

ABSTRACT
Candida albicans is an important opportunistic pathogen causing various human infections that are often treated with azole antifungals. The U.S. CDC now regards developing candidal antifungal resistance as a threat, creating a need for new and more effective antifungal treatments. Iron is an essential nutrient for all living cells, and there is growing evidence that interference with iron homeostasis of C. albicans can improve its response to antifungals. This study was aimed at establishing whether withholding iron by currently used medical iron chelators and the novel chelator DIBI could restrict growth and also enhance the activity of azoles against clinical isolates of C. albicans. DIBI, but not deferoxamine or deferiprone, inhibited the growth of C. albicans at relatively low concentrations in vitro, and this inhibition was reversed by iron addition. DIBI in combination with various azoles demonstrated stronger growth inhibition than the azoles alone and greatly prolonged the inhibition of cell multiplication. In addition, the administration of DIBI along with fluconazole (FLC) to mice inoculated with an FLC-sensitive isolate in a model of experimental C. albicans vaginitis showed a markedly improved clearance of infection. These results suggest that iron chelation by DIBI has the potential to enhance azole efficacy for the treatment of candidiasis.
KEYWORDS: Candida albicans, DIBI, antifungal agents, azoles, iron, synergy
INTRODUCTION
Candida spp. are commonly associated with health care-associated infections in the United States as well as worldwide (1). An estimated 46,000 Candida infections occur among hospitalized patients in the United States each year, and of the more than 20 species of Candida yeasts that can cause infection in humans, the most common is C. albicans (2). As Candida typically inhabits the skin and mucosa, candidiasis is often superficial in nature (3); these infections are incredibly common affecting millions of people worldwide (4). While this is rarely life threatening, it can significantly impact a patient's quality of life (4, 5) and may spread to other individuals or become invasive (4). Vulvovaginal candidiasis (VVC) is the most common gynecological infection in healthy women (4), as 70% to 75% of women will experience this mucosal fungal infection at least once during their lives (6), and VVC can become recurrent due in part to azole resistance (7). With the limited range of available antifungal drug classes (5, 8) and growing incidence of azole resistance (7, 9–11), agents that might enhance or broaden the efficacies of the azole antifungals would be attractive.
While C. albicans colonizes the skin and mucosal surfaces of healthy individuals, predisposing host factors are implicated in infection (12). It is becoming increasingly clear that the innate immune response is an important host defense against Candida infection (13), with iron availability and sequestration being important components of susceptibility and immunity (14–16). Interestingly, estrogen administration has been shown to increase iron availability in the host (17–19), with a correspondingly increased susceptibility to infection (17, 19). Estrogen administration is also a requirement to predispose mice to experimental candidal vaginitis (7, 20).
Iron acquisition mechanisms of C. albicans have also been related to its virulence (21, 22). Iron is an irreplaceable essential nutrient for all pathogenic bacteria and fungi, as it is integral for the activity of various key enzyme systems of metabolism, repair, and defense (23). Therefore, targeting iron acquisition provides a potential basis for a new approach to antimicrobials (24). Interference with microbial iron nutrition in vivo might also provide an enhancement of the activities of antifungal agents. In this regard, daughter cells from C. albicans iron-limited biofilms have been shown to be more sensitive to amphotericin B (25). Additionally, lactoferrin, a key iron-withholding defense protein of vertebrate animals and essential for restricting iron availability to pathogens in vivo (26), has been shown to improve the in vitro efficacies of various antifungal agents, including fluconazole (FLC) (27, 28).
The elucidation of the roles of iron supply and iron restriction in enhancing FLC and other azole activities might be aided by the use of iron-specific chelators that can withdraw and deny nutritional iron from C. albicans. DIBI, recently patented by Chelation Partners Incorporated (29), is a high-affinity iron chelator which has been found to provide strong iron-specific growth inhibition of C. albicans (30). DIBI is the developmental code name for a new hydroxypyridinone functional iron-chelating polymer shown in Fig. 1. DIBI has chemical similarities to clinically used deferiprone as shown, but DIBI provides a platform on which adjacent pyridinone groups can together bind iron with a higher avidity than that provided by deferiprone (M. T. C. Ang, R. Gumbau-Brisa, D. S. Allan, R. McDonald, M. J. Ferguson, B. E. Holbein, and M. Bierenstiel, submitted for publication). In this study, we investigated the influence of iron withdrawal by DIBI on the activity of azole antifungals with C. albicans clinical isolates in vitro and in vivo.
FIG 1.
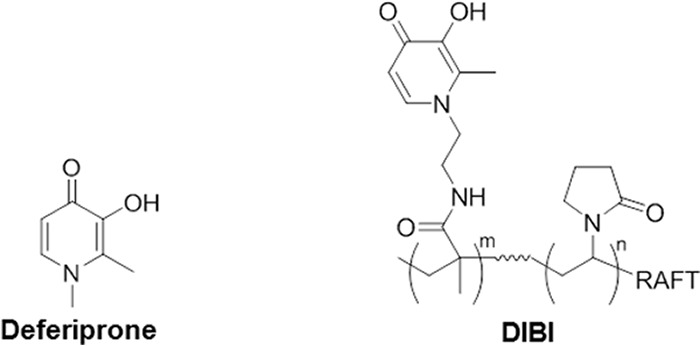
Structures of deferiprone and DIBI. Deferiprone is a hydroxypyridinone iron chelator (molecular mass, 139 Da) with similar chelating functionality to DIBI, an iron-chelating polymer (molecular mass, 9 kDa) containing 9 hydroxypyridinone groups per molecule.
RESULTS
Effect of DIBI on C. albicans growth.
DIBI inhibited C. albicans growth and was superior to the other chelators tested. The MIC80 (80% reduction) values for 3 iron chelators were determined for 3 strains of C. albicans (Table 1). MICs were read after 24 h (data not shown) and 48 h to examine prolonged inhibition from iron sequestration. While deferoxamine was noninhibitory at the concentrations tested, deferiprone and DIBI both inhibited the growth of these strains. However, deferiprone required 80 to 160 μg/ml for growth inhibition at 48 h, while DIBI was inhibitory at 2 μg/ml. These MICs corresponded to 0.57 to 1.15 mM for deferiprone and only 0.02 μM for DIBI.
TABLE 1.
MICs of iron chelators deferoxamine, deferiprone, and DIBI against 3 strains of C. albicans
| C. albicans strain | MIC (μg/ml)a |
||
|---|---|---|---|
| Deferoxamine | Deferiprone | DIBI | |
| SC5314 | >1,280 | 160 | 2 |
| Ca3969 | >1,280 | 80 | 2 |
| Ca5031 | >1,280 | 80 | 2 |
Values reported are 48-h results from 4 replicates within 2 independent experiments.
Iron dependence of DIBI inhibition.
DIBI provided dose-dependent and iron-reversible growth inhibition when tested against both a reference strain (SC5314, data not shown) and a clinical strain (Ca3969) (Fig. 2). The growth of C. albicans over 48 h was suppressed by DIBI addition compared to that in the control medium. This repression was dose dependent, as the inhibitory effect of DIBI at 10 μg/ml was greater than that observed by the addition of 1 μg/ml and persisted until 48 h. DIBI inhibition of growth was also reversed directly by iron addition, and the extent of reversal was iron concentration dependent (Fig. 2).
FIG 2.
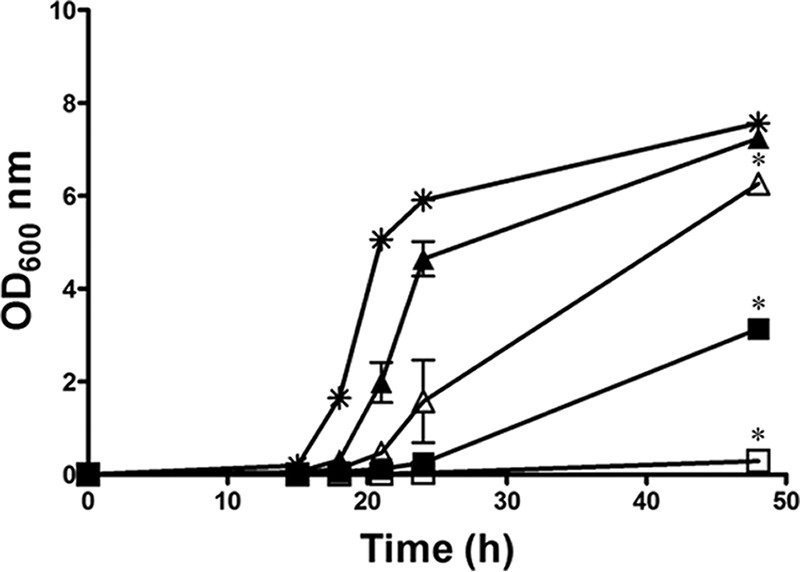
DIBI growth inhibition of C. albicans is iron reversible. RPMI (✳), RPMI with DIBI at 1 (△) or 10 μg/ml (□), and RPMI supplemented with 1 μM iron with DIBI at 1 (▲) or 10 μg/ml (■) were inoculated with C. albicans strain Ca3969. Growth was measured as OD at 600 nm over 48 h. Data points are means ± SEMs from 2 independent experiments. *, P < 0.05 for DIBI 1 or 10 μg/ml or RPMI plus iron plus DIBI 10 μg/ml versus RPMI, for DIBI 1 μg/ml versus DIBI 10 μg/ml, for DIBI 1 μg/ml versus RPMI plus iron plus DIBI 1 μg/ml, and for DIBI 10 μg/ml versus RPMI plus iron plus DIBI 10 μg/ml.
Improvement of azole activity with DIBI.
Azole MICs, including MIC80 and MIC50 values, for several C. albicans strains were determined in RPMI medium (Table 2). The majority of the strains shared similar sensitivity profiles: FLC sensitive (MIC50 = 0.12 to 0.25 μg/ml, MIC80 = 0.5 to 1 μg/ml) and susceptible or susceptible-dose dependent for itraconazole ([ITC] MIC50 = 0.03 to 0.06 μg/ml, MIC80 = 0.25 to 0.5 μg/ml). In contrast, strain LP1158-07 was resistant to both azoles (FLC, MIC80 and MIC50 > 8 μg/ml; ITC, MIC80 and MIC50 ≥ 1 μg/ml). In regard to clotrimazole (CLT), although there are no definitive interpretive breakpoints (31), most strains shared similar profiles (MIC50 < 0.005 μg/ml, MIC80 = 0.005 to 0.03 μg/ml), with MICs for strain LP1158-07 at 0.03 μg/ml (MIC50) and 0.5 to 1 μg/ml (MIC80).
TABLE 2.
Azole MICs against 5 strains of C. albicans
| C. albicans strain | MIC80 (MIC50) (μg/ml)a |
||
|---|---|---|---|
| FLC | ITC | CLT | |
| 96113 | 0.5 (0.25) | 0.5 (0.06) | 0.01 (<0.005) |
| SC5314 | 0.5 (0.12) | 0.5 (0.03) | 0.03 (<0.005) |
| Ca3969 | 1 (0.25) | 0.5 (0.06) | 0.03 (<0.005) |
| Ca5031 | 0.5 (0.25) | 0.25 (0.03) | 0.005 (<0.005) |
| LP1158-07 | >64 (16) | 8 (1) | 0.5–1 (0.03) |
Values reported are from at least 2 independent experiments.
Checkerboard assays were performed to assess DIBI synergy with FLC. For this investigation of two independent fungistatic agents, the more stringent MIC80 endpoint was used to more clearly interpret the interaction between the chelator and the azole. Furthermore, MIC80 was used for studies with DIBI alone. The results showed that DIBI was synergistic with FLC when tested against strain 96113, with fractional inhibitory concentration indices (FICIs) of 0.38 at 24 h and 0.3 to 0.38 at 48 h. In addition, spot plates were prepared from the wells on the basis of a method similar to that outlined by Fiori and Van Dijck (32). Recovery spots prepared from typical 24-h checkerboards (Fig. 3A) showed killing (reduced recovery of viable cells) for FLC alone at ≥0.5 μg/ml and with DIBI alone at ≥32 μg/ml. Increasing concentrations of DIBI together with FLC at 0.25 μg/ml resulted in decreasing recovery of viable CFU in a synergistic manner. Recovery spots prepared from 48-h checkerboard plates (Fig. 3B) indicated tolerance to FLC at 0.5 to 8 μg/ml with DIBI at 0 to 2 μg/ml, recoveries being greater from these wells than was observed for 24 h when comparing the outlined section in Fig. 3B to that in 3A. However, the combination of FLC at ≥0.25 μg/ml with DIBI at ≥4 μg/ml resulted in little to no recovery growth. Similar results were observed for spot plating of checkerboards testing Ca3969 with FLC, for both Ca5031 and Ca3969 with ITC, and via disk diffusion assays as a complementary visual assessment (data not shown).
FIG 3.

Recovery growth of C. albicans is inhibited after exposure to a combination of fluconazole (FLC) and DIBI. A 5-μl volume from each well of a checkerboard assay that had been inoculated with Ca5031 was spotted onto a YPD agar plate following 24-h (A) or 48-h (B) exposure to the agent. Recovery plates were incubated for 24 h before being visually inspected and photographed. Images are representative of results observed in 3 independent experiments.
The improvement of azole activity by DIBI was also assessed using a growth inhibition/kill type assay system as shown for four stains with FLC, ITC, or CLT (Fig. 4). Azole addition alone resulted in a partial growth inhibition of all strains, including the azole-resistant isolate LP1158-07. The DIBI concentrations tested provided either no (96113) or partial inhibition (all other strains) over 72 h. While the combination of DIBI with each of the azoles provided total growth inhibition for most strains tested (Fig. 4B to F), it resulted in a moderate fungicidal effect over 72 h for strain 96113 with FLC (Fig. 4A).
FIG 4.

Long-term growth inhibition of C. albicans by combinations of DIBI with fluconazole (FLC), itraconazole (ITC), and clotrimazole (CLT). RPMI ( ), RPMI with DIBI alone (△), RPMI with azole alone (○), and RPMI with DIBI and azole combined (◊) were inoculated with azole-susceptible strains (96113, Ca3969, Ca5031) or the FLC-resistant isolate (LP1158-07), as indicated. (A) 1 μg/ml DIBI, 1 μg/ml FLC; (B) 10 μg/ml DIBI, 128 μg/ml FLC; (C) 10 μg/ml DIBI, 0.5 μg/ml ITC; (D) 10 μg/ml DIBI, 0.25 μg/ml ITC; (E and F) 10 μg/ml DIBI, 0.03 μg/ml CLT. Viable cell counts were determined by plate counting over 72 h. Data points are means ± SEMs from 3 independent experiments. *, P < 0.01 for DIBI plus azole versus RPMI, DIBI alone, or azole alone.
), RPMI with DIBI alone (△), RPMI with azole alone (○), and RPMI with DIBI and azole combined (◊) were inoculated with azole-susceptible strains (96113, Ca3969, Ca5031) or the FLC-resistant isolate (LP1158-07), as indicated. (A) 1 μg/ml DIBI, 1 μg/ml FLC; (B) 10 μg/ml DIBI, 128 μg/ml FLC; (C) 10 μg/ml DIBI, 0.5 μg/ml ITC; (D) 10 μg/ml DIBI, 0.25 μg/ml ITC; (E and F) 10 μg/ml DIBI, 0.03 μg/ml CLT. Viable cell counts were determined by plate counting over 72 h. Data points are means ± SEMs from 3 independent experiments. *, P < 0.01 for DIBI plus azole versus RPMI, DIBI alone, or azole alone.
Improvement of FLC by DIBI in vivo.
Experimental vaginitis in estrogenized mice was examined with FLC-sensitive strain 96113 (Fig. 5). The treatments included DIBI (40 mg/kg) alone administered topically on days 3 to 6 postinoculation, FLC (25 mg/kg) alone administered by oral gavage on days 3 and 5 postinoculation, and DIBI plus FLC administered similarly. Vaginal fungal burden was evaluated on day 7 postinoculation. DIBI alone had no effect on vaginal fungal burden. The organism burden was highly variable in the FLC-treated animals but was significantly reduced compared to that of the vehicle control (P < 0.0001). The addition of DIBI with FLC did not reduce the fungal burden further when statistically compared to that of FLC-treated mice but did enhance the rate of clearance (39% of treated mice) compared to that with FLC alone (10% of treated mice) (P < 0.007). Higher concentrations of DIBI (80 mg/kg) had no additional effects (data not shown). The overall results were supported by the qualitative scoring of lavage fluid for yeast/hyphae of individual animals; cleared animals had no visible organisms present (score, 0), FLC or DIBI-plus-FLC-treated animals had scores ranging from 1 to 4, and vehicle- and DIBI-treated animals had scores ranging from 3 to 4. In other experiments, the FLC-resistant strain LP1158-07 was similarly tested. The results showed no effect of FLC alone at 25 or 50 mg/kg or in combination with DIBI at 40 mg/kg (data not shown).
FIG 5.

Improvement of fluconazole (FLC) by DIBI in vivo. Experimental murine vaginitis was established using FLC-sensitive strain 96113. Animals were given DIBI alone (40 mg/kg), FLC alone (25 mg/kg), or a combination of DIBI plus FLC. Vaginal fungal burden was evaluated at day 7 postinoculation. Results are expressed as CFU/100 μl vaginal lavage fluid. Percentages indicate rates of fungal clearance. Results are cumulative of 5 independent experiments.
DISCUSSION
This study investigated the roles that iron supply and its in situ sequestration by iron chelation have in relation to growth inhibition and azole sensitivity of C. albicans in vitro and in vivo. Deferoxamine and deferiprone are iron chelators used clinically in humans for the treatment of iron overload disorders. Deferoxamine was completely noninhibitory and deferiprone was only weakly inhibitory for clinical strains of C. albicans, a finding consistent with our previous results for the reference strain ATCC 10231 and also for Candida vini (30). DIBI is a relatively low-molecular-weight (9 kDa) hydroxypyridinone-containing polymeric chelator with strong selective iron-binding characteristics (Fig. 1), and it was found to strongly inhibit C. albicans in an iron-reversible manner. DIBI inhibition occurred at very low concentrations in comparison to the chemically related hydroxypyridinone, deferiprone. On a comparative molar concentration basis, DIBI was >10,000-fold more inhibitory than deferiprone. Thompson et al. (33) also demonstrated that deferoxamine was not inhibitory for a number of bacterial pathogens and reported that deferiprone (also known as Apo-L1) and the related Apo-6619, while exhibiting better activity than deferoxamine, were nevertheless only modest growth inhibitors. The deferiprone MICs observed for C. albicans were in the mM range, which is similar to those found for various bacterial pathogens. Given the >10,000-fold increased activity found for DIBI, it would appear to be a good candidate for a stand-alone growth inhibitor and also as an enhancing agent for antibiotics. Zarember et al. (34) demonstrated the synergy between deferiprone and azoles for Aspergillus fumigatus conidia using a checkerboard assay. Other agents that impact microbial essential iron and other metals have also been shown to affect azole sensitivity. Members of the tetracycline family have been shown to enhance FLC activity in C. albicans as well as chelate metal ions. Shi et al. (35) proposed that minocycline enhances FLC penetration due to Ca efflux, while Fiori and Van Dijck (32) concluded that doxycycline enhancement of FLC was related to its iron-binding activity. The tetracycline family of compounds, including minocycline and doxycycline, are known to bind iron, and this iron interaction can neutralize these agents, interfering with antibiotic uptake (36). However, the tetracyclines also bind Co and Ni (37) as well as Ca and Mg (38). Thus, it is possible the iron addition that negated the doxycycline enhancement of FLC activity, as observed by Fiori and Van Dijck (32), may have been in part related to iron neutralization of doxycycline, or the iron might have displaced other doxycycline-sequestered essential metals such as Co, Ni, Mg, or Ca, making these available to C. albicans. We have shown that DIBI is strongly iron selective and does not bind Ca, Mg, Co, or Ni (30); therefore, it is unlikely that the sequestration of these other essential metals was related to its observed activity. Prasad et al. (39) reported that iron limitation resulted in membrane leakiness of C. albicans in association with an increased sensitivity to various antifungals, including FLC. However, their studies employed the addition of bathophenanthroline sulfonate (BPS), a chelator that is known not to be iron specific and can also bind Ni, Cu, Co, and Zn (40). Therefore, BPS used at relatively high concentrations may have had other metal-sequestering activities in addition to that for iron.
We have demonstrated a strong ability of DIBI at low concentrations to improve the activities of various azoles, including fluconazole, itraconazole, and clotrimazole. DIBI at concentrations as low as 1 μg/ml in combination with an azole provided strong and prolonged inhibition of the growth of various clinical isolates of C. albicans in vitro, including one which is azole resistant. The in vivo results showing that DIBI administered intravaginally to infected mice along with oral treatment with FLC promoted an enhanced clearance of the C. albicans vaginitis provide further evidence of an improvement of the azoles by DIBI. The 96113 strain used was also more sensitive to FLC when combined with DIBI in vitro; fungistasis was observed in vitro. Two additional aspects of the infection results warrant further discussion. First, infection fungal burdens in FLC-treated mice (with both FLC alone and with FLC and DIBI) were quite variable compared to those found for either sham-treated controls or DIBI-treated mice. A likely explanation is that the FLC concentration used was suboptimal in an attempt to promote potentiation by DIBI. Additionally, FLC administered orally is highly bioavailable, rapidly entering the systemic circulation for delivery to tissues, including into vaginal secretions, but with >60% of the FLC load being eliminated nonmetabolized and intact via urinary excretion (41). It seems possible that an additional local delivery of FLC via urine at the opening of the vagina might be a source of variations in effective vaginal FLC concentrations in the infected mice. In this regard, it would be interesting to investigate intravaginal administration of FLC (or an alternate azole, especially CLT, which is typically used topically in humans), as this approach would possibly provide a less variable response to azole therapy. Second, DIBI alone in vivo had minimal effects on vaginal fungal burden in contrast to the in vitro results. The exception was the lack of in vitro activity against strain 96113 that was also used in vivo. However, it is notable that control growth for this strain in vitro was less robust than for other strains, which could account for the differences observed. Nevertheless, the in vivo results were reflective of the in vitro results. Further studies will be needed to determine if DIBI alone in vivo has any appreciable antimicrobial effects. However, when considering the synergistic activity of DIBI with the azoles, it is interesting to consider how the concentration of DIBI might impact the outcome. DIBI displays fungistatic activity on its own at higher concentrations in vitro, but at lower concentrations, it provides iron-restricted yet continued slowed growth as measured by CFU yield. This condition of iron-restricted growth resulting from DIBI exposure improved the response to FLC (and to the other azoles) in vitro. Our results suggest that DIBI is providing an iron stress by restricting the supply to C. albicans in vivo, resulting in an enhanced susceptibility to FLC, a result fully consistent with that observed in vitro. The finding that estrogen treatment increases available host iron for candidal and other urogenital infections (17–19) is noteworthy as well. Thus, DIBI may help to sequester estrogen-mobilized excess iron available in the vaginal sections of the mice during infection and create an iron-limiting condition.
Unfortunately, DIBI had no enhancing or potentiating effect on an FLC-resistant isolate in vivo, in contrast to the in vitro results. A possible explanation is that DIBI requires some effect of the azole in order to provide any potentiation. While FLC had a modest effect alone on the FLC-resistant isolate in vitro, there was no effect of FLC in vivo, even at high concentrations. Additional studies will be necessary to investigate this mechanism further.
Also noteworthy is that our studies showed no adverse toxic effects with intravaginal administration of DIBI alone in mice. More comprehensive testing of DIBI toxicity has been completed separately. Both acute and chronic systemic toxicity testing in male and female rats receiving daily intravenous (i.v.) administration of DIBI at up to 200 mg/kg (highest dosage tested) for 14 consecutive days showed no significant adverse effects. This included the rate of food consumption, modification in weight gain, and parameters of hematological, clinical chemistry, organ weight, and tissue histopathological changes (B. E. Holbein, unpublished results). Thus, DIBI appears safe for use in conjunction with azoles for the treatment of fungal infections.
Our results with DIBI provide stronger evidence for a direct role of iron in azole sensitivity. On this basis, we hypothesize a rational and direct physiological link between azole sensitivity and iron availability as centered on the cytochrome P450 14alpha-demethylase, which is the primary azole target and essential for ergosterol synthesis (39, 42). This is a heme (iron)-dependent enzyme system and therefore an iron-dependent target. The binding of the azoles and the subsequent inactivation of this enzyme as their known mode of action prevents demethylation of lanosterol (43–45) and correspondingly depletes the ergosterol content of membranes (46). This in turn leads to an accumulation of methylated sterols and induces changes in membrane physiology, such as leakiness and altered membrane-bound enzymes, resulting in the cessation of growth of C. albicans (44, 46).
We propose that iron chelation by DIBI results in reduced iron availability and uptake from the fungal growth environment, leading to the suppressed production of functional iron-dependent enzymes, including the cytochrome P450-dependent ergosterol demethylase. Reduced quantities of this azole target could then improve relative inactivation efficiency by azoles (reduced target content), while a loss of membrane integrity from impaired ergosterol synthesis could favor improved influx of additional azole. These complementary responses could act together to improve the overall azole inhibition of growth. Consistent with this proposed mode of action is the finding that exposure to ketoconazole induced an upregulation of various genes related to both P450 demethylase activity and iron uptake acquisition systems in C. albicans (47), indicating an increased need for iron in response to azole exposure.
The data presented here support the use of specific iron chelators that withhold iron necessary for fungal growth as a promising avenue for enhancing azole effectiveness in the treatment of fungal infections. Given its apparent lack of toxicity and activity at low concentrations, DIBI appears to have strong potential in this regard and warrants further investigation.
MATERIALS AND METHODS
Yeast strains.
The C. albicans strains used in this study are listed in Tables 1 and 2. The reference strain ATCC MYA-2876 (SC5314, clinical origin) was received from Malcom Whiteway (National Research Council, Montreal, Quebec, Canada); the reference strain ATCC 96113 and the fluconazole-resistant clinical strain LP1158-07 were from the Fidel lab culture collection. All other strains were clinical isolates obtained from the Capital District Health Authority, Halifax, Nova Scotia (Ca3969 blood specimen, Ca5031 oral specimen). The isolates were subcultured on a regular basis from frozen stock at −80°C and maintained at 4°C on Trypticase soy agar plates with 5% sheep blood (Becton Dickinson) or yeast extract-peptone-dextrose (YPD) agar plates (Difco BD).
Iron chelators and antifungals for in vitro susceptibility tests.
DIBI as previously reported (Chelation Partners Inc.) (30), deferoxamine (Novartis), and deferiprone (Sigma-Aldrich) were tested for their antifungal activity against C. albicans. All three chelators were dissolved in water. FLC (Sigma-Aldrich) was dissolved in water. ITC and CLT (Sigma-Aldrich) were both dissolved in dimethyl sulfoxide (DMSO). Stocks were filter-sterilized (0.2-μm filter) prior to dilution in water or medium.
Cultivation and assay media.
RPMI 1640 medium (with l-glutamine, without NaHCO3, and supplemented with 2% glucose; Sigma-Aldrich), pH 7.0, buffered with 0.165 M morpholinepropanesulfonic acid ([MOPS] Sigma-Aldrich) was routinely used. RPMI agar plates were prepared using 1.5% (wt/vol) agar (Bioshop Canada) addition. YPD agar plates used for checkerboard spot recovery plating contained 1% (wt/vol) yeast extract (Bacto; Becton Dickinson), 2% (wt/vol) peptone (Bacto; Becton Dickinson), 2% (wt/wt) glucose (Sigma-Aldrich), and 2% (wt/wt) agar (Acumedia; Neogen).
Susceptibility testing in broth microdilution assays.
MICs for the iron chelators were determined in RPMI medium. Serial dilutions of each chelator were tested using the following ranges: deferoxamine and deferiprone, 0.63 to 1280 μg/ml; DIBI, 0.06 to 128 μg/ml. Microtiter plates were inoculated with 0.5 × 103 to 2.5 × 103 CFU/ml C. albicans prepared from overnight cultures in RPMI medium and incubated at 35°C for 48 h. MIC endpoints were defined as the lowest concentration of chelator that inhibited growth by 80% (MIC80, visual read) compared to that of a positive control. All experiments were replicated two times.
Azole MICs were determined according to the CLSI standard reference method for broth dilution antifungal susceptibility testing of yeasts (48, 49), with the following modifications. Serial dilutions of azoles were assayed in the following ranges: FLC, 0.12 to 64 μg/ml; ITC, 0.01 to 8 μg/ml; and CLT, 0.005 to 4 μg/ml. Microtiter plates were inoculated with 0.5 × 103 to 2.5 × 103 CFU/ml C. albicans prepared from overnight cultures and incubated at 35°C for 24 h. The MIC80 and MIC50 endpoints were determined by visual observation as relative to the growth of an azole-free control. All experiments were repeated a minimum of two times.
Effect of RPMI iron concentration and DIBI on growth.
The effects of DIBI addition and RPMI iron concentration were determined for C. albicans strain Ca3969 in RPMI alone, RPMI plus DIBI 1 μg/ml, RPMI plus DIBI 10 μg/ml, RPMI plus DIBI 1 μg/ml plus 1 μM iron, and RPMI plus DIBI 10 μg/ml plus 1 μM iron. Aqueous ferric di-citrate (Sigma-Aldrich) was used as an exogenous iron source. Ten milliliters of each test medium was inoculated with Ca3969 at a final dilution of 1:2,000 from an overnight culture in RPMI medium first adjusted to an optical density (OD) of 0.12. Cultures were incubated at 35°C with shaking, and OD readings were taken over a 48-h period. The experiments were replicated two times.
Synergy testing by checkerboard assay.
DIBI and FLC were tested in a checkerboard assay in 96-well plates in a similar manner to that described by Fiori and Van Dijck (32), with the following modifications. The DIBI concentrations tested ranged from 0.5 to 128 μg/ml and the range of FLC was from 0.12 to 8 μg/ml. Microtiter plates were inoculated with strains 96113 and Ca5031 as described above for azole MIC tests and incubated at 35°C for 48 h. Synergy was defined as an FICI of ≤0.5 (50) using MIC80 values. For additional visualization of checkerboard results, a multichannel pipette was used to spot 5 μl from each well of the checkerboard assay plate onto a recovery YPD agar plate (32) at 24 and 48 h postexposure. Recovery plates were incubated at 35°C and were inspected after 24 h of incubation. The experiments were repeated a minimum of two times.
Azole improvement by measuring replicative growth.
The beneficial effects of DIBI with FLC, ITC, and CLT were examined using a growth curve procedure. Test media were inoculated with C. albicans prepared as described above to obtain an initial viable count of approximately 103 CFU/ml in the following medium combinations: RPMI alone, RPMI plus DIBI alone (1 μg/ml for 96113 and 10 μg/ml for LP1158-07, Ca3969, and Ca5031), RPMI plus azole alone (FLC at 1.0 μg/ml for 96113 and 128 μg/ml for LP1158-07; ITC at 0.25 μg/ml for Ca5031 and 0.5 μg/ml for Ca3969; CLT at 0.03 μg/ml for both Ca5031 and Ca3969), and RPMI plus DIBI plus azole (concentrations as above). The cultures were incubated for 72 h at 35°C with shaking. Viable counts were measured every 24 h. Each experiment was replicated 3 times.
Murine vaginitis model.
Female C3H/HeNCrl mice, 8 to 10 weeks old and weighing ∼20 g, were obtained from Charles River Laboratories (Wilmington, MA). All animals were housed and handled according to institutionally recommended guidelines. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the Louisiana State University Health Sciences Center (LSUHSC). The estrogen-dependent model has been described previously (51). Briefly, animals (n = 6/group) were injected subcutaneously with estradiol valerate (0.1 mg dissolved in 100 μl sesame oil) 3 days prior to and 4 days after vaginal inoculation. One day prior to inoculation, a blastospore culture of the C. albicans isolate(s) to be used in each study was prepared. On the day of inoculation, blastospores were collected and washed once with phosphate-buffered saline (PBS) and resuspended at 2.5 × 106/ml in PBS for an inoculum of 5 × 104 cells/20 μl PBS. For inoculation, animals were anesthetized “to effect” by isoflurane inhalation. To anesthetized animals, 5 × 104 blastospores in 20 μl PBS were pipetted directly into the vagina, using a Pipetman. Topical treatments of 5% polyvinylpyrrolidone (PVP) vehicle only or 4% or 8% DIBI (40 or 80 mg/kg, respectively) dissolved in vehicle (20 μl) were given once daily starting on day 3 and continued through day 6. Fluconazole (25 or 50 mg/kg) or PBS vehicle was given via oral gavage on days 3 and 5. Animals given both DIBI and fluconazole followed the same treatment regimens. On day 7, animals were sacrificed and the vaginal cavity was lavaged with 100 μl of PBS. The lavage fluid was examined microscopically for yeast/hyphae, qualitatively scored (0 to 4 based on volume of organisms present), and serially diluted and cultured for quantitative enumeration of organisms (expressed as CFU/100 μl lavage fluid).
Statistical analysis.
The data are reported as means ± standard errors. For in vitro studies, the data were analyzed using a two-way analysis of variance (ANOVA) with Bonferroni's posttest (GraphPad Prism software). For in vivo studies, the data were analyzed using the Mann-Whitney U test (GraphPad Prism software). Additional one-way ANOVA, Tukey, and chi-square analyses were performed for clearance data using SAS 9.4 software. Significant differences were defined as a P value of <0.05.
ACKNOWLEDGMENTS
This work was supported by funding from the Government of Canada (Industrial Research Assistance Program and Atlantic Canada Opportunities Agency, Productivity and Business Skills Initiative) and from BioTalent Canada.
We thank Jasmine Boudreau for assisting with the in vitro studies.
B.E.H. has a beneficial interest in Chelation Partners Inc., and K.A.S., M.D.C.P., and D.S.A. are employees of this company. None of the other authors have competing interests in relation to the work presented.
Footnotes
[This article was published on 27 July 2018 with the second author's surname incorrectly indexed as "del Carmen Parquet" instead of "Parquet." This has been corrected in the current version, posted on 7 December 2018.]
REFERENCES
- 1.Pfaller MA, Diekema DJ. 2007. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States, 2013. Publication C5239559-B Centers for Disease Control and Prevention, Atlanta, GA: https://www.cdc.gov/drugresistance/threat-report-2013/. [Google Scholar]
- 3.Jayatilake JAMS. 2011. A review of the ultrastructural features of superficial candidiasis. Mycopathologia 171:235–250. doi: 10.1007/s11046-010-9373-7. [DOI] [PubMed] [Google Scholar]
- 4.Garber G. 2001. An overview of fungal infections. Drugs 61(Suppl 1):1–12. doi: 10.2165/00003495-200161001-00001. [DOI] [PubMed] [Google Scholar]
- 5.Bondaryk M, Kurzątkowski W, Staniszewska M. 2013. Antifungal agents commonly used in the superficial and mucosal candidiasis treatment: mode of action and resistance development. Postepy Dermatol Alergol 30:293–301. doi: 10.5114/pdia.2013.38358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sobel JD. 2007. Vulvovaginal candidosis. Lancet 369:1961–1971. doi: 10.1016/S0140-6736(07)60917-9. [DOI] [PubMed] [Google Scholar]
- 7.Garvey EP, Hoekstra WJ, Schotzinger RJ, Sobel JD, Lilly EA, Fidel PL Jr. 2015. Efficacy of the clinical agent VT-1161 against fluconazole-sensitive and -resistant Candida albicans in a murine model of vaginal candidiasis. Antimicrob Agents Chemother 59:5567–5573. doi: 10.1128/AAC.00185-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cowen LE, Anderson JB, Kohn LM. 2002. Evolution of drug resistance in Candida albicans. Annu Rev Microbiol 56:139–165. doi: 10.1146/annurev.micro.56.012302.160907. [DOI] [PubMed] [Google Scholar]
- 9.Martel CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AGS, Kelly DE, Kelly SL. 2010. A clinical isolate of Candida albicans with mutations in ERG11 (encoding sterol 14alpha-demethylase) and ERG5 (encoding C22 desaturase) is cross resistant to azoles and amphotericin B. Antimicrob Agents Chemother 54:3578–3583. doi: 10.1128/AAC.00303-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oxman DA, Chow JK, Frendl G, Hadley S, Hershkovitz S, Ireland P, McDermott LA, Tsai K, Marty FM, Kontoyiannis DP, Golan Y. 2010. Candidaemia associated with decreased in vitro fluconazole susceptibility: is Candida speciation predictive of the susceptibility pattern? J Antimicrob Chemother 65:1460–1465. doi: 10.1093/jac/dkq136. [DOI] [PubMed] [Google Scholar]
- 11.Marchaim D, Lemanek L, Bheemreddy S, Kaye KS, Sobel JD. 2012. Fluconazole-resistant Candida albicans vulvovaginitis. Obstet Gynecol 120:1407–1414. doi: 10.1097/AOG.0b013e31827307b2. [DOI] [PubMed] [Google Scholar]
- 12.Arendrup MC. 2010. Epidemiology of invasive candidiasis. Curr Opin Crit Care 16:445–452. doi: 10.1097/MCC.0b013e32833e84d2. [DOI] [PubMed] [Google Scholar]
- 13.Netea MG, Brown GD, Kullberg BJ, Gow NAR. 2008. An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol 6:67–78. doi: 10.1038/nrmicro1815. [DOI] [PubMed] [Google Scholar]
- 14.Weinberg ED. 2009. Iron availability and infection. Biochim Biophys Acta 1790:600–605. doi: 10.1016/j.bbagen.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Skaar EP. 2010. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog 6:e1000949. doi: 10.1371/journal.ppat.1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caza M, Kronstad JW. 2013. Shared and distinct mechanisms of iron acquisition by bacterial and fungal pathogens of humans. Front Cell Infect Microbiol 3:80. doi: 10.3389/fcimb.2013.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamad M, Awadallah S. 2013. Estrogen-dependent changes in serum iron levels as a translator of the adverse effects of estrogen during infection: a conceptual framework. Med Hypotheses 81:1130–1134. doi: 10.1016/j.mehy.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Hou Y, Zhang S, Wang L, Li J, Qu G, He J, Rong H, Ji H, Liu S. 2012. Estrogen regulates iron homeostasis through governing hepatic hepcidin expression via an estrogen response element. Gene 511:398–403. doi: 10.1016/j.gene.2012.09.060. [DOI] [PubMed] [Google Scholar]
- 19.Jerse AE, Wu H, Packiam M, Vonck RA, Begum AA, Garvin LE. 2011. Estradiol-treated female mice as surrogate hosts for Neisseria gonorrhoeae genital tract infections. Front Microbiol 2:107. doi: 10.3389/fmicb.2011.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahman D, Mistry M, Thavaraj S, Challacombe SJ, Naglik JR. 2007. Murine model of concurrent oral and vaginal Candida albicans colonization to study epithelial host-pathogen interactions. Microbes Infect 9:615–622. doi: 10.1016/j.micinf.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mishra NN, Prasad T, Sharma N, Payasi A, Prasad R, Gupta DK, Singh R. 2007. Pathogenicity and drug resistance in Candida albicans and other yeast species: a review. Acta Microbiol Immunol Hung 54:201–235. doi: 10.1556/AMicr.54.2007.3.1. [DOI] [PubMed] [Google Scholar]
- 22.Noble SM. 2013. Candida albicans specializations for iron homeostasis: from commensalism to virulence. Curr Opin Microbiol 16:708–715. doi: 10.1016/j.mib.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neilands JB. 1974. Microbial iron metabolism. Academic Press, New York, NY. [Google Scholar]
- 24.Ballouche M, Cornelis P, Baysse C. 2009. Iron metabolism: a promising target for antibacterial strategies. Recent Pat Antiinfect Drug Discov 4:190–205. doi: 10.2174/157489109789318514. [DOI] [PubMed] [Google Scholar]
- 25.Baillie GS, Douglas LJ. 1998. Iron-limited biofilms of Candida albicans and their susceptibility to amphotericin B. Antimicrob Agents Chemother 42:2146–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bullen JJ, Rogers HJ, Spalding PB, Ward CG. 2006. Natural resistance, iron and infection: a challenge for clinical medicine. J Med Microbiol 55:251–258. doi: 10.1099/jmm.0.46386-0. [DOI] [PubMed] [Google Scholar]
- 27.Kuipers ME, de Vries HG, Eikelboom MC, Meijer DKF, Swart PJ. 1999. Synergistic fungistatic effects of lactoferrin in combination with antifungal drugs against clinical Candida isolates. Antimicrob Agents Chemother 43:2635–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi T, Kakeya H, Miyazaki T, Izumikawa K, Yanagihara K, Ohno H, Yamamoto Y, Tashiro T, Kohno S. 2011. Synergistic antifungal effect of lactoferrin with azole antifungals against Candida albicans and a proposal for a new treatment method for invasive candidiasis. Jpn J Infect Dis 64:292–296. [PubMed] [Google Scholar]
- 29.Holbein BE, Feng M, Huber AL, Kidby DK. June 2011. Polymeric metal chelating compositions and methods of preparing same for controlling growth and activities of living cells and organisms.International patent WO2012167368 A1.
- 30.Holbein BE, Mira de Orduña R. 2010. Effect of trace iron levels and iron withdrawal by chelation on the growth of Candida albicans and Candida vini. FEMS Microbiol Lett 307:19–24. doi: 10.1111/j.1574-6968.2010.01956.x. [DOI] [PubMed] [Google Scholar]
- 31.Pfaller MA, Diekema DJ. 2012. Progress in antifungal susceptibility testing of Candida spp. by use of clinical and laboratory standards institute broth microdilution methods, 2010 to 2012. J Clin Microbiol 50:2846–2856. doi: 10.1128/JCM.00937-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fiori A, Van Dijck P. 2012. Potent synergistic effect of doxycycline with fluconazole against Candida albicans is mediated by interference with iron homeostasis. Antimicrob Agents Chemother 56:3785–3796. doi: 10.1128/AAC.06017-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson MG, Corey BW, Si Y, Craft DW, Zurawski DV. 2012. Antibacterial activities of iron chelators against common nosocomial pathogens. Antimicrob Agents Chemother 56:5419–5421. doi: 10.1128/AAC.01197-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zarember KA, Cruz AR, Huang CY, Gallin JI. 2009. Antifungal activities of natural and synthetic iron chelators alone and in combination with azole and polyene antibiotics against Aspergillus fumigatus. Antimicrob Agents Chemother 53:2654–2656. doi: 10.1128/AAC.01547-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi W, Chen Z, Chen X, Cao L, Liu P, Sun S. 2010. The combination of minocycline and fluconazole causes synergistic growth inhibition against Candida albicans: an in vitro interaction of antifungal and antibacterial agents. FEMS Yeast Res 10:885–893. doi: 10.1111/j.1567-1364.2010.00664.x. [DOI] [PubMed] [Google Scholar]
- 36.Greenberger NJ. 1971. Absorption of tetracyclines: interference by iron. Ann Intern Med 74:792–793. doi: 10.7326/0003-4819-74-5-792. [DOI] [PubMed] [Google Scholar]
- 37.Baker WA Jr, Brown PM. 1966. Metal binding in tetracyclines. Cobalt(II) and nickel(II) complexes. J Am Chem Soc 88:1314–1317. doi: 10.1021/ja00958a041. [DOI] [PubMed] [Google Scholar]
- 38.Lambs L, Decock-Le Révérend B, Kozlowski H, Berthon G. 1988. Metal ion-tetracycline interactions in biological fluids. 9. Circular dichroism spectra of calcium and magnesium complexes with tetracycline, oxytetracycline, doxycycline, and chlortetracycline and discussion of their binding modes. Inorg Chem 27:3001–3012. doi: 10.1021/ic00290a022. [DOI] [Google Scholar]
- 39.Prasad T, Chandra A, Mukhopadhyay CK, Prasad R. 2006. Unexpected link between iron and drug resistance of Candida spp.: iron depletion enhances membrane fluidity and drug diffusion, leading to drug-susceptible cells. Antimicrob Agents Chemother 50:3597–3606. doi: 10.1128/AAC.00653-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yokoyama T, Nakano K, Zenki M. 1999. Specific separation of nickel ion with bathophenanthroline disulfonic acid by capillary zone electrophoresis. Anal Chim Acta 396:117–123. doi: 10.1016/S0003-2670(99)00504-8. [DOI] [Google Scholar]
- 41.Bellmann R, Smuszkiewicz P. 2017. Pharmacokinetics of antifungal drugs: practical implications for optimized treatment of patients. Infection 45:737–779. doi: 10.1007/s15010-017-1042-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Craven RJ, Mallory JC, Hand RA. 2007. Regulation of iron homeostasis mediated by the heme-binding protein Dap1 (damage resistance protein 1) via the P450 protein Erg11/Cyp51. J Biol Chem 282:36543–36551. doi: 10.1074/jbc.M706770200. [DOI] [PubMed] [Google Scholar]
- 43.Lamb DC, Kelly DE, White TC, Kelly SL. 2000. The R467K amino acid substitution in Candida albicans sterol 14α-demethylase causes drug resistance through reduced affinity. Antimicrob Agents Chemother 44:63–67. doi: 10.1128/AAC.44.1.63-67.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marichal P, Vanden Bossche H. 1995. Mechanisms of resistance to azole antifungals. Acta Biochim Pol 42:509–516. [PubMed] [Google Scholar]
- 45.Vanden Bossche H. 1997. Mechanisms of antifungal resistance. Rev Iberoam Micol 14:44–49. [PubMed] [Google Scholar]
- 46.Cannon RD, Lamping E, Holmes AR, Niimi K, Tanabe K, Niimi M, Monk BC. 2007. Candida albicans drug resistance another way to cope with stress. Microbiology 153:3211–3217. doi: 10.1099/mic.0.2007/010405-0. [DOI] [PubMed] [Google Scholar]
- 47.Liu TT, Lee REB, Barker KS, Lee RE, Wei L, Homayouni R, Rogers PD. 2005. Genome-wide expression profiling of the response to azole, polyene, echinocandin, and pyrimidine antifungal agents in Candida albicans. Antimicrob Agents Chemother 49:2226–2236. doi: 10.1128/AAC.49.6.2226-2236.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard—3rd ed CLSI document M27-A3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 49.Clinical and Laboratory Standards Institute. 2012. Reference method for broth dilution antifungal susceptibility testing of yeasts; 4th informational supplement. CLSI document M27-S4. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 50.Odds FC. 2003. Synergy, antagonism, and what the chequerboard puts between them. J Antimicrob Chemother 52:1. doi: 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- 51.Fidel PL Jr, Cutright JL, Sobel JD. 1997. Efficacy of D0870 treatment of experimental Candida vaginitis. Antimicrob Agents Chemother 41:1455–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]