

Mycobacterium kansasii pulmonary infection is a global problem. Standard combination therapy consists of isoniazid at 300 mg/day, rifampin at 600 mg/day, and ethambutol at 15 mg/kg of body weight/day for 18 months.
KEYWORDS: hollow-fiber system model, pharmacokinetics/pharmacodynamics, efflux pumps, benchmarking model, standard therapy
ABSTRACT
Mycobacterium kansasii pulmonary infection is a global problem. Standard combination therapy consists of isoniazid at 300 mg/day, rifampin at 600 mg/day, and ethambutol at 15 mg/kg of body weight/day for 18 months. Coincubation of M. kansasii with different clofazimine concentrations over 7 days in test tubes resulted in a maximal kill (maximum effect [Emax]) of 2.03 log10 CFU/ml below the day 0 bacterial burden. The concentration associated with Emax was 110 times the MIC. Next, the effects of human-like concentration-time profiles of clofazimine human-equivalent doses ranging from 0 to 200 mg daily for 21 days were examined in the hollow-fiber model of intracellular M. kansasii (HFS-Mkn). On day 14, when the clofazimine microbial effect was maximal, the Emax was 2.57 log10 CFU/ml, while the dose associated with Emax was 100 mg/day. However, no dose killed M. kansasii to levels below the day 0 bacterial burden. Thus, the antimicrobial effect of clofazimine monotherapy in the HFS-Mkn was modest. Human-equivalent concentration-time profiles of standard combination therapy and doses were used as comparators in the HFS-Mkn. On day 14, standard therapy killed to a level 2.32 log10 CFU/ml below the day 0 bacterial burden. The effect of standard therapy was consistent with a biexponential decline, with kill rate constants of 1.85 per day (half-life = 0.37 days) and 0.06 per day (half-life = 12.76 days) (r2 > 0.99). This means that standard therapy would take 9.3 to 12 months to completely eliminate M. kansasii in the model, which is consistent with clinical observations. This observation for standard therapy means that the modest to poor effect of clofazimine on M. kansasii identified here is likely to be the same in the clinic.
INTRODUCTION
In the United States, the Mycobacterium kansasii disease incidence is 2.4 cases per 100,000 adults per year in the general population and M. kansasii disease is the third most common pulmonary mycobacterial disease after Mycobacterium avium complex (MAC) infection and tuberculosis; in the subset of patients with AIDS, the incidence is 647 cases per 100,000 persons per year (1). The U.S. M. kansasii epidemiological pattern is similar to that in South American countries, Australia, and some European countries, such as the Netherlands, Italy, and Greece, while in African countries, M. kansasii is actually a more common cause of disease than MAC (2, 3). Thus, M. kansasii pulmonary disease is of global importance and classified as a neglected disease. The current recommendations for the treatment of M. kansasii lung disease are a combination regimen of isoniazid (300 mg/day), rifampin (600 mg/day), and ethambutol (15 mg/kg of body weight/day) given daily for 18 months; this has been associated with a sustained sputum conversion rate of 80% (4, 5). Accompanying the modest culture conversion rate are high rates of morbidity and mortality, as patients with M. kansasii disease present with a clinical picture more similar to severe tuberculosis (6). Therefore, there is considerable interest to develop new drug regimens to shorten the duration and to improve the efficacy of M. kansasii chemotherapy.
Clofazimine is used to treat tuberculosis and leprosy and was recently examined for use for the treatment of Mycobacterium abscessus infection (7–9). In prior studies by Wallace et al. (10) and others (11), 90% of M. kansasii isolates had clofazimine MICs (MIC90) of ≤0.5 mg/liter. The dosing regimen for clofazimine in clinical practice has varied, ranging from 100 mg daily for tuberculosis to 300 mg given intermittently for leprosy and even dose reductions to 50 mg daily for infections cause by other nontuberculous mycobacteria in response to patient tolerability. A clofazimine 1.6-mg/kg daily dose achieves a peak serum level of about 0.24 to 0.62 mg/liter, which is above the MIC90 (12). In order to make practical use of these findings, the pharmacokinetic (PK)/pharmacodynamic (PD) parameters (maximum concentration [Cmax]/MIC, area under the concentration-time curve [AUC]/MIC, the percentage of the time that the concentration persists above the MIC [%TMIC]) of clofazimine in the treatment of M. kansasii disease need to be identified. In the study described here we used our hollow-fiber system model of intracellular M. kansasii (HFS-Mkn), described before in this journal, to perform clofazimine PK/PD studies (13). We also aimed to determine if efflux pump inhibitors could enhance clofazimine efficacy and reduce the emergence of resistance (14, 15), given the antibiotic resistance arrow of time (16). We benchmarked these findings against the performance of standard combination therapy in the HFS-Mkn.
RESULTS
The clofazimine MIC of the M. kansasii (ATCC 12478) laboratory strain obtained using broth microdilution was 0.06 mg/liter on two separate occasions. The clofazimine MIC obtained using the resazurin colorimetric assay was also 0.06 mg/liter. Thus, the MIC of the laboratory strain was on the lower end of the MIC distribution of clinical isolates reported in the literature (10).
Given the good MIC results, we coincubated eight clofazimine concentrations with M. kansasii in triplicate test tubes for 7 days, washed the bacteria to remove carryover drug, and cultured the bacteria on Middlebrook 7H10 agar with 10% oleic acid, albumin, dextrose, and catalase (OADC) supplement for determination of CFU counts. Inhibitory sigmoid maximum effect (Emax) model results are shown in Fig. 1. The relationship was described by the equation effect (number of log10 CFU per milliliter) = 9.16 − 6.01 · C0.48/(0.370.48 + C0.48) (r2 = 0.96), where maximal kill (Emax) was 6.01 log10 CFU/ml; C is the concentration; C50 is the concentration mediating 50% of Emax, which was 0.37 mg/liter (which is 6.2 times the MIC); and the Hill factor was 0.48. The concentration mediating 80% of Emax (C80), considered optimal, was calculated as 110 times the MIC, which is about 6.6 times the peak concentration of the 200-mg/day dose in patients. The day 0 bacterial burden was 5.18 ± 0.06 log10 CFU/ml, which means that clofazimine killed at a level of 2.03 log10 CFU/ml below the day 0 bacterial burden (stasis) in just 7 days.
FIG 1.
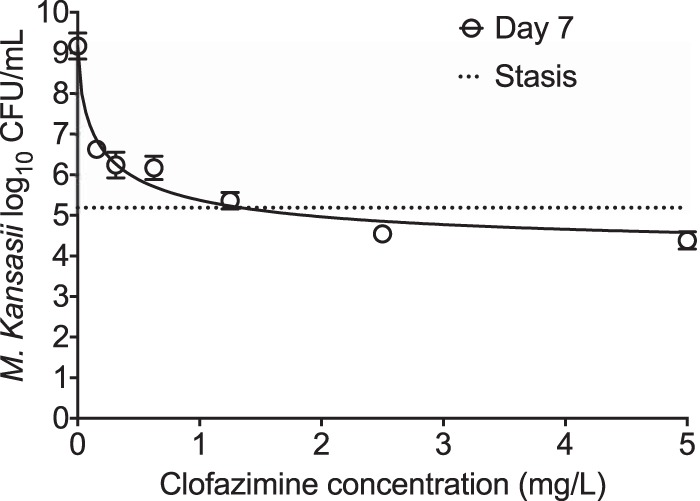
Clofazimine concentration-response. An inhibitory sigmoid Emax model was used to determine the relationship between the clofazimine concentration and the bacterial burden using GraphPad Prism software (v 7.0; GraphPad Software, La Jolla, CA, USA). Error bars indicate the standard deviations for triplicate test tubes. Different concentrations were coincubated with intracellular M. kansasii for 7 days. On day 7, the lowest clofazimine dose of 0.07 mg/liter showed a 3.60 ± 0.34-log10 CFU/ml kill of M. kansasii compared to that achieved for the nontreated controls on day 7. The two highest doses killed M. kansasii compared to the day 0 bacterial burden (5.18 log10 CFU/ml), or the stasis effect.
Next, we examined the efficacy of clofazimine in the intracellular HFS-Mkn. We mimicked and achieved a 12.5-h half-life of clofazimine, with the time to the maximum concentration (Tmax) equaling 6 h, identified in healthy volunteers during the first week of therapy (17, 18). Measurement of the clofazimine concentrations achieved in the HFS-Mkn revealed the PK/PD exposures shown in Table 1. A separate set of HFS-Mkn was treated with standard combination therapy at human-equivalent doses of isoniazid at 300 mg/daily and rifampin at 600 mg/daily, both at a half-life of 3 h, plus ethambutol at 15 mg/kg/day at a biphasic half-life of 3 h in the first 12 h and 8 h in the last 12 h. Figure 2 shows the time-kill curves of standard therapy and the different clofazimine doses in the HFS-Mkn. Standard therapy killed M. kansasii to levels below the day 0 bacterial burden (stasis). However, none of the clofazimine monotherapy doses killed enough bacteria to keep the bacterial burden below stasis, although they all performed better than no treatment in the nontreated controls. The maximal difference compared to the bacterial burden achieved with no treatment in the nontreated controls was on day 14.
TABLE 1.
Intended clofazimine dose and PK/PD exposures achieved in the HFS-Mkn
| Intended dose (mg/kg/day) | Cmax/MIC | AUC0–24/MIC | %TMIC |
|---|---|---|---|
| 0 | 0 | 0 | 0 |
| 12.5 | 0.20 | 2.08 | 0 |
| 25 | 0.40 | 4.17 | 0 |
| 50 | 0.80 | 8.33 | 0 |
| 75 | 1.25 | 12.50 | 25 |
| 100 | 1.60 | 16.67 | 33 |
| 200 | 3.30 | 33.33 | 63 |
FIG 2.
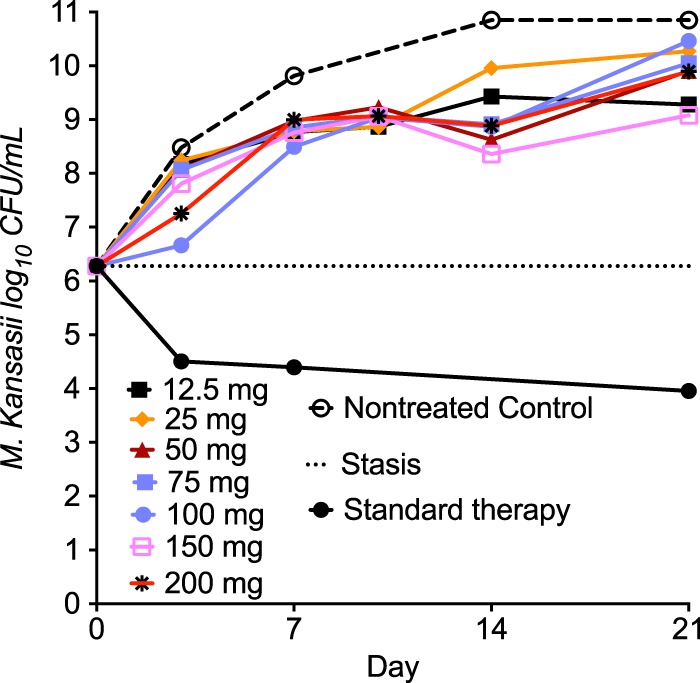
Clofazimine efficacy in the hollow-fiber model system. As opposed to the static concentration experiments in test tubes, none of the clofazimine doses were able to keep the bacterial burden below that at day 0 in the intracellular hollow-fiber system M. kansasii infection model. Thus, the static concentration studies overestimated the clofazimine efficacy; this is common to virtually all antibiotics tested in this system. On the other hand, the standard combination therapy performed relatively well during the 21-day experiment and killed to levels below stasis. GraphPad Prism software (v 7.0; GraphPad Software, La Jolla, CA, USA) was used for graphing.
Figure 3 shows the results obtained with the inhibitory sigmoid Emax models for each day of sampling. The r2 for the regression was 0.70 on day 3, 0.81 on day 7, and >0.99 on day 14; the regression on day 21 was poorer (r2 = 0.45). The Emax on day 14 was 1.97 log10 CFU/ml (95% confidence interval [CI], 1.87 to 2.09 log10 CFU/ml). Thus, efficacy with clofazimine monotherapy was modest, at best. It can be seen that the dose associated with 50% microbial kill (D50) and D80 differed on the basis of the sampling day; the highest 80% effective dose was 81 mg/day on day 3. Since the clofazimine capsule is scored as 50-mg tablets, this would be a dose of 100 mg/day (19). As such, we also compared the effect of the clofazimine dose of 100 mg/day to that of the same dose plus the efflux pump inhibitor reserpine. The results are shown in Fig. 4; the broad-spectrum efflux pump inhibitor did not improve the kill of M. kansasii by clofazimine in the HFS-Mkn.
FIG 3.
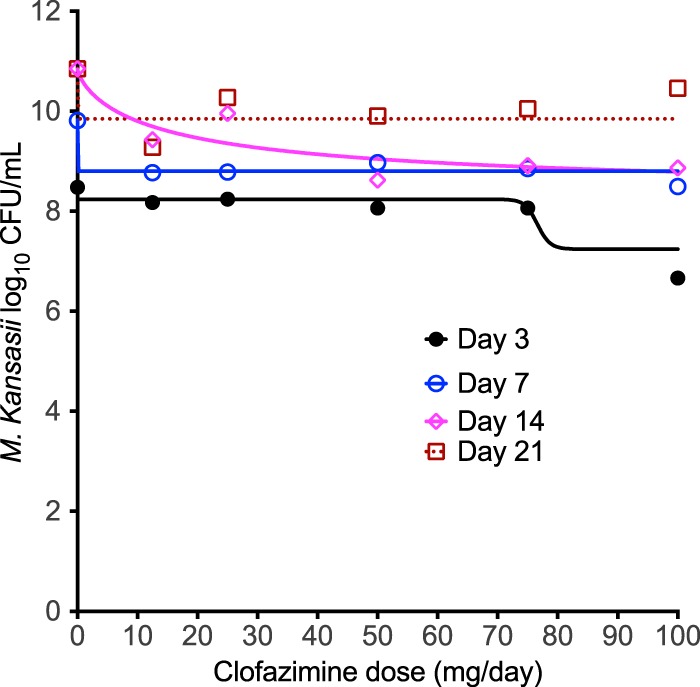
Clofazimine dose versus bacterial burden in the hollow-fiber system. Each color-coded line shows the inhibitory sigmoid Emax regression, determined with GraphPad Prism software (v 7.0; GraphPad Software, La Jolla, CA, USA), for a specific sampling day. There was one HFS-Mkn unit per dose. The best fit of the model to the data was on day 14 (magenta), and by day 21 (cayenne) it had begun to flatten out.
FIG 4.
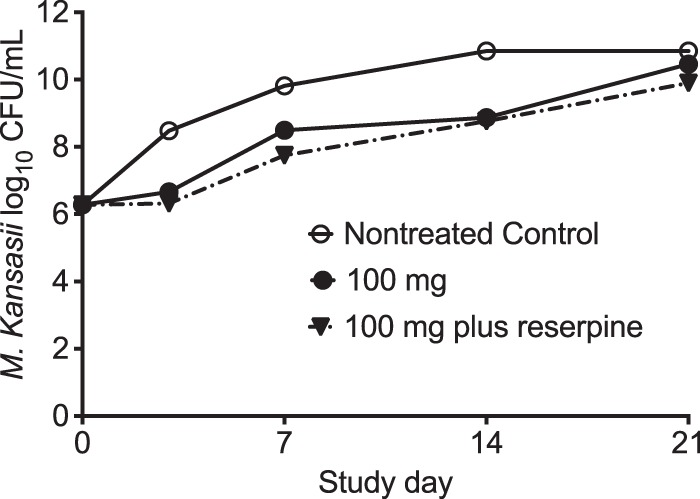
Effect of efflux pump blocker on clofazimine efficacy. The bacterial burden in HFS-Mkn treated with the human-equivalent clofazimine dose of 100 mg/day in combination with reserpine was 0.56-log10 CFU/ml lower than that achieved with clofazimine alone and 0.95-log10 CFU/ml lower than that achieved in the nontreated HFS-Mkn on day 21.
Figure 5 shows the effect of the standard combination therapy (positive control), with the number of CFU per milliliter being provided on a non-log scale, based on a biexponential decline model. For the exponential growth model in nontreated controls, the growth rate constant was 0.50 day−1 (95% CI, 0.49 to 0.51 day−1) and the doubling time was 1.38 days (95% CI, 1.37 to 1.40 days) (r2 = 0.99). Standard therapy microbial kill was described by an exponential decline rate constant of 1.61 ± 0.30 per day (which translates to a half-life of 0.43 days) with a bacterial burden at time zero of 1,895,501 CFU/ml, or 6.28 log10 CFU/ml, with an r2 value of >0.99 for the regression. However, on inspection the kill rate clearly plateaued off and could be broken into two curves, one with an exponential decline rate constant of 1.85 per day (half-life, 0.37 days), which is similar to the overall decline rate constant presented above, and a second terminal exponential decline rate constant of 0.06 per day (half-life, 12.76 days) with an r2 value of >0.99. With the latter rates, it would take 284 days (about 9.3 months) for the bacterial population to decline to 10−2 CFU/ml starting at this bacterial burden, which we have declined to bacterial population extinction elsewhere (37). If the starting bacterial burdens were increased 2.0 log10 CFU/ml, then this would take about 369 days, or just over 1 year.
FIG 5.
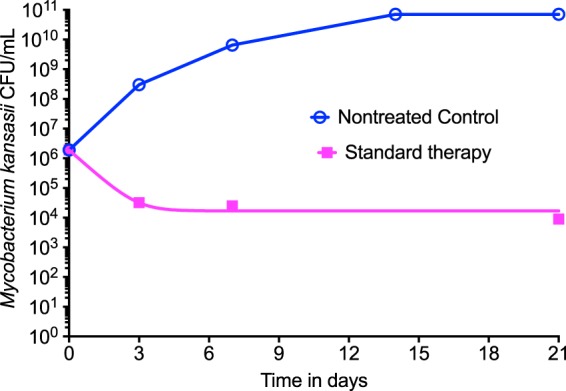
Performance of standard therapy. The standard therapy effect was compared to that of no treatment (nontreated control). Standard therapy showed a biexponential decline pattern on inspection: a quick 2-log10 CFU/ml drop followed by a slow decline in the terminal phase.
DISCUSSION
First, we show that clofazimine had efficacy against M. kansasii in test tubes and killed >2.0 log10 CFU/ml in just 7 days. However, in the HFS-Mkn, clofazimine monotherapy failed to keep the bacterial burden below stasis up to 21 days. It could be argued that we used a relatively short half-life of clofazimine of 12.5 h compared to that of up to 70 days in patients at steady state, hence providing a lower area under the concentration-time curve from 0 to 24 h (AUC0–24) and the percentage of the time that the concentration persisted above the MIC (%TMIC), which could therefore account for the lower kill rates. However, several doses achieved a tested %TMIC of 100%, while the peak concentration did not differ between test tubes and HFS-Mkn. Second, there is a massive accumulation of clofazimine inside macrophages and monocytes (20, 21), such that several hundred-fold higher concentrations would have been achieved in HFS-Mkn than in test tubes. Third, and incontrovertible, the inhibitory sigmoid Emax model achieved good convergence, especially on day 14, and thus, our results show that microbial kill reached maximal kill (Emax) with the exposure-effect strategy that we utilized, so that a longer half-life (greater AUC0–24/MIC or %TMIC) would not have added any more kill since a drug cannot kill at levels higher than its Emax, by definition. Thus, the relatively poor performance of clofazimine is likely due to the poor kill of intracellular M. kansasii.
We also show that addition of the efflux pump blocker reserpine did not improve the efficacy of clofazimine against M. kansasii. These findings are in contrast to those of our previous work in other mycobacteria, where we have shown that efflux pumps play an important role in the development of drug resistance in both M. tuberculosis (22) and MAC (16). This means that clofazimine's modest to poor activity against M. kansasii was unlikely due to efflux-induced tolerance, which we identified with moxifloxacin (13). This also means that we are unlikely to boost the efficacy of clofazimine by use of combination therapy with efflux pump inhibitors. However, our findings do not mean that clofazimine would not work if it had synergy with other (non-efflux inhibitor-based) drugs in a combination regimen. In the HFS-Mkn model, moxifloxacin at an optimal dose of 800 mg daily showed excellent efficacy as monotherapy (13). Promisingly, Brown-Elliot and Wallace (23) recently reported M. kansasii susceptibility to tedizolid, a novel oxazolidinone that carries an improved side effect profile compared to that of linezolid. In combination therapy, if there is synergy with these drugs, clofazimine could play a role but would not add much if there were strict additivity, as defined by Bliss independence. Thus, clofazimine's role in combination therapy remains to be established.
Finally, we benchmarked efficacy against standard therapy for M. kansasii. The kill curves, obtained using a biexponential decline model, revealed very low kill rates for standard therapy. It would take a year or more to achieve total sterilization of the lungs if initial bacterial burdens are assumed to be similar to those of M. tuberculosis at an average of 7.0 log10 CFU/ml. These kill rates are consistent with the long duration of therapy for standard therapy observed in the clinic and the relapse rates after treatment with therapy of <12 months' duration (24, 25). This means that the results obtained with our model have clinical relevance and suggest that future studies of drugs examined for the treatment of M. kansasii infection should all be compared or indexed to standard therapy. This also means that the modest to poor effect of clofazimine in this same model is likely to be reflective of what we may encounter in the clinic.
In summary, clofazimine has limited activity as monotherapy against M. kansasii in the HFS-Mkn. Despite a low MIC at currently recommended doses, clofazimine may not improve therapy outcomes. The standard regimen kill rates were consistent with current durations of therapy advocated in the clinic.
MATERIALS AND METHODS
Bacterial strain, cell line, and supplies.
M. kansasii strain ATCC 12478 was used in all the experiments. The growth medium and culture conditions were the same as those reported previously (13). The human monocyte-derived THP-1 monocyte cell line (ATCC TIB-202) was used in the HFS-Mkn experiments under culture conditions described elsewhere (13). Hollow-fiber cartridges were purchased from FiberCell Systems Inc. (MD, USA). Clofazimine (Sigma-Aldrich, St. Louis, MO, USA) was first dissolved in 100% dimethyl sulfoxide (DMSO) and then back diluted in DMSO and 100% ethanol at a 1:1 ratio. The final concentrations of DMSO and ethanol were 0.004% and 0.206% (vol/vol), respectively.
Determination of MIC and concentration-response at static concentration.
The clofazimine MIC was determined using broth microdilution and the resazurin colorimetric assay (13). Briefly, M. kansasii cultures were grown for 4 days to obtain the logarithmic-phase culture, and the inoculum was prepared to get a bacterial density of ∼1.5 × 105 CFU/ml. The concentrations of clofazimine tested were 0, 0.07, 0.15, 0.3, 0.6, 1.2, 2.5, and 5 mg/liter in 24-well plates. After 3 days of incubation at 37°C with 5% CO2, 50 μl of resazurin solution (final concentration, 0.001% [vol/vol]) was added to each well (13). The plates were incubated at 37°C for 24 h, and the lowest clofazimine concentration that prevented the color change from blue to pink was recorded as the MIC. For the broth microdilution method, the cultures were incubated for 7 days, and plates were inspected for the presence of a bacterial pellet. The clofazimine concentration that completely inhibited bacterial growth (the absence of a bacterial pellet) was recorded as the MIC.
For the concentration-response study, the culture conditions, inoculum preparation, and the concentration ranges were the same as those described above, except that the experiment was set in 15-ml screw-capped tubes with a total volume of 5 ml. M. kansasii cultures were coincubated with the different clofazimine concentrations for 7 days at 37°C under shaking conditions (120 rpm). On day 7, the culture was washed with normal saline to remove carryover drug, serially 10-fold diluted, and inoculated on Middlebrook 7H10 agar supplemented with 10% OADC. The number of M. kansasii CFU on the agar plates was counted after 7 to 10 days of incubation at 37°C.
Clofazimine dose-response in the HFS-Mkn.
The hollow-fiber system model has been used to establish the PK/PD relationships of drugs used to treat M. tuberculosis, MAC, and M. abscessus infections, as well as for M. kansasii (13, 26–35). In the HFS-Mkn dose-response studies, the human monocyte-derived THP-1 monocytes were infected with M. kansasii to get a multiplicity of infection of 10 bacteria per THP-1 cell (10:1). Twenty milliliters of infected THP-1 cells was inoculated into the peripheral compartment of each HFS-Mkn. In humans, clofazimine bioavailability varies from 42% to 62%, depending upon whether the drug is taken with or without food (17). In our HFS-Mkn experiments, we mimicked the clofazimine PKs seen in humans when the drug is taken with food. Clofazimine was administered via computer-controlled syringe pumps to achieve profiles that mimic the concentration-time profiles achieved with human-equivalent doses of 12.5, 25, 50, 75, 100, 150, and 200 mg daily administered with food. We mimicked a 12.5-h half-life of clofazimine, with the time to the maximum concentration (Tmax) being equal to 6 h (17, 18). In addition, one set of HFS-Mkn was also treated with clofazimine at a 100-mg daily dose plus reserpine at 10 mg/liter to determine if clofazimine efficacy can be improved with the use of an efflux pump blocker. Standard combination therapy with isoniazid at 300 mg/daily plus rifampin at 600 mg/daily plus ethambutol at 15 mg/kg/day was used to compare the kill curves of the different clofazimine doses in one set of HFS-Mkn. The peripheral compartment of each HFS-Mkn was sampled on days 0, 3, 7, 10, 14, and 21 to count the number of viable THP-1 cells as well as to determine the total bacillary burden in each system.
Drug concentration measurements.
We used previously published methods without any modifications to measure isoniazid, rifampin, and ethambutol concentrations (27, 36). Clofazimine and clofazimine-d7 (the internal standard [IS]) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Toronto Research Chemicals (Toronto, Canada), respectively. Liquid chromatography-tandem mass spectrometry analysis was performed using a Waters Acquity ultraperformance liquid chromatography (UPLC) system coupled with a Waters Xevo TQ mass spectrometer. Data were collected using MassLynx (v 4.1 SCN810) software. Separation was achieved by injecting 10 μl of sample on a Waters Acquity UPLC HSS T3 column (50 by 2.1 mm; 1.8 μm). Stock solutions of the standard and IS were prepared in DMSO at a concentration of 1 mg/ml. A calibration curve and low-quality-control (LQC) and high-quality-control (HQC) samples were prepared by diluting the stock solution in blank medium. Samples were diluted 1:20 with the IS solution in 0.1% aqueous formic acid (FA). The plates were vortexed and then centrifuged at 4,000 rpm for 15 min. Solvents for UPLC were 0.1% aqueous FA (solvent A) and 0.1% formic acid in acetonitrile (solvent B). The flow rate was 0.4 ml/min; the total run time was 6 min. Compounds were detected using positive electrospray ionization in the multiple reaction monitoring mode. The transitions used were m/z 473 to 431 (clofazimine) and m/z 480 to 432 (clofazimine-d7). The between-day percent coefficients of variation (CVs) for analysis of low and high quality controls were 9% and 5%, respectively. The intraday CVs for analysis of low and high quality controls were 3% and 1%, respectively. The lower limit of quantitation was 0.0025 μg/ml.
ACKNOWLEDGMENTS
This work was supported by National Institute of General Medical Sciences of the National Institutes of Health New Innovator Award DP2OD001886-01 and National Institute of Allergy and Infectious Diseases award R01AI079497 to Tawanda Gumbo.
T.G. designed the HFS-Mkn study, S.S. and T.G. performed the experiments, and S.S. and T.G. wrote the manuscript.
We have no conflicts of interest.
REFERENCES
- 1.Bloch KC, Zwerling L, Pletcher MJ, Hahn JA, Gerberding JL, Ostroff SM, Vugia DJ, Reingold AL. 1998. Incidence and clinical implications of isolation of Mycobacterium kansasii: results of a 5-year, population-based study. Ann Intern Med 129:698–704. doi: 10.7326/0003-4819-129-9-199811010-00004. [DOI] [PubMed] [Google Scholar]
- 2.Okoi C, Anderson STB, Antonio M, Mulwa SN, Gehre F, Adetifa IMO. 2017. Non-tuberculous mycobacteria isolated from pulmonary samples in sub-Saharan Africa—a systematic review and meta analyses. Sci Rep 7:12002. doi: 10.1038/s41598-017-12175-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prevots DR, Marras TK. 2015. Epidemiology of human pulmonary infection with nontuberculous mycobacteria: a review. Clin Chest Med 36:13–34. doi: 10.1016/j.ccm.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier K, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K. 2007. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 175:367–416. doi: 10.1164/rccm.200604-571ST. [DOI] [PubMed] [Google Scholar]
- 5.Diel R, Ringshausen F, Richter E, Welker L, Schmitz J, Nienhaus A. 2017. Microbiological and clinical outcomes of treating non-Mycobacterium avium complex nontuberculous mycobacterial pulmonary disease: a systematic review and meta-analysis. Chest 152:120–142. doi: 10.1016/j.chest.2017.04.166. [DOI] [PubMed] [Google Scholar]
- 6.Ahn CH, Lowell JR, Ahn SS, Ahn S, Hurst GA. 1981. Chemotherapy for pulmonary disease due to Mycobacterium kansasii: efficacies of some individual drugs. Rev Infect Dis 3:1028–1034. doi: 10.1093/clinids/3.5.1028. [DOI] [PubMed] [Google Scholar]
- 7.Dey T, Brigden G, Cox H, Shubber Z, Cooke G, Ford N. 2013. Outcomes of clofazimine for the treatment of drug-resistant tuberculosis: a systematic review and meta-analysis. J Antimicrob Chemother 68:284–293. doi: 10.1093/jac/dks389. [DOI] [PubMed] [Google Scholar]
- 8.Garrelts JC. 1991. Clofazimine: a review of its use in leprosy and Mycobacterium avium complex infection. DICP 25:525–531. doi: 10.1177/106002809102500513. [DOI] [PubMed] [Google Scholar]
- 9.Martiniano SL, Wagner BD, Levin A, Nick JA, Sagel SD, Daley CL. 2017. Safety and effectiveness of clofazimine for primary and refractory nontuberculous mycobacterial infection. Chest 152:800–809. doi: 10.1016/j.chest.2017.04.175. [DOI] [PubMed] [Google Scholar]
- 10.Wallace RJ Jr, Nash DR, Steele LC, Steingrube V. 1986. Susceptibility testing of slowly growing mycobacteria by a microdilution MIC method with 7H9 broth. J Clin Microbiol 24:976–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins FM, Klayman DL, Morrison NE. 1982. Activity of 2-acetylpyridine and 2-acetylquinoline thiosemicarbazones tested in vitro in combination with other antituberculous drugs. Am Rev Respir Dis 125:58–60. [DOI] [PubMed] [Google Scholar]
- 12.van Ingen J, Egelund EF, Levin A, Totten SE, Boeree MJ, Mouton JW, Aarnoutse RE, Heifets LB, Peloquin CA, Daley CL. 2012. The pharmacokinetics and pharmacodynamics of pulmonary Mycobacterium avium complex disease treatment. Am J Respir Crit Care Med 186:559–565. doi: 10.1164/rccm.201204-0682OC. [DOI] [PubMed] [Google Scholar]
- 13.Srivastava S, Pasipanodya J, Sherman CM, Meek C, Leff R, Gumbo T. 2015. Rapid drug tolerance and dramatic sterilizing effect of moxifloxacin monotherapy in a novel hollow-fiber model of intracellular Mycobacterium kansasii disease. Antimicrob Agents Chemother 59:2273–2279. doi: 10.1128/AAC.04441-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diacon AH, Pym A, Grobusch M, Patientia R, Rustomjee R, Page-Shipp L, Pistorius C, Krause R, Bogoshi M, Churchyard G, Venter A, Allen J, Palomino JC, De Marez T, van Heeswijk RP, Lounis N, Meyvisch P, Verbeeck J, Parys W, de Beule K, Andries K, McNeeley DF. 2009. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med 360:2397–2405. doi: 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- 15.Hartkoorn RC, Uplekar S, Cole ST. 2014. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:2979–2981. doi: 10.1128/AAC.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmalstieg AM, Srivastava S, Belkaya S, Deshpande D, Meek C, Leff R, van Oers NS, Gumbo T. 2012. The antibiotic resistance arrow of time: efflux pump induction is a general first step in the evolution of mycobacterial drug resistance. Antimicrob Agents Chemother 56:4806–4815. doi: 10.1128/AAC.05546-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaad-Lanyi Z, Dieterle W, Dubois JP, Theobald W, Vischer W. 1987. Pharmacokinetics of clofazimine in healthy volunteers. Int J Lepr Other Mycobact Dis 55:9–15. [PubMed] [Google Scholar]
- 18.Nix DE, Adam RD, Auclair B, Krueger TS, Godo PG, Peloquin CA. 2004. Pharmacokinetics and relative bioavailability of clofazimine in relation to food, orange juice and antacid. Tuberculosis (Edinb) 84:365–373. doi: 10.1016/j.tube.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Moodley R, Godec TR, STREAM Trial Team . 2016. Short-course treatment for multidrug-resistant tuberculosis: the STREAM trials. Eur Respir Rev 25:29–35. doi: 10.1183/16000617.0080-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baik J, Rosania GR. 2012. Macrophages sequester clofazimine in an intracellular liquid crystal-like supramolecular organization. PLoS One 7:e47494. doi: 10.1371/journal.pone.0047494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoon GS, Keswani RK, Sud S, Rzeczycki PM, Murashov MD, Koehn TA, Standiford TJ, Stringer KA, Rosania GR. 2016. Clofazimine biocrystal accumulation in macrophages upregulates interleukin 1 receptor antagonist production to induce a systemic anti-inflammatory state. Antimicrob Agents Chemother 60:3470–3479. doi: 10.1128/AAC.00265-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srivastava S, Musuka S, Sherman C, Meek C, Leff R, Gumbo T. 2010. Efflux-pump-derived multiple drug resistance to ethambutol monotherapy in Mycobacterium tuberculosis and the pharmacokinetics and pharmacodynamics of ethambutol. J Infect Dis 201:1225–1231. doi: 10.1086/651377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown-Elliott BA, Wallace RJ Jr. 2017. In vitro susceptibility testing of tedizolid against nontuberculous mycobacteria. J Clin Microbiol 55:1747–1754. doi: 10.1128/JCM.00274-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sauret J, Hernandez-Flix S, Castro E, Hernandez L, Ausina V, Coll P. 1995. Treatment of pulmonary disease caused by Mycobacterium kansasii: results of 18 vs 12 months' chemotherapy. Tuber Lung Dis 76:104–108. [DOI] [PubMed] [Google Scholar]
- 25.Anonymous. 1994. Mycobacterium kansasii pulmonary infection: a prospective study of the results of nine months of treatment with rifampicin and ethambutol. Research Committee, British Thoracic Society. Thorax 49:442–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gumbo T, Louie A, Deziel MR, Parsons LM, Salfinger M, Drusano GL. 2004. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis 190:1642–1651. doi: 10.1086/424849. [DOI] [PubMed] [Google Scholar]
- 27.Srivastava S, Pasipanodya JG, Ramachandran G, Deshpande D, Shuford S, Crosswell HE, Cirrincione KN, Sherman CM, Swaminathan S, Gumbo T. 2016. A long-term co-perfused disseminated tuberculosis-3D liver hollow fiber model for both drug efficacy and hepatotoxicity in babies. EBioMedicine 6:126–138. doi: 10.1016/j.ebiom.2016.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deshpande D, Srivastava S, Bendet P, Martin KR, Cirrincione KN, Lee PS, Pasipanodya JG, Dheda K, Gumbo T. 2018. Antibacterial and sterilizing effect of benzylpenicillin in tuberculosis. Antimicrob Agents Chemother 62:e02232-17. doi: 10.1128/AAC.02232-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deshpande D, Srivastava S, Gumbo T. 2017. A programme to create short-course chemotherapy for pulmonary Mycobacterium avium disease based on pharmacokinetics/pharmacodynamics and mathematical forecasting. J Antimicrob Chemother 72:i54–i60. doi: 10.1093/jac/dkx309. [DOI] [PubMed] [Google Scholar]
- 30.Deshpande D, Srivastava S, Meek C, Leff R, Hall GS, Gumbo T. 2010. Moxifloxacin pharmacokinetics/pharmacodynamics and optimal dose and susceptibility breakpoint identification for treatment of disseminated Mycobacterium avium infection. Antimicrob Agents Chemother 54:2534–2539. doi: 10.1128/AAC.01761-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deshpande D, Srivastava S, Musuka S, Gumbo T. 2016. Thioridazine as chemotherapy for Mycobacterium avium complex diseases. Antimicrob Agents Chemother 60:4652–4658. doi: 10.1128/AAC.02985-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferro BE, Srivastava S, Deshpande D, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2016. Tigecycline is highly efficacious against Mycobacterium abscessus pulmonary disease. Antimicrob Agents Chemother 60:2895–2900. doi: 10.1128/AAC.03112-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferro BE, Srivastava S, Deshpande D, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2016. Moxifloxacin's limited efficacy in the hollow-fiber model of Mycobacterium abscessus disease. Antimicrob Agents Chemother 60:3779–3785. doi: 10.1128/AAC.02821-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferro BE, Srivastava S, Deshpande D, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2016. Failure of the amikacin, cefoxitin, and clarithromycin combination regimen for treating pulmonary Mycobacterium abscessus infection. Antimicrob Agents Chemother 60:6374–6376. doi: 10.1128/AAC.00990-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferro BE, Srivastava S, Deshpande D, Sherman CM, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2015. Amikacin pharmacokinetics/pharmacodynamics in a novel hollow-fiber Mycobacterium abscessus disease model. Antimicrob Agents Chemother 60:1242–1248. doi: 10.1128/AAC.02282-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Srivastava S, Pasipanodya JG, Meek C, Leff R, Gumbo T. 2011. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J Infect Dis 204:1951–1959. doi: 10.1093/infdis/jir658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Magombedze G, Pasipanodya J, Srivastava S, Deshpande D, McIlleron H, Gumbo T. Transformation morphisms and time to extinction analysis that map therapy duration from pre-clinical models to patients with tuberculosis: translating from apples to oranges. Clin Infect Dis, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]