

CTX-M is the most prevalent family of extended-spectrum β-lactamases. We recently developed a tetrazole-derived noncovalent inhibitor of CTX-M-9.
KEYWORDS: antibacterial, β-lactamase, inhibitor
ABSTRACT
CTX-M is the most prevalent family of extended-spectrum β-lactamases. We recently developed a tetrazole-derived noncovalent inhibitor of CTX-M-9. Here, we present the biochemical and microbiological activity of this inhibitor across a representative panel of serine β-lactamases and Gram-negative bacteria. The compound displayed significant activity against all major subgroups of CTX-M, including CTX-M-15, while it exhibited some low-level inhibition of other serine β-lactamases. Complex crystal structures with the CTX-M-14 S237A mutant and CTX-M-27 illustrate the binding contribution of specific active-site residues on the β3 strand. In vitro pharmacokinetic studies revealed drug-like properties and positive prospects for further optimization. These studies suggest that tetrazole-based compounds can provide novel chemotypes for future serine β-lactamase inhibitor discovery.
INTRODUCTION
The production of β-lactamases is one of the main mechanisms of resistance to β-lactam antibiotics, particularly in Gram-negative bacteria (1). These enzymes hydrolyze and deactivate β-lactam compounds, which are covalent inhibitors of penicillin-binding proteins (PBPs), essential for bacterial cell wall synthesis (2). On the basis of sequence similarity and the catalytic mechanism, there are four classes of β-lactamases. Classes A, C, and D utilize a serine residue to catalyze the hydrolysis reaction, whereas class B β-lactamases are zinc-based metalloenzymes (1). Classes A and C are the most frequently observed in the clinic (3), whereas class B β-lactamases, such as NDM-1, and class D enzymes, such as OXA-48, have also emerged as serious health threats (4–6).
CTX-M class A β-lactamases are the most common extended-spectrum β-lactamases (ESBLs) with enhanced activity against third-generation cephalosporins, such as cefotaxime (7, 8). There are at least six subgroups of CTX-M enzymes: CTX-M-1, CTX-M-2, CTX-M-8, CTX-M-9, CTX-M-25, and KLUC (9). Among these, subgroups CTX-M-1 (particularly member CTX-M-15) and CTX-M-9 (including CTX-M-14 and CTX-M-27) are the most clinically prevalent (10). In an effort to combat the bacterial resistance caused by CTX-M ESBLs, we have recently applied a structure-based fragment approach to develop an aryl tetrazole-based noncovalent inhibitor of CTX-M-9 displaying a Ki of 89 nM (compound 1; Fig. 1) (11, 12). Although the inhibitor was optimized initially for CTX-M-9, the similarities shared by the active sites of CTX-M enzymes and other serine β-lactamases suggest that this compound may inhibit other clinically important β-lactamases. In addition, the contribution of active-site residues to inhibitor binding, particularly that involving several side chains displaying variations among different CTX-M enzymes and other serine β-lactamases, such as residues 237 and 240, was not well understood (13). To evaluate the potential of the aryl tetrazole scaffold for future development of β-lactamase inhibitors, particularly inhibitors of CTX-M enzymes, we have tested the activity of compound against a series of β-lactamases through biochemical, microbiological, and structural analyses, together with pharmacokinetic (PK) assays.
FIG 1.
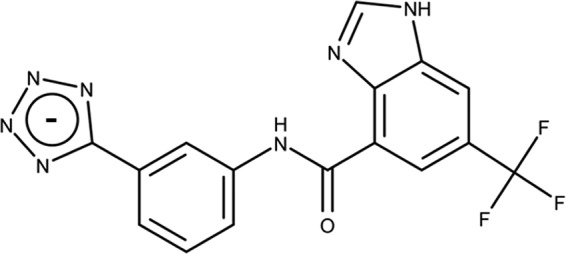
Chemical structure of an aryl tetrazole-based inhibitor of CTX-M-9 (compound 1).
RESULTS AND DISCUSSION
The apparent Ki values against purified enzymes were analyzed using nitrocefin hydrolysis assays. The biochemical testing included four clinically isolated CTX-M enzymes (three from the CTX-M-9 subgroup and CTX-M-15) and one CTX-M-14 S237A mutant. The active-site residues are highly conserved among CTX-M enzymes, with the one notable exception of residue 240, which is an aspartate in CTX-M-9 and CTX-M-14 and a glycine in CTX-M-15 and CTX-M-27 (14). Ser237, conserved in CTX-M enzymes and important for hydrogen bonding with compound 1, also displays some variation among class A β-lactamases and is replaced by an alanine in some narrow-spectrum β-lactamases, such as TEM-1 (13, 15). Compound 1 showed similar binding affinities to CTX-M-9 and CTX-M-14, as CTX-M-9 differs from CTX-M-14 only by a single substitution outside the active site (V231A). CTX-M-27 also differs from CTX-M-14 at a single residue (D240G). Even though CTX-M-15 and CTX-M-27 belong to two different subgroups, their active-site residues are identical, which is reflected by their similar binding affinities for compound 1 (Table 1). The D240G substitution decreased ligand binding by ∼3-fold. Meanwhile, the S237A mutation reduced the binding affinity more significantly by more than 10-fold, suggesting an important contribution to ligand binding.
TABLE 1.
Inhibition of serine β-lactamases in biochemical assays
| β-Lactamase | Ki (μM) |
|---|---|
| CTX-M-9 | 0.089a |
| CTX-M-14 | 0.085 ± 0.02 |
| CTX-M-27 | 0.28 ± 0.09 |
| CTX-M-15 | 0.25 ± 0.02 |
| CTX-M-14 S237A | 1.23 ± 0.27 |
| TEM-1 | No inbb |
| SHV-2 | 25.8 ± 1.9 |
| KPC-2 | 97.3 ± 1.7 |
| AmpC | 386.9 ± 43.3 |
| OXA-48 | 478.3 ± 166.6 |
As previously reported in reference 12.
No inb, no inhibition.
To understand the molecular interactions underlying the binding of the inhibitor to the different CTX-M enzymes, we determined complex crystal structures of compound 1 bound to the CTX-M-14 S237A mutant and CTX-M-27 (Fig. 2; see also Table S1 in the supplemental material). The compound binds the mutants in a fashion nearly identical to that for the CTX-M-14 wild-type (WT) enzyme, except that the hydrogen bond (HB) interactions with Ser237 and Asp240 in the CTX-M-14 WT are absent in the S237A mutant and CTX-M-27, respectively. The biochemical and structural results suggest that Ser237 plays an important role in inhibitor binding. It may play a similar role in enhancing interactions with substrates, particularly extended-spectrum β-lactam antibiotics, such as cefotaxime, as a serine or threonine residue is often observed at position 237 in class A enzymes with ESBL activity (13, 15). Meanwhile, the HB with Asp240 is less crucial. Previous experiments demonstrated that the replacement of a benzene ring by benzimidazole increases inhibitor binding for CTX-M-9 by 15-fold (12). Compared with the benzene ring, the benzimidazole moiety forms an additional HB with Asp240 and amide-π stacking interactions with the peptide backbone surrounding Gly238 on the β3 strand (Fig. 2). These new results suggest that the amide-π stacking interactions may have contributed more to the increase in binding affinity than the HB with Asp240, highlighting the π surfaces of the β3 strand amide bonds as a binding hot spot in CTX-M.
FIG 2.

Complex crystal structures of compound 1. (A and B) Compound 1 in the active sites of CTX-M-14 S237A (A) and CTX-M-27 (B). The mFo – DFc omit electron density map (green) and 2mFo – DFc omit electron density map (blue) are contoured at 2 and 1.5 σ, respectively. Hydrogen bonds are shown as black dashes. (C and D) Schematic diagram of protein-inhibitor interactions of compound 1 with CTX-M-14 S237A (C) and CTX-M-27 (D). The figures were generated using PyMOL and Discovery Studio Visualizer software.
In addition to CTX-M enzymes, compound 1 displayed inhibition of several other serine β-lactamases from all three classes, most notably, SHV-2, a class A enzyme that also has ESBL activity (1). However, we observed no inhibition of TEM-1, a narrow-spectrum β-lactamase. This is likely due to the steric clashes with Glu240 in TEM-1, as the benzimidazole group of compound 1 was engineered to form a HB with the smaller side chain of Asp240 in CTX-M-9. In comparison, the end of the β3 strand shifts away from the active site in SHV-2, allowing its Glu240 to avoid such steric clashes. However, these changes in the β3 strand reduce both nonpolar and HB interactions between SHV-2 and the inhibitor. Similarly, in KPC-2, a significant protrusion in the β3 strand, caused by the Cys69-Cys238 disulfide bond, can also affect the interactions with the benzimidazole group of compound 1. In addition, in comparison to Asn104 and Ser237 in CTX-M enzymes, other class A β-lactamases lack similar side chains at one or both of these positions, abolishing favorable HB contacts with the inhibitor and resulting in lower binding affinities (13). Taken together, these results suggest that this scaffold may be suitable for engineering cross-class inhibitors of serine β-lactamases, although additional design and synthesis will be needed to target conserved binding hot spots in different classes of β-lactamases.
We also tested the ability of compound 1 to synergize with cefotaxime across a panel of Gram-negative clinical strains expressing various β-lactamases (Table 2). The MICs were determined by the broth microdilution method according to Clinical and Laboratory Standards Institute (CLSI) recommendations, including quality control testing with Escherichia coli strain ATCC 25922 against cefotaxime (MIC value, 0.06 μg/ml). The most significant effect, an 8-fold reduction in the cefotaxime MIC, was observed in E. coli strains expressing CTX-M-9 and CTX-M-14, consistent with the high affinity of compound 1 for these two enzymes. A similar decrease in MIC was also achieved for Klebsiella pneumoniae expressing CTX-M-2, whose active-site residues are identical to those of CTX-M-9. This observation may reflect similarities in the permeability of compound 1 in E. coli and K. pneumoniae. In comparison, a 4-fold reduction was observed for E. coli expressing CTX-M-15, consistent with the slightly lower affinity of compound 1. Aside from the binding affinity of compound 1, other factors, such as protein expression level, might have also played a role in influencing the cell-based activity against bacteria expressing CTX-M-1 and particularly against those expressing CTX-M-8, both of which share the same active-site residues as CTX-M-9. In addition, the compound showed minimal (2-fold decrease) to no effect against other non-CTX-M β-lactamases, some of which were weakly inhibited by the inhibitor in vitro. Not surprisingly, clavulanate, a covalent inhibitor of class A β-lactamases with apparent Kis of ∼0.02 to 40 μM, was more active in reducing the MIC of cefotaxime in the control experiments (Table 2) (16). Overall, these results suggest that compound 1 can inhibit serine β-lactamases, particularly CTX-M enzymes, inside bacteria.
TABLE 2.
Reduction of cefotaxime MIC in inhibiting bacterial growtha
| β-Lactamase | CTX MIC (μg/ml) | CTX + Cpd1 MIC (μg/ml) | Cpd1 MIC reduction (μg/ml) | CTX + Cla MIC (μg/ml) | Cla MIC reduction (μg/ml) |
|---|---|---|---|---|---|
| E. coli CTX-M-1 | 256–128 | 64–32 | 4 | 0.25 | 1,024–512 |
| E. coli CTX-M-15 | 64 | 16 | 4 | 0.12 | 512 |
| K. pneumoniae CTX-M-2 | 512–256 | 64–32 | 8 | 0.5 | 1,024–512 |
| E. coli CTX-M-8 | 32–16 | 16–8 | 2 | 0.06 | 512–256 |
| E. coli CTX-M-9 | 128 | 16 | 8 | 0.12 | 1,024 |
| E. coli CTX-M-14 | 64 | 8 | 8 | 0.06 | 1,024 |
| E. coli SHV-5 | 16 | 16 | 0.06 | 256 | |
| E. coli TEM-24 | 4 | 2 | 2 | 0.06 | 64 |
| E. coli AmpC | 8 | 4 | 2 | 8 | |
| E. coli OXA-48 | 0.5 | 0.25 | 2 | 0.5 | |
| E. coli NDM-1 | 32 | 32 | 32 | ||
| K. pneumoniae KPC-2 | 16 | 16 | 0.5 | 32 |
CTX, cefotaxime; Cpd1, compound 1; Cla, clavulanate. Cefotaxime plus compound 1 and cefotaxime plus clavulanate were used at a 1:1 ratio.
The drug-like properties of compound 1 were evaluated using in vitro absorption, distribution, metabolism, and excretion (ADME) assays (Table 3). Compound 1 exhibited low predicted intrinsic clearance (CLint) in both rat and human, on the basis of in vitro liver microsome stability assays, and displayed excellent kinetic solubility but low permeability (A to B) in an MDCK cell monolayer assay. Importantly, compound 1 did not measurably inhibit the hERG channel at 10 μM in an in vitro assay, implying a low risk of hERG-mediated cardiotoxicity. Mammalian cytotoxicity studies with HEK293 cells were also carried out on compound 1 (data not shown). Compound 1 was incubated with HEK293 cells for 48 h at various concentrations (0 to 100 μg/ml). The results demonstrated that compound 1 had no deleterious effect on cell survival even at 100 μg/ml. To control for the potential effect of serum protein in the toxicity studies, we determined the MIC of cefotaxime using E. coli BL21(DE3) cells expressing the CTX-M-14 WT in the presence of compound 1 (10 μg/ml) and 10% heat-inactivated fetal bovine serum (FBS). The inhibitor reduced the MIC value of cefotaxime by 16-fold and 8-fold in the absence and presence of FBS, respectively. In addition, FBS did not have any effect on the MIC of cefotaxime alone without the inhibitor. These results suggest that the majority of compound 1 was not sequestered by serum protein in the cell culture medium and thus was able to exert its biological effect.
TABLE 3.
In vitro ADME profile of compound 1
| Parameter | Value for compound 1 |
|---|---|
| Human microsome stability | CLinta < 9.6 ml/min/kg |
| Rat microsome stability | CLint = 21.2 ml/min/kg |
| Kinetic aqueous solubility | 195 μM |
| MDCK cell permeability (A to B) | Pappb = 1.27 × 10−6 cm/s |
| hERG IC50 | >10 μM |
CLint = CL · 45 mg microsome/g liver · g liver weight/kg body weight.
Papp, apparent permeability.
On the basis of these in vitro data, a PK study was performed in male BALB/c mice with a single intravenous (i.v.) dose of compound 1 at 10 mg/kg of body weight (Fig. 3). In this study, compound 1 exhibited a low plasma clearance value of 12.9 ml/min/kg, which is ∼15% of the rate of hepatic blood flow in mice. The elimination half-life was 2.94 h, and the volume of distribution at steady state (Vss) was 0.38 liter/kg, less than the normal volume of total body water (0.7 liter/kg). The area under the concentration-time curve (AUC) was 12,748 ng · h/ml. Overall, the in vitro ADME and PK data indicated favorable drug-like properties for compound 1 and highlight the potential PK advantages that can be realized with nonelectrophilic, non-β-lactam-derived chemotypes. The PK/ADME data also revealed areas for improvement of the aryl tetrazole chemotype, such as improvement of the low membrane permeability, which could limit oral absorption.
FIG 3.
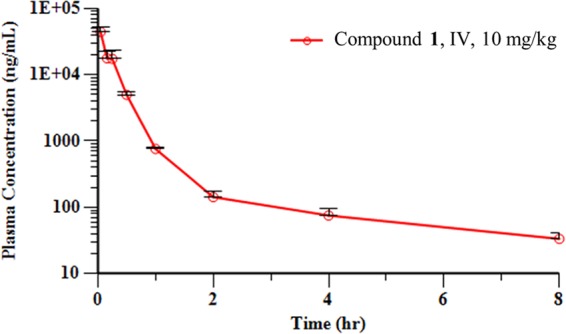
Mean plasma concentration-time profiles of compound 1 following a single intravenous (IV) administration (dose, 10 mg/kg) to male BALB/c mice.
In conclusion, the tetrazole-based inhibitor represents a promising novel scaffold for the development of inhibitors of serine β-lactamases, particularly the CTX-M family of ESBLs. These studies have also revealed important contributions of residues on the β3 strand for future efforts to discover drugs with activity against these enzymes.
MATERIALS AND METHODS
Construct designs.
Genes encoding CTX-M-9, CTX-M-14, and CTX-M-27 were cloned into the pET-9a vector. The CTX-M-14 S237A mutant was generated by site-directed mutagenesis using CTX-M-14 as a template and cloned into the pET-9a vector. The gene encoding CTX-M-15 was custom synthesized (DNA 2.0/ATUM) and cloned into the pJ411 vector. The genes encoding KPC-2, E. coli AmpC, SHV-2, and TEM-1 were cloned into the pET-GST vector modified with an N-terminal 6× His–glutathione S-transferase (GST) tag. The gene encoding OXA-48 was custom synthesized (DNA 2.0/ATUM) and cloned into the pD441-NH vector with an N-terminal 12× His tag. All the constructs were transformed into NEB 5-alpha competent E. coli cells (New England BioLabs) and plated onto LB agar containing 50 μg/ml kanamycin. Single colonies were isolated and grown overnight at 37°C in LB medium containing 50 μg/ml kanamycin. Cells were harvested, and plasmid DNA was obtained using a miniprep kit. The gene sequences of all the β-lactamases were verified.
Expression and purification.
All the β-lactamase-encoding genes were transformed into E. coli BL21(DE3) cells. Single colonies were grown overnight at 37°C in LB medium supplemented with 50 μg/ml kanamycin. The overnight culture was then diluted into 1 liter LB medium at 1:500 and incubated at 37°C until the optical density at 600 nm reached 0.6 to 0.8. Protein expression was initiated by the addition of 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside), and incubation was continued overnight at 20°C. The cells were harvested by centrifugation at 5,000 × g for 10 min. For CTX-M β-lactamases (12), the cell pellet was resuspended in buffer A (50 mM MES [morpholineethanesulfonic acid], pH 6.0, 2 mM EDTA). For E. coli AmpC, SHV-2, and TEM-1, the cell pellets were resuspended in buffer A (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 20 mM imidazole, 10% [vol/vol] glycerol). For KPC-2 (17) and OXA-48, the cell pellets were resuspended in buffer A (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 20 mM imidazole). After resuspension, cells were disrupted by sonication followed by ultracentrifugation at 35,000 rpm for 40 min to remove cellular debris. The supernatant containing CTX-M β-lactamases was loaded onto a CM Sepharose column. The proteins were eluted with a gradient of increasing NaCl concentrations. The supernatants containing KPC-2, E. coli AmpC, OXA-48, SHV-2, and TEM-1 were loaded onto a HisTrap affinity column. The proteins were eluted with a gradient of increasing imidazole concentrations. The fractions containing the β-lactamases were pooled and concentrated. All β-lactamase samples were further purified using a HiLoad 16/60 Superdex 75 column. The purity of the proteins was determined by SDS-PAGE to be >95%.
β-Lactamase inhibition assays.
The hydrolytic activities of the purified β-lactamases were measured using the β-lactamase substrate nitrocefin in 100 mM Tris-HCl, pH 7.0, 0.01% (vol/vol) Triton X-100, for all β-lactamases except OXA-48, which was measured in 100 mM Tris-H2SO4, pH 7.0, 50 mM NaHCO3, 0.01% (vol/vol) Triton X-100 (18). The absorbance at 486 nm was monitored using a BioTek Synergy microplate reader. The nitrocefin concentrations used in the assays were 40 μM for CTX-M β-lactamases, OXA-48, TEM-1, and E. coli AmpC and 20 μM for KPC-2 and SHV-2. The protein activity was measured in the presence of increasing amounts of compound 1. The 50% inhibitory concentrations (IC50s) were obtained from the sigmoidal concentration dependence curve generated using SigmaPlot software (version 12.5). The Ki values were calculated by the method described by Waley (19).
Determination of MIC values and compound 1 potentiation experiments.
The MICs were determined by the broth microdilution method according to the Clinical and Laboratory Standards Institute (CLSI) recommendations, including quality control testing with Escherichia coli strain ATCC 25922 (20). The potentiation of antibiotic activity by compound 1 in bacterial strains was performed as previously described (12).
Crystallization and structure determination.
Crystals of CTX-M-14 S237A were grown at 20°C using the hanging-drop vapor diffusion method. Protein solution (40 mg/ml) was mixed 1:2 (vol/vol) with a reservoir solution containing 1.0 M potassium phosphate, pH 8.3. Crystals of CTX-M-27 were grown at 20°C using the hanging-drop vapor diffusion method. Protein solution (22.8 mg/ml) was mixed 1:2 (vol/vol) with a reservoir solution containing 0.1 M sodium acetate, pH 8.3, and 1.6 M ammonium sulfate. Crystals of CTX-M-14 S237A were soaked for 48 h in 1.0 M potassium phosphate, pH 8.3, containing 2.5 mM compound 1 and then cryoprotected with 1.0 M potassium phosphate, pH 8.3, supplemented with 30% (wt/vol) sucrose before flash cooling with liquid nitrogen. Crystals of CTX-M-27 were soaked overnight in 0.1 M sodium acetate, pH 8.3, and 1.6 M ammonium sulfate containing 2.5 mM compound 1 and then cryoprotected with 0.1 M sodium acetate, pH 8.3, and 1.6 M ammonium sulfate supplemented with 30% (wt/vol) sucrose before flash cooling with liquid nitrogen. X-ray data for the CTX-M-14 S237A/compound 1 complex and CTX-M-27/compound 1 complex were collected using beamline 22-ID-D at the Advanced Photon Source (APS), Argonne, IL. Diffraction data were indexed and integrated with the iMOSFLM program (21) and scaled with the SCALA program (22) from the CCP4 suite (23). Phasing was performed using molecular replacement with the program Phaser (24) with the CTX-M-14 structure (PDB accession number 4UA6) (25) and CTX-M-27 structure (PDB accession number 1YLP) (26). Structure refinement was performed using the phenix.refine tool of the PHENIX software suite (27) and model building in the WinCoot program (28). The program eLBOW in PHENIX was used to obtain geometry restraint information for compound 1 (29). The mFo – DFc and 2mFo – DFc composite omit electron density maps (where Fo and Fc are the experimentally measured and model-based amplitudes, respectively, m is the figure of merit, and D is the sigma-A weighting factor) were generated with the composite omit map program of PHENIX (30). The final model qualities were assessed using the MolProbity web server (31). Figures were generated in PyMOL (version 2.0) software (Schrödinger). Discovery Studio Visualizer software was used to generate ligand-protein interactions.
Pharmacokinetic analysis of compound 1 in mice.
The mouse pharmacokinetic study was conducted at Sai Life Sciences Limited, Pune, India, in accordance with the Study Plan SAIDMPK/PK-13-12-449 and the guidelines of the Institutional Animal Ethics Committee (IAEC). Nine male BALB/c mice were weighed before dose administration and then dosed with the compound 1 solution formulation at 10 mg/kg of body weight. The dosing volume administered was 5 ml/kg for intravenous administration. Compound 1 was formulated as follows. An accurately weighed amount of 3.93 mg of compound 1 was taken into a labeled bottle; 0.098 ml of N-methyl-2-pyrrolidone, 0.098 ml of Solutol HS, and 1.768 ml of 20% (2-hydroxypropyl)-beta-cyclodextrin in water was added to the bottle. The bottle was vortexed for 2 min after each addition, and the final solution formulation was sonicated for 2 min to obtain a clear solution. Blood samples (approximately 60 μl) were collected from the retro-orbital plexus of three mice at predose and 0.048, 0.16, 0.25, 0.5, 1, 2, 4 and 8 h postdose. Samples were collected into labeled microtubes containing K2EDTA solution (20% K2EDTA solution) as an anticoagulant. Plasma was immediately harvested from the blood by centrifugation at 4,000 rpm for 10 min at 4 ± 2°C and stored at temperatures below −70°C until bioanalysis. The noncompartmental analysis module in Phoenix WinNonlin software (version 6.3) was used to assess the pharmacokinetic parameters. The areas under the concentration-time curves (the AUC from time zero to the last quantifiable concentration [AUClast] and the AUC from time zero to infinity [AUCinf]) were calculated by use of the linear trapezoidal rule. The terminal elimination rate constant (kel) was determined by regression analysis of the linear terminal portion of the log plasma concentration-time curve. The terminal half-life (t1/2) was estimated to be 0.693/kel, CLi.v. was equal to dose/AUCinf (where CLi.v. is clearance after i.v. administration), and Vss was equal to MRT × CLi.v. (where MRT is the mean residence time).
HEK293 cell cytotoxicity assays.
The cytotoxicity assays were performed using human embryonic kidney (HEK293) cells, and the methods were performed as previously described (32). Controls with solvent only were used as a baseline to compare treatments, and the results are represented as percent recovery.
Accession number(s).
The PDB accession numbers for the complex crystal structures of compound 1 bound to the CTX-M-14 S237A mutant and CTX-M-27 are 6BT6 and 6BU3, respectively.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH (AI103158).
For disclosure of competing financial interests, D.A.N., A.R.R., and Y.C. are listed as inventors on a published patent application describing compound 1 and analogs (WO2013103760 A1).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02563-17.
REFERENCES
- 1.Bush K, Jacoby GA. 2010. Updated functional classification of β-lactamases. Antimicrob Agents Chemother 54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang YH, Labgold MR, Richards JH. 1990. Altering enzymatic activity: recruitment of carboxypeptidase activity into an RTEM β-lactamase/penicillin-binding protein 5 chimera. Proc Natl Acad Sci U S A 87:2823–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Powers RA. 2016. Structural and functional aspects of extended-spectrum AmpC cephalosporinases. Curr Drug Targets 17:1051–1060. doi: 10.2174/1573399811666150615144707. [DOI] [PubMed] [Google Scholar]
- 4.Khan AU, Maryam L, Zarrilli R. 2017. Structure, genetics and worldwide spread of New Delhi metallo-β-lactamase (NDM): a threat to public health. BMC Microbiol 17:101. doi: 10.1186/s12866-017-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antunes NT, Lamoureaux TL, Toth M, Stewart NK, Frase H, Vakulenko SB. 2014. Class D β-lactamases: are they all carbapenemases? Antimicrob Agents Chemother 58:2119–2125. doi: 10.1128/AAC.02522-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacoby GA, Munoz-Price LS. 2005. The new β-lactamases. N Engl J Med 352:380–391. doi: 10.1056/NEJMra041359. [DOI] [PubMed] [Google Scholar]
- 7.Rossolini GM, D'Andrea MM, Mugnaioli C. 2008. The spread of CTX-M-type extended-spectrum β-lactamases. Clin Microbiol Infect 14(Suppl 1):S33–S41. [DOI] [PubMed] [Google Scholar]
- 8.Paterson DL, Hujer KM, Hujer AM, Yeiser B, Bonomo MD, Rice LB, Bonomo RA, International Klebsiella Study Group. 2003. Extended-spectrum β-lactamases in Klebsiella pneumoniae bloodstream isolates from seven countries: dominance and widespread prevalence of SHV- and CTX-M-type β-lactamases. Antimicrob Agents Chemother 47:3554–3560. doi: 10.1128/AAC.47.11.3554-3560.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonnet R. 2004. Growing group of extended-spectrum β-lactamases: the CTX-M enzymes. Antimicrob Agents Chemother 48:1–14. doi: 10.1128/AAC.48.1.1-14.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canton R, Gonzalez-Alba JM, Galan JC. 2012. CTX-M enzymes: origin and diffusion. Front Microbiol 3:110. doi: 10.3389/fmicb.2012.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Shoichet BK. 2009. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nat Chem Biol 5:358–364. doi: 10.1038/nchembio.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nichols DA, Jaishankar P, Larson W, Smith E, Liu G, Beyrouthy R, Bonnet R, Renslo AR, Chen Y. 2012. Structure-based design of potent and ligand-efficient inhibitors of CTX-M class A β-lactamase. J Med Chem 55:2163–2172. doi: 10.1021/jm2014138. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu-Ibuka A, Oishi M, Yamada S, Ishii Y, Mura K, Sakai H, Matsuzawa H. 2011. Roles of residues Cys69, Asn104, Phe160, Gly232, Ser237, and Asp240 in extended-spectrum β-lactamase Toho-1. Antimicrob Agents Chemother 55:284–290. doi: 10.1128/AAC.00098-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonnet R, Recule C, Baraduc R, Chanal C, Sirot D, De Champs C, Sirot J. 2003. Effect of D240G substitution in a novel ESBL CTX-M-27. J Antimicrob Chemother 52:29–35. doi: 10.1093/jac/dkg256. [DOI] [PubMed] [Google Scholar]
- 15.Adamski CJ, Cardenas AM, Brown NG, Horton LB, Sankaran B, Prasad BV, Gilbert HF, Palzkill T. 2015. Molecular basis for the catalytic specificity of the CTX-M extended-spectrum β-lactamases. Biochemistry 54:447–457. doi: 10.1021/bi501195g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hecker SJ, Reddy KR, Totrov M, Hirst GC, Lomovskaya O, Griffith DC, King P, Tsivkovski R, Sun D, Sabet M, Tarazi Z, Clifton MC, Atkins K, Raymond A, Potts KT, Abendroth J, Boyer SH, Loutit JS, Morgan EE, Durso S, Dudley MN. 2015. Discovery of a cyclic boronic acid β-lactamase inhibitor (RPX7009) with utility vs class A serine carbapenemases. J Med Chem 58:3682–3692. doi: 10.1021/acs.jmedchem.5b00127. [DOI] [PubMed] [Google Scholar]
- 17.Pemberton OA, Zhang X, Chen Y. 2017. Molecular basis of substrate recognition and product release by the Klebsiella pneumoniae carbapenemase (KPC-2). J Med Chem 60:3525–3530. doi: 10.1021/acs.jmedchem.7b00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chow C, Xu H, Blanchard JS. 2013. Kinetic characterization of hydrolysis of nitrocefin, cefoxitin, and meropenem by β-lactamase from Mycobacterium tuberculosis. Biochemistry 52:4097–4104. doi: 10.1021/bi400177y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waley SG. 1982. A quick method for the determination of inhibition constants. Biochem J 205:631–633. doi: 10.1042/bj2050631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balouiri M, Sadiki M, Ibnsouda SK. 2016. Methods for in vitro evaluating antimicrobial activity: a review. J Pharm Anal 6:71–79. doi: 10.1016/j.jpha.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 23.Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 24.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nichols DA, Hargis JC, Sanishvili R, Jaishankar P, Defrees K, Smith EW, Wang KK, Prati F, Renslo AR, Woodcock HL, Chen Y. 2015. Ligand-induced proton transfer and low-barrier hydrogen bond revealed by X-ray crystallography. J Am Chem Soc 137:8086–8095. doi: 10.1021/jacs.5b00749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Delmas J, Sirot J, Shoichet B, Bonnet R. 2005. Atomic resolution structures of CTX-M β-lactamases: extended spectrum activities from increased mobility and decreased stability. J Mol Biol 348:349–362. doi: 10.1016/j.jmb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 29.Moriarty NW, Grosse-Kunstleve RW, Adams PD. 2009. Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr 65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Adams PD, Read RJ, Zwart PH, Hung LW. 2008. Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias. Acta Crystallogr D Biol Crystallogr 64:515–524. doi: 10.1107/S0907444908004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB III, Snoeyink J, Richardson JS, Richardson DC. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fleeman R, Van Horn KS, Barber MM, Burda WN, Flanigan DL, Manetsch R, Shaw LN. 2017. Characterizing the antimicrobial activity of N2,N4-disubstituted quinazoline-2,4-diamines toward multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 61:e00059-17. doi: 10.1128/AAC.00059-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.