

Fatty acid hydroperoxides are involved in host-pathogen interactions. In both plants and mammals, polyunsaturated fatty acids are liberated during infection and enzymatically oxidized to the corresponding toxic hydroperoxides during the defensive oxidative burst that is designed to thwart the infection.
KEYWORDS: organic hydroperoxide, Ohr, gene regulation, DNase I footprinting, MarR, ROS, transcription factors
ABSTRACT
Fatty acid hydroperoxides are involved in host-pathogen interactions. In both plants and mammals, polyunsaturated fatty acids are liberated during infection and enzymatically oxidized to the corresponding toxic hydroperoxides during the defensive oxidative burst that is designed to thwart the infection. The bacterial transcription factor OhrR (organic hydroperoxide reductase regulator) is oxidized by organic hydroperoxides, as a result of which the ohr gene encoding organic hydroperoxide reductase is induced. This enzyme converts the hydroperoxides to less toxic alcohols. We show here that OhrR from Burkholderia thailandensis represses expression of ohr. Gene expression is induced by cumene hydroperoxide and to a lesser extent by inorganic oxidants; however, Ohr contributes to degradation only of the organic hydroperoxide. B. thailandensis OhrR, which binds specific sites in both ohr and ohrR promoters, as evidenced by DNase I footprinting, belongs to the 2-Cys subfamily of OhrR proteins, and its oxidation leads to reversible disulfide bond formation between conserved N- and C-terminal cysteines in separate monomers. Oxidation of the N-terminal Cys is sufficient for attenuation of DNA binding in vitro, with complete restoration of DNA binding occurring on addition of a reducing agent. Surprisingly, both overexpression of ohr and deletion of ohr results in enhanced survival on exposure to organic hydroperoxide in vitro. While Δohr cells are more virulent in a Caenorhabditis elegans model of infection, ΔohrR cells are less so. Taken together, our data suggest that B. thailandensis OhrR has several unconventional features and that both OhrR and organic hydroperoxides may contribute to virulence.
INTRODUCTION
Molecular oxygen is essential to the cellular respiration of aerobic organisms. However, incomplete electron transfer to O2 gives rise to reactive oxygen species (ROS), which may in turn react with cellular macromolecules to cause cellular injury or death if they are not inactivated by antioxidant defenses (1, 2). However, ROS also perform important signaling roles, and they are produced by both plants and animals in the initial oxidative burst designed to combat an infection. The enzyme NADPH oxidase, which is integral to this response, transfers electrons from cytoplasmic NADPH to molecular oxygen to generate superoxide anion, which is in turn rapidly converted to peroxynitrite or to the more stable H2O2 (3, 4). H2O2 may then react with myeloperoxidase in neutrophils to form hypochlorous acid (HOCl; also the active ingredient in bleach), and it can react with transition metals, such as Fe2+, to generate hydroxyl radicals. Plants also generate fatty acid (mostly linoleic acid) hydroperoxides, which are important determinants of successful interactions between the plant host and would-be pathogens or symbionts (5, 6). In mammals, bacterial infection can lead to activation of phospholipase A2; this leads to the release of the polyunsaturated fatty acid arachidonic acid, which is subsequently oxidized to hydroperoxyeicosatetranoic acid (HpETE) by lipoxygenase enzymes (7–9). Organic hydroperoxides are more lethal, as they not only react with intracellular macromolecules, forming toxic adducts, but also cause free radical generation, prolonging cellular damage (10). Pathogenic bacteria have evolved numerous strategies to defend themselves against this oxidative burst. In addition to proteins that sense or directly detoxify compounds, such as superoxide anion and H2O2, enzymes that are dedicated to inactivation of organic hydroperoxides have been identified. These include alkylhydroperoxide reductase (AhpC) and organic hydroperoxide reductase (Ohr), which both reduce the toxic organic hydroperoxides to the corresponding alcohols; AhpC also degrades H2O2 (11, 12). The gene encoding Ohr is repressed by OhrR (organic hydroperoxide reductase regulator) (13, 14), a transcription factor that belongs to the multiple antibiotic resistance regulator (MarR) protein family (15). OhrR is found in both Gram-negative and Gram-positive bacterial species, and it senses changes in the environment by means of cysteine residues. A highly conserved Cys near the N terminus is oxidized by organic hydroperoxides to generate an unstable sulfenic acid (16). In some OhrR proteins, such as OhrR from Bacillus subtilis, this oxidation leads to the formation of a mixed disulfide with low-molecular-weight thiols or to further oxidation to sulfinic or sulfonic acid (17). In OhrR from other bacterial species, including Xanthomonas campestris, a second Cys closer to the C terminus is required for disulfide bond formation with the oxidized N-terminal Cys and to produce the protein conformational changes required for altered DNA binding (18, 19). In addition, an autoregulatory circuit may exist in which OhrR also controls ohrR expression (20–22).
Nosocomial infections are on the rise and a major cause of concern. In recent years, bacteria belonging to the Burkholderia cepacia complex (Bcc) have emerged as dangerous opportunistic pathogens, frequently infecting immunocompromised individuals or persons living with cystic fibrosis (23). Burkholderia spp. are generally characterized by inherent resistance not just to antibiotics but also to disinfectants (chemical oxidants) (24). We show here that Burkholderia thailandensis OhrR, which is a member of the 2-Cys OhrR subfamily, functions as a transcriptional repressor of ohr and that it responds to both organic and inorganic oxidants. The N-terminal cysteine is the main target for oxidation, as expected, and a disulfide bond with the C-terminal Cys is not required for attenuation of DNA binding in vitro. While bacteria lacking a functional Ohr were, surprisingly, more virulent in Caenorhabditis elegans than the wild type (WT), ΔohrR cells overexpressing ohr were less so. Our data suggest that oxidation of B. thailandensis OhrR is associated with induction of ohr expression and degradation of organic hydroperoxides, as expected, but that the relationship between cellular levels of organic hydroperoxides and virulence may be complex.
RESULTS
Conservation of ohr-ohrR.
The genes encoding Ohr and OhrR are typically in close proximity, and they may be oriented either in the same or in different orientations. Among Burkholderia species, ohrR is in the same orientation and upstream of ohr and in a conserved genomic context (Fig. 1A, top; see also Fig. S1 in the supplemental material). Previously characterized OhrR proteins were shown to bind an AT-rich site in gene promoters (with the consensus sequence TACAATTNAATTGTA [20]), and examination of predicted B. thailandensis ohr and ohrR promoters reveals the presence of AT-rich regions that might serve as OhrR binding sites. B. thailandensis OhrR shares 55% identity with X. campestris OhrR, and it conserves the two Cys residues implicated in oxidant sensing, the oxidant-sensing C16/C16′ in α1/α1′ and the C-terminal C121/C121′ in α5/α5′ (Fig. 1A, bottom, and S2).
FIG 1.

B. thailandensis OhrR is a dimer. (A) (Top) The genomic locus encoding ohr and ohrR. The inverted triangles indicate the positions of transposon insertion. (Bottom) The model is based on the structure of reduced X. campestris OhrR-C22S (PDB accession number 2PEX [19]; global model quality estimate [GMQE] = 0.72). One subunit is in magenta with helices identified, and the other is in brown. Cysteine residues are in yellow space-filling representation, with C16′ in α1′ (brown subunit) and C121 in α5 (magenta subunit) being identified. The illustration was generated with the PyMOL program. (B) Far-UV CD spectrum reflecting the expected secondary structure composition. Ellipticity (θ) is reported in millidegrees (mdeg). (C) Elution of reduced and oxidized OhrR from SEC column. mAU, milli-absorbance units. (D) Standard curve reflecting MW standards eluting from the SEC column. Both reduced (blue) and oxidized (red) OhrR eluted corresponding to an MW of ∼34 kDa.
B. thailandensis OhrR is oxidized in vitro.
The gene encoding OhrR was amplified from B. thailandensis E264 genomic DNA and expressed in Escherichia coli with an N-terminal His6 tag. The protein was purified to apparent homogeneity (Fig. S3). The secondary structure composition (54% α-helices, 12% β-sheets, and 34% random coils) was estimated from far-UV circular dichroism (CD) spectra using the K2D algorithm on DichroWeb and found to be similar to that expected for MarR (OhrR) proteins (Fig. 1B) (19). Size exclusion chromatography (SEC) confirmed that OhrR exists as a dimer in both the reduced and the oxidized states, eluting as a single species with a molecular weight (MW) of ∼34 kDa (Fig. 1C and D). Single and double Cys-to-Ala mutants (OhrR-C16A, OhrR-C121A, and OhrR-C16AC121A) were generated and also shown to elute from the SEC column as dimers (Fig. S3 and Table S1), verifying that OhrR exists as a dimer regardless of Cys oxidation.
Sequence conservation suggests that B. thailandensis OhrR belongs to the 2-Cys family of OhrR proteins (Fig. 1A and S2), predicting that oxidation with organic hydroperoxides will lead to C16 oxidation, followed by disulfide bond formation with C121′ from the other monomer. Wild-type and mutant OhrR were incubated with oxidants, and the effect of oxidants on protein stability was assessed by measuring the fluorescence of SYPRO Orange dye bound to unfolded protein as a function of temperature. As shown in Fig. 2A, OhrR was quite stable with a melting temperature (Tm) of ∼57°C; the Cys-to-Ala substitutions resulted in a further stabilization (Tm, 63 to 66°C; Table 1). Incubation with cumene hydroperoxide (CHP) resulted in a particularly marked destabilization of WT OhrR and OhrR-C121A (Tms, 39 and 44°C, respectively, with 2 mM CHP), a modest destabilization of OhrR-C16A (Tm, ∼55°C), and no effect on the stability of OhrR-C16AC121A (Table 1 and Fig. S4). These observations point to CHP-mediated oxidation of both Cys residues, with the oxidation of C16 being associated with the largest changes in protein stability. Similarly, addition of 2 mM H2O2 resulted in the most significant destabilization of WT OhrR and OhrR-C121A (Tms, 51 and 54°C, respectively), with the reduction in the Tm for oxidized OhrR-C16A being ∼4°C and with no effect on the Tm of the double mutant. Incubation with higher concentrations of NaOCl modestly destabilized all protein variants (Table 1).
FIG 2.

In vitro oxidation and stability of OhrR variants. (A) Thermal stability of OhrR variants determined by fluorescence of SYPRO Orange bound to unfolded protein as a function of temperature. (B to D) Oxidant-treated OhrR variants separated on 12% nonreducing SDS-PAGE gels. Migration corresponding to the monomer and dimer, identified at the right. Lanes marked kDa, MW markers; lanes 1 to 3, WT OhrR; lanes 4 to 6, OhrR-C16A; lanes 7 to 9, OhrR-C121A; lanes 10 to 12, OhrR-C16AC121A. The addition of CHP (0.1 or 5.0 mM; B), H2O2 (0.1 or 5.0 mM; C), or NaOCl (7 or 70 μM; D) is identified below each gel.
TABLE 1.
Thermal stability of reduced and oxidized OhrR variants
| Protein | Tma (°C) ± SD in air | Oxidant | Avg Tma (°C) ± SD at: |
|
|---|---|---|---|---|
| 100 μM | 2 mM | |||
| OhrR | 56.8 ± 0.5 | CHP | 53.0 ± 0.6 | 39.3 ± 1.6 |
| H2O2 | 53.4 ± 0.7 | 51.1 ± 0.9 | ||
| NaOCl | 57.2 ± 0.5 | 55.3 ± 0.6 | ||
| OhrR-C16A | 65.0 ± 0.4 | CHP | 62.4 ± 0.8 | 54.7 ± 0.6 |
| H2O2 | 59.0 ± 1.2 | 60.6 ± 1.0 | ||
| NaOCl | 65.8 ± 0.04 | 56.3 ± 1.0 | ||
| OhrR-C121A | 63.0 ± 0.4 | CHP | 62.9 ± 0.9 | 43.6 ± 1.0 |
| H2O2 | 62.3 ± 0.4 | 54.0 ± 0.8 | ||
| NaOCl | 61.4 ± 1.3 | 56.7 ± 0.4 | ||
| OhrR-C16AC121A | 66.4 ± 0.7 | CHP | 66.5 ± 0.6 | 65.3 ± 1.1 |
| H2O2 | 66.4 ± 1.0 | 65.0 ± 0.5 | ||
| NaOCl | 63.0 ± 1.2 | 62.3 ± 1.2 | ||
The Tm of reduced protein exposed to air during assembly of the reaction mixtures or incubated for 15 min with the indicated oxidants prior to measurement of thermal stability.
The effects of oxidants were also determined by electrophoresis on SDS-PAGE gels. Incubation of WT OhrR with 100 μM CHP resulted in the essentially complete conversion to a dimeric species, represented as a doublet (Fig. 2B). In the absence of C121, no dimer formation was observed, whereas oxidation of OhrR-C16A did yield dimeric species, albeit very modestly so and only on addition of 5 mM CHP. These data indicate that intersubunit disulfide bond formation involves both C16 and C121. Disulfide bond formation was completely reversed on addition of excess dithiothreitol (DTT) (Fig. S5A). A similar pattern was observed on oxidation with H2O2, except that WT OhrR primarily formed the faster-migrating (and reversible) dimeric species on oxidation and that a higher concentration of H2O2 was required for complete conversion to the dimer (Fig. 2C and S5B). In contrast, incubation with NaOCl resulted in the comparable formation of dimeric species for WT OhrR and OhrR-C16A as well as some smearing in the gel for all protein variants, suggesting protein degradation. Based on these observations, we infer that OhrR may be oxidized comparably by CHP and H2O2 in vitro, with disulfide bond formation involving C16 and C121′ from separate subunits within the OhrR dimer, whereas NaOCl-oxidized OhrR also appears to be able to form disulfide bonds between C121 residues (likely involving separate protein dimers, given the predicted location of C121; Fig. 1A) with a likelihood comparable to that between C16 and C121′.
OhrR binds upstream of the ohr and ohrR coding regions.
OhrR is expected to control expression of both ohr and ohrR. To address this expectation, electrophoretic mobility shift assays (EMSAs) were performed using ohrO and ohrRO, representing DNA upstream of the ohr and ohrR coding regions, respectively. Reduced OhrR bound ohrO, forming multiple complexes with an overall apparent dissociation constant (Kd) of 2.8 ± 0.8 nM, while a single complex was observed with ohrRO, with the Kd being equal to 128 ± 25 nM (Fig. S6 and Table 2; note that all EMSAs included a >400-fold excess of nonspecific DNA, indicating that binding was specific). All Cys-to-Ala mutant proteins had a lower affinity for ohrO than WT OhrR, whereas their affinity for ohrRO was not markedly altered (Table 2). While the binding of WT OhrR to ohrO DNA exhibited modest negative cooperativity (Hill coefficient [nH], ∼0.7), this negative cooperativity was not seen for mutant proteins, and no cooperativity of binding to ohrRO was observed.
TABLE 2.
Affinity of OhrR variants for ohrO and ohrROa
| Protein |
ohrO |
ohrRO |
||
|---|---|---|---|---|
| Kd (nM) | nH | Kd (nM) | nH | |
| OhrR | 2.8 ± 0.8 | 0.7 ± 0.2 | 128 ± 25 | 1.0 ± 0.04 |
| OhrR-C16A | 38 ± 4 | 1.0 ± 0.1 | 215 ± 19 | 1.0 ± 0.2 |
| OhrR-C121A | 27 ± 10 | 0.9 ± 0.3 | 122 ± 14 | 1.0 ± 0.1 |
| OhrR-C16AC121A | 89 ± 10 | 0.9 ± 0.1 | 134 ± 18 | 1.0 ± 0.1 |
Data represent the average ± SD from at least three experiments.
DNase I footprinting was used for identification of specific sites. For this purpose, longer DNA duplexes encompassing ohrO and ohrRO as well as the flanking sequence were 5′ end labeled with 6-carboxyfluorescein (6-FAM). The amplified products were incubated in the absence or presence of the identified OhrR variant, followed by DNase I digestion and analysis of the resulting fragments on a genetic analyzer (25). At a DNA/protein ratio of 1:4, reduced WT OhrR protected a single region upstream of the ohr coding region, extending from positions −95 to −57 relative to the translational start, followed by subtle hypersensitive cleavage at position −56 (Fig. 3A). A sequence with similarity to the reported OhrR box consensus sequence could be identified within this region (Fig. 3A, bottom) (20). The length of the protected DNA combined with the requirement for a stoichiometric excess of protein and the detection of multiple complexes in EMSA gels suggests that full occupancy of this site requires binding of more than one OhrR dimer. All mutant proteins protected equivalent regions upstream of the ohr coding region, the main distinction being the presence of more extensive hypersensitive cleavage downstream of the protected sequence for OhrR-C121A and OhrR-C16AC121A (Fig. S7A to D and S8).
FIG 3.

OhrR binds specifically to ohr and ohrR promoter DNA. (A) Electropherogram traces of DNase I-digested ohr promoter DNA. (B) Electropherogram traces of DNase I-digested ohrR promoter DNA. Red, DNA only; blue, DNA incubated with OhrR at a stoichiometric DNA/protein ratio of 1:4. The protected regions are expanded in the lower panels, with the sequence being identified at the bottom. Numbering is relative to the translational start, defined as position +1. For ohrR promoter DNA, the ATG codon is underlined. For ohr promoter DNA, a sequence resembling the published OhrR consensus sequence is identified by a dashed underline; the asterisk marks a hypersensitive site. The results are representative of those from at least three replicates.
Upstream of the ohrR coding region, protection by WT OhrR extended from positions −61 to −9 relative to the translational start (Fig. 3B). While the protected DNA is AT rich, a sequence matching the reported consensus sequence could not be readily identified. Another distinction from the protection upstream of the ohr coding region was the partial protection at the upstream edge of the footprint and around positions −22 and −18. The patterns of protection resulting from binding of mutant proteins were similar to the pattern observed for WT OhrR (Fig. S7E to H).
To investigate the effect of oxidants on DNA binding, OhrR variants were oxidized and then incubated with ohr DNA, following which the reaction mixtures were treated with DNase I. As shown in Fig. 4B, incomplete oxidation of OhrR by CHP attenuated the protection from DNase I, restoring partial DNase I cleavage (magenta line), while fully oxidized OhrR did not protect ohr DNA at all (Fig. 4C). The CHP-mediated oxidation was reversible, as illustrated by the recovery of the DNase I footprint by OhrR that was first oxidized with 70 μM CHP and then rereduced (Fig. 4D, gray line). Consistent with the formation of C16-C121′ disulfide bonds on oxidation with both CHP and H2O2, H2O2-oxidized OhrR also failed to protect DNA upstream of the ohr coding region (Fig. 4E, green line). In contrast, oxidation with 7 μM NaOCl did not change the DNA protection, but it resulted in the appearance of hypersensitive cleavage downstream of the protected region, suggesting a slightly altered mode of binding (Fig. 4F and S8).
FIG 4.

Effect of OhrR oxidation on binding to ohr promoter DNA. (A) Electropherogram traces of DNase I-digested ohr promoter DNA and DNA incubated with reduced OhrR. (B) DNA incubated with reduced OhrR or with OhrR oxidized with 7 μM CHP. (C) DNA incubated with reduced OhrR or with OhrR oxidized with 70 μM CHP. (D) DNA incubated with OhrR oxidized with 70 μM CHP or with 70 μM CHP-oxidized OhrR subsequently rereduced with DTT. (E) DNA incubated with reduced OhrR or with OhrR oxidized with 0.7 mM H2O2. (F) Free ohr promoter DNA and DNA incubated with OhrR oxidized with 7 μM NaOCl. Asterisks mark hypersensitive sites (positions −56 and −49). The results are representative of those from at least three replicates.
A similar response to oxidants was observed for OhrR-C121A; CHP-oxidized protein did not protect ohrO DNA (Fig. S9D, magenta line), and this CHP-mediated oxidation was reversible (Fig. S9F, gray line). H2O2-oxidized OhrR-C121A likewise failed to protect this DNA (Fig. S9E, green line), whereas oxidation by NaOCl only subtly changed the binding, as reflected in the appearance of hypersensitive sites (Fig. S8). In contrast, oxidation of OhrR-C16A by CHP, H2O2, or NaOCl had no effect on protection from DNase I; the only observed changes were the appearance of hypersensitive sites, particularly for CHP- and NaOCl-oxidized protein (Fig. S8 and S9A to C). As expected, OhrR-C16AC121A footprints were unaffected by addition of oxidant to the protein (data not shown). Taken together, these observations reinforce the inference that oxidation of C16 by CHP or H2O2, even in the absence of the disulfide-bonding partner C121′, causes conformational changes, and they suggest that those conformational changes suffice for attenuation of DNA binding.
OhrR controls gene expression in response to oxidants.
OhrR is expected to repress expression of ohr (13, 14, 21). To verify this expectation, strains in which the ohrR or ohr gene was inactivated by transposon insertion into the open reading frame (ORF) (Fig. 1A, top) were obtained from the B. thailandensis E264 transposon mutant library (26) and verified by PCR. Expression of ohr was determined by quantitative reverse transcription-PCR (qRT-PCR). As shown in Fig. 5A, expression of ohr was increased almost 30-fold in the ΔohrR strain compared to the WT, consistent with OhrR functioning as a repressor. This phenotype was verified by complementation with ohrR carried by plasmid pBBR1-MCS5 (generating the ΔohrRc strain), in which repression of ohr expression was fully restored; as shown in Fig. 5B (orange hatched bar), ohrR expression was much higher in this strain than in the WT. Since complemented strains were grown with gentamicin, ohr expression was verified in the WT carrying empty pBBR1-MCS5 (WTe; grown with gentamicin) and found to be similar to that in the WT, indicating that gentamicin had no effect on gene expression.
FIG 5.

OhrR represses ohr expression. (A) Expression of ohr in WT and WTe (the WT strain containing the empty plasmid) cells and in ΔohrR and ΔohrRc (the ΔohrR strain complemented with ohrR) cells. The hatched green bar confirms abundant ohr expression in Δohr cells complemented with ohr (Δohrc). (B) Expression of ohrR in WT and WTe cells and in Δohr and Δohrc (the Δohr strain complemented with ohr) cells. The hatched orange bar confirms abundant ohrR expression in ΔohrR cells complemented with ohrR. Transcript levels are reported relative to the level of transcription of the reference gene (glutamate synthase) and were determined using the 2−ΔCT method. Error bars represent standard errors from three independent experiments. Asterisks directly above the bars denote statistically significant differences in expression compared to that in WT cells, as determined by an unpaired Student t test (**, P < 0.001).
Expression of ohrR was low in WT (and WTe) cells (Fig. 5B, blue bars), and it remained low in the Δohr strain and in the Δohr strain complemented with ohr (the Δohrc strain, green bars). Expression of ohr was very high in the Δohrc strain (Fig. 5A, green hatched bar); since the construct used for complementation contained the ohr open reading frame preceded by the sequence downstream of ohrR, this indicates that ohr is expressed under the control of its own promoter. Evidently, ohrR expression was unaffected by the levels of ohr expression.
To determine responses to oxidants, exponentially growing B. thailandensis was incubated with oxidants for 15 min, following which RNA was isolated for analysis of transcript levels by qRT-PCR. In WT cells, incubation with CHP (0.2 or 1 mM) resulted in a >200-fold induction of ohr expression and in modestly increased expression of ohrR (∼2-fold; Fig. 6A and B, light blue bars). A qualitatively similar pattern of induction was seen in WTe (Fig. 6A and B, dark blue bars). In ΔohrR cells, 0.2 mM CHP had no effect on ohr expression (Fig. 6A, orange bar), whereas CHP-dependent induction was restored in ΔohrRc cells (Fig. 6A, brown bar; that CHP was less efficient at inducing expression in ΔohrRc cells than in WT cells is likely due to the elevated expression of ohrR from the multicopy plasmid); these observations are consistent with CHP inducing gene expression by an OhrR-dependent mechanism. An ∼3-fold increase in ohr expression was observed in ΔohrR cells incubated with 1 mM CHP, possibly reflecting repression of ohr by a second, modestly CHP-sensitive transcription factor in the absence of OhrR. In Δohrc cells, CHP effected only an insignificant increase in ohr expression; however, in these cells, ohr expression from the multicopy plasmid was already very high (Fig. 5A), likely resulting in rapid degradation of CHP as well as reflecting the fact that cellular levels of OhrR may have been insufficient to repress multiple copies of the ohr gene.
FIG 6.

Gene expression is induced by oxidants. (A) Fold change in ohr expression after incubation of cells with 0.2 or 1 mM CHP relative to that in unsupplemented cultures. (B) Fold change in ohrR expression after incubation of cells with 0.2 or 1 mM CHP. (C, E) Fold change in ohr expression after incubation of cells with 0.2 or 1 mM H2O2 (C) or NaOCl (E) relative to that in unsupplemented cultures. (D, F) Fold change in ohrR expression after incubation of cells with 0.2 or 1 mM H2O2 (D) or NaOCl (F). Color coding is defined in the inset to panel B and is identical for all panels. Transcript levels were reported using the 2−ΔΔCT method. Error bars represent standard errors from at least two independent experiments. Asterisks directly above the bars denote a statistically significant difference in expression compared to that in the corresponding unsupplemented culture, as determined by an unpaired Student t test (*, P < 0.05; **, P < 0.001).
Expression of ohrR was modestly increased in almost all strains on addition of CHP; combined with the binding of OhrR to the ohrR promoter DNA (Fig. 3B), these data are consistent with a repression of ohrR by OhrR that is relieved on protein oxidation. One exception was Δohrc cells, in which induction of ohrR was observed only with 1 mM CHP, most likely on account of the overproduction of Ohr causing efficient degradation of CHP.
Incubation with H2O2 resulted in a modest but concentration-dependent increase in ohr expression in WT cells (Fig. 6C, light blue bars), whereas ohr expression was unaltered in ΔohrR cells after addition of H2O2 (orange bars); complementation with ohrR (in ΔohrRc cells; Fig. 6C, brown bars) did not restore significant sensitivity to 1 mM H2O2, likely due to excess cellular levels of OhrR. These data suggest that OhrR-mediated repression of ohr expression may be slightly relieved on exposure to H2O2. In H2O2-treated WTe cells, ohr expression was greater than that in WT cells, an effect that appears to be linked to the combined presence of oxidant and gentamicin. Expression of ohrR was unaffected by the addition of H2O2 to either of the strains, except for the Δohrc strain, in which addition of 1 mM H2O2 resulted in an ∼9-fold increase in expression (Fig. 6D).
A modest and variable increase in ohr expression in WT and WTe cells was also seen on addition of NaOCl (Fig. 6E, blue bars), and this increase was less pronounced in ΔohrR cells, also indicating that OhrR-mediated repression of ohr may be slightly relieved by NaOCl. The observation that ohr expression remained low after addition of NaOCl to Δohrc cells may reflect the already elevated expression. Expression of ohrR was increased ∼2-fold in WT cells on addition of 1 mM NaOCl, and this effect was attenuated on expression of excess ohrR (in ΔohrRc cells; Fig. 6F). Taken together, our data indicate that higher concentrations of the inorganic oxidants H2O2 and NaOCl can modestly relieve OhrR-mediated repression of ohr, while only NaOCl can induce ohrR expression.
Degradation of CHP in vivo correlates with ohr expression.
A ferrous oxidation-xylenol orange (FOX) assay was used to estimate the ability of B. thailandensis strains to degrade CHP. In this assay, ferrous iron is rapidly oxidized to ferric ions, which in turn form a colored complex with xylenol orange. Accordingly, spectrophotometric measurement of this colored complex allows determination of residual CHP (27). CHP was added to B. thailandensis cultures, and residual oxidant was determined at regular intervals. As shown in Fig. 7A and B, no CHP degradation was observed in Luria-Bertani (LB) medium (black), whereas the oxidant was gradually degraded by WT cells (and WTe cells carrying the empty vector; blue), with little oxidant remaining 40 min after CHP addition. As expected, Δohr cells were unable to degrade appreciable amounts of CHP within 40 min (Fig. 7A, light green). In contrast, Δohr cells complemented with ohr very efficiently degraded CHP, with no oxidant being detectable after 10 min (Δohrc cells; Fig. 7A, dark green), consistent with the observed high level of ohr expression in Δohrc cells (Fig. 5A). CHP degradation was also more efficient in ΔohrR cells (Fig. 7B, orange) than in WT cells, whereas complementation with ohrR (brown) restored CHP degradation to the level observed in WT cells. Taken together, these data show that CHP is primarily degraded by Ohr and that OhrR represses its synthesis. In contrast, H2O2 was degraded within a few minutes in WT cells (Fig. 7C, blue), and deletion of ohr or ohrR did not affect degradation of this oxidant, consistent with Ohr specifically degrading organic hydroperoxides.
FIG 7.

Determination of residual oxidant in cell culture medium. The relative levels of oxidants added at 0 min were determined using the FOX assay. (A, B) 200 μM CHP; (C) 200 μM H2O2. Oxidant-containing LB medium was used as a control. Where error bars (representing the SD from three experiments) are missing, they are smaller than the symbol sizes.
Degradation of CHP was more efficient in ΔohrR cells than in WT cells, with Δohr cells being unable to degrade appreciable amounts of the oxidant within the 40-min period of observation, and we expected survival to parallel the efficiency with which CHP was degraded. To address this prediction, cells were exposed to CHP for 15 min, following which serial dilutions were spotted on LB agar plates. Consistent with expectations, CHP was toxic to WT cells, with little growth being detected when cells were exposed to 0.8 mM oxidant. The ΔohrR cells were much more resistant than WT cells, consistent with the overexpression of ohr (Fig. 8, top row). However, Δohr cells did not exhibit enhanced sensitivity to CHP but instead exhibited a modestly increased resistance. The increased viability of both Δohr and ΔohrR cells compared to that of WT cells on exposure to CHP was verified by counting the number of CFU after exposure to 1 mM CHP (Table 3).
FIG 8.

Effect of ohr and ohrR deletions on survival and colony morphology. WT, ΔohrR, and Δohr cells were exposed to the identified concentrations of CHP (top row) or H2O2 (middle row) for 15 min, following which 10-fold serial dilutions were spotted on agar plates. The results are representative of those from at least three experiments. The corresponding colony morphologies, captured after ∼72 h, are illustrated at the bottom.
TABLE 3.
Viable cell count
| Strain | Viable cell count (CFU/ml)a |
|||
|---|---|---|---|---|
| Untreated | 1 mM CHP | 1 mM H2O2 | 1 mM NaOCl | |
| WT | TNTC | 7 × 103 | 27 × 105 | TNTC |
| Δohr | TNTC | 2 × 105 | 25 × 105 | TNTC |
| ΔohrR | TNTC | 26 × 105 | 30 × 105 | TNTC |
TNTC, too numerous to count. The data are representative of those from two experiments.
While WT cells exhibited the expected wrinkled colony morphology with an irregular edge, both Δohr and ΔohrR cells formed smoother colonies (Fig. 8, bottom row); for Δohr cells, a modest decrease in biofilm formation (as determined by crystal violet staining of the biofilm adhering to polystyrene tubes; Fig. S10) was also observed. These observations suggest that the absence of either gene affects bacterial physiology even under conditions that do not involve exposure to exogenous organic hydroperoxides. Consistent with the observation that H2O2 degradation was unaffected by deletion of either ohr or ohrR (Fig. 7C), survival was not altered either (Fig. 8, middle row, and Table 3).
Pathogenicity of B. thailandensis.
To investigate the role of Ohr and OhrR in virulence, the ability of the WT or mutant (Δohr, ΔohrR) strains to infect Caenorhabditis elegans was examined. Twelve hours after worms were deposited on plates inoculated with the various bacterial strains, the fraction of dead worms (as judged by a failure to move when touched) was determined. As expected, WT B. thailandensis was observed to be virulent, resulting in 16% worm death (Fig. 9). In contrast, the ΔohrR strain was less virulent, resulting in only 7% death, whereas 36% worm death was seen on the plates with the Δohr strain. Since both Δohr and ΔohrR cells were more resistant to CHP than WT cells, our data suggest that virulence in the C. elegans model does not simply correlate with such resistance.
FIG 9.
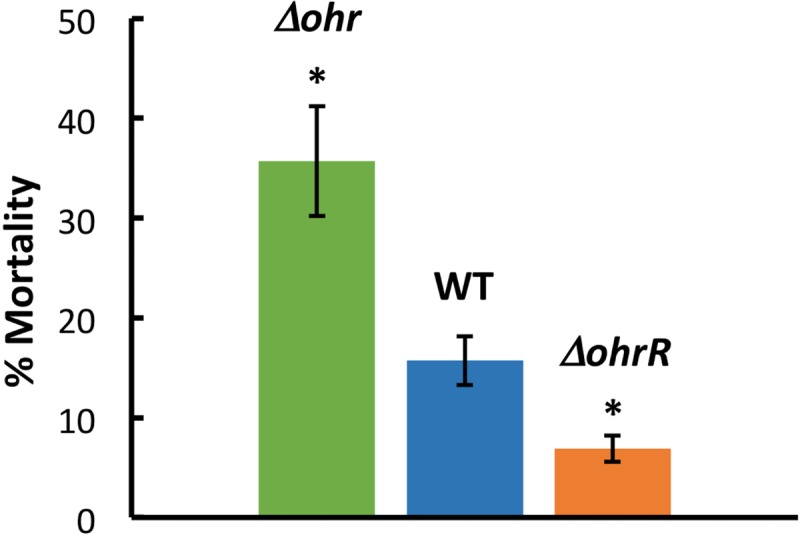
Pathogenicity of B. thailandensis in C. elegans. Percentage of dead worms 12 h after depositing worms (L4 larval stage) on plates inoculated with the identified B. thailandensis strain. Asterisks denote a P value of <0.05 compared to the WT, based on an unpaired t test. Data are from three independent experiments, with error bars indicating standard errors.
DISCUSSION
The oxidant-sensing cysteine.
B. thailandensis OhrR belongs to the 2-Cys subfamily of OhrR proteins, which sense oxidative changes in the environment via a conserved N-terminal cysteine (18, 20–22, 28). OhrR proteins generally respond preferentially or exclusively to organic hydroperoxides, and while B. thailandensis OhrR was also more sensitive to CHP, it did respond to inorganic (H2O2 and NaOCl) oxidants. C16 and C121 were essential for forming dimeric oxidative species (Fig. 2), with C16 being crucial for sensing oxidants, as evidenced by marked changes in the thermal stability of WT OhrR and OhrR-C121A on oxidation (Table 1).
Oxidants react differently with cysteines depending on their accessibility within the protein and the reactivity of the thiol. NaOCl and H2O2 may be less specific than the hydrophobic CHP, which preferentially interacts with nonpolar residues (19), and a concentration of CHP lower than the concentration of inorganic oxidants was sufficient to oxidize OhrR. As evinced by DNase I footprinting, oxidation of OhrR-C121A by both CHP and H2O2 rendered a protein that was unable to protect the ohr promoter, whereas oxidation of OhrR-C16A had more subtle effects on DNA binding (see Fig. S9 in the supplemental material), suggesting that CHP and H2O2 oxidized OhrR comparably, with attenuated DNA binding occurring as a consequence of C16 oxidation. In contrast, oxidation by NaOCl appeared to be nonspecific, and it did not lead to a loss of DNA binding but instead led to an altered binding mode, as demonstrated by the appearance of hypersensitive sites (Fig. 4F and S8). Similarly, HOCl released from professional phagocytes was reported to lead to nonspecific thiol oxidation (29).
As seen in the structure of X. campestris OhrR and the corresponding model of B. thailandensis OhrR, a small hydrophobic pocket that may allow CHP binding exists in the vicinity of the N-terminal cysteines. What is also conserved is a hydrogen-bonding network involving C16 and neighboring tyrosines (Y30′ and Y41′; Fig. S11), a network previously shown to be important for maintaining the reduced protein conformation (19, 30, 31). Indeed, the equivalent of Y30 in X. campestris OhrR was inferred to be the sensor of sulfenic acid formation. Such sensitivity may rationalize why the C16 oxidation of B. thailandensis OhrR-C121A induces a conformation that does not bind DNA despite the absence of a disulfide-bonding partner (Fig. S9D and E). For the Chromobacterium violaceum and Pseudomonas aeruginosa OhrR proteins, oxidation of the mutant protein that is missing the disulfide-bonding partner likewise produces a protein that cannot bind DNA in vitro (21, 28). However, DNA binding by the C. violaceum OhrR mutant protein is only partially recovered on addition of a reducing agent, while DNA binding by the P. aeruginosa OhrR variant is not recovered at all, perhaps reflecting an irreversible overoxidation of the N-terminal Cys. In contrast, DNA binding by oxidized B. thailandensis OhrR-C121A was recovered by addition of DTT, as evidenced by complete restoration of the DNA footprint (Fig. S9F).
The extent of thiol oxidation depends on the local environment and the potency of the oxidant. The initial oxidation event in OhrR proteins results in the formation of a sulfenic acid that subsequently forms a reversible mixed disulfide with a free thiol. Absent the latter, either the sulfenic acid may react with nitrogen nucleophiles to form a reversible sulfenamide or it may get irreversibly overoxidized to sulfonic/sulfinic acids (32). That oxidation of OhrR-C121A was fully reversible argues against sulfonic/sulfinic acid formation. Since C16 oxidation appears to be sufficient to induce a conformation that is incompatible with DNA binding in vitro, we also speculate that disulfide bond formation with C121′ may be necessary in vivo to prevent overoxidation of C16-sulfenic acid, requiring the costly de novo synthesis of functional OhrR (as previously suggested for other OhrR proteins [21, 32]), and/or to induce more substantive conformational changes that kinetically trap OhrR in the DNA-off state longer.
Formation of a C16-C121′ disulfide bond is likely to cause marked changes in the protein structure, as described for X. campestris OhrR (19). Specifically, a kink is induced in α5, which connects the dimerization and DNA-binding regions. Such kinking results in a placement of DNA-binding lobes in a position that is unfavorable for DNA binding. Similar kinking of α5 and the attendant loss of DNA binding were reported for Staphylococcus aureus MepR on the mutagenesis of several residues associated with multidrug-resistant infection, reinforcing the importance of α5 in controlling DNA binding (33). Consistent with these observations, substitution of C121, which resides in α5, resulted in a markedly lower affinity of B. thailandensis OhrR-C121A for ohr DNA (Table 2).
Differential induction of ohr and ohrR genes.
B. thailandensis OhrR repressed ohr, and gene expression was induced not only in the presence of organic oxidants but also on addition of inorganic oxidants, albeit variably and much less efficiently. The latter is in congruence with the findings of a study in sessile Burkholderia cenocepacia for which microarray data revealed an approximately 30-fold upregulation of ohr in the presence of high concentrations of either H2O2 or NaOCl (2 mM) (34). In contrast, in several other species, ohr expression is not induced by H2O2 (20, 21, 28). B. thailandensis Ohr did not participate in the degradation of H2O2 (Fig. 7C), yet H2O2 induced the upregulation of ohr (Fig. 6C). A possible explanation is that H2O2 (and NaOCl) may contribute to the accumulation of organic hydroperoxides and that upregulation of ohr prepares the cell for this eventuality.
Expression of ohr was more efficiently induced on addition of CHP (∼200-fold; Fig. 6A) to WT cells than on deletion of ohrR (∼30-fold; Fig. 5). We also observed a CHP concentration-dependent increase in ohr expression in ΔohrR cells, with a 3-fold increase in expression on addition of 1 mM CHP (Fig. 6A). These observations suggest the existence of a secondary repressor that is induced only at very high concentrations (≥1 mM) of CHP. An alternate regulator of ohr expression has been reported in P. aeruginosa, which encodes the CHP-sensitive OspR (a homolog of which does not appear to be encoded by the B. thailandensis genome) (35).
OhrR bound the ohr and ohrR promoters differently (Fig. 3). The protection of both the ohr and ohrR promoters was extensive, and EMSAs revealed the formation of multiple complexes with ohr DNA as well as modest negative cooperativity, which suggests that full occupancy requires binding of more than one OhrR dimer. The OhrR footprint in the ohr gene promoter was continuous, whereas the protection of ohrR DNA extended over a longer segment within which sections remained accessible to DNase I cleavage, perhaps reflecting a more open OhrR-DNA complex in which OhrR dimers are not as closely associated. In addition, the affinity of OhrR for the ohr promoter was much higher than that for the ohrR promoter (Table 2); the expectation is therefore for efficient repression of ohr expression even when cellular concentrations of OhrR are insufficient for repression of ohrR. This would ensure that ohrR remains expressed and that cellular levels of OhrR stay within an optimal range. It also suggests that ohr may remain in a repressed state unless organic hydroperoxide levels exceed the threshold at which both genes may be induced. Conversely, P. aeruginosa OhrR has an ∼4-fold higher affinity for the ohrR promoter than for ohr DNA, conditions that would favor ohr expression, even if the levels of inducing hydroperoxide are insufficient for complete oxidation of OhrR. Accordingly, P. aeruginosa OhrR may ensure that organic hydroperoxide levels remain low, whereas B. thailandensis OhrR may prevent hydroperoxide levels from getting too low, suggesting that accumulation of some organic hydroperoxide may be beneficial to the bacterium.
The affinity for ohr DNA was markedly decreased by OhrR mutations, whereas binding to ohrR DNA was not (Table 2). This suggests that binding to ohr DNA is more sensitive to conformational changes in the protein and that induction of ohr expression may be more responsive to partial or alternate protein oxidation. Induction of ohr is greater than that of ohrR, regardless of the oxidant (Fig. 6), which might be due to a combination of differential promoter strengths, different abilities of promoter-bound OhrR to compete with RNA polymerase (as suggested by the dissimilar footprints), and/or distinct mRNA stabilities; this would ensure that the cellular concentrations of OhrR do not exceed a range within which the control of ohr expression is exquisitely sensitive to oxidant levels.
Growth with both gentamicin and H2O2 resulted in the greater production of ohr mRNA than growth with H2O2 alone, whereas gentamicin alone had no effect on gene expression (Fig. 5 and 6A and B). Gentamicin has been reported to induce the formation of ROS (36); we therefore surmise that H2O2 in combination with gentamicin-induced ROS leads to the nonenzymatic production of inducing hydroperoxides. In Δohrc cells, however, exposure to 1 mM H2O2 resulted in a particularly marked increase (∼9-fold) in ohrR expression (Fig. 6D), whereas overexpression of ohr had no effect on ohrR expression in the absence of H2O2 (Fig. 5B). This also points to a combined effect of gentamicin and H2O2. With the site of Ohr synthesis in Δohrc cells being spatially disconnected from the ohr-ohrR locus, we speculate that it may take a while for the Ohr enzyme to reach the ohrR promoter, as a result giving ample time for inducing hydroperoxides to oxidize OhrR, resulting in induced ohrR expression.
OhrR plays a role in B. thailandensis-mediated killing of C. elegans.
Ohr functions as the primary organic hydroperoxide scavenger in B. thailandensis (Fig. 7A and B); while higher concentrations of CHP were lethal to WT cells, ΔohrR cells were less sensitive as a result of constitutive ohr expression (Fig. 8 and 10). An intriguing observation was the increased resistance of Δohr cells to CHP compared to that of WT cells (Fig. 8 and Table 3). Organic hydroperoxide levels are expected to be higher in Δohr cells than in WT cells, a consequence of which may be the more efficient induction of other hydroperoxide-sensitive genes that promote survival (Fig. 10). This hypothesis is supported by a determination of the CHP regulon in C. violaceum, in which the upregulation of numerous genes encoding proteins such as efflux pumps and DNA repair enzymes was observed (37). This suggests that recovery from the toxic effects of oxidants is induced by CHP. As noted above, the higher affinity of OhrR for ohr DNA than for the site in the ohrR promoter suggests that expression of ohr is not induced unless the levels of inducing hydroperoxide are high enough to efficiently oxidize OhrR. Combined with the increased survival of Δohr cells on exposure to CHP and the enhanced virulence in C. elegans, we suggest that organic hydroperoxides contribute to virulence by upregulating the expression of genes that promote fitness in an oxidative (host) environment (Fig. 10) and that ohr-ohrR expression is fine-tuned to ensure that hydroperoxide levels remain high enough for such induction. Consistent with this interpretation, we note that production of AhpC as a backup mechanism for CHP degradation was not indicated, as little degradation of CHP was seen in Δohr cells (Fig. 7).
FIG 10.

Deletion of ohr or ohrR differentially affects survival on exposure to organic hydroperoxides in vitro and killing of C. elegans. Ovals represent ΔohrR, WT, and Δohr cells in which the levels of organic hydroperoxide (OHP) are low, medium, and high, respectively (colored bar graphs), as determined by the FOX assay (Fig. 7). Genes in the OHP and/or OhrR regulons (including ohr, in the case of WT cells) are expected to be the most efficiently induced in WT and Δohr cells. Boxes below the ovals represent items that may promote (+) or counter (−) survival on exposure to OHP in vitro and C. elegans killing. n/a, not applicable.
OhrR and Ohr have previously been implicated in virulence, however, with different outcomes (21, 37, 38). In Mycobacterium tuberculosis, deletion of ohrR resulted in increased virulence in mice as a consequence of the constitutive expression of ohr and greater resistance to organic hydroperoxides (38). Consistent with this observation, OhrR was reported to respond to intracellular organic hydroperoxides (39). However, a Δohr strain exhibited the same virulence as WT cells, suggesting that alternate enzymes that can degrade the toxic hydroperoxides are induced. In contrast, deletion of neither ohr nor ohrR altered virulence in Brucella abortus (40). Similarly, both C. violaceum and P. aeruginosa Δohr strains exhibit a level of virulence similar to that of WT cells (in mouse and C. elegans, respectively) (21, 37). However, deletion of ohrR reduced virulence in both cases, an effect that was attributed to expression of other genes in the OhrR regulon. Such reduced virulence was also observed for B. thailandensis ΔohrR in the C. elegans model (Fig. 9 and 10); however, an increased virulence of Δohr cells has not been previously reported. We therefore speculate that carefully controlled ohr expression at the time of infection ensures that hydroperoxides produced by the host remain at nontoxic levels yet are sufficient for inducing virulence factors. The decreased virulence of ΔohrR cells supports this theory.
MATERIALS AND METHODS
Sequence alignment and structural modeling of OhrR.
Amino acid sequences were aligned using the MUSCLE sequence alignment server (41). Amino acid residues were shaded using the BOXSHADE (v3.21) server. A structural model of OhrR was generated using homology modeling by the SWISS-MODEL server in the automated mode (42). The model was built using the structure of X. campestris OhrR as a template (PDB accession number 2PFB), based on 55% identity.
Cloning and mutagenesis.
B. thailandensis E264 (ATCC 700388D-5) was cultured in Luria-Bertani (LB) broth at 37°C. Genomic DNA was extracted from an overnight culture and used as a template to amplify ohrR (BTH_II0598) with flanking NdeI and HindIII sites (for the primer sequences, see Table S2 in the supplemental material). The PCR product was cloned into the NdeI/HindIII sites of the pET28b expression vector, resulting in addition of a His6 tag at the N terminus. The recombinant plasmid was sequenced for verification. The plasmid harboring WT ohrR (pOhrR) was transformed into E. coli BL21(DE3)pLysS and plated on LB medium-kanamycin plates (50 μg/ml).
Plasmid pOhrR was used as the template for synthesizing constructs in which Cys residues were mutated to alanine using a whole-plasmid amplification approach, wherein PCR primers contained the requisite mismatch(es) (for the primer sequences, see Table S2). Single and double mutants were created, generating plasmids pC16A, pC121A, and pC16AC121A carrying the genes C16AohrR, C121AohrR, and C16AC121AohrR, respectively. The accuracy of the mutagenesis was verified by sequencing, and plasmids were transformed into E. coli BL21(DE3)pLysS.
Expression and purification of OhrR variants.
Individual colonies were used to inoculate LB medium-kanamycin (50 μg/ml); the cultures were grown overnight and then diluted 1:100 and grown at 37°C until the optical density at 600 nm (OD600) was ∼0.5. Overexpression of protein was induced by adding 0.5 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG) and allowing the cells to grow for 2 h. The induced cultures were then cooled on ice, cells were collected by centrifugation at 4°C, and the cell pellets were stored at −80°C.
Cell pellets obtained from a 1-liter culture were thawed on ice, and the cells were resuspended in 12 ml ice-cold lysis buffer (300 mM NaCl, 50 mM sodium phosphate buffer [pH 8.0], 5% [vol/vol] glycerol, 1 mM dithiothreitol [DTT], 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [PMSF], 5 mM imidazole). Resuspended cells were disrupted on ice by sonication. Lysozyme (300 μg/ml) and 0.05% (vol/vol) Triton X-100 were then added to the cell suspension. After allowing cell lysis to proceed on ice for 1 h, the sample was centrifuged at 9,000 rpm and 4°C for 1 h. The supernatant was mixed with preequilibrated HIS-Select nickel affinity beads (Sigma) and incubated with gentle agitation for 1 h at 4°C; the equilibration buffer contained 300 mM NaCl, 50 mM sodium phosphate buffer (pH 8.0), 5% glycerol. The suspension was loaded onto a column and washed using 15 column volumes of wash buffer (300 mM NaCl, 50 mM sodium phosphate buffer [pH 8.0], 5% glycerol, 10 mM imidazole, 1 mM DTT), followed by 10 column volumes of wash buffer with 20 mM imidazole. Protein was eluted with 6 column volumes each of wash buffer containing increasing concentrations of imidazole (50 mM-100 mM-250 mM imidazole). Pure eluates were pooled and dialyzed against dialysis buffer (300 mM NaCl, 50 mM sodium phosphate buffer [pH 8.0], 5% glycerol, 1 mM DTT) overnight at 4°C. Dialyzed protein was concentrated using a centrifugal filter unit, flash frozen using liquid nitrogen, and stored at −80°C. The concentration was calculated using a micro-bicinchoninic acid (BCA) protein assay kit (Pierce) and bovine serum albumin (BSA) as the standard and by measuring the absorbance at 280 nm using a calculated extinction coefficient of 13,000 M−1 cm−1.
Size exclusion chromatography.
A Superose 12 10/300 GL (GE Healthcare) column was preequilibrated at 4°C using gel filtration buffer (150 mM NaCl, 50 mM sodium phosphate [pH 8.0], 2% glycerol). Oxidized (with 20 mM cumene hydroperoxide [CHP]) and reduced (with 20 mM DTT) OhrR protein samples were run in gel filtration buffer at a flow rate of 0.8 ml/min at 4°C. A standard curve in which the Kav values of the protein markers (Bio-Rad) were plotted as a function of the log10 of their molecular weights was used (43). The equation Kav = (VE − VO)/(VT − VO), where VE, VO, and VT represent the retention volume of the protein, the void volume of the column, and the geometric bed volume of the column, respectively, was used. Markers were bovine serum albumin (66.0 kDa), ovalbumin (44.0 kDa), myoglobin (17.0 kDa), and vitamin B12 (1,350 Da).
CD spectroscopy.
The secondary structure composition of OhrR was estimated by circular dichroism (CD) spectroscopy using a Jasco J-815 CD spectrometer (Jasco Inc.). The far-UV CD spectrum of 10 μM OhrR was measured in CD buffer (20 mM NaCl, 12.5 mM sodium phosphate buffer [pH 8.0], 2.5% glycerol, 0.5 mM β-mercaptoethanol) at 20°C. Spectrometric readings were conducted in triplicate at 1-nm intervals using a quartz cuvette with a 0.1-cm path length. The secondary structure composition was calculated using the K2D program from Dichroweb (44). The goodness of fit was determined from the normalized root mean square deviation value to be 0.08 with a maximum error of 0.182.
Effect of oxidants measured using nonreducing SDS-PAGE.
Samples were prepared by incubating 15 μM WT and mutant OhrR proteins (already containing 1 mM DTT) with increasing concentrations (10 μM to 10 mM) of CHP, H2O2, and sodium hypochlorite (NaOCl) for 15 min at room temperature. Reactions were terminated by addition of SDS-PAGE sample loading buffer without the reducing agent. The reversibility of oxidation was determined by treating oxidized protein samples with 20 mM DTT for 15 min prior to adding sample buffer. Protein samples were subjected to electrophoresis on a 12% SDS-polyacrylamide gel and observed using Coomassie brilliant blue stain.
Thermal stability assay.
The thermal stability of OhrR was determined by measurement of the fluorescence of SYPRO Orange dye bound to unfolded protein using an Applied Biosystems (ABI) 7500 real-time PCR system (43). The reaction mixtures were assembled on ice in a 96-well plate. Sixty-microliter samples containing 8 μM WT and mutant OhrR proteins were prepared in thermal stability assay (TSA) buffer (100 mM Tris [pH 8.0], 100 mM NaCl, 5× SYPRO Orange [Invitrogen] dye). Protein samples were incubated with the concentrations of oxidant (CHP, H2O2, or NaOCl) indicated above for 15 min before being added to the TSA buffer. SYPRO Orange fluorescence emission was measured over a temperature range of 5°C to 94°C in 1°C increments. A SYBR green filter was used for fluorescence detection. The total fluorescence yield was corrected for the fluorescence from buffer blanks and then normalized to the maximal fluorescence value. The data were plotted using SigmaPlot (v9) software and fit to a four-parameter sigmoidal equation to obtain the melting temperature (Tm). The Tm values from technical triplicates for each sample were averaged for each experiment, and Tm values are reported as the average (±standard deviation [SD]) from three separate experiments.
DNA binding measured using electrophoretic mobility shift assay (EMSA).
Two operator DNA segments, 73-bp ohrO and 105-bp ohrRO segments, representing the sequences upstream of the ohr (BTH_II0597) and ohrR (BTH_II0598) open reading frames, respectively, were amplified from the B. thailandensis E264 genome using PCR (for the primer sequences, see Table S2). DNA was labeled at the 5′ ends with [32P]ATP and T4-polynucleotide kinase. Binding reaction mixtures contained 0.1 nM radiolabeled ohrO or ohrRO and increasing concentrations of the OhrR protein in binding buffer (25 mM Tris [pH 8.0], 50 mM NaCl, 0.1 mM Na2EDTA, 5 mM DTT, 0.05% Brij58, 50 μg/ml bovine serum albumin [BSA], 0.8% glycerol) and nonspecific DNA (0.8 nM linearized pET28b). Considering that pET28b is 5,368 bp, this corresponds to a >400-fold excess of nonspecific DNA compared to the 105-bp ohrRO DNA. The reaction mixtures were incubated at room temperature (25°C) for 30 min and then loaded on prerun 8% (wt/vol) polyacrylamide gels (39:1 acrylamide-bisacrylamide) in 0.5× Tris-borate EDTA (TBE) buffer with the power on and electrophoresed at 10 V cm−1 for 1 h. The gels were dried, exposed to phosphor screens, and visualized using a Typhoon 8600 phosphorimager (GE Healthcare). ImageQuant (v5.1) software was used for quantitation. Half-maximal saturation of ohrO and ohrRO DNA was calculated using the Kaleidagraph (v4.0) program (Synergy Software) by fitting the data to the equation f = fmax · [OhrR]nH/(Kd + [OhrR]nH), where nH is the Hill coefficient (which is 1 for DNA with a single binding site), Kd is the apparent equilibrium dissociation constant reflecting half-maximal saturation of the DNA, and fmax is the maximal fractional saturation (45).
The specificity of the interaction of OhrR with ohrO and ohrRO was determined by a competition assay in which OhrR was titrated with increasing concentrations of unlabeled nonspecific plasmid DNA (linearized pET28b) or unlabeled ohrO or ohrRO DNA in the presence of labeled ohrO or ohrRO in the binding buffer described above at room temperature for 30 min. Both DNAs were mixed prior to adding the protein. The remainder of the procedure was as described above.
DNase I footprinting.
To identify the specific DNA binding sites for OhrR, DNase I footprinting was carried out (25). Two fluorescently labeled PCR products representing sequences upstream of the ohr (272 bp) and ohrR (280 bp) open reading frames were amplified using B. thailandensis E264 genomic DNA as the template. The forward primer was 5′ end labeled with 6-carboxyfluorescein (6-FAM) (for the primer sequences, see Table S2). For each reaction, 30 ng (17 nM) of fluorescently labeled DNA was incubated without or with protein (70 nM) in binding buffer for 30 min at room temperature. For reactions involving the DNA sequence upstream of ohrRO, 0.4 nM nonspecific DNA (linearized pET28b) was included. This was followed by DNase I treatment (3 min, room temperature) by adding 1 μl 10× DNase I buffer and 1 μl of 0.2 U DNase I. Reactions were terminated by adding 10 μl of 100 mM EDTA, and the DNA was extracted with phenol-chloroform and ethanol precipitated. To determine the effect of oxidants on the DNA binding of OhrR, OhrR was first oxidized with increasing concentrations of CHP, H2O2, or sodium hypochlorite before incubating it with DNA.
For fragment analysis, 5 to 8 ng of DNA was resuspended in 10 μl Hi-Di formamide and mixed with 1 μl of a 1:10-diluted GeneScan 500 LIZ size standard. Prior to injection, samples were heat denatured by boiling and then loaded onto an ABI 3130 automated capillary sequence analyzer. Electropherograms were processed using GeneMapper (v4.1) software. Electropherograms representing reaction mixtures containing OhrR were overlaid with the electropherogram obtained from DNase I-digested DNA without protein added (25). Each experiment was repeated at least three times.
Confirmation of transposon insertion mutants.
Transposon mutants in which transposon T8 (ISlacZ hah-Tc) was inserted in B. thailandensis ohrR (at genomic position 699,144, open reading frame [ORF] location 172) and ohr (at genomic position 698,454, ORF location 247) were procured from the Manoil lab (26). B. thailandensis strains tnbt1_2r100315p02q176 (with interrupted ohrR) and tnbt1_2r100315p08q125 (with interrupted ohr) were grown on LB agar plates with 80 μg/ml tetracycline overnight at 37°C. Genomic DNA was isolated and used as the template for PCR verification of transposon mutants using both gene-specific and transposon-specific primers (Table S2).
Plasmid construction and conjugative transfer for complementation.
Plasmids for complementation contained the ohr or ohrR genes under the control of their native promoters. A 697-bp product containing full-length ohrR preceded by a 236-bp upstream sequence and a 665-bp product containing a 233-bp DNA sequence followed by full-length ohr were amplified from B. thailandensis E264 genomic DNA, with restriction sites HindIII-BamHI and KpnI-EcoRI, respectively, being introduced into the amplicons (for the primer sequences, see Table S2). The PCR products were cloned into the gentamicin-resistant broad-host-range cloning vector pBBR1-MCS5 (46). Empty plasmids were used as a control. All plasmids were verified by sequencing.
Complementation plasmids were transformed into B. thailandensis E264 strains using triparental mating. Overnight cultures of donor (E. coli Top10 with pBBR1-MCS5 derivative), recipient (B. thailandensis WT, Δohr, or ΔohrR), and helper [HB101(pRK2013::Tn7)] cells were grown at 37°C in LB medium, with the LB medium being supplemented with 60 μg/ml gentamicin for donor and recipient cells. Cells were mixed in 2:1:2 ratios of donor-recipient-helper strains, pelleted down, washed 3 to 4 times with LB medium, and resuspended in 60 μl LB medium. The suspension was then spotted on prewarmed LB agar plates and incubated overnight at 37°C. The cells were then scraped off and resuspended in 1.0 ml LB medium. Serial dilutions of this suspension were plated on LB agar plates containing the antibiotics tetracycline (80 μg/ml), gentamicin (1.0 mg/ml), and chloramphenicol (8 μg/ml) for selection of transconjugants, followed by PCR verification using primers Veri_pBBR_XbaI Fw and Rev (Table S2).
RNA isolation.
Overnight cultures of B. thailandensis WT, Δohr, and ΔohrR were grown in LB medium-gentamicin (30 μg/ml), and their respective complements (the We, Δohrc, and ΔohrRc strains) were grown in LB medium with 1 mg/ml gentamicin, all at 37°C. Cultures were diluted 1:100 in the same medium and grown at 37°C to an OD600 of ∼0.6. To assess the effect of oxidants on gene expression, cultures were incubated with 0.2 or 1 mM CHP, H2O2, or NaOCl for 15 min at 37°C. Cells (1.0 ml culture) were centrifuged at 4°C, washed with ice-cold diethyl pyrocarbonate (DEPC)-treated water, repelleted, and stored at −80°C. Total RNA was isolated using an Illustra RNAspin minikit (GE Healthcare). DNA contamination was removed using a Turbo DNase kit (Ambion), and the absence of DNA was verified by PCR. The integrity and quality of the RNA were determined spectrophotometrically by using a NanoDrop spectrophotometer (Thermo Scientific) and by running the RNA samples on a 1.2% agarose gel followed by staining with ethidium bromide.
In vivo gene expression analysis using qRT-PCR.
RNA was first converted into cDNA by mixing 600 ng RNA with 0.6 μM reverse primer for the reference gene (BTH_I3014, encoding the glutamate synthase [GS] large subunit), ohr, or ohrR (for the primer sequences, see Table S2), followed by incubating the mixture for 5 min at 65°C and then promptly placing it on ice. To this mixture, 8.5 μl of the master mix (containing 1× avian myeloblastosis virus [AMV] reverse transcriptase buffer, 1 mM MgCl2, 1 mM deoxynucleoside triphosphate mix, 10 U AMV reverse transcriptase [New England BioLabs]) was added, making a total reaction volume of 25 μl, followed by incubation for 1 h at 42°C. To compare differences in gene expression qualitatively, the cDNA was used as a template for PCR with gene-specific primers. PCR products were electrophoresed on a 1.2% agarose gel and observed after staining with ethidium bromide.
For quantifying mRNA transcripts, the cDNA was used as the template for qRT-PCR. Gene-specific forward and reverse primers were added along with Luna Universal quantitative PCR (qPCR) master mix (New England BioLabs) for performing qPCR on a ViiA 7 real-time PCR system (Applied Biosystems). Estimation of transcript levels was carried out using the 2−ΔCT method. For analysis of the effect of oxidation on gene expression levels, the comparative threshold cycle (CT) method (2−ΔΔCT) was used (47). Expression of the reference gene was constant, as reflected in consistent CT values under the various conditions. qPCR data are represented as the mean (±standard error of the mean) for at least two biological replicates.
Hydroperoxide degradation and resistance.
The ferrous oxidation-xylenol orange (FOX) assay was performed to assess the hydroperoxide reductase activity of the bacterial strains (27). Overnight cultures of B. thailandensis WT, Δohr, and ΔohrR and their respective complements were grown at 37°C in LB broth (containing gentamicin at the concentrations noted above). Overnight cultures were diluted 1:100 in LB medium-gentamicin and grown until the OD600 was ∼0.6. A 10-ml culture was then treated with CHP (200 μM) for 40 min; 110-μl aliquots were removed at intervals of 10 min after CHP addition, and the supernatant was collected by centrifugation. At each time point, the amount of residual CHP was calculated by mixing 100 μl of the supernatant with 400 μl of 25 mM sulfuric acid and incubating the mixture with 500 μl freshly prepared reaction buffer (25 mM sulfuric acid, 0.2 mM xylenol orange, 0.2 mM ferrous ammonium sulfate) for 10 min at room temperature in the dark. The absorbance at 540 nm was measured. LB medium was used as a blank. Residual CHP concentrations were determined by comparison to a CHP standard curve. The amount of residual H2O2 (200 μM) was measured similarly, except that it was measured at 3-min time points.
The extent of resistance to oxidants was assessed using a plate assay. Overnight cultures of B. thailandensis WT, Δohr, and ΔohrR were grown at 37°C in LB medium containing 30 μg/ml gentamicin. Cultures were diluted 1:100 in the same medium and grown until the OD600 was ∼0.6. One milliliter of culture was incubated with 0.2 or 0.8 mM CHP, H2O2, or NaOCl for 15 min at 37°C, followed by the spotting of 10 μl of serial dilutions on LB medium-gentamicin agar plates. The plates were incubated at 37°C for 18 to 22 h.
To determine viability, overnight cultures of B. thailandensis strains grown in LB medium with 30 μg/ml gentamicin were diluted 1:100 in the same medium and grown at 37°C to an OD600 of ∼0.6. One milliliter of each culture was then treated for 15 min with 1 mM oxidant, following which they were serially diluted. One hundred microliters of each serial dilution was spread on LB agar plates (30 μg/ml gentamicin), the plates were incubated overnight at 37°C, and the number of CFU was counted.
Colony morphology.
Overnight cultures grown in LB medium with 30 μg/ml gentamicin were diluted 1:100 in the same medium and grown at 37°C to an OD600 of ∼0.6. One milliliter of each culture was then pelleted, resuspended in 20 μl LB medium, and spotted on prewarmed LB medium-gentamicin plates. After incubating the plates at 37°C for 72 h, images of bacterial colony morphologies were captured using a Zeiss SteREO Lumar.V12 microscope.
Pathogenicity in Caenorhabditis elegans.
Nematode growth medium (NGM) agar plates were inoculated with 12 μl of a B. thailandensis overnight culture (OD600, ∼1.2). The plates were incubated for 24 h at 37°C, followed by incubation for ∼18 h at room temperature. About 10 to 15 C. elegans (Bristol strain N2, L4 larval stage) worms were picked using a loop and placed on each assay plate. After 12 h, worm death was scored using a dissecting microscope (Olympus SZ-ST). Only worms that failed to move, even on touch, were scored as dead.
Supplementary Material
ACKNOWLEDGMENTS
Support from the National Science Foundation is gratefully acknowledged (MCB-1515349 and MCB-1714219 to A.G.).
We thank R. Lu and F. Meng for providing worms and for helpful discussions about their care.
We have no conflicts of interest with this study.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00322-18.
REFERENCES
- 1.Sies H, Berndt C, Jones DP. 2017. Oxidative stress. Annu Rev Biochem 86:715–748. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- 2.Knoefler D, Leichert LI, Thamsen M, Cremers CM, Reichmann D, Gray MJ, Wholey WY, Jakob U. 2014. About the dangers, costs and benefits of living an aerobic lifestyle. Biochem Soc Trans 42:917–921. doi: 10.1042/BST20140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen H, Crowley JR, Heinecke JW. 2002. Human neutrophils use the myeloperoxidase-hydrogen peroxide-chloride system to chlorinate but not nitrate bacterial proteins during phagocytosis. J Biol Chem 277:30463–30468. doi: 10.1074/jbc.M202331200. [DOI] [PubMed] [Google Scholar]
- 4.Nathan C, Ding A. 2010. SnapShot: reactive oxygen intermediates (ROI). Cell 140:951–951.e952. doi: 10.1016/j.cell.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Klomsiri C, Panmanee W, Dharmsthiti S, Vattanaviboon P, Mongkolsuk S. 2005. Novel roles of ohrR-ohr in Xanthomonas sensing, metabolism, and physiological adaptive response to lipid hydroperoxide. J Bacteriol 187:3277–3281. doi: 10.1128/JB.187.9.3277-3281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prost I, Dhondt S, Rothe G, Vicente J, Rodriguez MJ, Kift N, Carbonne F, Griffiths G, Esquerre-Tugaye MT, Rosahl S, Castresana C, Hamberg M, Fournier J. 2005. Evaluation of the antimicrobial activities of plant oxylipins supports their involvement in defense against pathogens. Plant Physiol 139:1902–1913. doi: 10.1104/pp.105.066274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guillemot L, Medina M, Pernet E, Leduc D, Chignard M, Touqui L, Wu Y. 2014. Cytosolic phospholipase A2alpha enhances mouse mortality induced by Pseudomonas aeruginosa pulmonary infection via interleukin 6. Biochimie 107(Pt A):95–104. doi: 10.1016/j.biochi.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 8.Leslie CC. 2015. Cytosolic phospholipase A(2): physiological function and role in disease. J Lipid Res 56:1386–1402. doi: 10.1194/jlr.R057588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newcomer ME, Brash AR. 2015. The structural basis for specificity in lipoxygenase catalysis. Protein Sci 24:298–309. doi: 10.1002/pro.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Refsgaard HH, Tsai L, Stadtman ER. 2000. Modifications of proteins by polyunsaturated fatty acid peroxidation products. Proc Natl Acad Sci U S A 97:611–616. doi: 10.1073/pnas.97.2.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poole LB. 2005. Bacterial defenses against oxidants: mechanistic features of cysteine-based peroxidases and their flavoprotein reductases. Arch Biochem Biophys 433:240–254. doi: 10.1016/j.abb.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Lesniak J, Barton WA, Nikolov DB. 2002. Structural and functional characterization of the Pseudomonas hydroperoxide resistance protein Ohr. EMBO J 21:6649–6659. doi: 10.1093/emboj/cdf670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sukchawalit R, Loprasert S, Atichartpongkul S, Mongkolsuk S. 2001. Complex regulation of the organic hydroperoxide resistance gene (ohr) from Xanthomonas involves OhrR, a novel organic peroxide-inducible negative regulator, and posttranscriptional modifications. J Bacteriol 183:4405–4412. doi: 10.1128/JB.183.15.4405-4412.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuangthong M, Atichartpongkul S, Mongkolsuk S, Helmann JD. 2001. OhrR is a repressor of ohrA, a key organic hydroperoxide resistance determinant in Bacillus subtilis. J Bacteriol 183:4134–4141. doi: 10.1128/JB.183.14.4134-4141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deochand DK, Grove A. 2017. MarR family transcription factors: dynamic variations on a common scaffold. Crit Rev Biochem Mol Biol 52:595–613. doi: 10.1080/10409238.2017.1344612. [DOI] [PubMed] [Google Scholar]
- 16.Fuangthong M, Helmann JD. 2002. The OhrR repressor senses organic hydroperoxides by reversible formation of a cysteine-sulfenic acid derivative. Proc Natl Acad Sci U S A 99:6690–6695. doi: 10.1073/pnas.102483199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JW, Soonsanga S, Helmann JD. 2007. A complex thiolate switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proc Natl Acad Sci U S A 104:8743–8748. doi: 10.1073/pnas.0702081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panmanee W, Vattanaviboon P, Poole LB, Mongkolsuk S. 2006. Novel organic hydroperoxide-sensing and responding mechanisms for OhrR, a major bacterial sensor and regulator of organic hydroperoxide stress. J Bacteriol 188:1389–1395. doi: 10.1128/JB.188.4.1389-1395.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newberry KJ, Fuangthong M, Panmanee W, Mongkolsuk S, Brennan RG. 2007. Structural mechanism of organic hydroperoxide induction of the transcription regulator OhrR. Mol Cell 28:652–664. doi: 10.1016/j.molcel.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 20.Chuchue T, Tanboon W, Prapagdee B, Dubbs JM, Vattanaviboon P, Mongkolsuk S. 2006. ohrR and ohr are the primary sensor/regulator and protective genes against organic hydroperoxide stress in Agrobacterium tumefaciens. J Bacteriol 188:842–851. doi: 10.1128/JB.188.3.842-851.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atichartpongkul S, Fuangthong M, Vattanaviboon P, Mongkolsuk S. 2010. Analyses of the regulatory mechanism and physiological roles of Pseudomonas aeruginosa OhrR, a transcription regulator and a sensor of organic hydroperoxides. J Bacteriol 192:2093–2101. doi: 10.1128/JB.01510-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oh SY, Shin JH, Roe JH. 2007. Dual role of OhrR as a repressor and an activator in response to organic hydroperoxides in Streptomyces coelicolor. J Bacteriol 189:6284–6292. doi: 10.1128/JB.00632-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drevinek P, Mahenthiralingam E. 2010. Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin Microbiol Infect 16:821–830. doi: 10.1111/j.1469-0691.2010.03237.x. [DOI] [PubMed] [Google Scholar]
- 24.Rhodes KA, Schweizer HP. 2016. Antibiotic resistance in Burkholderia species. Drug Resist Updat 28:82–90. doi: 10.1016/j.drup.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sivapragasam S, Pande A, Grove A. 2015. A recommended workflow for DNase I footprinting using a capillary electrophoresis genetic analyzer. Anal Biochem 481:1–3. doi: 10.1016/j.ab.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 26.Gallagher LA, Ramage E, Patrapuvich R, Weiss E, Brittnacher M, Manoil C. 2013. Sequence-defined transposon mutant library of Burkholderia thailandensis. mBio 4:e00604-. doi: 10.1128/mBio.00604-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang ZY, Hunt JV, Wolff SP. 1992. Ferrous ion oxidation in the presence of xylenol orange for detection of lipid hydroperoxide in low density lipoprotein. Anal Biochem 202:384–389. doi: 10.1016/0003-2697(92)90122-N. [DOI] [PubMed] [Google Scholar]
- 28.da Silva Neto JF, Negretto CC, Netto LE. 2012. Analysis of the organic hydroperoxide response of Chromobacterium violaceum reveals that OhrR is a Cys-based redox sensor regulated by thioredoxin. PLoS One 7:e47090. doi: 10.1371/journal.pone.0047090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Degrossoli A, Muller A, Xie K, Schneider JF, Bader V, Winklhofer KF, Meyer AJ, Leichert LI. 2018. Neutrophil-generated HOCl leads to non-specific thiol oxidation in phagocytized bacteria. Elife 7:e32288. doi: 10.7554/eLife.32288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong M, Fuangthong M, Helmann JD, Brennan RG. 2005. Structure of an OhrR-ohrA operator complex reveals the DNA binding mechanism of the MarR family. Mol Cell 20:131–141. doi: 10.1016/j.molcel.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 31.Soonsanga S, Fuangthong M, Helmann JD. 2007. Mutational analysis of active site residues essential for sensing of organic hydroperoxides by Bacillus subtilis OhrR. J Bacteriol 189:7069–7076. doi: 10.1128/JB.00879-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soonsanga S, Lee JW, Helmann JD. 2008. Oxidant-dependent switching between reversible and sacrificial oxidation pathways for Bacillus subtilis OhrR. Mol Microbiol 68:978–986. doi: 10.1111/j.1365-2958.2008.06200.x. [DOI] [PubMed] [Google Scholar]
- 33.Birukou I, Tonthat NK, Seo SM, Schindler BD, Kaatz GW, Brennan RG. 2013. The molecular mechanisms of allosteric mutations impairing MepR repressor function in multidrug-resistant strains of Staphylococcus aureus. mBio 4:e00528-. doi: 10.1128/mBio.00528-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peeters E, Sass A, Mahenthiralingam E, Nelis H, Coenye T. 2010. Transcriptional response of Burkholderia cenocepacia J2315 sessile cells to treatments with high doses of hydrogen peroxide and sodium hypochlorite. BMC Genomics 11:90. doi: 10.1186/1471-2164-11-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atichartpongkul S, Vattanaviboon P, Wisitkamol R, Jaroensuk J, Mongkolsuk S, Fuangthong M. 2016. Regulation of organic hydroperoxide stress response by two OhrR homologs in Pseudomonas aeruginosa. PLoS One 11:e0161982. doi: 10.1371/journal.pone.0161982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CT, Lobritz MA, Braff D, Schwarz EG, Ye JD, Pati M, Vercruysse M, Ralifo PS, Allison KR, Khalil AS, Ting AY, Walker GC, Collins JJ. 2014. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci U S A 111:E2100–E2109. doi: 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Previato-Mello M, Meireles DA, Netto LES, da Silva Neto JF. 2017. Global transcriptional response to organic hydroperoxide and the role of OhrR in the control of virulence traits in Chromobacterium violaceum. Infect Immun 85:e00017-17. doi: 10.1128/IAI.00017-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saikolappan S, Das K, Dhandayuthapani S. 2015. Inactivation of the organic hydroperoxide stress resistance regulator OhrR enhances resistance to oxidative stress and isoniazid in Mycobacterium smegmatis. J Bacteriol 197:51–62. doi: 10.1128/JB.02252-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garnica OA, Das K, Dhandayuthapani S. 2017. OhrR of Mycobacterium smegmatis senses and responds to intracellular organic hydroperoxide stress. Sci Rep 7:3922. doi: 10.1038/s41598-017-03819-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caswell CC, Baumgartner JE, Martin DW, Roop RM II. 2012. Characterization of the organic hydroperoxide resistance system of Brucella abortus 2308. J Bacteriol 194:5065–5072. doi: 10.1128/JB.00873-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arnold K, Bordoli L, Kopp J, Schwede T. 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 43.Grove A, Kushwaha AK, Nguyen KH. 2015. Determining the role of metal binding in protein cage assembly. Methods Mol Biol 1252:91–100. doi: 10.1007/978-1-4939-2131-7_9. [DOI] [PubMed] [Google Scholar]
- 44.Whitmore L, Wallace BA. 2004. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res 32:W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilkinson SP, Grove A. 2004. HucR, a novel uric acid-responsive member of the MarR family of transcriptional regulators from Deinococcus radiodurans. J Biol Chem 279:51442–51450. doi: 10.1074/jbc.M405586200. [DOI] [PubMed] [Google Scholar]
- 46.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM II, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 47.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.