

Abstract
Work on the electronic structures of metal–oxo complexes began in Copenhagen over 50 years ago. This work led to the prediction that tetragonal multiply bonded transition metal–oxos would not be stable beyond the iron–ruthenium–osmium oxo wall in the periodic table and that triply bonded metal–oxos could not be protonated, even in the strongest Brønsted acids. In this theory, only double bonded metal–oxos could attract protons, with basicities being a function of the electron donating ability of ancillary ligands. Such correlations of electronic structure with reactivity have gained importance in recent years, most notably owing to the widespread recognition that high-valent iron–oxos are intermediates in biological reactions critical to life on Earth.
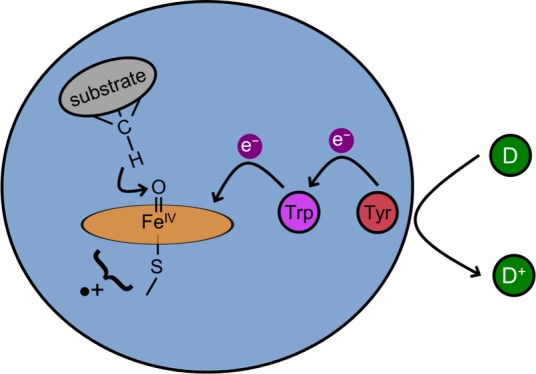
In this Account, we focus attention on the oxygenations of inert organic substrates by cytochromes P450, as these reactions involve multiply bonded iron–oxos. We emphasize that P450 iron–oxos are strong oxidants, so strong that they would destroy nearby amino acids if substrates are not oxygenated rapidly; it is our view that these high-valent iron–oxos are such dangerous reactive oxygen species that Nature surely found ways to disable them. Looking more deeply into this matter, mainly by examining many thousands of structures in the Protein Data Bank, we have found that P450s and other enzymes that require oxygen for function have chains of tyrosines and tryptophans that extend from active-site regions to protein surfaces. Tyrosines are near the heme active sites in bacterial P450s, whereas tryptophan is closest in most human enzymes. High-valent iron–oxo survival times taken from hole hopping maps range from a few nanoseconds to milliseconds, depending on the distance of the closest Trp or Tyr residue to the heme. In our proposed mechanism, multistep hole tunneling (hopping) through Tyr/Trp chains guides the damaging oxidizing hole to the protein surface, where it can be quenched by soluble protein or small molecule reductants. As the Earth’s oxygenic atmosphere is believed to have developed about 2.5 billion years ago, the increase in occurrence frequency of tyrosine and tryptophan since the last universal evolutionary ancestor may be in part a consequence of enzyme protective functions that developed to cope with the environmental toxin, O2.
Introduction
Human life on our planet could not exist were it not for two redox reactions: water oxidation to oxygen in photosynthesis and oxygen reduction to water in respiration. Also of enormous importance is the oxygenation of organic molecules by cytochrome P450. Years of research have shown that key reaction intermediates in these processes are multiply bonded oxo complexes of high-valent metals: cysteine-bound heme iron–oxos (compound I) in P450,1 a manganese–oxo (or oxyl) in the oxygen evolving complex (OEC) of photosystem II,2 and a histidine-bound heme iron–oxo in cytochrome c oxidase, the terminal enzyme in respiration.3 As it also is known that these metal–oxos are very powerful oxidants, we might expect that living cells full of organic molecules would suffer from oxidative destruction. The million dollar question is then: how do we live happily on a planet bathed in oxygen?
Our story started in Copenhagen, nearly 60 years ago, when one of us (H.B.G.) began an investigation of metal–oxo bonding. Employing a modified Mulliken molecular orbital (MO) theory for calculations of orbital energies, the d1 vanadyl ion was shown to possess a VIV–oxo triple bond (V≡O), with a dxz,dyz(π*) level well above a singly occupied dxy(nonbonding) orbital.4 In this axially compressed tetragonal ligand field, the dipole-allowed dxy → dxz,dyz(π*) absorption accounted for the blue color of the triply bonded oxo ion. Related work published soon after predicted that absorptions attributable to analogous low energy transitions would be observed in all multiply bonded (d1–d4) metal–oxo complexes.5
The Oxo Wall
The first two electrons in a C4v M(O)L5 complex (Figure 1) will occupy the dxy orbital: the π-bond order will be 2 in d0, d1, and d2 cases, and it will decrease to 3/2, 1, and 1/2 for d3, d4, and d5 complexes, respectively, as energetic dxz,dyz π* orbitals are populated.4 It follows that d4 metal–oxo bonds will be considerably weaker [ν(M–O) ≈ 800 cm–1]6 than those of d1,2 mono-oxos [ν(M–O) ≈ 900–1000 cm–1].7,8 Three π* electrons cannot be tolerated, so multiply bonded tetragonal d5 oxos are not stable. With a d4 limit, then, an oxo wall separates groups Fe–Ru–Os and Co–Rh–Ir in the periodic table (Figure 2).9
Figure 1.
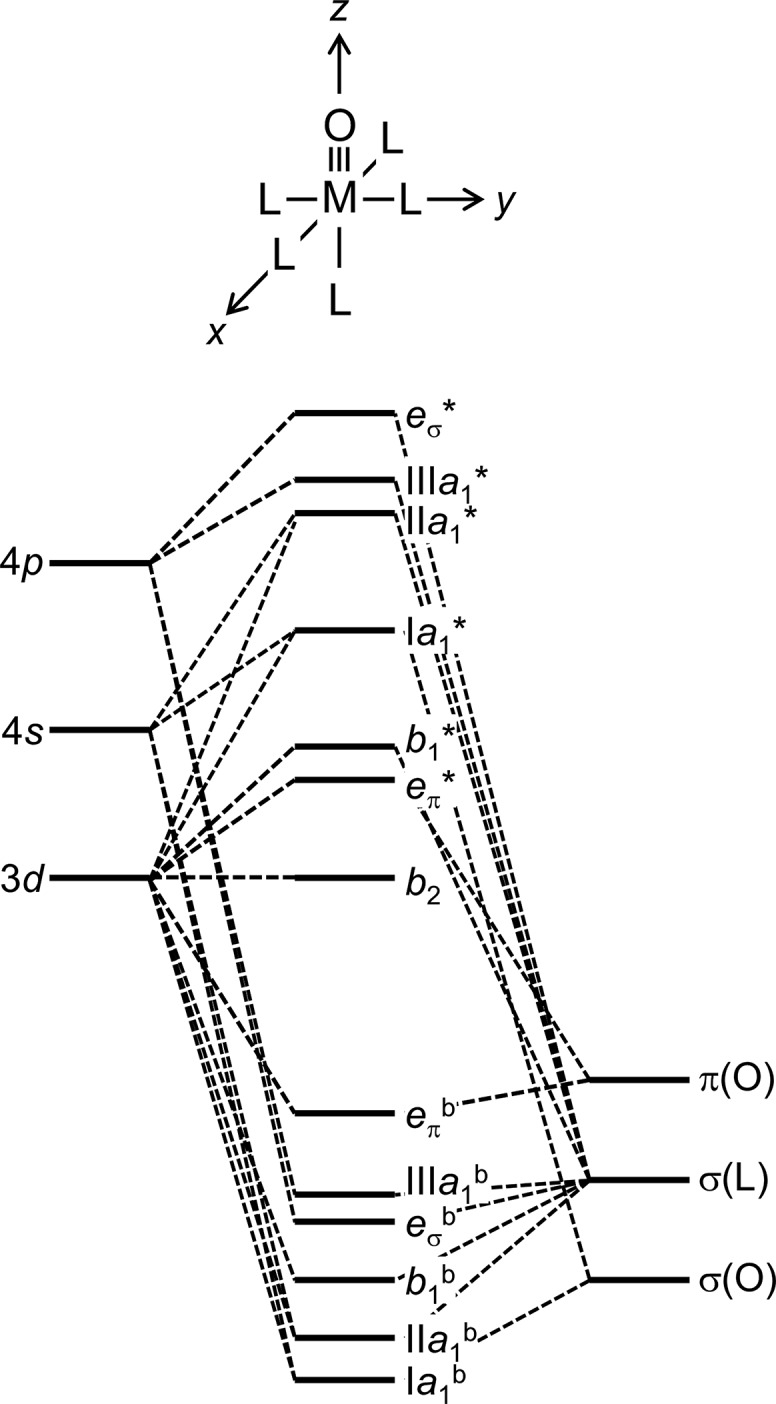
Molecular orbital model for a tetragonal M(O)L5 complex (superscript “b” indicates a bonding orbital, * indicates an antibonding orbital). Strong π-bonding between the metal and the terminal oxo ligand leads to a large energy gap (>10 000 cm–1) between the nonbonding b2(dxy) orbital and the M–oxo π-antibonding eπ*(dxz,yz) orbital.
Figure 2.
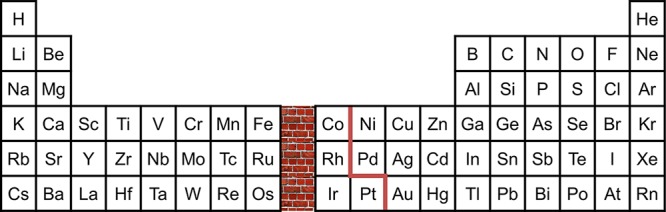
The d-orbital splitting pattern for tetragonal metal–oxo complexes (Figure 1) leads to an oxo wall between periodic table groups 8 and 9. The highest d-orbital occupation compatible with metal–oxo multiple bonding in these complexes is d4 (M=O double bond). On the left side of the wall, d4 corresponds to formal oxidation states less than or equal to IV. To the right of the wall, formal oxidation states of V or greater are necessary to form M–oxo multiple bonds. These high-oxidation-state metals are typically unstable with respect to elimination of peroxide or dioxygen. The location of the oxo-wall depends on the symmetry of the metal–oxo complex. For 4- or 5-coordinate trigonal (C3v), 3-coordinate planar (C2v), or 2-coordinate linear (C∞v) complexes, d6 configurations can support M–oxo multiple bonding, leading to a shift of the wall to the right (orange line).
Metal–Oxo pKa Values
In the tetragonal metal–oxo complexes described by the molecular orbital model depicted in Figure 1,4 the M–oxo bond order and, hence, the pKa of the conjugate acid (MOH+) are sensitive functions of the population of the eπ*(dxz,dyz) pair of orbitals (Figure 3). In complexes with 1 or 2 d electrons, the M–oxo π-bond order is two. In d3 configurations, however, at least one of those electrons must populate eπ*(dxz,dyz), leading to a reduction in M–oxo π-bond order, an increase in the M–oxo bond length, and a reduction in the energy gap between eπ* and b2 (ΔEπ). The reduction in ΔEπ as electrons populate eπ*(dxz,dyz) creates the possibility of a high-spin (S = 3/2) d3 ground state with an M–oxo π-bond order of 1. High-spin complexes with (b2)(eπ*)2 configurations must have smaller values of ΔEπ than those with low-spin d3 [(b2)2(eπ*)] ground states, owing to the presence of an extra M–oxo π-antibonding electron. Even in complexes where the high-spin and low-spin states are degenerate, the high-spin complex will have longer M–oxo bonds and smaller ΔEπ than the low-spin complex. A forbidden zone of ΔEπ values emerges from this coupling between electronic configuration and M–oxo bond length.9 This ambiguity is not present in d4 configurations unless population of higher lying σ* orbitals becomes feasible.
Figure 3.
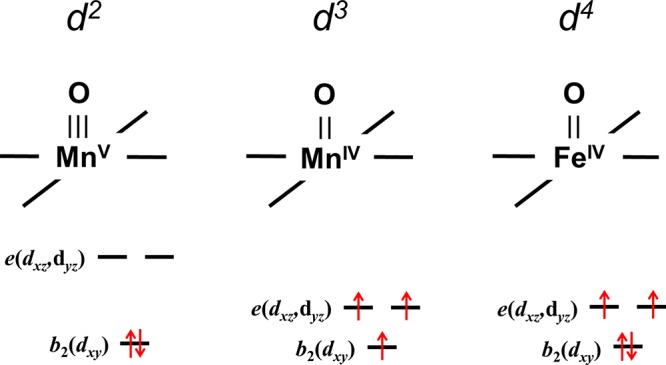
ΔEπ, the energy gap between the eπ*(dxz,dyz) and b2(dxy) orbitals, is a function of the electronic configuration: as electrons are added to eπ*(dxz,dyz), the M–oxo bond distance increases and ΔEπ decreases. The smaller energy gap, coupled with the relief of electron–electron repulsion upon electron unpairing, often produces a high-spin S = 3/2 ground state in the d3 configuration. The reduced M–oxo π-bond order in d3 and d4 configurations increases the basicity of M–oxo complexes.
Nature knew about the oxo wall when she picked metals for the generation and reduction of oxygen. In their reaction cycles, manganese (PS II) and iron (cytochrome c oxidase and P450) form multiply bonded metal–oxos with dramatically different properties (Figure 3). As we would predict, metal–oxo electronic structure is key: the triply bonded MnV–oxo is the conjugate base of a superacid, with a pKa less than −10. Such a highly electron-deficient metal–oxo could attract an oxygen donor, which in turn would promote redox-coupled O–O bond formation as proposed in one (but not the only) candidate mechanism for oxygen generation from water.10
The FeIV–oxo is much more basic than a MnV–oxo, owing to the presence of two Fe–oxo π* electrons. Ferryl species of this type are found in compounds I and II of the heme peroxidases. A key determinant of metal–oxo basicity, in addition to the M–oxo π* electron count, is the nature of the ligand trans to the oxo. Strongly donating ligands produce more basic oxos than weak donors. The proximal imidazole ligand in peroxidases is an intermediate strength ligand, and there has long been a question of whether compound II in peroxidases was PFeIV–oxo (P = porphyrin) or PFeIV–OH. X-ray and neutron crystallographic studies on ascorbate peroxidase compound II strongly indicate that the preferred configuration is PFeIV–OH.11,12 Peroxidase compounds I, however, are best described as P•+FeIV–oxo.11 The fact that peroxidase compounds II are likely PFeIV–OH species explains their relatively rapid formation from P•+FeIII–(OH2) precursors.13,14 Formation of M–oxo species by oxidation of M–(OH2) is extremely sluggish,15−17 presumably owing to the necessity to transfer two protons as well as one electron. The transfer of a single proton and electron to form PFeIV–OH makes for a more facile transformation but does not preclude the intermediacy of P•+FeIII–(OH).13−17
Cytochrome P450
Two π* electrons and an axial cysteine thiolate combine to make the doubly bonded FeIV–oxo of P450 compound II very basic, with a pKa of about 12.18,19 It is now widely recognized that the generation of such a basic FeIV–oxo is an essential step in the catalytic cycle of P450. Indeed, Green pointed out that compound I of P450 could extract an electron from a C–H bond only if coupled to proton transfer to the newly formed FeIV–oxo of compound II.19 The high reduction potential of compound I and the basicity of compound II together provide the driving force for the reaction (Figure 4).20
Figure 4.
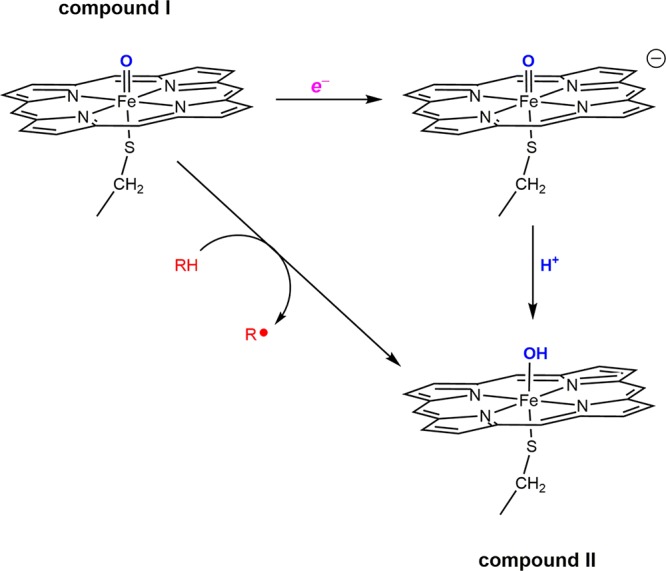
The capacity of cytochrome P450 to abstract H atoms from organic substrates depends on the strength of the FeO–H bond in compound II. Thermodynamic analyses demonstrate that this bond strength is a function of the reduction potential of compound I (E°) and the Brønsted acidity of compound II (pKa).
Reactive Oxygen Species from Uncoupled P450 Turnover
If powerfully oxidizing compounds I and II are generated in the absence of substrate, what keeps them from destroying the proteins they occupy? In looking for answers, let us first revisit the P450 reaction cycle (Figure 5).21 The heme iron in the resting enzyme is six-coordinate (axial ligands Cys–S–, H2O), low-spin, in the FeIII oxidation state (P(Cys–S)FeIII(OH2), P = porphyrin). Substrate binding in the heme pocket displaces the axial H2O ligand, prompting a transition to high-spin and a positive shift in the FeIII/II reduction potential. The increased E° favors electron delivery from NAD(P)H via cytochrome P450 reductase (CPR) to produce the FeII state, followed by oxygen binding to yield P(Cys–S)FeIII(O2–). Delivery of a second electron from CPR induces O–O bond scission, producing H2O and the critical hydroxylating agent, ferryl compound I ([P(Cys–S)]•+FeIV(O)). Hydrogen atom abstraction (HAT) from the substrate generates compound II (P(Cys–S)FeIV(OH)) and a substrate radical; hydroxyl rebound produces the product, and H2O ligation regenerates the enzyme resting state.
Figure 5.

Cytochrome P450 catalytic cycle begins with substrate (RH) binding and water displacement in the distal pocket of the ferriheme. Sequenced delivery of electrons, dioxygen, and protons leads to an active intermediate (compound I) responsible for substrate hydroxylation. Three short circuits (dashed arrows) lead to nonproductive dioxygen consumption via autoxidation (A), peroxide shunt (P), and oxidase (O) pathways.
The idealized stoichiometry of cytochrome P450 catalysis predicts that one molecule each of NAD(P)H and O2 are consumed for each molecule of oxidized substrate produced. Indeed, the prototypical prokaryotic enzyme from Pseudomonas putida (P450cam, CYP101) catalyzes hydroxylation of camphor at the 5-exo position in 94% yield relative to NADH consumed.22 Replace camphor with dehydrocamphor, however, and the yield of 5-exo-epoxide drops to 75% (relative to NADH). Substrate deuteration also lowers the product yield in CYP101, with the excess electrons producing H2O from O2.23,24 This loss of coupling of between NADH/O2 consumption and product formation has been observed in many P450 enzymes, even with “natural” substrates. The human liver enzyme CYP3A4, found on the endoplasmic reticulum membrane and responsible for a broad range of substrate hydroxylation reactions, including the metabolism of roughly half of therapeutic drugs,25 couples NADH and O2 consumption to substrate oxidation with barely 10% efficiency.26 Sligar and co-workers identified three branching points in the canonical P450 mechanism that could lead to uncoupling.26,27 The first branch point occurs at the FeIII(O2–) intermediate, involving competition between delivery of a second electron and O2– loss (autoxidation). The second branch point occurs at the FeIII(O2H) intermediate: O–O bond scission leads to ferryl compound I in competition with loss of H2O2 (peroxide shunt).The final branch point reflects the competition between substrate oxidation by compound I and two electron transfers to compound I to produce water (oxidase pathway).
We have suggested that reductants internal to the enzyme could protect the enzyme from damage by compound I when reaction with substrate is not possible.28 Intraprotein electron transfer (ET) to ferryl compound I will be a first-order kinetics process with a time constant that is independent of the concentrations of external reductants. The survival time of compound I will depend only on the placement and reduction potential of this antioxidant residue. Redox chains comprised of tyrosine and tryptophan residues (Tyr/Trp chains) could guide oxidizing equivalents (i.e., holes) away from the critical active site, steering them to the enzyme surface where they could be scavenged by soluble reductants (e.g., glutathione, ascorbate, ferrocytochrome b5).
Tyr/Trp Chains in P450
We have examined over 90 000 X-ray crystal structures in the RCSB Protein Data Bank (PDB) to identify Tyr/Trp chains in proteins.28 We found that long chains (≥3 residues) occur with highest frequency in the glycosylases and oxidoreductases. The survival time of P450 compound I will depend on the proximity of the nearest Tyr or Trp residue. We determined the shortest heme–Tyr/Trp distances in 134 cytochrome P450 structures with less than 90% sequence identity available in the PDB in April 2018 (Figure 6). The values range from 3.4 to 17.2 Å with a mean of 8.4 Å and standard deviation of 2.9 Å. Although the results should be interpreted cautiously, owing to distortions, owing to the limited sample size, some interesting trends appear in these data. Tyrosine is the redox-active residue closest to the heme in 81% of the bacteria and archaea, whereas in the eukaryotes 71% are tryptophans. Green’s analysis of the high pKa of FeIV(OH) in P450 points to the fact that the Gibbs free-energy change for nonproductive Tyr oxidation by compound I is comparable to that for productive activation of an aliphatic C–H bond.19 The energetics are evenly split between the two pathways, and the net reaction flux will be determined by their relative rates. Productive C–H activation requires positioning of a suitable substrate with the target H atom close to the compound I FeIV–oxo.20 We suggest that the nonproductive antioxidant pathway involves long-range electron transfer from Tyr or Trp. Neglecting small variations in reaction energetics, the key determinant of the internal antioxidant time constant will be the distance from the heme to the nearest Tyr/Trp residue.29−31 If the ferryl fails to oxidize substrate within the prescribed time limit, reducing equivalents from Tyr/Trp residues will rescue the enzyme by regenerating the ferric resting state. The estimated compound I survival time for a Tyr or Trp residue 8.4 Å from the heme (ΔG° = 0.2 eV, λ = 0.8 eV) is about 10 μs; across the full range of heme–Tyr/Trp distances, estimated survival times are as short as 30 ns (3.4 Å) and as long as 140 ms (17.2 Å). Regeneration of the ferric enzyme requires the delivery of a second electron and proton to compound II. The reduction potential of compound II is likely to be near that of compound I,32 so Tyr or Trp residues could again provide the reducing equivalents, although with more sluggish kinetics.13,14
Figure 6.
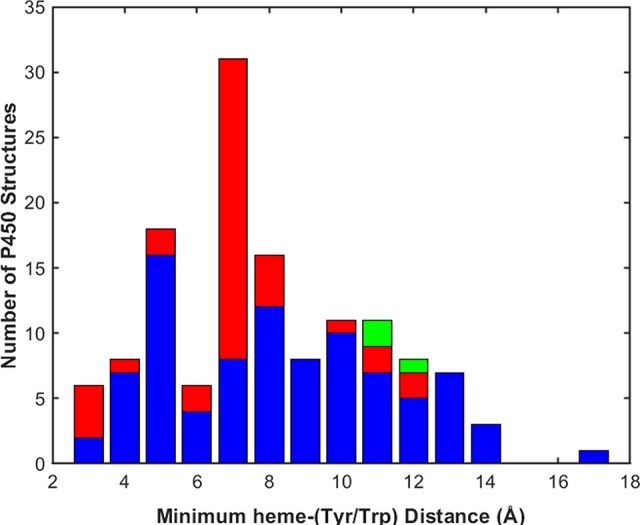
Structural database of the RCSB Protein Data Bank contains X-ray crystal structures of 134 cytochromes P450 with sequence identity less than 90%. The histogram illustrates the distribution of the shortest distances between the heme (Fe or members of the porphyrin π-system) and side chain π-system atoms of the redox-active Tyr or Trp residues within this set of P450 structures. The colors indicate the contributions from the three domains: archaea (green), bacteria (blue), eukaryotes (red).
Protection Pathways
Our analysis of heme–Tyr/Trp distances (Figure 6) found that values of 7–8 Å are the most common. A large fraction of the 41 structurally characterized eukaryotic P450s are mammalian enzymes that have a Trp residue hydrogen bonded to one of the heme-propionates. This position is occupied by a His residue in many of the prokaryotic enzymes. The corresponding residue in human CYP3A4 is Trp126, located 7.2 Å from the heme, and 4.7 Å from surface exposed Tyr99 (PDB ID 1TQN, Figure 7).33 We have analyzed a protection pathway involving hole transfer from the heme via Trp126 and terminating at Tyr99. A hopping map predicts time constants for migration of the hole from compound I to Tyr99 as functions of the driving forces for the two reactions.34 Taking values of −ΔG1° = −0.1 eV and −ΔG2 = 0.1 eV, we estimate a CYP3A4 compound I survival time of 200 ns. This estimate could be in error by an order of magnitude or more, but it illustrates the tight tolerance for productive substrate oxidation. Once the hole reaches Tyr99, it can be scavenged by the NADH/cytochrome b5 redox system or by soluble reductants such as glutathione.
Figure 7.
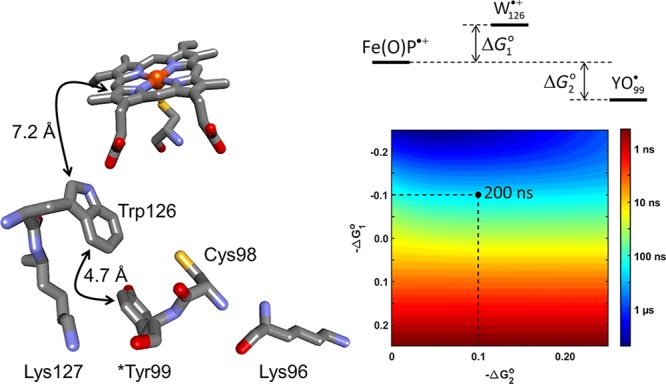
A route for hole migration from the CYP3A4 (PDB ID 1TQN) heme to the enzyme surface could involve multistep electron tunneling from surface exposed Tyr99 via Trp126. The color map illustrates the driving-force dependence of kinetics modeling results for this pathway. If reduction of the compound I heme by Trp126 is endergonic by 100 meV (ΔG1°), and reduction of intermediate Trp126•+ is exergonic by 200 meV (ΔG2 – ΔG1°), the hole-transfer time constant (τh+) is estimated to be 200 ns. Hence, the heme–Trp126–Tyr99 pathway is an efficient route for enzyme protection by migration of oxidizing equivalents to the surface for scavenging by reductants such as cytochrome b5. Cross-linking studies have implicated Lys126 and Lys96 as potential cytochrome b5 binding sites on CYP3A4.
A primary function of CYP3A4 is xenobiotic metabolism, and its active site binds a broad range of organic substrates. Poor coupling is expected from substrate diversity, and antioxidant rescue pathways are essential for enzyme survival. In this regard, it is noteworthy that cytochrome b5 has been reported to exhibit protective effects for CYP3A4 expressed in E. coli cells.35 Chemical cross-linking studies with human CYP3A4 and cytochrome b5 indicate that P450 residues Lys96 and Lys126 interact with Glu56 on cytochrome b5.36 The two P450 lysine residues span the location of Tyr99 on the CYP3A4 surface (Figure 7), suggesting a route for electron transport along a nonproductive oxidase pathway.
Another human P450 enzyme, CYP11A1, found on the inner mitochondrial membrane, catalyzes the first step in steroid biosynthesis, the conversion of cholesterol to pregnenolone in three turnovers, requiring one O2 and one NAD(P)H molecule per step. This coupling between NAD(P)H consumption and pregnenolone formation is high (>90%),37 and the enzyme is unreactive toward xenobiotics.38 As in CYP102A1 and CYP3A4, one of the heme propionates in CYP11A1 is hydrogen bonded to a tryptophan residue – Trp108, located 7.6 Å from the closest heme atom (PDB ID 3N9Y).39 In contrast to the situation for Trp126 in CYP3A4, no redox-active residues are within 10 Å of Trp108. A second tryptophan residue, Trp87, lies above the distal face of the heme at a distance 8.1 Å. Trp87 is the first member of a five-residue chain (Trp87, Trp231, Tyr90, Tyr94, Tyr93) that extends to the enzyme surface with a 3.7 Å average separation distance between residues (Figure 8). Kinetics modeling of this hole transfer pathway suggests that the compound I survival time is about 0.5 μs. Beratan and co-workers examined the electronic coupling strengths between residue pairs in this pathway, reporting values in the range 10–500 cm–1.40 They calculated a 1 μs mean first passage time for this pathway.
Figure 8.
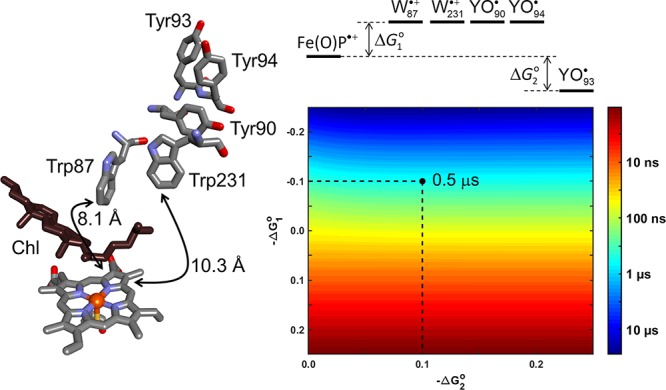
A route for hole migration from the CYP11A1 (PDB ID 3N9Y) heme to the enzyme surface could involve multistep electron tunneling from surface exposed Tyr93 via Tyr94, Tyr90, Trp231, and Trp87. The color map illustrates the driving-force dependence of kinetics modeling results for this pathway. If reduction of the compound I heme by Trp87 is endergonic by 100 meV (ΔG1°), hole migration through Trp231, Tyr90, and Tyr94 is ergoneutral, and reduction of intermediate Tyr94• is exergonic by 200 meV (ΔG2 – ΔG1°), the hole-transfer time constant (τh+) is estimated to be 0.5 μs.
Hole Hopping in CYP102A1 (P450BM3) and CYP119
The P450 from Bacillus megaterium (P450-BM3, CYP102A1) differs from most prokaryotic enzymes in that tryptophan (Trp96) is the closest redox-active residue to the heme. The CYP102A1 X-ray crystal structure reveals that Trp96 is H-bonded to the heme propionate at a distance of 7.2 Å (PDB ID 2IJ2) in a position closely analogous to that found in many eukaryotic enzymes (Figure 9).41 This enzyme has been the focus of our efforts to study hole hopping reactions in P450s.42,43 We attached a Ru(diimine)32+ residue to a mutant Cys97 (RuC97(CYP102A1)W96) and demonstrated that flash-quench generated RuIIIC97(CYP102A1)W96 will oxidize P(Cys–S)FeIII(OH2) to P(Cys–S)FeIV(OH) with a time constant of about 300 μs.43 The kinetics of this reaction indicate that the initial reaction product, ([P(Cys–S)]•+FeIII(OH2)), relaxes by internal proton and electron transfer to yield P(Cys–S)FeIV(OH).13,14,43
Figure 9.
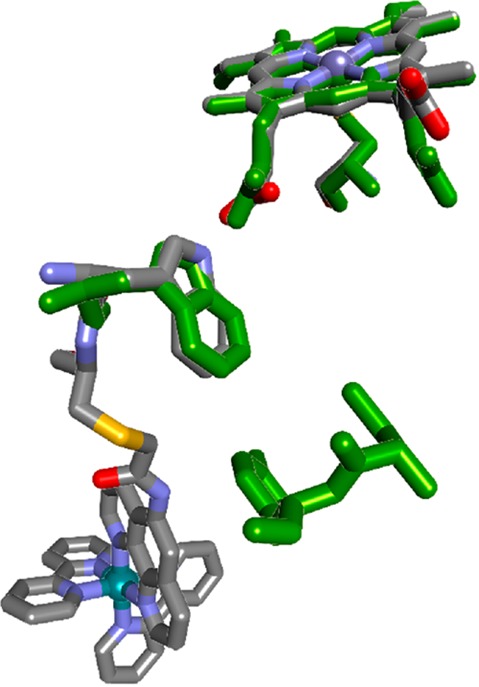
Illustration of the structural similarity of Ru-modified CYP102A1 (PDB ID 3NPL) and CYP3A4 (green, PDB ID 1TQN). Trp96 in CYP102A1 mediates high potential hole transfer from the heme to photochemically generated Ru(diimine)33+. Trp126 in CYP3A4 occupies the same site as Trp96 in CYP102A1; Tyr99 in CYP3A4 is adjacent to the Ru binding site in CYP102A1.
The archaeal P450 enzyme from Sulfolobus acidocaldarius (CYP119, PDB ID 1IO7),44,45 like most prokaryotic P450s, has a His residue (His76) at the same location as Trp96 in CYP102A1. We prepared a conjugate with Ru(diimine)32+ bound to a mutant Cys77 residue in CYP119 and found that flash-quench generated RuIII(CYP119)H76 does not oxidize P(Cys–S)FeIII(OH2).42 The critical distinction between the CYP102A1 and CYP119 Ru conjugates is the intervening aromatic residue: W96 in CYP102A1; His76 in CYP119. Mutants in which this residue is exchanged for the alternative display inverse ET behavior: heme oxidation fails in CYP102A1W96H but succeeds in CYP119H76W. Low-potential ET reactions, however, are insensitive to the identity of the intervening residue. Flash-quench generation of Ru(diimine)3+ in the resting enzyme is followed by ∼10 μs intraprotein ET to produce P(Cys–S)FeII(OH2) in wild-type and mutant CYP102A1 and CYP119.
The experiments with Ru-modified enzymes indicate that Trp residues (CYP102A1W96 and CYP119H76W) create an efficient hole-hopping pathway for high-potential electron transport between enzyme surfaces and the hemes. That the hole injected from RuIII lands on the heme rather than the intervening Trp residue suggests that E°(Trp•+/0) lies above both E°([P(Cys–S)]•+FeIII(OH2)/P(Cys–S)FeIII(OH2)) and E°(P(Cys–S)FeIV(OH)/P(Cys–S)FeIII(OH2)). The relative heme and Trp potentials are consistent with values used to model compound I survival in CYP3A4 wherein the higher potential of the proximal Trp residue creates a kinetic barrier to compound I hole migration to the enzyme surface. If E°(Trp•+/0) were lower than heme potentials, the compound I survival time would be reduced by an order of magnitude or more, possibly preventing productive substrate reaction.
Are Multicopper Oxidases Protected by Tyr/Trp Chains?
Many different metalloenzymes catalyze reactions involving O2, typically generating high-potential reactive intermediates.46 Tyrosine and tryptophan residues often lie within 5 Å of the metal cofactors in the active sites of these enzymes.28 An interesting case in point is provided by the cupredoxins and their evolutionary descendants, the multicopper oxidases.47
Single-domain cupredoxins are ET proteins characterized by an eight-stranded β-barrel fold with a type 1 copper binding site near one end of the barrel.48 Cu is coordinated in a trigonal plane by two histidine imidazole ligands and a cysteine thiolate; a longer distance axial interaction with a methionine sulfur atom is commonly, although not exclusively, found. This coordination environment produces the intense blue color of the CuII forms, and leads to CuII/I formal potentials ranging from 184 mV vs NHE for stellacyanin to 680 mV for rusticyanin.48 Of 30 structurally characterized cupredoxins, none has Tyr or Trp residues within 5 Å of the copper center. The only cupredoxin with a Tyr or Trp residue near the active site is the iso-2 azurin from Methylomonas sp. (strain J) (PDB ID 1CUO);49 Tyr114 is 5.03 Å from Cu.50
Multicopper oxidase enzymes (MCO) are constructed from 2 or 3 cupredoxin domains, one of which retains a type 1 copper. A trinuclear Cu (TNC) active site is at the interface between two of the domains, and it is here that oxygen is reduced to water.51 A survey of 25 X-ray crystal structures of 3-domain MCOs reveals that a Trp or Tyr/Trp pair is found adjacent to the TNC in all but two of these enzymes. In the Thermus thermophilus enzyme, Trp133 is just 3.6 Å from one of the Cu centers in the TNC (PDB ID 2YAE, Figure 10).52 This residue could play a protective role for the enzyme, analogous to that postulated for the cytochromes P450. Under normal circumstances, the four electrons required to reduce oxygen to water can be provided by four CuI centers. If the enzyme were to react with O2 and not have a full complement of reduced Cu sites, reactive oxygen species (ROS) such as superoxide, hydrogen peroxide, and hydroxyl radical might be formed. Electron delivery from Trp133 could provide an electron to the TNC during reactions with O2, preventing formation of ROS. Notably, Trp133 is the first residue in a 4-member Tyr/Trp chain that leads to Trp118 residue on the enzyme surface.
Figure 10.
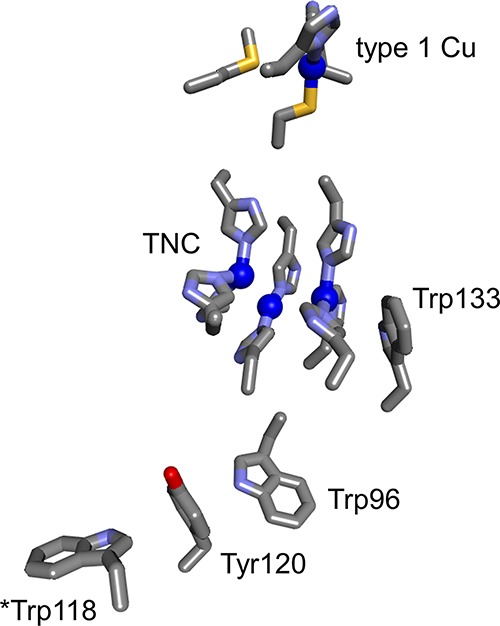
Trp133 is positioned 3.6 Å from a TNC Cu center in the MCO from T. thermophilus (PDB ID 2YAE). Reduction of oxygen to water requires four electrons. Under normal circumstances, those reducing equivalents could be delivered by four CuI centers. In cases where four electrons are not available, Trp133 could supply an electron to prevent formation of damaging reactive oxygen species.
Concluding Remarks
The oxygen-rich atmosphere that appeared on Earth about 2.5 billion years ago forced living organisms to evolve mechanisms to protect themselves from this new toxin in their environment.53 Oxygenases and oxidases exploit the oxidizing power of O2, but at the risk of damage from the highly reactive intermediates formed in their catalytic cycles.54 The high-potential redox reactivities of tyrosine, tryptophan, cysteine, and possibly methionine offer a likely protection mechanism. Proper alignment of these residues can create a conduit that directs oxidizing equivalents away from easily damaged active sites toward the enzyme surface. The increase in occurrence frequency of these amino acids compared to that estimated for the last universal ancestor may reflect a redox function that developed as the atmospheric O2 concentration increased.55,56 Regardless of the evolutionary exigencies, it seems clear that the side chains of Tyr, Trp, Cys, and Met should be considered as potential redox cofactors in proteins that react with O2 and its congeners.
Acknowledgments
Research reported in this Account was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01DK019038. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support was provided by the National Science Foundation (CHE-1305124) and the Arnold and Mabel Beckman Foundation.
Biographies
Harry B. Gray completed a doctoral thesis on inorganic reaction mechanisms at Northwestern, worked on ligand field theory as a postdoctoral researcher in Copenhagen, then joined the chemistry faculty at Columbia University, where in the early 1960s he investigated the electronic structures of metal complexes. He moved to Caltech in 1966, where he is the Arnold O. Beckman Professor of Chemistry and Founding Director of the Beckman Institute. He works with students and postdoctoral researchers on problems in inorganic photochemistry and biological inorganic chemistry.
Jay R. Winkler received a B.S. in chemistry from Stanford University in 1978 and a Ph.D. from Caltech in 1984, where he worked with Harry Gray. After postdoctoral work with Norman Sutin and Thomas Netzel at Brookhaven National Laboratory, he was appointed to the scientific staff in the Brookhaven Chemistry Department. In 1990, he moved to Caltech, where he is currently the Director of the Beckman Institute Laser Resource Center, Member of the Beckman Institute, and Faculty Associate in Chemistry.
The authors declare no competing financial interest.
References
- Krest C. M.; Onderko E. L.; Yosca T. H.; Calixto J. C.; Karp R. F.; Livada J.; Rittle J.; Green M. T. Reactive Intermediates in Cytochrome P450 Catalysis. J. Biol. Chem. 2013, 288, 17074–17081. 10.1074/jbc.R113.473108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askerka M.; Brudvig G. W.; Batista V. S. The O2-Evolving Complex of Photosystem II: Recent Insights from Quantum Mechanics/Molecular Mechanics (QM/MM), Extended X-ray Absorption Fine Structure (EXAFS), and Femtosecond X-ray Crystallography Data. Acc. Chem. Res. 2017, 50, 41–48. 10.1021/acs.accounts.6b00405. [DOI] [PubMed] [Google Scholar]
- Wikström M.; Krab K.; Sharma V. Oxygen Activation and Energy Conservation by Cytochrome c Oxidase. Chem. Rev. 2018, 118, 2469–2490. 10.1021/acs.chemrev.7b00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballhausen C. J.; Gray H. B. The Electronic Structure of the Vanadyl Ion. Inorg. Chem. 1962, 1, 111–122. 10.1021/ic50001a022. [DOI] [Google Scholar]
- Gray H. B.; Hare C. R. The Electronic Structures and Spectra of Chromyl and Molybdenyl Ions. Inorg. Chem. 1962, 1, 363–368. 10.1021/ic50002a034. [DOI] [Google Scholar]
- Pinakoulaki E.; Daskalakis V.; Ohta T.; Richter O.-M. H.; Budiman K.; Kitagawa T.; Ludwig B.; Varotsis C. The Protein Effect in the Structure of Two Ferryl-Oxo Intermediates at the Same Oxidation Level in the Heme Copper Binuclear Center of Cytochrome c Oxidase. J. Biol. Chem. 2013, 288, 20261–20266. 10.1074/jbc.M113.468488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto K.Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part A: Theory and Applications in Inorganic Chemistry, 6th ed.; John Wiley & Sons: Hoboken, NJ, 2009. [Google Scholar]
- Nakamoto K.Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, 6th ed.; John Wiley & Sons: Hoboken, NJ, 2009. [Google Scholar]
- Winkler J. R.; Gray H. B.. Electronic Structures of Oxo-Metal Ions. In Molecular Electronic Structures of Transition Metal Complexes I; Mingos D. M. P., Day P., Dahl J. P., Eds.; Springer: Berlin, Heidelberg, 2012; pp 17–28. [Google Scholar]
- Hunter B. M.; Gray H. B.; Müller A. M. Earth-Abundant Heterogeneous Water Oxidation Catalysts. Chem. Rev. 2016, 116, 14120–14136. 10.1021/acs.chemrev.6b00398. [DOI] [PubMed] [Google Scholar]
- Gumiero A.; Metcalfe C. L.; Pearson A. R.; Raven E. L.; Moody P. C. E. Nature of the Ferryl Heme in Compounds I and II. J. Biol. Chem. 2011, 286, 1260–1268. 10.1074/jbc.M110.183483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.; Basran J.; Casadei C. M.; Fielding A. J.; Schrader T. E.; Ostermann A.; Devos J. M.; Aller P.; Blakeley M. P.; Moody P. C. E.; Raven E. L. Direct visualization of a Fe(IV)–OH intermediate in a heme enzyme. Nat. Commun. 2016, 7, 13445. 10.1038/ncomms13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund J.; Pascher T.; Winkler J. R.; Gray H. B. Photoinduced Oxidation of Horseradish Peroxidase. J. Am. Chem. Soc. 1997, 119, 2464–2469. 10.1021/ja961026m. [DOI] [Google Scholar]
- Low D. W.; Winkler J. R.; Gray H. B. Photoinduced Oxidation of Microperoxidase-8 - Generation of Ferryl and Cation-Radical Porphyrins. J. Am. Chem. Soc. 1996, 118, 117–120. 10.1021/ja9530477. [DOI] [Google Scholar]
- Higginson W. C. E.; Sykes A. G. Kinetic studies of the oxidation of vanadium(III) by iron(III) in solution in aqueous perchloric acid. J. Chem. Soc. 1962, 2841–2851. 10.1039/jr9620002841. [DOI] [Google Scholar]
- Dobson J. C.; Meyer T. J. Redox properties and ligand loss chemistry in aqua/hydroxo/oxo complexes derived from cis- and trans-[(bpy)2RuII(OH2)2]2+. Inorg. Chem. 1988, 27, 3283–3291. 10.1021/ic00292a008. [DOI] [Google Scholar]
- Costentin C.; Robert M.; Savéant J.-M.; Teillout A.-L. Concerted proton-coupled electron transfers in aquo/hydroxo/oxo metal complexes: Electrochemistry of [OsII(bpy)2py(OH2)]2+ in water. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 11829–11836. 10.1073/pnas.0905020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M. T.; Dawson J. H.; Gray H. B. Oxoiron(IV) in Chloroperoxidase Compound II is Basic: Implications for P450 Chemistry. Science 2004, 304, 1653–1656. 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- Yosca T. H.; Rittle J.; Krest C. M.; Onderko E. L.; Silakov A.; Calixto J. C.; Behan R. K.; Green M. T. Iron(IV)hydroxide pK(a) and the Role of Thiolate Ligation in C-H Bond Activation by Cytochrome P450. Science 2013, 342, 825–829. 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer J. M. Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res. 2011, 44, 36–46. 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov I. G.; Makris T. M.; Sligar S. G.; Schlichting I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- Gelb M. H.; Malkonen P.; Sligar S. G. Cytochrome P450cam catalyzed epoxidation of dehydrocamphor. Biochem. Biophys. Res. Commun. 1982, 104, 853–858. 10.1016/0006-291X(82)91327-4. [DOI] [PubMed] [Google Scholar]
- Atkins W. M.; Sligar S. G. Metabolic switching in cyctochrome P-450cam: deuterium isotope effects on regiospecificity and the monooxygenase/oxidase ratio. J. Am. Chem. Soc. 1987, 109, 3754–3760. 10.1021/ja00246a038. [DOI] [Google Scholar]
- Atkins W. M.; Sligar S. G. Deuterium isotope effects in norcamphor metabolism by cytochrome P-450cam: kinetic evidence for the two-electron reduction of a high-valent iron-oxo intermediate. Biochemistry 1988, 27, 1610–1616. 10.1021/bi00405a033. [DOI] [PubMed] [Google Scholar]
- Wienkers L. C.; Heath T. G. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discovery 2005, 4, 825. 10.1038/nrd1851. [DOI] [PubMed] [Google Scholar]
- Grinkova Y. V.; Denisov I. G.; McLean M. A.; Sligar S. G. Oxidase uncoupling in heme monooxygenases: Human cytochrome P450 CYP3A4 in Nanodiscs. Biochem. Biophys. Res. Commun. 2013, 430, 1223–1227. 10.1016/j.bbrc.2012.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loida P. J.; Sligar S. G. Molecular recognition in cytochrome P-450: Mechanism for the control of uncoupling reactions. Biochemistry 1993, 32, 11530–11538. 10.1021/bi00094a009. [DOI] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10920–10925. 10.1073/pnas.1512704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B. Electron Flow through Metalloproteins. Chem. Rev. 2014, 114, 3369–3380. 10.1021/cr4004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B. Electron flow through biological molecules: does hole hopping protect proteins from oxidative damage?. Q. Rev. Biophys. 2015, 48, 411–420. 10.1017/S0033583515000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B. Long-Range Electron Tunneling. J. Am. Chem. Soc. 2014, 136, 2930–2939. 10.1021/ja500215j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhangrazi A. S.; Fossett M. E.; Powers L. S.; Ellis W. R. Jr. Variable-Temperature Spectroelectrochemical Study of Horseradish Peroxidase. Biochemistry 1995, 34, 2866–2871. 10.1021/bi00009a017. [DOI] [PubMed] [Google Scholar]
- Yano J. K.; Wester M. R.; Schoch G. A.; Griffin K. J.; Stout C. D.; Johnson E. F. The Structure of Human Microsomal Cytochrome P450 3A4 Determined by X-ray Crystallography to 2.05-Å Resolution. J. Biol. Chem. 2004, 279, 38091–38094. 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Ener M. E.; Vlček A.; Winkler J. R.; Gray H. B. Electron hopping through proteins. Coord. Chem. Rev. 2012, 256, 2478–2487. 10.1016/j.ccr.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voice M. W.; Zhang Y.; Wolf C. R.; Burchell B.; Friedberg T. Effects of Human Cytochrome b5 on CYP3A4 Activity and Stability in Vivo. Arch. Biochem. Biophys. 1999, 366, 116–124. 10.1006/abbi.1999.1192. [DOI] [PubMed] [Google Scholar]
- Zhao C.; Gao Q.; Roberts A. G.; Shaffer S. A.; Doneanu C. E.; Xue S.; Goodlett D. R.; Nelson S. D.; Atkins W. M. Cross-Linking Mass Spectrometry and Mutagenesis Confirm the Functional Importance of Surface Interactions between CYP3A4 and Holo/Apo Cytochrome b5. Biochemistry 2012, 51, 9488–9500. 10.1021/bi301069r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanukoglu I.; Rapoport R.; Weiner L.; Sklan D. Electron Leakage from the Mitochondrial NADPH-Adrenodoxin Reductase-Adrenodoxin-P450scc (Cholesterol Side Chain Cleavage) System. Arch. Biochem. Biophys. 1993, 305, 489–498. 10.1006/abbi.1993.1452. [DOI] [PubMed] [Google Scholar]
- Omura T. Mitochondrial P450s. Chem.-Biol. Interact. 2006, 163, 86–93. 10.1016/j.cbi.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Strushkevich N.; MacKenzie F.; Cherkesova T.; Grabovec I.; Usanov S.; Park H.-W. Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 10139–10143. 10.1073/pnas.1019441108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polizzi N. F.; Migliore A.; Therien M. J.; Beratan D. N. Defusing redox bombs?. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10821–10822. 10.1073/pnas.1513520112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girvan H. M.; Seward H. E.; Toogood H. S.; Cheesman M. R.; Leys D.; Munro A. W. Structural and spectroscopic characterization of P450BM3 mutants with unprecedented P450 heme iron ligand sets - New heme ligation states influence conformational equilibria in P450BM3. J. Biol. Chem. 2007, 282, 564–572. 10.1074/jbc.M607949200. [DOI] [PubMed] [Google Scholar]
- Ener M. E.; Gray H. B.; Winkler J. R. Hole Hopping through Tryptophan in Cytochrome P450. Biochemistry 2017, 56, 3531–3538. 10.1021/acs.biochem.7b00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ener M. E.; Lee Y. T.; Winkler J. R.; Gray H. B.; Cheruzel L. Photooxidation of cytochrome P450-BM3. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18783–18786. 10.1073/pnas.1012381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittle J.; Green M. T. Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science 2010, 330, 933–937. 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- Park S.-Y.; Yamane K.; Adachi S.-i.; Shiro Y.; Weiss K. E.; Maves S. A.; Sligar S. G. Thermophilic cytochrome P450 (CYP119) from Sulfolobus solfataricus: high resolution structure and functional properties. J. Inorg. Biochem. 2002, 91, 491–501. 10.1016/S0162-0134(02)00446-4. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Gray H. B.; Stiefel E. I.; Valentine J. S.. Biological Inorganic Chemistry - Structure and Reactivity; University Science Books: Sausalito, CA, 2007. [Google Scholar]
- Nakamura K.; Go N. Function and molecular evolution of multicopper blue proteins. Cell. Mol. Life Sci. 2005, 62, 2050–2066. 10.1007/s00018-004-5076-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennison C. Investigating the structure and function of cupredoxins. Coord. Chem. Rev. 2005, 249, 3025–3054. 10.1016/j.ccr.2005.04.021. [DOI] [Google Scholar]
- Inoue T.; Suzuki S.; Nishio N.; Yamaguchi K.; Kataoka K.; Tobari J.; Yong X.; Hamanaka S.; Matsumura H.; Kai Y. The Significance of the Flexible Loop in the Azurin (Az-iso2) from the Obligate Methylotroph Methylomonas sp. Strain J. J. Mol. Biol. 2003, 333, 117–124. 10.1016/j.jmb.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. The Rise of Radicals in Bioinorganic Chemistry. Isr. J. Chem. 2016, 56, 640–648. 10.1002/ijch.201600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S. M.; Solomon E. I. Electron transfer and reaction mechanism of laccases. Cell. Mol. Life Sci. 2015, 72, 869–883. 10.1007/s00018-014-1826-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Posada H.; Centeno-Leija S.; Rojas-Trejo S. P.; Rodríguez-Almazán C.; Stojanoff V.; Rudiño-Piñera E. X-ray-induced catalytic active-site reduction of a multicopper oxidase: structural insights into the proton-relay mechanism and O2-reduction states. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015, 71, 2396–2411. 10.1107/S1399004715018714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons T. W.; Reinhard C. T.; Planavsky N. J. The rise of oxygen in Earth’s early ocean and atmosphere. Nature 2014, 506, 307–315. 10.1038/nature13068. [DOI] [PubMed] [Google Scholar]
- Holtmann D.; Hollmann F. The Oxygen Dilemma: A Severe Challenge for the Application of Monooxygenases?. ChemBioChem 2016, 17, 1391–1398. 10.1002/cbic.201600176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks D. J.; Fresco J. R.; Lesk A. M.; Singh M. Evolution of amino acid frequencies in proteins over deep time: Inferred order of introduction of amino acids into the genetic code. Mol. Biol. Evol. 2002, 19, 1645–1655. 10.1093/oxfordjournals.molbev.a003988. [DOI] [PubMed] [Google Scholar]
- Brooks D. J.; Fresco J. R.; Singh M. A novel method for estimating ancestral amino acid composition and its application to proteins of the Last Universal Ancestor. Bioinformatics 2004, 20, 2251–2257. 10.1093/bioinformatics/bth235. [DOI] [PubMed] [Google Scholar]