

Neutrophil extracellular traps (NETs) are produced by neutrophils as an innate immune defense mechanism to trap and kill microbial pathogens. NETs are comprised of ejected chromatin that forms a lattice structure enmeshed with numerous antimicrobial proteins.
KEYWORDS: Pseudomonas aeruginosa, deoxyribonuclease, neutrophil extracellular trap, phosphatase
ABSTRACT
Neutrophil extracellular traps (NETs) are produced by neutrophils as an innate immune defense mechanism to trap and kill microbial pathogens. NETs are comprised of ejected chromatin that forms a lattice structure enmeshed with numerous antimicrobial proteins. In addition to forming the structural backbone of NETs, extracellular DNA (eDNA) has membrane-disrupting antimicrobial activity that contributes to NET killing. Many pathogens produce secreted extracellular DNases to evade the antimicrobial activity of NETs. Pseudomonas aeruginosa encodes an operon of two secreted enzymes, a predicted alkaline phosphatase and a DNase. The DNase (eddB) degrades eDNA to use as a nutrient source. Here we report that both eDNA and NETs are potent inducers of this DNase-phosphatase operon. Furthermore, the secreted DNase contributes to degrading NET DNA and defends P. aeruginosa against NET-mediated killing. We demonstrate that EddA has both alkaline phosphatase and phosphodiesterase (PDase) activities and also protects against the antimicrobial activity of NETs. Although the phosphatase does not cause DNA degradation similar to that of the DNase, its protective function is likely a result of removing the cation-chelating phosphates from the eDNA phosphodiester backbone. Therefore, both the DNase and PDase contribute to defense against NET killing of P. aeruginosa, highlighting the role of DNA-manipulating enzymes in targeting the eDNA in neutrophil extracellular traps.
INTRODUCTION
As an opportunistic pathogen, Pseudomonas aeruginosa causes chronic infections in immunocompromised individuals, including those with the heritable genetic disease cystic fibrosis (CF). However, P. aeruginosa is also capable of infecting burn and diabetic foot wounds and causing nosocomial infections as well as eye and urinary tract infections. During chronic infection in the CF lung, the immune response is dominated by an influx and activation of neutrophils, in an effort to contain and clear P. aeruginosa (1, 2). The antimicrobial strategies employed by neutrophils include the phagocytosis of bacterial invaders, the release of toxic effectors, and the production of neutrophil extracellular traps (NETs) (3, 4). As recently described innate immune effectors, NETs are capable of ensnaring viruses, fungi, and both Gram-positive and Gram-negative bacteria (3, 4).
The deployment of NETs (a process termed NETosis) can result from a lytic (3) or a nonlytic (vital) (5, 6) pathway in neutrophils and consists of an ejected DNA lattice that is enmeshed with numerous neutrophil granule proteins. Lytic NETosis can be chemically stimulated with phorbol myristate acetate (PMA) treatment of isolated neutrophils, and vital NETosis has been reported in neutrophils with intact membranes, which are still capable of phagocytosis and chemotaxis (5). Extracellular traps with a DNA backbone have also been observed for other innate immune cells, including mast cells, eosinophils, and macrophages (7). NETs have been observed in P. aeruginosa eye (8) and skin (9) infection models as well as in sputum recovered from CF lungs (10–12). Although they perform the innate immune function of trapping and killing bacteria, excessive NET production contributes to airway obstruction, inflammation, and pathophysiology and has been observed under numerous noninfectious conditions (13, 14).
NET antimicrobial activity is generally attributed to the neutrophil granular and chromatin proteins that are localized to the DNA lattice (4). Antibodies against the neutrophil-bound proteins, such as histones or cathepsin G, have been used to reduce bacterial killing, demonstrating their direct role in killing (3, 15). We previously showed that DNA has a potent antimicrobial activity by chelating surface-bound cations, disrupting outer and inner membrane integrity, which results in rapid, lytic cell death (16). DNA also contributes to bacterial killing by NETs and can be neutralized with treatment of NETs with excess cations or by alkaline phosphatase (AP) treatment (9). The cation-chelating phosphodiester backbone of DNA is responsible for the antibacterial nature of DNA, and we hypothesize a model where phosphatase treatment blocks NET killing by removing phosphates from DNA but does not alter the NET structure or the function or localization of some NET effector proteins (9).
Several microbial pathogens have been shown to defend against NETs through diverse strategies. Streptococcus pneumoniae produces a capsule and d-alanylated lipoteichoic acid on the bacterial surface that reduces the trapping and killing of the bacteria in NETs (17). The secretion of extracellular nucleases that degrade the chromatin backbone of NETs is a conserved evasion strategy. A growing list of bacteria that employ nucleases includes the Gram-positive bacteria Staphylococcus aureus, S. pneumoniae, group A Streptococcus, and Streptococcus suis and, recently, the Gram-negative bacteria Vibrio cholerae, Neisseria gonorrhoeae, and Prevotella intermedia (18–22). DNase production allows pathogens to evade killing by degrading the NET backbone and diluting the concentration of toxic NET-bound effector proteins while also directly neutralizing the antimicrobial activity of DNA (9, 16). S. aureus produces a nuclease that degrades DNA and RNA to 5′-phosphomononucleotides and dinucleotides, which are subsequently converted to 2′-deoxyadenosine (dAdo) through the AdsA enzyme (23). The dAdo product is cytotoxic to macrophages and therefore protects S. aureus by restricting these innate immune cells from entering staphylococcal abscesses (23).
An early observation of CF patient pathology was that the CF sputum is highly enriched in DNA (24). Given the strong recruitment of neutrophils in the lungs of individuals suffering from cystic fibrosis (1, 2) and the direct observation of NETs in CF sputum (10–12), this suggests that much of the sputum DNA is derived from neutrophils. P. aeruginosa is a dominant pathogen in CF infections and demonstrates greater resistance to NET killing than S. aureus and Escherichia coli (9). P. aeruginosa promotes the tolerance of NETs through the modification of the outer surface lipopolysaccharide (LPS) charge by the addition of aminoarabinose to lipid A or by the production of surface spermidine, both of which mask the negatively charged surface groups (9, 25). These surface modifications also contribute to resistance to antimicrobial peptides and aminoglycosides, both cationic antibiotics that bind to bacterial surfaces with a negative charge, as a first step in disrupting the membrane or entering the cell (16, 25–27).
We previously identified a secreted DNase that is required for DNA degradation and the utilization of DNA as a phosphate source (28). The DNase EddB (extracellular DNA degradation protein B) is downstream of a predicted alkaline phosphatase (16, 29). Since the operon is induced by phosphate limitation (28), it is likely that these two enzymes contribute to phosphate acquisition. Here we test the hypothesis that these secreted enzymes are involved in defense against the DNA component of neutrophil extracellular traps.
RESULTS
Phosphodiesterase-DNase operon induced by eDNA and NETs.
We previously demonstrated that the P. aeruginosa xcp type II secretion system is required for the secretion of a DNase capable of degrading extracellular DNA (eDNA) (28). The DNase EddB, encoded by PA3909, promotes P. aeruginosa growth when DNA is supplied as the sole phosphate source, and DNA can be used as a source of carbon and nitrogen (28). Interestingly, the upstream open reading frame (ORF) PA3910 is predicted to encode an alkaline phosphatase, EddA, and shares a promoter with eddB, forming a bicistronic operon (16, 29). EddA contains a predicted signal peptide for the twin-arginine translation (TAT) pathway, which transports folded proteins across the inner membrane, and uses the Xcp pathway to cross the outer membrane (30). Alkaline phosphatase proteins fall into 3 groups: PhoX, PhoA, and PhoD-type phosphatases (31). EddA shares ∼49% homology with Bacillus subtilis PhoD, the archetypal secreted alkaline phosphatase (32). Interestingly, PhoD phosphatases possess both monoesterase as well as phosphodiesterase (PDase) activities (30, 31). Moreover, EddA homologs, such as the Zymomonas mobilis CP4 PhoD alkaline phosphatase, have been shown to hydrolyze nucleotides more effectively than sugar phosphates (33).
Given that eddAB are both likely involved in utilizing DNA as a nutrient, we assessed whether their expression is regulated by the presence of extracellular DNA (Fig. 1). Quantitative reverse transcription-PCR (qRT-PCR) analysis of eddB expression revealed that this gene is highly induced in the presence of low levels of eDNA after coincubation for 60 and 120 min (Fig. 1A) (28). During infection, P. aeruginosa encounters and must defend against neutrophil extracellular traps, particularly in the CF lung. We sought to extend our analysis to determine whether NETs were also capable of inducing the expression of these genes. We used PMA treatment to induce the production of NETs in human neutrophils, which also induced the expression of eddAB to comparable levels when exposed to pure DNA (Fig. 1A). There was no observable growth (optical density at 600 nm [OD600]) during these quantitative RT-PCR (qRT-PCR) experiments, as P. aeruginosa was suspended in buffers that lack all nutrients. Gene expression was normalized to the expression of the 16S rRNA genes, which can normalize for any subtle differences in growth in the presence of DNA or NETs.
FIG 1.
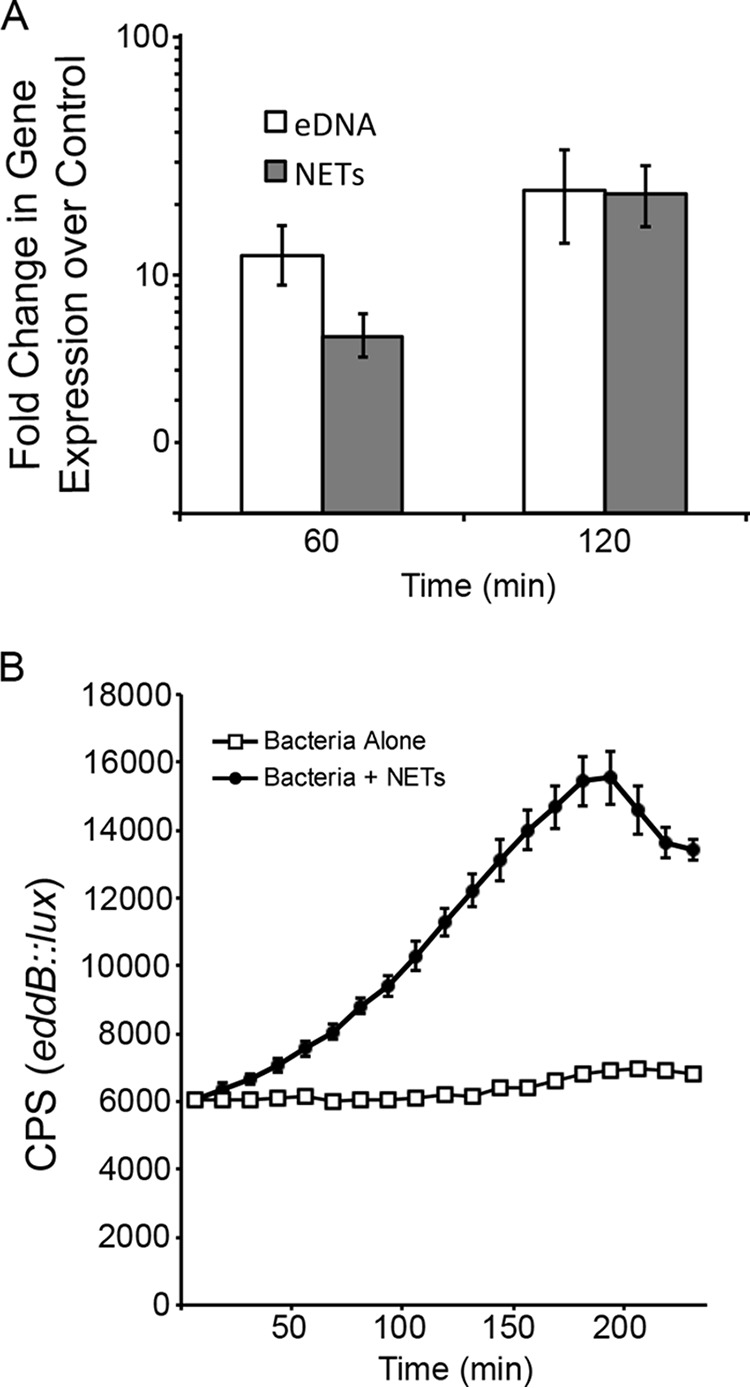
Expression of the eddAB operon is induced by eDNA and exposure to neutrophil extracellular traps. (A) qRT-PCR was used to measure eddB expression during exposure of P. aeruginosa to 0.2% eDNA or NETs. Bacteria were resuspended in 10 mM Tris buffer (pH 7) with 0.2% DNA or in HBSS with human neutrophils (MOI of 4:1) that were PMA stimulated to induce NET formation. After incubation, total bacterial RNA was extracted, converted to cDNA, and used as the template in qRT-PCR analyses, where values shown are the means and standard deviations from 8 replicates. Expression levels under the test conditions were compared to those under control conditions without eDNA or NETs and normalized to the expression level of the 16S rRNA housekeeping gene. (B) The P. aeruginosa eddB::lux reporter strain was grown in the absence or presence of PMA-activated human neutrophils (MOI of 10:1), and gene expression (counts per second) was measured every 20 min. The values shown are the means and standard deviations from 6 replicates.
We confirmed the dynamic nature of the NET-induced gene expression pattern by utilizing the P. aeruginosa eddB::lux reporter strain in coincubation experiments with neutrophils that were PMA stimulated to produce NETs (Fig. 1B). P. aeruginosa appears to sense the DNA in a NET and increase the gene expression level of an operon that produces a secreted phosphatase and DNase.
Secreted DNase and phosphodiesterase activities.
We wanted to confirm the predicted enzymatic function of EddA and EddB, both of which are secreted extracellularly by P. aeruginosa. EddB was previously shown to degrade bacterial genomic DNA in the presence of excess Ca2+ and Mg2+ (28). To quantitate DNA degradation, we used a fluorescence-based DNA degradation assay using bacterial supernatants as the source of bacterial enzymes. Bacterial supernatants from P. aeruginosa lacking either the Xcp type II secretion system or PA3909 (eddB) were strongly impaired for the degradation of Sytox green-labeled extracellular salmon sperm DNA, relative to the control (Fig. 2A). Complementation with pEddB restored the capacity of P. aeruginosa eddB::lux to degrade eDNA, with a slight delay in the degradation kinetics (Fig. 2A).
FIG 2.
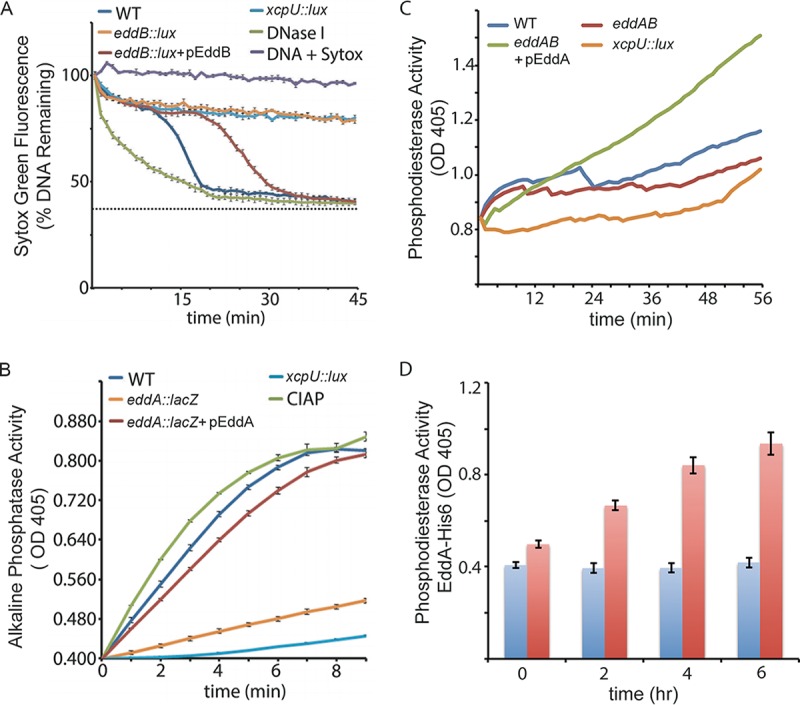
DNase and phosphatase activities of EddB and EddA. (A) DNase activity measured in culture supernatants that were incubated with 1 μg DNA and 2.5 μM Sytox green. Supernatants were isolated from cultures grown in limiting-phosphate BM2 medium. Green fluorescence was measured every minute from black-well, clear-bottom, 96-well microplates. Values shown are the means and standard deviations from quadruplicate samples. WT, wild type. (B) Alkaline phosphatase activity measured in culture supernatants incubated with the NBT/BCIP monoesterase substrate in alkaline phosphatase buffer. The colored AP reaction product was measured by monitoring the OD405. (C and D) Phosphodiesterase activity measured in culture supernatants (C) or from purified EddA-His6 (red bars) incubated with 25 mM BNPP compared to the negative-control sample with no EddA-His6 (blue bars) (D). Values shown are means and standard deviations from triplicate samples.
To assess the phosphatase activity of EddA, we utilized a colorimetric assay in which bacterial supernatants were added to the AP (monoesterase) substrate nitroblue tetrazolium–5-bromo-4-chloro-3-indolylphosphate (NBT/BCIP). Coincubation of wild-type P. aeruginosa bacterial supernatants with NBT/BCIP led to a rapid increase in the amount of the cleaved substrate (Fig. 2B). Monoesterase activity was strongly dependent on the presence of a functional Xcp type II secretion system and on PA3910/EddA. Complementation with pEddA restored the capacity of P. aeruginosa eddA::lacZ to cleave the AP substrate (Fig. 2B).
We wanted to determine if EddA also had phosphodiesterase activity, which would support the cleavage of phosphates from DNA. To detect secreted phosphodiesterase activity, cell-free culture supernatants were incubated with the phosphodiesterase substrate bis(p-nitrophenyl)phosphate (BNPP) (31). The type II secretion mutant had low levels of phosphodiesterase activity (Fig. 2C), which were similar to those of wild-type PAO1 grown under phosphate-rich (1,600 μM) conditions (data not shown). The eddAB double mutant showed a modest defect for phosphodiesterase activity, which was restored to higher-than-parental levels when overexpressing pEddA from a plasmid (Fig. 2C). In both the DNase and phosphatase assays, there appeared to be low levels of activity remaining in the supernatants of eddA and eddB mutants, suggesting the presence of other enzymes with these activities (Fig. 2A to C).
EddA was cloned as a C-terminally His-tagged protein (EddA-His6), which permitted the purification of protein using HisPur cobalt resin. Purified EddA-His6 was incubated with the phosphodiesterase substrate BNPP and demonstrated enzyme activity (Fig. 2D). Consistent with other PhoD-type phosphatases, EddA has both monoesterase and phosphodiesterase activities, which supports the hypothesis that EddA is capable of the cleavage of phosphates from the DNA backbone.
DNase but not phosphatase dissolves neutrophil extracellular traps.
NET killing of P. aeruginosa can be neutralized with treatments that specifically target the DNA component of the NETs (9). Exogenous DNase is frequently used to dissolve NETs and block bacterial killing (3, 9). Therefore, in an attempt to neutralize the cation-chelating activity of DNA but to retain the overall NET structure, NETs were treated with calf intestinal alkaline phosphatase (CIAP) (9). Phosphatase treatment blocked NET killing of P. aeruginosa and E. coli and blocked bacterial killing with pure DNA but did not alter NET structure or function or the localization of NET effectors (9). These experiments confirmed that the phosphates of DNA were responsible for the antimicrobial activity of DNA in a neutrophil extracellular trap. Therefore, we hypothesized that the EddA phosphodiesterase might also protect P. aeruginosa from NETs.
To assess whether these enzymes differed in their capacities to degrade the DNA backbone of the NETs, we performed a NET degradation assay using the same Sytox green-based approach used with pure DNA (Fig. 2A) (20). We stimulated NETosis by the use of PMA and monitored NET production using fluorescence measurements of Sytox green incorporation into NETs. After 200 min of PMA induction, NETs were formed, and bacteria were introduced for coincubation with NETs to assess their effects on the total DNA fluorescence (Fig. 3). As predicted, wild-type P. aeruginosa was capable of the full degradation of NETs within 7 h after addition (Fig. 3). Disruption of either the xcp type II secretion system or the eddAB enzymes resulted in the stability of NETs (Fig. 3). Complementation of the ΔeddAB double mutant with plasmid-encoded pEddB restored the NET degradation capacity of the ΔeddAB mutant to a level comparable to that of the wild-type parental strain (Fig. 3). Exogenous DNase was the most effective treatment for degrading NETs. However, complementation of the ΔeddAB double mutant with the plasmid-encoded pEddA phosphodiesterase had no effect on degrading the DNA from PMA-stimulated neutrophils (Fig. 3). We conclude that only the DNase can cause significant damage to and degradation of the NET structure.
FIG 3.
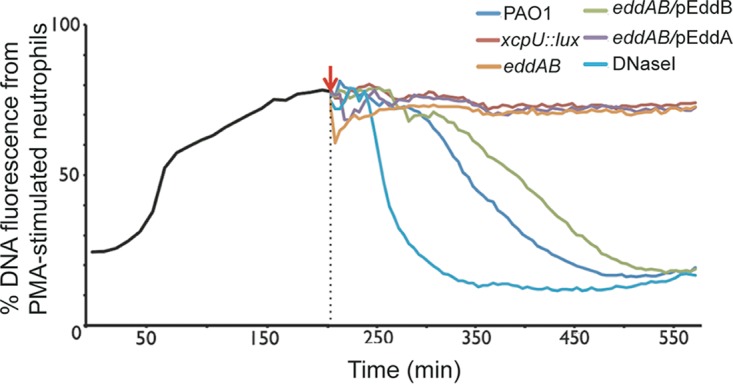
Degradation of DNA in neutrophil extracellular traps by secreted DNase from P. aeruginosa. DNA release from 1 × 106 neutrophils was stimulated with PMA, and the resulting NET DNA was stained with the cell-impermeant fluorescent DNA dye Sytox green. An increase in NET formation was seen up to 200 min, at which time P. aeruginosa cultures were added to the NETs to determine their effect on DNA integrity (red arrow). Staining of NET DNA with Sytox green was measured in 10-min intervals, and the values shown are the means and standard deviations from 8 replicates. The percentages of DNA fluorescence are relative to the value for the Triton X-100-treated neutrophil positive control (100% lysis) (n = 3).
Phosphatase and DNase are required to defend against NETs.
Since it is common among bacteria to produce secreted DNases that degrade and disarm NETs (18–22), we tested the role of the EddA phosphatase and EddB DNase in protecting P. aeruginosa from neutrophil extracellular traps. Neutrophils were stimulated with PMA and exposed to P. aeruginosa wild-type and mutant cultures, and viable counts were determined to monitor bacterial viability. Interestingly, the xcp type II secretion system mutant demonstrated reduced tolerance to PMA-stimulated NETs (Fig. 4A). This result suggests that enzymes secreted by the type II secretion system are required to protect P. aeruginosa from NETs. The eddAB double mutant showed increased susceptibility to NET killing, and complementation with either pEddA or pEddB expressed in trans restored P. aeruginosa survival (Fig. 4B). While neither enzyme alone restored wild-type levels of NET tolerance, the enzymes likely work together in wild-type strain PAO1. Interestingly, the bacterial phosphodiesterase EddA significantly increased the survival of P. aeruginosa when exposed to NETs; however, it seems incapable of robust NET degradation relative to EddB (Fig. 3 and 4B). This protective function is congruent with the previous observation that NETs are no longer antimicrobial after treatment with CIAP (9).
FIG 4.
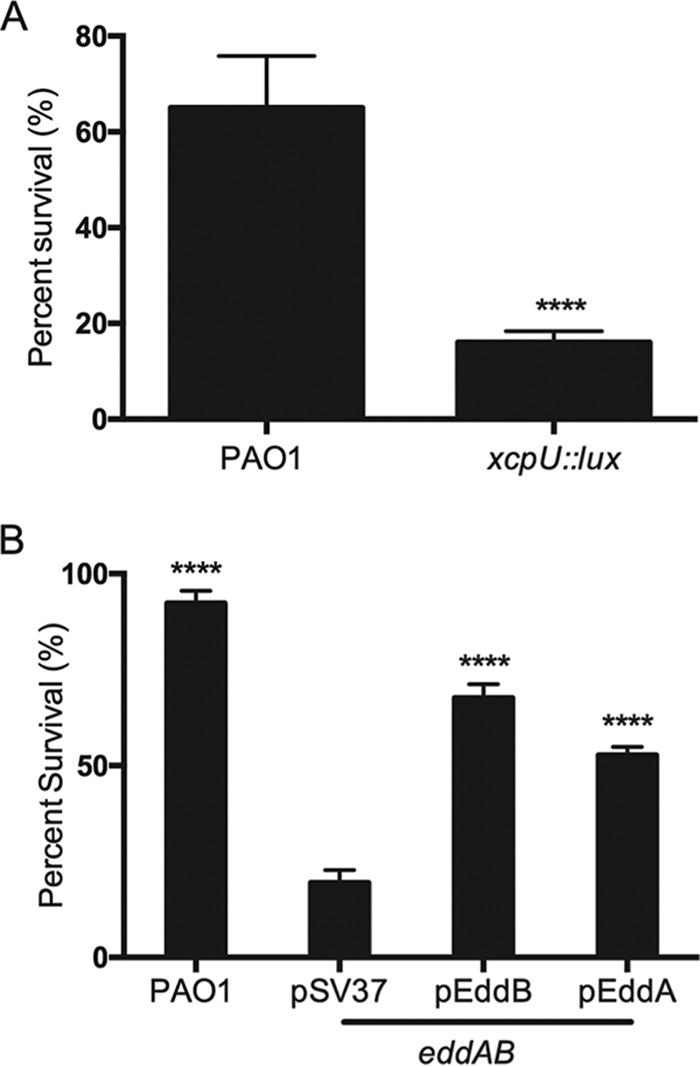
P. aeruginosa survival after exposure to neutrophil extracellular traps. A total of 1 × 107 CFU of P. aeruginosa bacteria were incubated with PMA-treated neutrophils (MOI of 10:1), and bacterial viability was determined by direct plate counts (CFU per milliliter) before and after 4 h of incubation with NETs. Bacterial counts were normalized to counts in the absence of neutrophils. DNase I was added exogenously 30 min prior to the end of the experiment to degrade NETs and ensure accurate counts of recoverable colonies. Data are presented as the average percentages of bacterial survival, and error bars represent the standard deviations from 6 replicates (n = 3). (A) Wild-type PAO1 survival compared to that of the xcpU::lux type II secretion mutant. Significant differences were determined by using the unpaired t test (****, P < 0.0001). (B) Bacterial survival of wild-type PAO1 and the eddAB double mutant, which was complemented with pEddA or pEddB. Significant differences compared to the double mutant were determined by one-way ANOVA with Bonferroni posttests (****, P < 0.0001).
DNase activity in culture supernatants of cystic fibrosis isolates.
The CF lung is highly enriched in eDNA of neutrophil origin, which is the reason why inhaled recombinant DNase is used to thin CF sputum and alleviate infection symptoms (34). Therefore, we sought to characterize whether P. aeruginosa epidemic strains isolated from cystic fibrosis patients shared the capacity to degrade DNA. We obtained a panel of transmissible P. aeruginosa isolates from the Adult Cystic Fibrosis Clinic culture collection at the University of Calgary (35) and assessed their capacity to degrade DNA using the Sytox green-labeled DNA assay (Fig. 5). CF isolates produced a spectrum of activity ranging from high to low levels of DNA degradation (Fig. 5). However, 12 out of the 36 CF isolates tested did not have the capacity to degrade extracellular DNA. This phenotypic diversity is common among CF isolates, despite relatively similar genetic backgrounds, and is due to the complex selection pressure in the CF lung and the accumulation of loss-of-function mutations.
FIG 5.
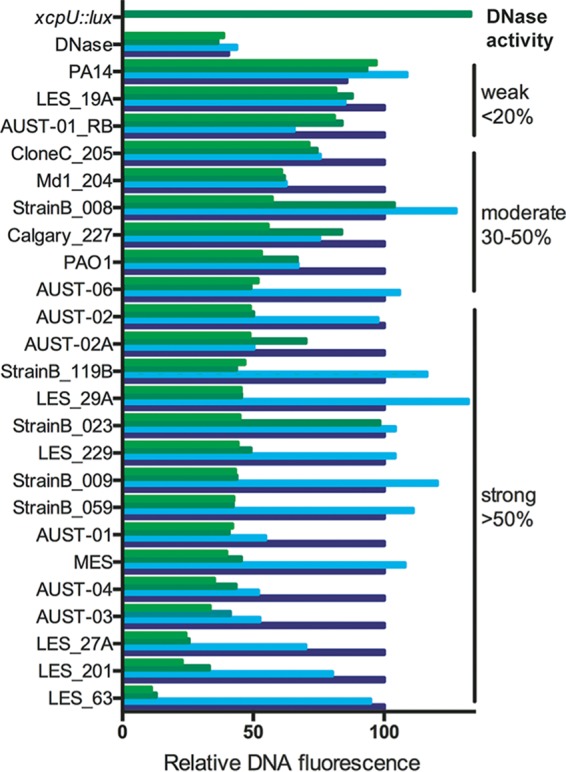
Clinical CF Pseudomonas aeruginosa isolates produce extracellular DNase activity under phosphate-limiting conditions. Cell-free supernatants were collected from cultures grown in BM2 medium with a range of phosphate concentrations, including 200 μM (light green), 400 μM (dark green), 800 μM (light blue), and 1,600 μM (dark blue). Supernatants were coincubated with Sytox green-stained DNA, and green fluorescence was measured in 10-min intervals. Relative DNA fluorescence values were derived by calculating the decrease in DNA fluorescence compared that under phosphate-rich conditions of 1,600 μM phosphate (t = 3 h). Pure DNase I (3 U) was used as a positive control, and the xcpU::lux mutant was used as a negative-control strain.
DISCUSSION
In order to combat bacterial infection, NETs are deployed by neutrophils in numerous infection sites, including the CF lung (10–12). Although many proteins enmeshed in NETs (histones, cathepsin G, and calprotectin) have demonstrated antimicrobial properties (3, 4), here we focus on bacterial defense against the antimicrobial activity of DNA. The DNA backbone itself also contributes to the broad-spectrum antibacterial mechanism of NETs, through a mechanism of disrupting membrane integrity by chelating bacterial membrane-bound divalent cations (9, 16). The concentration of DNA in CF sputum can reach as high as 20 mg/ml, a level sufficient to promote membrane destabilization and rapid lysis of P. aeruginosa (16, 36, 37). This toxic effect exerted by eDNA may explain why multiple species, including CF P. aeruginosa isolates, share a virulence strategy of secreting DNA-degrading enzymes (Fig. 5).
Here we demonstrate that the Gram-negative, opportunistic pathogen P. aeruginosa employs two DNA-modifying type II secreted enzymes, the phosphodiesterase EddA and the DNase EddB, to promote bacterial evasion of NETs. Although we demonstrate that secreted EddA possesses alkaline phosphatase and phosphodiesterase activities (Fig. 2) and can promote bacterial viability (Fig. 4), the enzyme does not degrade NETs in our fluorescence-based assay (Fig. 3). The bacterial phosphatase EddA behaves similarly to calf intestinal alkaline phosphatase, which also blocks NET killing without disrupting the NET structure (9). Therefore, we propose that the bacterial phosphodiesterase EddB cleaves phosphates from DNA in a NET and neutralizes its cation-chelating, antimicrobial activity.
The DNase-PDase operon seems to have a dual role in phosphate acquisition (28) and in protection against neutrophil extracellular traps. It is also possible that secreted phosphatases cleave phosphates from the bacterial cell surface, as some enzymes can be surface localized (38). The removal of cell surface phosphates might limit DNA binding and therefore protect the outer membrane from DNA-mediated killing, which normally sequesters surface-bound cations and disturbs outer and inner membrane integrity (9).
Phosphate limitation is a nutrient cue shared by multiple Gram-negative bacteria to produce phosphate acquisition pathways, including phosphatases and phosphate uptake systems (39). The pho regulon is not only required for phosphate homeostasis but is also an important stress response pathway that contributes to bacterial virulence and cytotoxicity (39, 40). The DNase was originally shown to be regulated under phosphate limitation (28); however, limiting phosphate levels does not solely regulate the expression of this operon. We also detected expression or secreted enzyme activity in rich brain heart infusion (BHI) medium (Fig. 4B) and in the presence of pure eDNA or NETs, respectively (Fig. 1). Others have suggested a virulence role for the P. aeruginosa phosphodiesterase-DNase operon, due to its induced expression upon exposure to human airway epithelial cells (41). These observations highlight that multiple host cues can induce the expression of these virulence factors, and they are likely expressed in the CF lung.
Although the lower airways during chronic CF infections are colonized by complex multispecies communities referred to as the CF lung microbiome, P. aeruginosa remains a dominant pathogen (42). Chronic infection leads to a sustained inflammatory immune response and the heavy recruitment of NET-producing neutrophils (10–12). The capacity of P. aeruginosa to tolerate this NET-rich environment suggests that the bacterium can detoxify and tolerate the eDNA and NETs present in CF sputum. Recombinant human DNase (rhDNase) is used as an inhaled treatment in CF patients, and its beneficial effects are attributed to reducing sputum viscosity, facilitating airway clearance, improving lung function, and reducing the risk of exacerbation (24). Bacterial DNase and PDase may favor the bacterium by protecting P. aeruginosa from NET killing but may also promote its own clearance by thinning the CF sputum. Overall, the effects of bacterial DNA-modifying enzymes are likely insufficient to promote clearance, hence the need for exogenous rhDNase treatment. We demonstrate a virulence role for two extracellular DNA-modifying enzymes in defending against NET-mediated killing, which reinforces the concept that eDNA is an important innate immune effector that bacteria directly target to promote survival during infections.
MATERIALS AND METHODS
Bacterial strains, plasmids, and primers.
The bacterial strains utilized in this study are summarized in Table 1 and in Fig. 5. Pseudomonas aeruginosa PAO1 was used as the wild-type strain. The mini-Tn5-lux mutants eddB::lux and xcpU::lux function as both transposon insertion mutants and transcriptional luxCDABE fusions that were constructed previously (29). P. aeruginosa was cultured at 37°C in defined BM2 minimal medium (0.1 M HEPES [pH 7], 7 mM ammonium chloride, 20 mM sodium succinate [pH 6.7], 2 mM magnesium sulfate, 10 μM iron sulfate, 1,600 μM phosphate buffer [pH 7.2], 1.62 μM manganese sulfate, 2.45 μM calcium chloride, 13.91 μM zinc chloride, 4.69 μM boric acid, 0.67 μM cobalt chloride) supplemented with 10 mM MgCl2. Limiting-phosphate BM2 medium for culture or DNase and phosphatase supernatant assays contained 400 μM phosphate buffer (pH 7.2), rather than 1,600 μM. To assess protective effects of DNase or phosphodiesterase activity, P. aeruginosa was cultured in BHI medium (Difco) overnight at 37°C, subcultured 1:1,000 in fresh medium, and grown to mid-log phase. When added, salmon sperm DNA was used at the concentrations indicated (catalog number UB14405S2; USB).
TABLE 1.
Bacterial strains, plasmids, and primers
| Strain, plasmid, or primer | Description or sequence | Source or reference |
|---|---|---|
| Strains | ||
| PAO1 | Wild type | R. E. Hancock |
| eddB::lux | Mini-Tn5-lux insertion mutant in PA3909 (EddB); transcriptional lux fusion | 29 |
| eddA::lacZ | Mini-Tn5-lacZ insertion mutant in PA3910 (EddA) | 46 |
| ΔeddAB | Unmarked deletion mutant of eddAB | This study |
| xcpU::lux | Mini-Tn5-lux insertion mutant in xcpU; nontranscriptional lux fusion | 45 |
| Plasmids | ||
| pEddA | EddA as an EcoRI-HindIII fragment in pPSV37 with a C-terminal His tag (EddA-His6) | This study |
| pEddB | EddB cloned as a BamHI-XhoI fragment into pUCP22 | This study |
| pPSV37 | IPTG-inducible expression vector; Genr | 44 |
| pUCP22 | Multicopy shuttle cloning vector; Ampr Genr | 47 |
| Primers | ||
| 16SrtF | GAAATCCCCGGGCTCAACCTG | 45 |
| 16SrtR | CCCCACGCTTTCGCACCTCA | 45 |
| EddB-F | GAGGATCCATGCACCCCTTGCGTAACGC | This study |
| EddB-R | GACTCGAGCTACCTGCGGTGCTTCTTCATCG | This study |
| EddA-soe-F | AAGCCTGAAACTCACTGAAG | This study |
| EddA-soe-R | ATACGCATGCCGCCGATATCCGGATTGATCCCGAAAC | This study |
| EddB-soe-F | CGGCGGCATGCGTATAGGAAAGGGCTATTCCTACG | This study |
| EddB-soe-R | TGGAGTGACCTCCGTTCC | This study |
| EddA-RBS-F | ATGCGAATTCAGGAGGAAACGATGAGTGGGATGGACCTCAAGCGC | This study |
| EddA-6×His-R | ATGCAAGCTTTCAGTGGTGATGGTGATGATGGGCGCCGTCGGGCTGCAGTTCCTG | This study |
Mutant construction and complementation.
The generation of the P. aeruginosa eddAB double mutant was conducted by allelic exchange as described previously (43). Briefly, wild-type PAO1 genomic DNA flanking the eddAB operon was amplified by using sequence-specific primers that added complementary regions to each amplified product. Stitching (splicing by overhang extension [SOE]) PCR was used to combine each flanking region into one PCR product that was complementary to the flanking regions of the eddAB operon but lacked the coding sequence. This SOE PCR product was gateway cloned (BP reaction) into a gateway-compatible vector backbone overnight, and the reaction was halted with the addition of proteinase K. Gateway constructs were transformed into electrocompetent E. coli DH10B cells. The isolated plasmid was then transformed into S17.1λpir and then mated with wild-type P. aeruginosa via puddle mating. Single-crossover events were selected for by growing P. aeruginosa on Pseudomonas isolation agar plus gentamicin (100 μg/ml). Counterselection for double recombination events was achieved by plating colonies on medium containing 15% sucrose.
The construction of the pEddA expression plasmid was completed by PCR amplifying the coding sequence from P. aeruginosa PAO1 genomic DNA. The amplified PCR product was cloned as an EcoRI-HindIII fragment with a C-terminal His6 tag into the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible pPSV37 vector (44). The pEddB expression plasmid contained eddB cloned as a BamHI-XhoI product into the pUCP22 vector. Digested PCR products were gel purified, ligated, and directly transformed into P. aeruginosa PAO1 ΔeddAB, selecting for gentamicin resistance (30 μg/ml).
Neutrophil isolation.
Neutrophils were isolated from healthy donors as described previously (9). Briefly, whole blood was collected and mixed 5:1 in acid citrate dextrose, followed by the removal of red blood cells using dextran sedimentation and hypotonic lysis with KCl. After all red blood cells were lysed, the cell pellet was subjected to Ficoll-Histopaque density centrifugation. The subsequent pellet was resuspended in 2 ml of HBSS (Hanks' balanced salt solution, no cations) (catalog number 14175-095; Invitrogen) or BHI medium for NET killing assays. The viable neutrophil concentration was determined by using a hemocytometer and trypan blue staining.
Bactericidal NET assays.
Quantification of bacterial viability was performed by direct plate counting methods, where a reduction in cell number indicates bacterial killing (9). NET killing experiments were performed in HBSS (Fig. 4A) or BHI medium (Fig. 4B). Isolated human neutrophils were mixed with mid-log-phase bacteria in black, clear-bottom, 96-well plates with 2.0 × 107 CFU bacteria and 2.0 × 106 neutrophils (multiplicity of infection [MOI] of 10:1). After 1 to 4 h of incubation, 50 μl of a DNase I solution (430 kU/ml; VWR) was added to every well, mixed, and incubated for 30 min at 37°C, in order to release bacteria trapped in NETs for accurate plate counts. Fifteen microliters of the suspension was serially diluted (1/10) in a 0.9% NaCl solution in a sterile 96-well plate, and 5 μl from each well was stamped onto LB agar plates to obtain bacterial plate count data at time zero (T0) and after 4 h (T4). CFU per milliliter values from the T4 and T0 time points were used to calculate the percent survival by subtracting the T4 plate counts from the T0 plate counts and dividing the value under “bacterium and neutrophil” conditions by the value under “bacterium-alone” conditions and multiplying this value by 100.
Gene expression assays.
Gene expression assays were carried out with a method similar to the one described previously (9, 16, 28). In brief, fresh subcultures of PAO1 eddB::lux were normalized and diluted to a final concentration of 4 × 107 CFU/well into BM2–400 μM phosphate buffer (a threshold concentration of phosphate that neither induces nor suppresses gene expression) or BM2–400 μM phosphate supplemented with 0.2% DNA (2 mg/ml) or 3-h PMA-stimulated NETs generated by 1 × 106 neutrophils. Cultures were overlaid with mineral oil to prevent evaporation. The OD600 and counts per second were monitored every 10 min over time in the Wallac Victor3 luminescence plate reader (Perkin-Elmer) at 37°C.
Quantitative reverse transcription-PCR was performed as described previously (45). The gene expression levels of eddB (PA3909) in triplicate 200-μl samples of wild-type PAO1, under the conditions outlined above, were measured after 60 and 120 min of coincubation. RNA was extracted according to the manufacturer's instructions (Bacteria Protect and RNeasy; Qiagen). Pseudomonas gene expression levels were measured by using the iQ SYBR green supermix (Bio-Rad) and bacterium-specific primers for eddB and the 16S rRNA housekeeping gene. For qRT-PCR, quantification and melting curve analyses were performed with an iQ5 instrument (Bio-Rad) according to the manufacturer's instructions. Each reaction is done in triplicate, and standard deviations were used to calculate a range of fold activation values by using the 2−ΔΔCT method (45).
DNase and phosphatase assays.
To assess the production of EddA and EddB, PAO1 bacteria were cultured overnight in BM2 medium under limited-phosphate conditions (400 μM phosphate buffer [pH 7.2], 10 mM MgCl2) or in BHI medium (Difco) overnight at 37°C. Epidemic CF strains were cultured in BM2 medium under a range of phosphate conditions (200, 400, 800, and 1,600 μM phosphate buffer, pH 7.2). Cultures of P. aeruginosa strains grown overnight were normalized to an OD600 of 1, and supernatants were collected by centrifugation at 8,000 rpm for 10 min. To assess DNase activity, 100 μl of the supernatant was incubated with 100 μl of 0.2 μg/μl salmon sperm DNA or 106 PMA-stimulated neutrophils with 2.5 μM Sytox green (as outlined above) in a black-walled, clear-bottom, 96-well plate and placed into a Wallac3 plate reader, and the plate was read every minute for 120 min.
For alkaline phosphatase activity, the substrate NBT/BCIP (Sigma-Aldrich) was used. For phosphodiesterase activity, 0.25 mM the substrate BNPP was used (Sigma-Aldrich). For enzyme assays, 100 μl of the clarified bacterial supernatants was added to 100 μl of 2× alkaline phosphatase buffer (1.0 M diethanolamine buffer with 0.50 mM magnesium chloride [pH 9.8]) supplemented with NBT/BCIP or BNPP in a 96-well, black-walled, clear-bottom plate, and the OD405 was measured every minute for 30 min.
Purification of C-terminally His-tagged EddA.
Mid-log-phase cultures of P. aeruginosa/pPSV37 expressing EddA-His6 under the control of an IPTG-inducible promoter were grown in LB supplemented with 30 μg/ml gentamicin until an OD600 of ∼0.5 was reached, when IPTG was added to the culture (0.2 mM) for a subsequent 3-h induction at 37°C. Cells were pelleted, the supernatant was removed, and the pellet was frozen prior to cell lysis and protein purification. The frozen pellet was gently resuspended in 5 ml 4°C wash buffer (0.5× phosphate-buffered saline [PBS] [pH 7.1], 300 mM NaCl, 5 mM imidazole) and sonicated for 15 min (5 s on and 10 s off) to lyse the resuspended pellet on ice. The lysate was centrifuged in a 4°C Beckman Coulter Avanti JA-20 rotor at 15,000 rpm for 30 min. The soluble fraction supernatant was removed and stored on ice. One milliliter of HisPur (ThermoFisher Scientific) cobalt Sepharose was equilibrated with 10 ml of 4°C wash buffer. The soluble lysate was then passed over the protein column twice, and the slurry was then washed by the addition of 10 ml wash buffer to the protein column. The recombinant soluble protein was eluted by the addition of 1 ml elution buffer (0.5× PBS [pH 7.1], 300 mM NaCl, 50 mM NaPO4, 250 mM imidazole) and dialyzed (molecular weight cutoff [MWCO] of 4,000 Da) into the appropriate buffer for 18 h at 4°C.
Western blot detection of His-tagged EddA.
Ten microliters of the eluted protein was boiled for 10 min in SDS-PAGE loading buffer. The protein was loaded onto a 10% SDS-PAGE gel, electrophoresed for 45 min at 180 V, and then transferred to nitrocellulose by using the Trans-Blot turbo transfer system (Bio-Rad). After the transfer, the nitrocellulose membrane was incubated in 1× Tris-buffered saline plus Tween (TBST) with 5% (wt/vol) skim milk powder for 18 h at 4°C. After incubation, anti-His (Sigma-Aldrich) was added at a 1:10,000 dilution for 1 h at room temperature, with rocking. The blot was then washed 3 times using 20 ml fresh TBST. The secondary anti-mouse antibody (Cell Signaling Technology) was added at a 1:10,000 dilution in 5% skim milk plus TBST, and the mixture was rocked at room temperature for an additional hour. The blot was washed with TBST as described above and then developed by using chemiluminescent Western blotting detection (Pierce ECL).
Statistical analysis.
Statistical analysis was performed by using GraphPad Prism v4.0 software. One-way analyses of variance (ANOVA) with Bonferroni posttests and unpaired t tests were used to calculate significant differences for bacterial survival and gene expression. Significant differences refer to P values of ≤0.05 or as otherwise indicated.
ACKNOWLEDGMENTS
This research was supported by a Cystic Fibrosis Canada operating grant and a research incentive grant from Athabasca University. M.W. was supported by a Cystic Fibrosis Canada postdoctoral fellowship, and T.W.R.H. was supported by a Cystic Fibrosis Canada studentship. M.D.P. and the collection of clinical P. aeruginosa isolates were supported by funds from the Canadian Institutes of Health Research and Cystic Fibrosis Canada.
We thank Marina Tom and Barbara Waddell for technical support and Joe Harrison for help constructing the allelic exchange mutant.
REFERENCES
- 1.Konstan MW, Hilliard KA, Norvell TM, Berger M. 1994. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am J Respir Crit Care Med 150:448–454. doi: 10.1164/ajrccm.150.2.8049828. [DOI] [PubMed] [Google Scholar]
- 2.Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. 1995. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med 151:1075–1082. doi: 10.1164/ajrccm.151.4.7697234. [DOI] [PubMed] [Google Scholar]
- 3.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 4.Brinkmann V, Zychlinsky A. 2012. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol 198:773–783. doi: 10.1083/jcb.201203170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, Malawista SE, de Boisfleury Chevance A, Zhang K, Conly J, Kubes P. 2012. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 18:1386–1393. doi: 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yipp BG, Kubes P. 2013. NETosis: how vital is it? Blood 122:2784–2794. doi: 10.1182/blood-2013-04-457671. [DOI] [PubMed] [Google Scholar]
- 7.Goldmann O, Medina E. 2013. The expanding world of extracellular traps: not only neutrophils but much more. Front Immunol 3:420. doi: 10.3389/fimmu.2012.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shan Q, Dwyer M, Rahman S, Gadjeva M. 2014. Distinct susceptibilities of corneal Pseudomonas aeruginosa clinical isolates to neutrophil extracellular trap-mediated immunity. Infect Immun 82:4135–4143. doi: 10.1128/IAI.02169-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halverson TW, Wilton M, Poon KK, Petri B, Lewenza S. 2015. DNA is an antimicrobial component of neutrophil extracellular traps. PLoS Pathog 11:e1004593. doi: 10.1371/journal.ppat.1004593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young RL, Malcolm KC, Kret JE, Caceres SM, Poch KR, Nichols DP, Taylor-Cousar JL, Saavedra MT, Randell SH, Vasil ML, Burns JL, Moskowitz SM, Nick JA. 2011. Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS One 6:e23637. doi: 10.1371/journal.pone.0023637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manzenreiter R, Kienberger F, Marcos V, Schilcher K, Krautgartner WD, Obermayer A, Huml M, Stoiber W, Hector A, Griese M, Hannig M, Studnicka M, Vitkov L, Hartl D. 2012. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J Cyst Fibros 11:84–92. doi: 10.1016/j.jcf.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Papayannopoulos V, Staab D, Zychlinsky A. 2011. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS One 6:e28526. doi: 10.1371/journal.pone.0028526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jorch SK, Kubes P. 2017. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med 23:279–287. doi: 10.1038/nm.4294. [DOI] [PubMed] [Google Scholar]
- 14.Cortjens B, van Woensel JBM, Bem RA. 2017. Neutrophil extracellular traps in respiratory disease: guided anti-microbial traps or toxic webs? Paediatr Respir Rev 21:54–61. doi: 10.1016/j.prrv.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 15.Lappann M, Danhof S, Guenther F, Olivares-Florez S, Mordhorst IL, Vogel U. 2013. In vitro resistance mechanisms of Neisseria meningitidis against neutrophil extracellular traps. Mol Microbiol 89:433–449. doi: 10.1111/mmi.12288. [DOI] [PubMed] [Google Scholar]
- 16.Mulcahy H, Charron-Mazenod L, Lewenza S. 2008. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog 4:e1000213. doi: 10.1371/journal.ppat.1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, Henriques-Normark B. 2007. Capsule and d-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol 9:1162–1171. doi: 10.1111/j.1462-5822.2006.00857.x. [DOI] [PubMed] [Google Scholar]
- 18.Buchana JT, Simpson JA, Aziz RK, Liu GY, Kristian SA, Kotb M, Feramisco J, Nizet V. 2006. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol 16:396–400. doi: 10.1016/j.cub.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 19.Berends ETM, Horswill AR, Haste NM, Monestier M, Nizet V, von Köckritz-Blickwede M. 2010. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun 2:576–586. doi: 10.1159/000319909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seper A, Hosseinzade A, Gorkiewicz G, Lichtenegger S, Roier S, Leitner DR, Rohm M, Grutsch A, Reidl J, Urgan CF, Schild S. 2013. Vibrio cholerae evades neutrophil extracellular traps by the activity of two extracellular nucleases. PLoS Pathog 9:e1003614. doi: 10.1371/journal.ppat.1003614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juneau RA, Stevens JS, Apicella MA, Criss AK. 2015. A thermonuclease of Neisseria gonorrhoeae enhances bacterial escape from killing by neutrophil extracellular traps. J Infect Dis 212:316–324. doi: 10.1093/infdis/jiv031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doke M, Fukamachi H, Morisaki H, Arimoto T, Kataoka H, Kuwata H. 2017. Nucleases from Prevotella intermedia can degrade neutrophil extracellular traps. Mol Oral Microbiol 32:288–300. doi: 10.1111/omi.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thammavongsa V, Missiakas DM, Schneewind O. 2013. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 342:863–866. doi: 10.1126/science.1242255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pressler T. 2008. Review of recombinant human deoxyribonuclease (rhDNase) in the management of patients with cystic fibrosis. Biol Targets Ther 2:611–617. doi: 10.2147/BTT.S3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson L, Mulcahy H, Kanevets U, Shi Y, Lewenza S. 2012. Surface-localized spermidine protects the Pseudomonas aeruginosa outer membrane from antibiotic treatment and oxidative stress. J Bacteriol 194:813–826. doi: 10.1128/JB.05230-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewenza S. 2013. Extracellular DNA-induced antimicrobial peptide resistance mechanisms in Pseudomonas aeruginosa. Front Microbiol 4:21. doi: 10.3389/fmicb.2013.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilton M, Charron-Mazenod L, Moore R, Lewenza S. 2015. Extracellular DNA acidifies biofilms and induces aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 60:544–553. doi: 10.1128/AAC.01650-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulcahy H, Charron-Mazenod L, Lewenza S. 2010. Pseudomonas aeruginosa produces an extracellular deoxyribonuclease that is required for utilization of DNA as a nutrient source. Environ Microbiol 12:1621–1629. doi: 10.1111/j.1462-2920.2010.02208.x. [DOI] [PubMed] [Google Scholar]
- 29.Lewenza S, Falsafi RK, Winsor G, Gooderham WJ, McPhee JB, Brinkman FS, Hancock RE. 2005. Construction of a mini-Tn5-luxCDABE mutant library in Pseudomonas aeruginosa PAO1: a tool for identifying differentially regulated genes. Genome Res 15:583–589. doi: 10.1101/gr.3513905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ball G, Antelmann H, Imbert PRC, Gimenez MR, Voulhoux R, Ize B. 2016. Contribution of the twin arginine translocation system to the exoproteome of Pseudomonas aeruginosa. Sci Rep 6:27675. doi: 10.1038/srep27675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kageyama H, Tripathi K, Rai AK, Cha-um S, Waditee-Sirisattha R, Takabe T. 2011. An alkaline phosphatase/phosphodiesterase, PhoD, induced by salt stress and secreted out of the cells of Aphanothece halophytica, a halotolerant cyanobacterium. Appl Environ Microbiol 77:5178–5183. doi: 10.1128/AEM.00667-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eder S, Shi L, Jensen K, Yamane K, Hulett FM. 1996. A Bacillus subtilis secreted phosphodiesterase/alkaline phosphatase is the product of a Pho regulon gene, phoD. Microbiology 142(Part 8):2041–2047. doi: 10.1099/13500872-142-8-2041. [DOI] [PubMed] [Google Scholar]
- 33.Gomez PF, Ingram LO. 1995. Cloning, sequencing and characterization of the alkaline phosphatase gene (phoD) from Zymomonas mobilis. FEMS Microbiol Lett 125:237–245. doi: 10.1111/j.1574-6968.1995.tb07364.x. [DOI] [PubMed] [Google Scholar]
- 34.Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, Rosenstein BJ, Smith AL, Wohl ME. 1994. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. N Engl J Med 331:637–642. doi: 10.1056/NEJM199409083311003. [DOI] [PubMed] [Google Scholar]
- 35.Parkins MD, Glezerson BA, Sibley CD, Sibley KA, Duong J, Purighalla S, Mody CH, Workentine ML, Storey DG, Surette MG, Rabin HR. 2014. Twenty-five-year outbreak of Pseudomonas aeruginosa infecting individuals with cystic fibrosis: identification of the Prairie epidemic strain. J Clin Microbiol 52:1127–1135. doi: 10.1128/JCM.03218-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shak S, Capon DJ, Hellmiss R, Marsters SA, Baker CL. 1990. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc Natl Acad Sci U S A 87:9188–9192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ranasinha C, Assoufi B, Shak S, Christiansen D, Fuchs H, Empey D, Geddes D, Hodson M. 1993. Efficacy and safety of short-term administration of aerosolised recombinant human DNase I in adults with stable stage cystic fibrosis. Lancet 342:199–202. doi: 10.1016/0140-6736(93)92297-7. [DOI] [PubMed] [Google Scholar]
- 38.Tran AX, Whittimore JD, Wyrick PB, McGrath SC, Cotter RJ, Trent MS. 2006. The lipid A 1-phosphatase of Helicobacter pylori is required for resistance to the antimicrobial peptide polymyxin. J Bacteriol 188:4531–4541. doi: 10.1128/JB.00146-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lamarche MG, Dozois CM, Daigle F, Caza M, Curtiss R III, Dubreuil JD, Harel J. 2005. Inactivation of the pst system reduces the virulence of an avian pathogenic Escherichia coli O78 strain. Infect Immun 73:4138–4145. doi: 10.1128/IAI.73.7.4138-4145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bains M, Fernandez L, Hancock RE. 2012. Phosphate starvation promotes swarming motility and cytotoxicity of Pseudomonas aeruginosa. Appl Environ Microbiol 78:6762–6768. doi: 10.1128/AEM.01015-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frisk A, Schurr JR, Wang G, Bertucci DC, Marrero L, Hwang SH, Hassett DJ, Schurr MJ. 2004. Transcriptome analysis of Pseudomonas aeruginosa after interaction with human airway epithelial cells. Infect Immun 72:5433–5438. doi: 10.1128/IAI.72.9.5433-5438.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Surette MG. 2014. The cystic fibrosis lung microbiome. Ann Am Thorac Soc 11(Suppl 1):S61–S65. doi: 10.1513/AnnalsATS.201306-159MG. [DOI] [PubMed] [Google Scholar]
- 43.Hmelo LR, Borlee BR, Almblad H, Love ME, Randall TE, Tseng BS, Lin C, Irie Y, Storek KM, Yang JJ, Siehnel RJ, Howell PL, Singh PK, Tolker-Nielsen T, Parsek MR, Schweizer HP, Harrison JJ. 2015. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat Protoc 10:1820–1841. doi: 10.1038/nprot.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee P-C, Stopford CM, Svenson AG, Rietsch A. 2010. Control of effector export by the Pseudomonas aeruginosa type III secretion proteins PcrG and PcrV. Mol Microbiol 75:924–941. doi: 10.1111/j.1365-2958.2009.07027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mulcahy H, Sibley CD, Surette MG, Lewenza S. 2011. Drosophila melanogaster as an animal model for the study of Pseudomonas aeruginosa biofilm infections in vivo. PLoS Pathog 7:e1002299. doi: 10.1371/journal.ppat.1002299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Held K, Ramage E, Jacobs M, Gallagher L, Manoil C. 2012. Sequence-verified two-allele transposon mutant library for Pseudomonas aeruginosa PAO1. J Bacteriol 194:6387–6389. doi: 10.1128/JB.01479-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.West SE, Schweizer HP, Dall C, Sample AK, Runyen-Janecky LJ. 1994. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene 148:81–86. doi: 10.1016/0378-1119(94)90237-2. [DOI] [PubMed] [Google Scholar]