the conditions described in a previous study (39) (see Table S1 in the supplemental material). The primer pairs used to generate each codon replacement are provided in Table S1 (Integrated DNA Technology, IA). Each PCR-generated EmrE variant was sequenced by Sanger sequencing (Eurofins Genomics, Toronto, ON, Canada) using upstream Ptac promoter primer Ptac to verify the accuracy of single-codon EmrE mutations. The full sequence of every emrE variant gene as well as the parental plasmid included in this analysis was sequenced by Sanger sequencing to verify that the correct codon mutation was generated after PCR. Plasmids were sequenced a second time from JW0451 transformants to verify that the emrE mutations were unchanged. No compensatory mutations were identified in any variant emrE sequences. The majority of EmrE residues (31 of 33) targeted for mutation were selected on the basis of their high degree of conservation (≥70% residue identity) in protein alignments of 290 taxonomically diverse EmrE homologues from a previous bioinformatic study (9) (Table 2). The W31A and W76A variants, exhibiting moderate conservation (50 to 60% identity; Table 2), were also selected for statistical comparisons of conserved residues. All targeted residues were replaced with either cysteine or alanine, since both of these amino acids have the advantage of maintaining overall protein hydrophobicity and neither has a strict localization preference when considering the amino acid distributions in membrane protein structures (40–42). Cys residues were selected for the majority of amino acid replacements in this study on the basis of their side group neutrality, their lack of conservation at any particular position (9), their potential to generate the most severe loss of activity alterations in the EmrE variants due to transient disulfide bond cross-linkages, and the lowest frequency of occurrence of Cys in EmrE homologues (the number of cysteines present in 290 EmrE homologues was the lowest of all amino acids, 0.9 ± 0.9) (9). Ala residues were also selected to generate short hydrophobic residue replacements for all tryptophan and tyrosine aromatic residues; in addition, variants with changes to the G17 and L7 positions were selected. The replacement of the active-site residue E14 by an aspartate (E14D) or alanine (E14A) was selected on the basis of previous studies demonstrating its deleterious effect on drug resistance, and these variants served as an important internal resistance control (10, 19).
To verify the expression and protein accumulation of EmrE variants in E. coli membranes, whole membranes were isolated from each JW0531 ΔemrE plasmid transformant culture and EmrE variant proteins were purified using an organic extraction reverse-phase gel filtration method and visualized by sodium dodecyl sulfate (SDS)-Tricine polyacrylamide gel electrophoresis as described in the data file “Supplementary Materials and Methods” and Fig. S5 in the supplemental material.
Agar spot dilution QCC susceptibility testing experiments for MIC determination.
A total of 18 QCCs were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Novo Nordisk Pharmatech A/S (Køge, Denmark). Each QCC was selected on the basis of its use in previous studies involving EmrE and chemical properties as a disinfectant or industrial surfactant (Table 1 and Fig. S1). The concentration ranges used for QCC susceptibility testing experiments are listed in Table 1. Expression of EmrE variants was accomplished by leaky Ptac promoter induction, which has been shown to be less toxic to E. coli strains than IPTG (isopropyl-β-d-thiogalactopyranoside) induction, which produced detectable protein within the membrane, and which conferred a resistant phenotype, as previously shown (39, 43). Agar spot dilution experiments were performed as described previously (44) with the following modifications: all overnight (16-h) LB broth cultures were standardized to an optical density at 600 nm of 1.5 units prior to being diluted 10−1 and 10−2 into fresh medium in a 96-well microtiter plate. One microliter from each diluted culture was spotted with a 48-pin replicator (Boekel Scientific, PA) onto LB-AMP agar plates containing a defined QCC concentration (Table 1). Culture spotting was randomized to account for any plate location bias on MIC determination. The plates were incubated for 16 h, and MICs were calculated as the lowest QCC concentration that resulted in a total lack of colony/spot formation. Experiments were repeated in triplicate (see the data set in Fig. S4), and the mean MICs from these studies are reported in Table S2.
Statistical analyses.
Statistical analyses were performed on MIC data derived from a 10−2 spot dilution since this dilution achieved significant and reproducible ≤±2-fold MIC differences between pEMR-11 and pMS119EH transformants. The QCC STAC was excluded from analysis since it failed to meet a minimum ±2-fold MIC cutoff. MIC values were used in hierarchical clustering analyses and were scaled according to the equation fold changeQCC = variantQCC/WTQCC, where fold changeQCC is the fold change in the MIC for the QCC, which is the ratio of the mean EmrE variant MIC value (variantQCC) divided by the mean WT EmrE MIC value (WTQCC) for the same QCC (Table S3). Hierarchical agglomerative clustering analysis was performed on the MIC data set using R statistics software (45) and the heatmap.plus package (46). Values listed on each dendrogram show the approximately unbiased (AU) and bootstrap probability (BP) P values obtained after 100 bootstrapped replicates using the R statistics pvclust package and function (47). k-means cluster analysis was used to estimate the significance of various distance and linkage metrics, and from this analysis, a Euclidean distance method using Ward linkage resulted in the most significant number of clusters. Fourfold changes in the MIC value were determined to be the most significant (P ≤ 0.001) when those values were compared to 2-fold changes in the MIC values (P ≤ 0.05) (Fig. S2), on the basis of MIC data obtained for EmrE E14D/E14A (Table S3); hence, the results for 11 QCCs were selected for final assessment (Fig. 1). Venn diagrams of correlated QCC chemicals or properties and EmrE variants were generated using the R statistics VennDiagram package and function (48), using the selection criteria described in the Fig. 3 legend.
TM helical wheel analysis.
Helical wheel projections derived from the highest resolved (3.8-Å) X-ray diffraction crystal structure of the EmrE dimer bound to TPP (PDB accession number 3B5D) (6) were selected for EmrE variant mapping (Fig. 2). It is important to note that this model has insufficient resolution to precisely assign residues; therefore, all conclusions drawn from helical wheel projections are primarily interpreted from helical positioning. Helical wheel projections were assembled from assigned TM regions using the program DrawColi (version 1.0) (49).
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Taylor and L. El Houjeiri for their assistance in generating EmrE variants, M. Beketskaia and R. Pushpker for their technical assistance, and C. Slipski assistance with manuscript revisions.
This work was funded by a National Science and Engineering Research Council (NSERC) operating grant to R.J.T.
We all declare that we do not have any financial or scientific relationship that is considered a conflict of interest in any of the findings reported herein.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00461-18.
REFERENCES
- 1.Bay DC, Rommens KL, Turner RJ. 2008. Small multidrug resistance proteins: a multidrug transporter family that continues to grow. Biochim Biophys Acta 1778:1814–1838. doi: 10.1016/j.bbamem.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 2.Buffet-Bataillon S, Tattevin P, Bonnaure-Mallet M, Jolivet-Gougeon A. 2012. Emergence of resistance to antibacterial agents: the role of quaternary ammonium compounds—a critical review. Int J Antimicrob Agents 39:381–389. doi: 10.1016/j.ijantimicag.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 3.Hegstad K, Langsrud S, Lunestad BT, Scheie AA, Sunde M, Yazdankhah SP.. 2010. Does the wide use of quaternary ammonium compounds enhance the selection and spread of antimicrobial resistance and thus threaten our health? Microb Drug Resist 16:91–104. doi: 10.1089/mdr.2009.0120. [DOI] [PubMed] [Google Scholar]
- 4.Schuldiner S. 2007. When biochemistry meets structural biology: the cautionary tale of EmrE. Trends Biochem Sci 32:252–258. doi: 10.1016/j.tibs.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Schuldiner S. 2007. Controversy over EmrE structure. Science 317:748–751. doi: 10.1126/science.317.5839.748d. [DOI] [PubMed] [Google Scholar]
- 6.Chen YJ, Pornillos O, Lieu S, Ma C, Chen AP, Chang G. 2007. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci U S A 104:18999–19004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korkhov VM, Tate CG. 2009. An emerging consensus for the structure of EmrE. Acta Crystallogr D Biol Crystallogr 65:186–192. doi: 10.1107/S0907444908036640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrison EA, Dekoster GT, Dutta S, Vafabakhsh R, Clarkson MW, Bahl A, Kern D, Ha T, Henzler-Wildman KA. 2011. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature 481:45–52. doi: 10.1038/nature10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bay DC, Turner RJ. 2009. Diversity and evolution of the small multidrug resistance protein family. BMC Evol Biol 9:140. doi: 10.1186/1471-2148-9-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muth TR, Schuldiner S. 2000. A membrane-embedded glutamate is required for ligand binding to the multidrug transporter EmrE. EMBO J 19:234–240. doi: 10.1093/emboj/19.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yerushalmi H, Schuldiner S. 2000. A common binding site for substrates and protons in EmrE, an ion-coupled multidrug transporter. FEBS Lett 476:93–97. doi: 10.1016/S0014-5793(00)01677-X. [DOI] [PubMed] [Google Scholar]
- 12.Lytvynenko I, Brill S, Oswald C, Pos KM. 2016. Molecular basis of polyspecificity of the small multidrug resistance efflux pump AbeS from Acinetobacter baumannii. J Mol Biol 428:644–657. doi: 10.1016/j.jmb.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Brill S, Sade-Falk O, Elbaz-Alon Y, Schuldiner S. 2015. Specificity determinants in small multidrug transporters. J Mol Biol 427:468–477. doi: 10.1016/j.jmb.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 14.Brill S, Falk OS, Schuldiner S. 2012. Transforming a drug/H+ antiporter into a polyamine importer by a single mutation. Proc Natl Acad Sci U S A 109:16894–16899. doi: 10.1073/pnas.1211831109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Son MS, Del Castilho C, Duncalf KA, Carney D, Weiner JH, Turner RJ. 2003. Mutagenesis of SugE, a small multidrug resistance protein. Biochem Biophys Res Commun 312:914–921. doi: 10.1016/j.bbrc.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 16.Amadi ST, Koteiche HA, Mishra S, McHaourab HS. 2010. Structure, dynamics, and substrate-induced conformational changes of the multidrug transporter EmrE in liposomes. J Biol Chem 285:26710–26718. doi: 10.1074/jbc.M110.132621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qazi SJS, Chew R, Bay DC, Turner RJ. 2015. Structural and functional comparison of hexahistidine tagged and untagged forms of small multidrug resistance protein, EmrE. Biochem Biophys Rep 1:22–32. doi: 10.1016/j.bbrep.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodall NB, Yin Y, Bowie JU. 2015. Dual-topology insertion of a dual-topology membrane protein. Nat Commun 6:8099. doi: 10.1038/ncomms9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yerushalmi H, Schuldiner S. 2000. An essential glutamyl residue in EmrE, a multidrug antiporter from Escherichia coli. J Biol Chem 275:5264–5269. doi: 10.1074/jbc.275.8.5264. [DOI] [PubMed] [Google Scholar]
- 20.Robinson AE, Thomas NE, Morrison EA, Balthazor BM, Henzler-Wildman KA. 2017. New free-exchange model of EmrE transport. Proc Natl Acad Sci U S A 114:E10083–E10091. doi: 10.1073/pnas.1708671114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrison EA, Henzler-Wildman KA. 2014. Transported substrate determines exchange rate in the multidrug resistance transporter EmrE. J Biol Chem 289:6825–6836. doi: 10.1074/jbc.M113.535328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soskine M, Adam Y, Schuldiner S. 2004. Direct evidence for substrate-induced proton release in detergent-solubilized EmrE, a multidrug transporter. J Biol Chem 279:9951–9955. doi: 10.1074/jbc.M312853200. [DOI] [PubMed] [Google Scholar]
- 23.Korkhov VM, Tate CG. 2008. Electron crystallography reveals plasticity within the drug binding site of the small multidrug transporter EmrE. J Mol Biol 377:1094–1103. doi: 10.1016/j.jmb.2008.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ong YS, Lakatos A, Becker-Baldus J, Pos KM, Glaubitz C. 2013. Detecting substrates bound to the secondary multidrug efflux pump EmrE by DNP-enhanced solid-state NMR. J Am Chem Soc 135:15754–15762. doi: 10.1021/ja402605s. [DOI] [PubMed] [Google Scholar]
- 25.Winstone TL, Duncalf KA, Turner RJ. 2002. Optimization of expression and the purification by organic extraction of the integral membrane protein EmrE. Protein Expr Purif 26:111–121. doi: 10.1016/S1046-5928(02)00525-9. [DOI] [PubMed] [Google Scholar]
- 26.Elbaz Y, Steiner-Mordoch S, Danieli T, Schuldiner S. 2004. In vitro synthesis of fully functional EmrE, a multidrug transporter, and study of its oligomeric state. Proc Natl Acad Sci U S A 101:1519–1524. doi: 10.1073/pnas.0306533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soskine M, Steiner-Mordoch S, Schuldiner S. 2002. Crosslinking of membrane-embedded cysteines reveals contact points in the EmrE oligomer. Proc Natl Acad Sci U S A 99:12043–12048. doi: 10.1073/pnas.192392899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dutta S, Morrison EA, Henzler-Wildman KA. 2014. Blocking dynamics of the SMR transporter EmrE impairs efflux activity. Biophys J 107:613–620. doi: 10.1016/j.bpj.2014.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mordoch SS, Granot D, Lebendiker M, Schuldiner S. 1999. Scanning cysteine accessibility of EmrE, an H+-coupled multidrug transporter from Escherichia coli, reveals a hydrophobic pathway for solutes. J Biol Chem 274:19480–19486. doi: 10.1074/jbc.274.27.19480. [DOI] [PubMed] [Google Scholar]
- 30.Morrison EA, Robinson AE, Liu Y, Henzler-Wildman KA. 2015. Asymmetric protonation of EmrE. J Gen Physiol 146:445–461. doi: 10.1085/jgp.201511404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rapp M, Drew D, Daley DO, Nilsson J, Carvalho T, Melen K, De Gier JW, Von Heijne G. 2004. Experimentally based topology models for E. coli inner membrane proteins. Protein Sci 13:937–945. doi: 10.1110/ps.03553804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ninio S, Schuldiner S. 2003. Characterization of an archaeal multidrug transporter with a unique amino acid composition. J Biol Chem 278:12000–12005. doi: 10.1074/jbc.M213119200. [DOI] [PubMed] [Google Scholar]
- 33.Elbaz Y, Tayer N, Steinfels E, Steiner-Mordoch S, Schuldiner S. 2005. Substrate-induced tryptophan fluorescence changes in EmrE, the smallest ion-coupled multidrug transporter. Biochemistry 44:7369–7377. doi: 10.1021/bi050356t. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Rath A, Deber CM. 2014. Functional response of the small multidrug resistance protein EmrE to mutations in transmembrane helix 2. FEBS Lett 588:3720–3725. doi: 10.1016/j.febslet.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 35.Adam Y, Tayer N, Rotem D, Schreiber G, Schuldiner S. 2007. The fast release of sticky protons: kinetics of substrate binding and proton release in a multidrug transporter. Proc Natl Acad Sci U S A 104:17989–17994. doi: 10.1073/pnas.0704425104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elbaz Y, Salomon T, Schuldiner S. 2008. Identification of a glycine motif required for packing in EmrE, a multidrug transporter from Escherichia coli. J Biol Chem 283:12276–12283. doi: 10.1074/jbc.M710338200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tal N, Schuldiner S. 2009. A coordinated network of transporters with overlapping specificities provides a robust survival strategy. Proc Natl Acad Sci U S A 106:9051–9056. doi: 10.1073/pnas.0902400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu H, Naismith JH. 2008. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 8:91. doi: 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bay DC, Turner RJ. 2012. Small multidrug resistance protein EmrE reduces host pH and osmotic tolerance to metabolic quaternary cation osmoprotectants. J Bacteriol 194:5941–5948. doi: 10.1128/JB.00666-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dunten RL, Sahin-Toth M, Kaback HR. 1993. Cysteine scanning mutagenesis of putative helix XI in the lactose permease of Escherichia coli. Biochemistry 32:12644–12650. [DOI] [PubMed] [Google Scholar]
- 41.Frillingos S, Sahin-Toth M, Wu J, Kaback HR. 1998. Cys-scanning mutagenesis: a novel approach to structure function relationships in polytopic membrane proteins. FASEB J 12:1281–1299. doi: 10.1096/fasebj.12.13.1281. [DOI] [PubMed] [Google Scholar]
- 42.Morrison KL, Weiss GA. 2001. Combinatorial alanine-scanning. Curr Opin Chem Biol 5:302–307. doi: 10.1016/S1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- 43.Beketskaia MS, Bay DC, Turner RJ. 2014. Outer membrane protein OmpW participates with small multidrug resistance protein member EmrE in quaternary cationic compound efflux. J Bacteriol 196:1908–1914. doi: 10.1128/JB.01483-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiegand I, Hilpert K, Hancock REW. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc 3:163–175. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- 45.R Development Core Team. 2008. R: a language and environment for statistical computing. The Comprehensive R Archive Network (CRAN), R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 46.Day A. 2012. heatmap.plus.package: heatmap with more sensible behavior 1.3. The Comprehensive R Archive Network (CRAN), R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 47.Suzuki R. 2014. Package “pvclust”: hierarchical clustering with P-values via multiscale bootstrap resampling 1.3-2. The Comprehensive R Archive Network (CRAN), R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 48.Chen H. 2014. Generate high-resolution Venn and Euler plots: package “VennDiagram” 1.6.9. The Comprehensive R Archive Network (CRAN), R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 49.Grigoryan G, Keating AE. 2008. Structural specificity in coiled-coil interactions. Curr Opin Struct Biol 18:477–483. doi: 10.1016/j.sbi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gutman N, Steiner-Mordoch S, Schuldiner S. 2003. An amino acid cluster around the essential Glu-14 is part of the substrate- and proton-binding domain of EmrE, a multidrug transporter from Escherichia coli. J Biol Chem 278:16082–16087. doi: 10.1074/jbc.M213120200. [DOI] [PubMed] [Google Scholar]
- 51.Rotem D, Steiner-Mordoch S, Schuldiner S. 2006. Identification of tyrosine residues critical for the function of an ion-coupled multidrug transporter. J Biol Chem 281:18715–18722. doi: 10.1074/jbc.M602088200. [DOI] [PubMed] [Google Scholar]
- 52.Sharoni M, Steiner-Mordoch S, Schuldiner S. 2005. Exploring the binding domain of EmrE, the smallest multidrug transporter. J Biol Chem 280:32849–32855. doi: 10.1074/jbc.M504910200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.