

Abstract
Aims
β1- and β2-adrenergic receptors (β-ARs) produce different acute contractile effects on the heart partly because they impact on different cytosolic pools of cAMP-dependent protein kinase (PKA). They also exert different effects on gene expression but the underlying mechanisms remain unknown. The aim of this study was to understand the mechanisms by which β1- and β2-ARs regulate nuclear PKA activity in cardiomyocytes.
Methods and results
We used cytoplasmic and nuclear targeted biosensors to examine cAMP signals and PKA activity in adult rat ventricular myocytes upon selective β1- or β2-ARs stimulation. Both β1- and β2-AR stimulation increased cAMP and activated PKA in the cytoplasm. Although the two receptors also increased cAMP in the nucleus, only β1-ARs increased nuclear PKA activity and up-regulated the PKA target gene and pro-apoptotic factor, inducible cAMP early repressor (ICER). Inhibition of phosphodiesterase (PDE)4, but not Gi, PDE3, GRK2 nor caveolae disruption disclosed nuclear PKA activation and ICER induction by β2-ARs. Both nuclear and cytoplasmic PKI prevented nuclear PKA activation and ICER induction by β1-ARs, indicating that PKA activation outside the nucleus is required for subsequent nuclear PKA activation and ICER mRNA expression. Cytoplasmic PKI also blocked ICER induction by β2-AR stimulation (with concomitant PDE4 inhibition). However, in this case nuclear PKI decreased ICER up-regulation by only 30%, indicating that other mechanisms are involved. Down-regulation of mAKAPβ partially inhibited nuclear PKA activation upon β1-AR stimulation, and drastically decreased nuclear PKA activation upon β2-AR stimulation in the presence of PDE4 inhibition.
Conclusions
β1- and β2-ARs differentially regulate nuclear PKA activity and ICER expression in cardiomyocytes. PDE4 insulates a mAKAPβ-targeted PKA pool at the nuclear envelope that prevents nuclear PKA activation upon β2-AR stimulation.
Keywords: Adrenergic receptors • cAMP-dependent protein kinase • Compartmentation • Nucleus
1. Introduction
Acute stimulation of β-adrenergic receptors (β-ARs) is essential for the adaptation of cardiac performance to physiological needs, but persistent activation exerts deleterious effects that ultimately result in heart failure (HF). The normal heart mainly expresses β1- and β2-AR subtypes, and although both couple to the Gs/cAMP/protein kinase (PKA) cascade, their functional effects differ markedly. β1- but not β2-ARs elicit strong inotropic and lusitropic responses associated with concerted phosphorylation of excitation–contraction coupling (ECC) proteins,1 induce HF upon moderate expression,2,3 and promote cardiomyocyte apoptosis.4
These diverse effects are explained by a functional compartmentation model which integrates alternative coupling of β2-ARs to Gi,4,5 distinct location of receptors in specialized microdomains of the plasma membrane,6–10 differential coupling of β1- and β2-ARs to adenylyl cyclases Types V and VI,11 and differential regulation by cyclic-nucleotide phosphodiesterases (PDEs) Types 3 and 4.12 Additional differences between β1- and β2-ARs include receptor desensitization, with β2-ARs being more rapidly internalized than β1-ARs.13
Another critical organizer of β-AR signalling integration is constituted by A-kinase anchoring proteins (AKAPs). In cardiomyocytes, AKAPs target PKA to the plasma membrane, the sarcoplasmic reticulum and the myofilaments for local regulation of major ECC proteins.14 AKAPs also target PKA to other intracellular organelles such as the nucleus to regulate gene expression.14,15 In cardiomyocytes, the scaffold protein muscle AKAPβ (mAKAPβ) organizes a complete cAMP signalling module including PKA and PDE4D3 at the nuclear envelope. Because PDE4D3 is phosphorylated and activated by PKA16 this constitutes a negative feedback loop modulating local cAMP level and PKA activity.17 Several subsequent studies showed that mAKAPβ assemble a much larger signalosome which major function is to modulate pathological hypertrophy.18–20 Upon β-AR stimulation and cAMP elevation, PKA activation also drives CREB-dependent transcriptional activation of inducible cAMP early repressor (ICER), a potent pro-apoptotic factor in cardiomyocytes.21,22 In addition, PKA regulates Class II histone deacetylases 4 and 5 in the nucleus.23–25 However, thus far the mechanisms that control the dynamics of nuclear PKA activity in cardiomyocytes remain poorly understood. We and others have used genetically-encoded PKA biosensors targeted to the cytoplasm and the nucleus to show temporal segregation of PKA responses in both compartments in neonatal and adult cardiomyocytes.26,27 Such organization provides a mechanism by which acute β-AR stimulation may regulate contractility independently of gene expression. Our previous results also identified PDE4 as an important upstream regulator of nuclear PKA activity upon β-AR stimulation. However, how cAMP/PKA signalling generated by β1- and β2-ARs are integrated in the nucleus of cardiomyocytes and whether this participates in functional differences observed upon stimulation of these receptors remains elusive. Here, we reveal that β1- and β2-ARs differentially regulate nuclear PKA activity and ICER expression in adult cardiomyocytes. We provide evidence that PDE4 prevents activation by β2-ARs of a mAKAPβ-targeted PKA pool at the nuclear envelope which is required for PKA activation inside the nucleus and contributes to ICER induction.
2. Methods
An expanded methods section is provided in the Data Supplementary material online.
2.1 Experimental animals
All procedures were performed in accordance with the European Community guiding principles in the Care and Use of Animals (2010/63/UE), the local Ethics Committee (CREEA Ile-de-France Sud) guidelines and the French decree no. 2013-118 on the protection of animals used for scientific purposes. Male Wistar rats were anaesthetized by intraperitoneal injection of pentobarbital (0.1 mg/g).
2.2 FRET-based cAMP and PKA measurements in adult rat ventricular myocytes
After transduction of freshly isolated adult rat ventricular myocytes (ARVMs) with the appropriate adenoviruses, cells were subjected to Förster resonance energy transfer (FRET) measurements as described in.26
3. Results
3.1 β1- and β2-ARs differentially regulate nuclear PKA activity in adult cardiomyocytes
To analyse the dynamics of PKA activity in the bulk cytoplasm and in the nucleus, we used genetically encoded A-kinase activity reporters (AKAR3) targeted to these compartments by the addition of a nuclear export sequence (NES), and a nuclear localizing sequence (NLS), respectively.28 As shown previously in26 and in the pseudocolour images of Figure 1, adenoviral transfer allowed robust and compartment-specific expression of these biosensors after 24 h in ARVMs.
Figure 1.

Stimulation of β1- and β2-ARs induce differential activation of cytoplasmic and nuclear PKA activity in ARVMs. A–D Representative time course of cytoplasmic and nuclear PKA activities reported by the normalized yellow fluorescent protein (YFP)/cyan fluorescent protein (CFP) ratio in ARVMs transduced with Ad.AKAR3-NES (A and C) or Ad.AKAR3-NLS (B and D) for 24 h at a multiplicity of infection (MOI) of 1000 active viral particles/cell. β1-AR stimulation was achieved using a combination of 10 nM Iso and 10 nM ICI 118, 551 (ICI) (A and B); β2-AR stimulation using 100 nM Iso in combination with 100 nM CGP 20712A (CGP) (C and D). Pseudo-colour images of the YFP/CFP ratio were recorded at the times indicated by the letters on the graphs. Scale bars represent 20 µm. (E and F) Mean variation (±S.E.M.) of the YFP/CFP ratio in ARVMs expressing either AKAR3-NES or AKAR3-NLS upon β1-AR stimulation using 1, 3 and 10 nM Iso in combination with 10 nM ICI (E) or β2-AR stimulation using 10, 30, and 100 nM Iso in combination with 100 nM CGP (F). Number of cells/animals is indicated in brackets. Statistical significance is indicated as ***P < 0.001 vs. ICI+Iso 1 nM or CGP+Iso 10 nM in the cytoplasm, $$P < 0.01 vs. ICI+Iso 1 nM in the nucleus, ###P < 0.001 by nested ANOVA with Tukey’s post hoc test. (G) Nuclear PKA activation (% increase in YFP/CFP ratio in ARVMs expressing AKAR3-NLS) is plotted as a function of cytoplasmic PKA activation (% increase in YFP/CFP ratio in ARVMs expressing AKAR3-NES) for β1- and β2-AR stimulations. Values (±S.E.M.) of (E and F) were used for this graph.
To determine how β1- and β2-ARs regulate cytoplasmic and nuclear PKA activities in ARVMs, we selectively stimulated these two receptors using a combination of isoprenaline (Iso) and either the β2-AR antagonist ICI 118, 551 (ICI, 10 nM) or the β1-AR antagonist CGP 20712 A (CGP, 100 nM), respectively. Stimulation of cytoplasmic PKA activity by β1- and β2-ARs were completely abolished by 100 nM CGP and 10 nM ICI, respectively, indicating that the chosen concentrations of ICI and CGP in combination with Iso were appropriate for selective stimulation of β-AR subtypes (see Supplementary material online, Figure S1). β1-AR stimulation led to a fast increase in cytoplasmic PKA activity and a robust, but slower increase in nuclear PKA activity (Figure 1A and B). In contrast, β2-AR stimulation increased cytoplasmic PKA activity but had negligible effects on nuclear PKA activity (Figure 1C and D). As shown in Figure 1E, β1-AR stimulation increased PKA activity in a concentration-dependent manner in both compartments, whereas this occurred only in the cytoplasm with β2-AR stimulation (Figure 1F). In Figure 1G, nuclear PKA activation was plotted as a function of cytoplasmic PKA activation for the two receptors. The steeper slope of the linear regression further illustrates that β1-ARs are more efficient than β2-ARs to increase nuclear PKA activity, even when cytoplasmic PKA is activated at the same level.
3.2 β1- and β2-ARs elevate cAMP in the nucleus
Because cAMP generated by β2-ARs was reported to be locally confined,29 we hypothesized that cAMP may not reach the nucleus upon β2-AR stimulation, hence explaining the lack of PKA activation in this compartment. To test this hypothesis, we generated a nuclear-targeted version of the cytoplasmic cAMP sensor Epac-SH187 by addition of 3 NLS at the C-terminus (Epac-SH187-3NLS).30 Adenoviral vectors allowed robust and compartment-specific expression of Epac-SH187 and Epac-SH187-3NLS in ARVMs at 24 h (see Supplementary material online, Figure S2A). As shown by the individual traces in Figure 2A–D and summarized in Figure 2E and F, stimulation of both β1- and β2-ARs increased cAMP in a concentration-dependent manner in the two compartments. Regardless of the receptor type or the Iso concentration employed, the FRET responses recorded in the nucleus were systematically higher than in the cytoplasm. This difference apparently reflected a greater sensitivity of the 3NLS version of the sensor to cAMP and not higher cAMP levels in the nucleus. When the changes in FRET induced by Iso in the cytoplasm were determined for both sensors following expression for 48 h, a condition for which the Epac-SH187-3NLS sensor became expressed not only in the nucleus, but also in the cytoplasm (see Supplementary material online, Figure S2B), the cytoplasmic response of the mislocalized Epac-SH187-3NLS sensor to Iso was higher than that measured with Epac-SH187 (see Supplementary material online, Figure S2C). Regardless of the difference in sensitivity of the Epac-SH187 and Epac-SH187-3NLS sensors, the relative response in the cytoplasmic and nuclear compartments did not differ between β1- and β2-ARs (Figure 2G), in contrast to what was observed with the PKA sensors (Figure 1G). Thus the lack of nuclear PKA activation cannot be attributed to the absence of a global nuclear cAMP elevation upon β2-AR stimulation.
Figure 2.

Both β1- and β2-AR increase cytoplasmic and nuclear cAMP levels in ARVMs. Representative time course of the normalized CFP/YFP ratio upon selective β1-AR stimulation with Iso (1, 3, and 10 nM) in combination with 10 nM ICI (A and B) or β2-AR stimulation with Iso (10, 30 and 100 nM) in combination with 100 nM CGP (C and D) in ARVMs transduced with Ad.Epac-SH187(A and C) or Ad.Epac-SH187-3NLS (B and D) for 24 h at a MOI of 1000 active viral particles/cell. Pseudo-colour images of the CFP/YFP ratio were recorded at the times indicated by the letters on the graphs. Scale bars represent 20 µm. (E and F) Mean variation (±S.E.M.) of the CFP/YFP ratio in ARVMs expressing either Epac-SH187 or Epac-SH187-3NLS upon β1-AR stimulation (E) or β2-AR stimulation (F). Number of cells/animals is indicated in brackets. Statistical significance is indicated as * P < 0.05; ** P < 0.01; *** P < 0.001 vs. ICI+Iso 1 nM or CGP+Iso 10 nM in the cytoplasm, $$P < 0.01, $$$P < 0.001 vs. ICI+Iso 1 nM or CGP+Iso 10 nM in the nucleus, #P< 0.05, ###P < 0.001, £££P < 0.001 by nested ANOVA with Tukey’s post-hoc test. (G) Nuclear cAMP elevation (% increase in CFP/YFP ratio in ARVMs expressing Epac-SH187-3NLS) is plotted as a function of cytoplasmic cAMP elevation (% increase in CFP/YFP ratio in ARVMs expressing Epac-SH187) in response to either β1- or β2-AR stimulation. Values (±S.E.M.) of (E and F) were used for this graph.
3.3 Mechanisms that prevent nuclear PKA activation by β2-ARs
Multiple mechanisms have been proposed to compartmentalize β2-AR activation of PKA, by acting not only on cAMP generation and propagation but also downstream. In particular, receptor coupling to Gi may not only temper cAMP synthesis, but also activate alternative signalling pathways through βγ to circumvent the concurrent PKA activation.31,32 To test this hypothesis, we inhibited Gi with pertussis toxin (PTX, 1.5 µg/ml, 2 h). As a control, we verified that PTX effectively blocked the anti-adrenergic effect of acetylcholine on the β1-AR-induced cytoplasmic PKA activation (see Supplementary material online, Figure S3A and B). Under β2-AR stimulation, PTX-treated cells exhibited higher cytoplasmic PKA activity but no difference in nuclear PKA activation (Figure 3A and B). Similarly, co-expression of AKAR3-NES or AKAR3-NLS with a C-terminal fragment of GRK2 (βARK-ct, see Supplementary material online, Figure S3C), that scavenges βγ subunits of heterotrimeric G proteins and prevents GRK2 phosphorylation and desensitization of the receptor, led to a potentiation of cytoplasmic PKA activity, but was without effect on nuclear PKA activity (Figure 3C and D).
Figure 3.

Gi proteins, caveolae and GRK2 regulate cytoplasmic but not nuclear PKA activation in response to β2-ARs stimulation. Average time course of the YFP/CFP ratio upon β2-AR stimulation in ARVMs expressing AKAR3-NES (A, C, E, and G) or AKAR3-NLS (B, D, F, and H). (A and B) ARVMs treated or not with PTX (1.5 µg/ml, 2 h) were exposed to 10 nM Iso plus 100 nM CGP to stimulate β2-ARs. In all other protocols, β2-ARs were stimulated with 30 nM Iso plus 100 nM CGP. (C and D) ARVMs were treated or not with 2 mM MβCD for 1 h. (E) ARVMs were co-transduced with Ad.AKAR3-NES (MOI 200) and Ad.β-Galactosidase (β-Gal, MOI 2000) or Ad.AKAR3-NES and an adenovirus encoding a dominant-negative Cav3 mutant (Ad. Cav3DN, MOI 2000) for 48 h. (F) ARVMs were co-transduced with Ad.AKAR3-NLS and Ad. β-Gal or Ad.AKAR3-NLS and Ad.Cav3DN for 48 h. (G) ARVMs were co-transduced with Ad.AKAR3-NES (MOI 200) and Ad.β-Gal (MOI 1000) or Ad.AKAR3-NES (MOI 200) and Ad.βARK-ct (MOI 1000) for 48 h. (H) ARVMs were co-transduced with Ad.AKAR3-NLS (MOI 200) and Ad.β-Gal (MOI 1000) or Ad.AKAR3-NLS (MOI 200) and Ad.βARK-ct (MOI 1000) for 48 h. In each panel, the number of cells/animals is indicated in brackets for the different experimental conditions. Statistical significance is indicated as *P < 0.05; **P , 0.01; ***P < 0.001 by nested ANOVA with Tukey’s post-hoc test.
Another related mechanism for β2-AR compartmentalization is receptor localization to caveolae.6,8,10 To address this possibility, caveolae were disrupted by treating the cells with methyl-β-cyclodextrin (MβCD, 2 mM, 1 h) to deplete cholesterol, or by co-expressing a dominant negative caveolin-3 mutant (Cav3DN) together with AKAR3-NES or AKAR3-NLS.10 The efficiency of MβCD to deplete cholesterol was verified by filipin staining, whereas expression of Cav3DN was demonstrated by immunocytochemistry and western blot (see Supplementary material online, Figure S4A–C). In both cases, caveolae-disrupted cells showed potentiation of cytoplasmic but not nuclear PKA activity upon β2-AR stimulation (Figure 3E–H). Conversely, overexpression of wild type Cav3 induced a small decrease in β2-AR-stimulated cytoplasmic PKA activity, consistent with its previously reported effect on β2-AR-generated cytoplasmic cAMP (see Supplementary material online, Figure S4D).10
Thus, inhibition of Gi, of GRK2, and caveolae disruption potentiated β2-AR stimulation of PKA activity in the cell cytoplasm as expected, but did not confer PKA activation in the nucleus. Because recent studies emphasized the role of PDE3 and PDE4 in controlling the activation of discrete PKA pools by β1- and β2-ARs in cardiomyocytes,33 we investigated the contribution of these enzymes to the regulation of cytoplasmic and nuclear PKA. We have shown previously in26 that neither the PDE3 inhibitor cilostamide (Cil, 1 µM) alone nor the PDE4 inhibitor Ro-201724 (Ro, 10 µM) alone has an effect on basal PKA activity. Under β1-AR stimulation, PDE4 inhibition potentiated both cytoplasmic and nuclear PKA activities whereas PDE3 inhibition had no significant effect (Figure 4A and B). In comparison, under β2-AR stimulation PDE3 inhibition led to a large potentiation of cytoplasmic PKA activity but was without effect on nuclear PKA activity (Figure 4C and D). Interestingly, PDE4 inhibition had a similar potentiating effect on cytoplasmic PKA activity under β2-AR stimulation but in this case it generated a strong nuclear PKA activation (Figure 4C and D). These results unveiled a critical role of PDE4 in controlling nuclear PKA activation.
Figure 4.

PDE4 is predominant for regulation of β1- and β2-AR induced cytoplasmic and nuclear PKA activation. (A and B) Average variation of the YFP/CFP ratio upon β1-AR stimulation using 1 nM Iso plus 10 nM ICI alone or in the presence of 1 µM cilostamide (Cil), a PDE3 inhibitor, or 10 µM Ro-201724 (Ro) a PDE4 inhibitor in ARVMs transduced with Ad.AKAR3-NES (A) or Ad.AKAR3-NLS (B) at MOI 1000 for 24 h. (C and D) Average variation of the YFP/CFP ratio upon β2-AR stimulation using 30 nM Iso plus 100 nM CGP alone or in the presence of 1 µM Cil or 10 µM Ro in ARVMs transduced with Ad.AKAR3-NES (C) or Ad.AKAR3-NLS (D) at MOI 1000 for 24 h. Number of cells/animals is indicated in brackets. Statistical significance is indicated as *P < 0.05; **P < 0.01; ***P < 0.001; $$P < 0.01 vs. β1- or β2-AR by nested ANOVA with Tukey’s post-hoc test. (E and F) ICER mRNA expression in ARVMs in primary culture for 24 h and stimulated or not by β1-ARs (100 nM Iso plus 10 nM ICI during 2 h) or β2-ARs (100 nM Iso plus 100 nM CGP during 2 h) alone or in combination with 10 µM Ro or 1 µM Cil. Number of animals is indicated in brackets. Statistical significance is indicated as ***P < 0.001 vs. control; $$P < 0.01, $$$P < 0.001 by Kruskal-Wallis test with Dunn’s post-hoc test (E) or one-way ANOVA with Tukey’s post-hoc test (F).
3.4 β1- and β2-ARs differentially regulate the PKA target gene and pro-apoptotic factor, ICER
Although β-AR stimulation may regulate the expression of a considerable number of genes in cardiomyocytes, their identity and the mechanisms that couple receptor activation to gene expression remain, in most cases, poorly characterized. However, ICER, a splice variant of the CREM gene, stands as one of the few exceptions to this statement. Indeed, it was shown that ICER is up-regulated by β-AR stimulation in a PKA and CREB-dependent manner in cardiomyocytes.21,22,34 Thus, in the following, we investigated the regulation of ICER by β1-ARs and β2-ARs in ARVMs. As shown in Figure 4E, β1-AR stimulation of isolated ARVMs led to a strong induction of ICER mRNA. This effect was potentiated by concomitant PDE4 inhibition, but not by PDE3 inhibition (Figure 4E). In comparison, β2-AR stimulation alone had no significant effect on ICER expression, but concomitant PDE4 inhibition resulted in ICER induction (Figure 4F). In the absence of β-AR stimulation, neither PDE3 nor PDE4 inhibition had effect on ICER mRNA expression (see Supplementary material online, Figure S5A). β1-AR stimulation of ICER mRNA expression was not observed in cells transduced with an adenovirus encoding the selective PKA inhibitor peptide (PKI) (see Supplementary material online, Figure S5B). To examine the specific contribution of nuclear vs. cytoplasmic PKA in ICER regulation, we fused the first 25 amino acids of PKI with the red fluorescent protein mCherry and appended either a 3 NLS or a NES at the C-terminus for nuclear or cytoplasmic targeting, respectively. As shown in the images and intensity profiles of Supplementary material online, Figure S6A and B, the resulting constructs localized specifically in the compartment of interest in ARVMs. In cardiomyocytes co-expressing nuclear PKI with AKAR3-NLS, nuclear PKA activation by β1-ARs was abolished as expected (see Supplementary material online, Figure 6 C). To verify the efficiency of PKA inhibition by cytoplasmic PKI, β-AR stimulation of Ca2+ transients was compared in Fura2-loaded and electrically paced ARVMs expressing cytoplasmic PKI, nuclear PKI or β-Gal. As shown in Supplementary Fig. 7, β-AR stimulation with 10 nM Iso increased Ca2+ transient amplitude to a similar extent in ARVMs expressing β-Gal or PKI-3NLS, whereas these effects were absent in ARVMs expressing PKI-NES. Interestingly, the cytoplasmic PKI completely blocked nuclear PKA activation in response to β1-ARs stimulation (see Supplementary material online, Figure S6D). Both cytoplasmic and nuclear PKI also blocked ICER mRNA induction by β1-ARs (Figure 5A). These results show that upon β1-AR stimulation, PKA activation outside the nucleus is a prerequisite to enhanced nuclear PKA activity and ICER induction. The augmentation of ICER mRNA by β1-AR stimulation in combination with PDE4 inhibition was also suppressed by nuclear PKI expression (Figure 5B). Similarly, nuclear PKI completely blocked nuclear PKA activation by β2-ARs with concomitant PDE4 inhibition (see Supplementary material online, Figure S6E). However, ICER mRNA induction by β2-ARs with concomitant PDE4 inhibition was only partially blocked by nuclear PKI, suggesting that alternative mechanisms contribute to this effect (see Supplementary material online, Figure 5C).35,36 Because Epac1 was localized in the perinuclear region in cardiomyocytes,18,37 similar experiments were conducted in cells pre-incubated with 10 µM of the Epac1 inhibitor, CE3F4.38 However, as shown in Figure 5D, CE3F4 did not modify the stimulatory effect of β2-ARs + Ro on ICER mRNA expression. In contrast, overexpression of PKI-NES abrogated ICER induction by β2-ARs with concomitant PDE4 inhibition, indicating that cytoplasmic PKA is required for ICER regulation by this receptor (Figure 5E).
Figure 5.

β1-ARs and β2-ARs differentially regulate the PKA target gene, ICER in cardiomyocytes. (A) ICER mRNA expression in ARVMs transduced during 24 h with Ad.β-Gal (MOI 250) with or without β1-AR stimulation (100 nM Iso plus 10 nM ICI during 2 h), Ad. PKI-NES (MOI 250) with β1-AR stimulation (100 nM Iso plus 10 nM ICI during 2 h) and Ad.PKI-3NLS (MOI 250) with β1-AR stimulation (100 nM Iso plus 10 nM ICI during 2 h). (B) Effects of β1-AR stimulation (100 nM Iso plus 10 nM ICI during 2 h) with concomitant PDE4 inhibition (Ro, 10 µM) on ICER mRNA expression in ARVMs transduced during 24 h with Ad.β-Gal (MOI 250) or Ad.PKI-3NLS (MOI 250). (C) Effects of β2-AR stimulation (100 nM Iso plus 100 nM CGP during 2 h) with concomitant PDE4 inhibition (Ro, 10 µM) on ICER mRNA expression in ARVMs transduced during 24 h with Ad.β-Gal (MOI 250) or Ad.PKI-3NLS (MOI 250). (D) Effects of β2-AR stimulation (100 nM Iso plus 100 nM CGP during 2 h) with concomitant PDE4 inhibition (Ro, 10 µM) on ICER mRNA expression in ARVMs transduced during 24 h with Ad.β-Gal (MOI 250) and pre-treated or not with 10 µM CE3F4 during 15 min at 37°C. (E) Effects of β2-AR stimulation (100 nM Iso plus 100 nM CGP during 2 h) with concomitant PDE4 inhibition (Ro, 10 µM) on ICER mRNA expression in ARVMs transduced during 24 h with Ad.β-Gal (MOI 250) or Ad.PKI-NES (MOI 250). Number of rats is indicated in the respective panels. Statistical significance is indicated as **P < 0.01; ***P < 0.001 vs. β-Gal; $$$P < 0.001 by Kruskal-Wallis test with Dunn’s post-hoc test.
3.5 Role of mAKAP in the activation of nuclear PKA by β1- and β2-ARs
In cardiomyocytes, mAKAPβ has been shown to organize a cAMP-responsive network containing PKA and PDE4 at the perinuclear membrane.17 To study the contribution of mAKAPβ in shaping nuclear PKA responses under β1- or β2-AR stimulation, we used adenoviruses expressing a short hairpin RNA (shRNA) to reduce its expression.39 As shown in Figure 6A and B, this led to ∼50% decrease in mAKAPβ perinuclear staining 72 h post-transduction. Downregulation of mAKAPβ had no effect on the bulk cytoplasmic PKA activity under either β1- or β2-AR stimulation in the presence of Ro (see Supplementary material online, Figure S8). However, mAKAPβ silencing induced a small decrease in β1-ARs stimulation of nuclear PKA activity (Figure 6C) which was not observed when PDE4 was inhibited (Figure 6D). Decreasing mAKAPβ failed to unmask a nuclear PKA response to β2-AR stimulation (Figure 6E) but induced a ∼60% reduction of β2-AR + Ro stimulation (Figure 6F). These results indicate that when PDE4 is inhibited, stimulation of β2-ARs activates a specific pool of PKA maintained by mAKAPβ at the nuclear envelope to increase PKA activity inside the nucleus. This specific mAKAPβ-dependent PKA pool may also be mobilized upon β1-AR stimulation, albeit contributing to total nuclear PKA activity to a much lesser extent.
Figure 6.

The scaffold protein mAKAPβ controls β2-AR induced nuclear PKA activity when PDE4 is inhibited. (A) Immunocytochemical detection of mAKAPβ in ARVMs 72 h after sequential infection with adenoviruses encoding either a scrambled shRNA (Ad.Control shRNA MOI 2000) or a shRNA against mAKAPβ (Ad-mAKAPβ shRNA MOI 2000) for 48 h followed by infection with Ad.AKAR3-NLS (MOI 1000) for 24 h. Scale bars represent 20 µm. (B) Quantification of mAKAPβ fluorescence in ARVMs co-transduced with Ad.AKAR3-NLS and Ad.Control shRNA or Ad-mAKAPβ shRNA at 72 h. (C) Mean variation of the YFP/CFP ratio upon β1-AR stimulation with 3 nM Iso plus 10 nM ICI in ARVMs co-transduced with Ad.AKAR3-NLS and Ad.Control shRNA or Ad.AKAR3-NLS and Ad.mAKAPβ shRNA. (D) Mean variation of the YFP/CFP ratio upon β1-AR stimulation with 1 nM Iso plus 10 nM ICI in the presence of 10 µM Ro 201724 (Ro) to block PDE4. ARVMs were co-transduced with Ad.AKAR3-NLS and Ad.Control shRNA or with Ad.AKAR3-NLS and Ad.mAKAPβ shRNA. Mean variation of the YFP/CFP ratio upon β2-AR stimulation (using 30 nM Iso plus 100 nM CGP) alone (E) or in the presence of 10 µM Ro (F) in ARVMs co-transduced with Ad.AKAR3-NLS and Ad.Control shRNA or with Ad.AKAR3-NLS Ad-mAKAPβ shRNA. Number of cells/rats is indicated in brackets. Statistical significance is indicated as *P < 0.05; **P < 0.01 by nested ANOVA with Tukey’s post-hoc test.
4. Discussion
Numerous studies have emphasized the importance of spatiotemporal control of cAMP/PKA pools in specific subcellular compartments for physiological regulation of cardiomyocyte contractile function. By comparison, the mechanisms that control nuclear PKA activity are less well understood, despite their critical importance for modulation of gene expression and long term modification of cell growth and apoptosis by β-ARs. Here, we provide the first evidence that β1- and β2-ARs differentially activate nuclear PKA and gene expression in adult cardiomyocytes. As illustrated in Figure 7, we show that β1-AR stimulation engages PKA pools located outside the nucleus to enhance nuclear PKA activity and ICER expression, and that PDE4 is an important modulator of these responses. In the case of β2-ARs, our results are consistent with a model in which PDE4 insulates a mAKAPβ-targeted PKA pool at the nuclear envelope that prevents nuclear PKA activation and ICER induction upon β2-AR stimulation. However, even if PDE4 inhibition unmasks an activation of PKA in the nucleus upon β2-AR stimulation, other mechanisms that depend on cytoplasmic PKA contributes to ICER regulation by β2-AR stimulation when PDE4 is inhibited.
Figure 7.
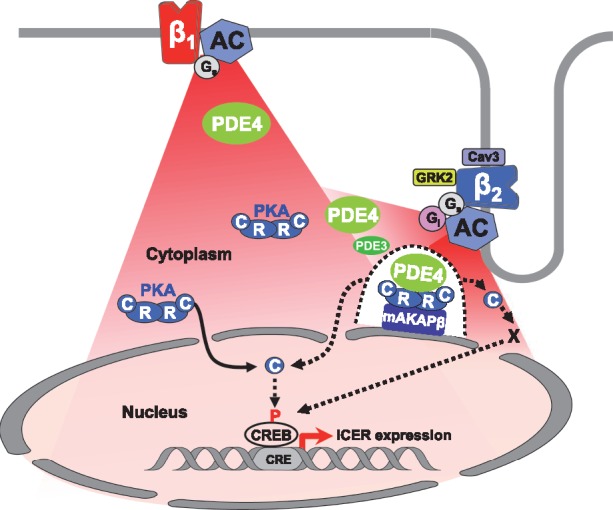
Proposed model for β1- and β2-AR regulation of cytoplasmic and nuclear PKA activity and ICER expression in adult cardiac myocytes. Stimulation of β1-ARs generate cAMP signals (in red) diffusing in the cytoplasm and the nucleus. Upon β1-ARs stimulation, PDE4 (in green) regulates cAMP levels to control PKA activity in the cytoplasm. A fraction of catalytic subunits (C) of PKA dissociate from regulatory subunits (R) and translocate inside the nucleus to increase nuclear PKA activity. Elevation of nuclear PKA activity allows induction of ICER transcription, presumably through CREB phosphorylation, which may be direct or indirect (dotted arrow between C and CREB). Stimulation of β2-ARs also generates cAMP elevation in the cytoplasm and the nucleus resulting in activation of cytoplasmic PKA. Cytoplasmic PKA activation upon β2-AR stimulation is restricted by caveolin3, Gi, GRK2, PDE3, and PDE4. In addition, PDE4 prevents activation by β2-ARs of a specific pool of PKA tethered by mAKAPβ at the perinuclear membrane (illustrated by the dotted line surrounding the mAKAPβ-PKA-PDE4 complex) which controls access of C subunits to the nucleus (dotted arrow) and nuclear PKA activation by β2-ARs. When PDE4 is inhibited, nuclear PKA activation contributes to ICER up-regulation by β2-ARs, although other mechanisms depending on cytoplasmic PKA activity are involved.
In this study we found that under selective β1-AR stimulation, PKA was activated in both the cytoplasmic and nuclear compartments, whereas β2-AR stimulation was less efficient to activate cytoplasmic PKA and failed to activate nuclear PKA. The slower kinetics of nuclear vs. cytoplasmic PKA activation observed upon β1-AR stimulation are consistent with previous observations using Iso in cardiomyocytes26,27 and suggest that the PKA holoenzyme is first activated outside the nucleus and then the catalytic subunits translocate inside the nucleus by diffusion, which is a slow process.40 The fact that cytoplasmic PKI abolished PKA phosphorylation of the nuclear-targeted PKA biosensor and induction of ICER upon β1-AR stimulation supports this model and argues against the contribution of a nuclear resident pool of PKA as demonstrated in HEK293 cells.15 These results also offer a complementary view to the local-activation/local-action of PKA signalling which prevails in other subcellular compartments of cardiomyocytes.
The robust increase in cytoplasmic PKA activity observed here for β1-ARs compared with the smaller β2-AR effect is consistent with previous real-time monitoring of PKA activity in mouse ventricular myocytes.41 However, this difference is not sufficient to explain the lack of nuclear PKA activation by β2-ARs since for a similar increase in cytoplasmic PKA activity, β1- but not β2-ARs activate nuclear PKA (Figure 1G). Thus, compartmentalization rather than intensity of β2-AR signals must explain their inability to induce nuclear PKA activation. However, our cAMP measurements with a fourth-generation cAMP FRET sensor harboring a superior dynamic range30 clearly showed that β2-AR stimulation elevated cAMP in the nucleus. This result was not expected given a previous report that β2-AR stimulation generates locally confined cAMP signals.29 However, a recent study using the same biosensor suggests that cAMP diffusivity is equivalent upon non-selective β-AR or selective β2-AR stimulation.42 Thus, one possibility is that the cAMP generated by β2-AR stimulation is able to diffuse to the nucleus even if the receptor and organelle are located at some distance from each other. But alternatively, β2-ARs could be localized in close proximity to the nucleus in ARVMs. Indeed, β2-ARs were shown to be located in T-tubules,9 and T-tubules are known to extend from the cell surface to the nuclear envelope, where they establish close contacts with the nucleus.43,44
Previous studies have proposed that alternative coupling of β2-ARs to Gi and their localization to caveolae circumvent β2-AR signalling by acting not only at the level of cAMP, but also downstream, by activating phosphatases.8,31,32 Accordingly, inhibition of Gi or disruption of caveolae potentiated β2-AR responses in the cytoplasm. However, these manoeuvres failed to unmask an effect of β2-AR stimulation on nuclear PKA activity, making it unlikely that phosphatase activation would explain the lack of β2-AR response in this compartment. Similarly, we show that βARK-ct overexpression potentiates β2-AR-induced cytoplasmic but not nuclear PKA activity. The former is consistent with inhibition of β2-AR desensitization, and consequent increased Gs signalling, but could also be explained by a decrease in Gi signalling, since it has been shown that GRK2-mediated phosphorylation of β2-ARs is important for β2-AR coupling to Gi.45,46 In addition, βARK-ct should also prevent the recruitment of PDE4D5 to β2-ARs47 as recently demonstrated.48 Based on our results, failure of β2-ARs to increase nuclear PKA activity was not due to coupling of β2-ARs to PDE4D5. Altogether, these data demonstrate that nuclear PKA activity can be dissociated from the bulk cytoplasmic PKA activity upon β2-AR stimulation, and that none of the above mechanisms is sufficient to explain the lack of nuclear PKA activation upon β2-AR stimulation.
It is undisputed that higher rates of local cAMP degradation by PDEs participate in curtailing cAMP signal.49 For this reason, we investigated the role of PDE3 and PDE4, the two major cAMP-hydrolyzing PDEs expressed in rat cardiomyocytes,12 in tuning cytoplasmic and nuclear PKA responses to β-AR stimulation. Our findings show that PDE3 plays a minor role in regulating β1-AR stimulation, but controls cytoplasmic PKA activity in response to β2-AR stimulation, whereas PDE4 acts as the main modulator of PKA activity under both β1- and β2-AR stimulation. These data are consistent with earlier work showing that PDE4 regulates cAMP levels under both β1- and β2-AR stimulation, whereas PDE3 preferentially regulates cAMP generated under β2-AR stimulation.29 Strikingly, PDE4 inhibition unmasked nuclear PKA activation under β2-AR stimulation. Although several PDE4 isoforms were shown to be associated with β2-ARs,47,50 our cAMP measurements are not compatible with PDE4 preventing cAMP generated by β2-AR stimulation to access the nucleus. Hence, we reasoned that PDE4 may act as a sink, isolating a discrete PKA pool from cAMP influx generated upon β2-AR stimulation.51 In cardiomyocytes, a perinuclear pool of PKA is maintained together with PDE4D3 by the scaffolding protein mAKAPβ, localized at the external membrane of the nuclear envelope,17 a strategic position for nuclear regulation. Our results show that disrupting this PKA pool by mAKAPβ knockdown drastically reduced nuclear PKA activation by β2-ARs with concomitant PDE4 inhibition. This identifies mAKAPβ-associated PKA as the main route for β2-AR control of nuclear PKA. The co-localization of PDE4D3 and PKA within the mAKAP complex allows efficient activation of PDE4D3 by PKA, rapid degradation of cAMP and restoration of basal PKA activity.17 In line with recent studies suggesting that PKA activation within an AKAP complex does not involve dissociation of the catalytic subunits,52,53 PDE4D3 within the mAKAP complex may prevent catalytic subunit dissociation and their translocation inside the nucleus, hence explaining the lack of nuclear PKA activation by β2-ARs. Upon PDE4 inhibition, abnormally elevated cAMP levels are reached in the complex, allowing PKA catalytic subunit dissociation and transfer to the nucleus. We have previously demonstrated that mAKAPβ selectively binds Type 5 AC (AC5) in cardiomyocytes54 and others have shown that this AC5 is localized in T-tubules and is critical for β2-AR enhancement of ICa, L which is revealed by application of PDE3 and PDE4 inhibitors.11 This parallel with our results further supports the existence of a pool of β2-ARs localized at the interface between the T-tubular membrane and the nuclear envelope (Figure 7). The small impact of mAKAPβ silencing on β1-AR stimulation of nuclear PKA activity suggests that other PKA pools are involved, which may be controlled by distinct AKAPs. One possible candidate is AKAP-Lbc, which is localized in a relatively broad perinuclear region in neonatal myocytes.55 Indeed, AKAP-Lbc was recently shown to bind a PDE4 long isoform, the activation of which led to reduced forskolin-induced nuclear PKA activation and attenuated hypertrophic response to β-AR stimulation.56
Recent studies have emphasized the critical role of PKA in the pro-apoptotic effects of β-ARs.57 We have shown that nuclear PKA activation by β1-ARs is concentration-dependent (Figure 1), requires sustained receptor stimulation,26 and induces ICER expression (Figures 4E and 5A), which is a strong mediator of apoptosis by decreasing Bcl-2 in cardiomyocytes.21 In contrast, β2-ARs do not increase nuclear PKA activity (Figure 1) and fail to increase ICER expression (Figure 4F). These results are consistent with a previous study showing that cardiomyocyte fate is switched from survival to death depending on the strength of β-AR stimulation and the balance between a β2-AR/Gi-mediated ERK1/2 pathway leading to Bcl-2 induction at a low concentration range of Iso, and a β1-AR-mediated PKA/CREB/ICER pathway leading to Bcl-2 repression at higher concentrations of Iso.34 Thus, segregated PKA activation between the cytoplasm and the nucleus, together with activation of surviving signalling such as ERK1/2 is likely to contribute to the differential effects of low vs. high levels of β-AR stimulation as well as subtype-specific effects. However, we further show that β2-ARs can increase ICER expression when PDE4 is inhibited (Figure 4F). At variance with β1-ARs, this effect was only partially inhibited by nuclear PKI. It was not modified by the Epac1 inhibitor CE3F4 (Figure 5D), but could be almost completely abrogated by cytoplasmic PKI (Figure 5E). These results suggest that an alternative pathway depending on cytoplasmic but not nuclear PKA activity contributes to ICER induction by β2-ARs when PDE4 is inhibited. In neurons, PKA is required for nuclear translocation of ERK and ribosomal S6 kinase 2 and subsequent stimulation of CREB-dependent transcription.58 Further studies are needed to determine if a similar cross talk exists in cardiomyocytes. Nevertheless, the present results underline the fact that β1-ARs and β2-ARs trigger divergent signalling mechanisms to regulate gene expression in cardiomyocytes.
In a previous study, it was shown that in neonatal cardiomyocytes, PDE3 but not PDE4 inhibition increased CREB phosphorylation and ICER expression and thus induced Bcl-2 downregulation.22 These results contrast with the lack of effect of the PDE3 inhibitor cilostamide on basal ICER expression observed here (see Supplementary material online, Figure S5A). This might be explained by differences in experimental conditions or the cell type used (adult vs. neonatal myocytes).
In conclusion, our study unveils the molecular mechanisms by which β1- and β2-ARs differentially regulate nuclear PKA activity and ICER expression in terminally differentiated adult cardiomyocytes, and identify mAKAPβ and PDE4 as critical organizers of β2-AR nuclear signalling. Many questions remain, however, that will need to be addressed in future studies. The extent to which the findings obtained here for ICER can be generalized to the β-AR transcriptome, and the place of PKA in the control of gene expression by β-ARs vs. other cAMP effectors will deserve further investigations. The contribution of intracellular β-ARs located at the nuclear envelope,59,60 in endosomes,61 and at the Golgi61,62 should be evaluated in intact adult cardiac myocytes. In a translational perspective, it will also be important to determine whether the signalling routes linking β-ARs to nuclear PKA activation identified here in rat are conserved in larger mammals and humans. Finally, in hypertrophy and HF, there is a profound remodelling of the β-AR/cAMP/PKA pathway. Interestingly, while the expression of β1-ARs is reduced,63 β2-ARs and PDEs are redistributed, leading to enhanced β2-ARs signalling and loss of compartmentation.9,33,64 Future studies should aim to elucidate how nuclear PKA activity is controlled by β-ARs in diseased cardiomyocytes, which may be important for identification of new therapeutic targets in HF.
Supplementary Material
Acknowledgements
We thank Jean-Piero Margaria for technical advices and Dr Jérôme Leroy for helpful discussion on the paper. We thank Valérie Nicolas (Imaging platform) and Claudine Delomenie (Transcriptomic platform), from Institut Paris Saclay d’Innovation Thérapeutique (UMS IPSIT), for technical assistance.
Conflict of interest: none declared.
Funding
I.B. was supported by a PhD fellowship from the Région Ile-de-France (CORDDIM) and Fondation pour la Recherche Médicale, grant FDT20160736469. MSK was supported by NIH Grant HL126825. Our laboratory is a member of the Laboratory of Excellence LERMIT supported by a grant from ANR (ANR-10-LABX-33) under the programme ‘Investissements d’Avenir’ ANR-11-IDEX-0003-01.
Footnotes
Time for primary review: 41 days
References
- 1. Xiao RP, Lakatta EG.. Beta 1-adrenoceptor stimulation and beta 2-adrenoceptor stimulation differ in their effects on contraction, cytosolic Ca2+, and Ca2+ current in single rat ventricular cells. Circ Res 1993;73:286–300. [DOI] [PubMed] [Google Scholar]
- 2. Milano CA, Allen LF, Rockman HA, Dolber PC, McMinn TR, Chien KR, Johnson TD, Bond RA, Lefkowitz RJ.. Enhanced myocardial function in transgenic mice overexpressing the beta 2-adrenergic receptor. Science 1994;264:582–586. [DOI] [PubMed] [Google Scholar]
- 3. Engelhardt S, Hein L, Wiesmann F, Lohse MJ.. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A 1999;96:7059–7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Communal C, Singh K, Sawyer DB, Colucci WS.. Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein. Circulation 1999;100:2210–2212. [DOI] [PubMed] [Google Scholar]
- 5. Xiao RP. Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci STKE 2001;2001:re15. [DOI] [PubMed] [Google Scholar]
- 6. Rybin VO, Xu X, Lisanti MP, Steinberg SF.. Differential targeting of beta -adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway. J Biol Chem 2000;275:41447–41457. [DOI] [PubMed] [Google Scholar]
- 7. Xiang Y, Rybin VO, Steinberg SF, Kobilka B.. Caveolar localization dictates physiologic signaling of beta 2-adrenoceptors in neonatal cardiac myocytes. J Biol Chem 2002;277:34280–34286. [DOI] [PubMed] [Google Scholar]
- 8. Macdougall DA, Agarwal SR, Stopford EA, Chu H, Collins JA, Longster AL, Colyer J, Harvey RD, Calaghan S.. Caveolae compartmentalise beta2-adrenoceptor signals by curtailing cAMP production and maintaining phosphatase activity in the sarcoplasmic reticulum of the adult ventricular myocyte. J Mol Cell Cardiol 2012;52:388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE, Gorelik J.. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010;327:1653–1657. [DOI] [PubMed] [Google Scholar]
- 10. Wright PT, Nikolaev VO, O’Hara T, Diakonov I, Bhargava A, Tokar S, Schobesberger S, Shevchuk AI, Sikkel MB, Wilkinson R, Trayanova NA, Lyon AR, Harding SE, Gorelik J.. Caveolin-3 regulates compartmentation of cardiomyocyte beta2-adrenergic receptor-mediated cAMP signaling. J Mol Cell Cardiol 2014;67:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Timofeyev V, Myers RE, Kim HJ, Woltz RL, Sirish P, Heiserman JP, Li N, Singapuri A, Tang T, Yarov-Yarovoy V, Yamoah EN, Hammond HK, Chiamvimonvat N.. Adenylyl cyclase subtype-specific compartmentalization: differential regulation of L-type Ca2+ current in ventricular myocytes. Circ Res 2013;112:1567–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, Fischmeister R, Vandecasteele G.. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res 2006;98:1081–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xiang Y, Devic E, Kobilka B.. The PDZ binding motif of the beta 1 adrenergic receptor modulates receptor trafficking and signaling in cardiac myocytes. J Biol Chem 2002;277:33783–33790. [DOI] [PubMed] [Google Scholar]
- 14. Diviani D, Dodge-Kafka KL, Li J, Kapiloff MS.. A-kinase anchoring proteins: scaffolding proteins in the heart. Am J Physiol Heart Circ Physiol 2011;301:H1742–H1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sample V, Dipilato LM, Yang JH, Ni Q, Saucerman JJ, Zhang J.. Regulation of nuclear PKA revealed by spatiotemporal manipulation of cyclic AMP. Nat Chem Biol 2012;8:375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sette C, Conti M.. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J Biol Chem 1996;271:16526–16534. [DOI] [PubMed] [Google Scholar]
- 17. Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, Hill EV, Houslay MD, Langeberg LK, Scott JD.. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. Embo J 2001;20:1921–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD.. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 2005;437:574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kritzer MD, Li J, Dodge-Kafka K, Kapiloff MS.. AKAPs: the architectural underpinnings of local cAMP signaling. J Mol Cell Cardiol 2012;52:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Passariello CL, Li J, Dodge-Kafka K, Kapiloff MS.. mAKAP-a master scaffold for cardiac remodeling. J Cardiovasc Pharmacol 2015;65:218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tomita H, Nazmy M, Kajimoto K, Yehia G, Molina CA, Sadoshima J.. Inducible cAMP early repressor (ICER) is a negative-feedback regulator of cardiac hypertrophy and an important mediator of cardiac myocyte apoptosis in response to beta-adrenergic receptor stimulation. Circ Res 2003;93:12–22. [DOI] [PubMed] [Google Scholar]
- 22. Ding B, Abe J, Wei H, Xu H, Che W, Aizawa T, Liu W, Molina CA, Sadoshima J, Blaxall BC, Berk BC, Yan C.. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci U S A 2005;102:14771–14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Backs J, Worst BC, Lehmann LH, Patrick DM, Jebessa Z, Kreusser MM, Sun Q, Chen L, Heft C, Katus HA, Olson EN.. Selective repression of MEF2 activity by PKA-dependent proteolysis of HDAC4. J Cell Biol 2011;195:403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ha CH, Kim JY, Zhao J, Wang W, Jhun BS, Wong C, Jin ZG.. PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A 2010;107:15467–15472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang CW, Lee L, Yu D, Dao K, Bossuyt J, Bers DM.. Acute beta-Adrenergic Activation Triggers Nuclear Import of Histone Deacetylase 5 and Delays Gq-induced Transcriptional Activation. J Biol Chem 2013;288:192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haj Slimane Z, Bedioune I, Lechene P, Varin A, Lefebvre F, Mateo P, Domergue-Dupont V, Dewenter M, Richter W, Conti M, El-Armouche A, Zhang J, Fischmeister R, Vandecasteele G.. Control of cytoplasmic and nuclear protein kinase A by phosphodiesterases and phosphatases in cardiac myocytes. Cardiovasc Res 2014;102:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang JH, Polanowska-Grabowska RK, Smith JS, Shields CWt, Saucerman JJ.. PKA catalytic subunit compartmentation regulates contractile and hypertrophic responses to beta-adrenergic signaling. J Mol Cell Cardiol 2013;66C:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Allen MD, Zhang J.. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun 2006;348:716–721. [DOI] [PubMed] [Google Scholar]
- 29. Nikolaev VO, Bunemann M, Schmitteckert E, Lohse MJ, Engelhardt S.. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ Res 2006;99:1084–1091. [DOI] [PubMed] [Google Scholar]
- 30. Klarenbeek J, Goedhart J, van Batenburg A, Groenewald D, Jalink K.. Fourth-Generation Epac-Based FRET Sensors for cAMP Feature Exceptional Brightness, Photostability and Dynamic Range: characterization of Dedicated Sensors for FLIM, for Ratiometry and with High Affinity. PLoS One 2015;10:e0122513.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuschel M, Zhou YY, Cheng H, Zhang SJ, Chen Y, Lakatta EG, Xiao RP.. G(i) protein-mediated functional compartmentalization of cardiac beta(2)-adrenergic signaling. J Biol Chem 1999;274:22048–22052. [DOI] [PubMed] [Google Scholar]
- 32. Jo SH, Leblais V, Wang PH, Crow MT, Xiao RP.. Phosphatidylinositol 3-kinase functionally compartmentalizes the concurrent G(s) signaling during beta2-adrenergic stimulation. Circ Res 2002;91:46–53. [DOI] [PubMed] [Google Scholar]
- 33. Barbagallo F, Xu B, Reddy GR, West T, Wang Q, Fu Q, Li M, Shi Q, Ginsburg KS, Ferrier W, Isidori AM, Naro F, Patel HH, Bossuyt J, Bers D, Xiang YK.. Genetically Encoded Biosensors Reveal PKA Hyperphosphorylation on the Myofilaments in Rabbit Heart Failure. Circ Res 2016;119:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shin SY, Kim T, Lee HS, Kang JH, Lee JY, Cho KH, Kim do H.. The switching role of beta-adrenergic receptor signalling in cell survival or death decision of cardiomyocytes. Nat Comms 2014;5:5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lezoualc’h F, Fazal L, Laudette M, Conte C.. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ Res 2016;118:881–897. [DOI] [PubMed] [Google Scholar]
- 36. Bobin P, Varin A, Lefebvre F, Fischmeister R, Vandecasteele G, Leroy J.. Calmodulin kinase II inhibition limits the pro-arrhythmic Ca2+ waves induced by cAMP-phosphodiesterase inhibitors. Cardiovasc Res 2016;110:151–161. [DOI] [PubMed] [Google Scholar]
- 37. Pereira L, Rehmann H, Lao DH, Erickson JR, Bossuyt J, Chen J, Bers DM.. Novel Epac fluorescent ligand reveals distinct Epac1 vs. Epac2 distribution and function in cardiomyocytes. Proc Natl Acad Sci U S A 2015;112:3991–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Courilleau D, Bisserier M, Jullian JC, Lucas A, Bouyssou P, Fischmeister R, Blondeau JP, Lezoualc'h F.. Identification of a Tetrahydroquinoline Analog as a Pharmacological Inhibitor of the cAMP-binding Protein Epac. J Biol Chem 2012;287:44192–44202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS.. The mAKAP complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. J Cell Sci 2005;118:5637–5646. [DOI] [PubMed] [Google Scholar]
- 40. Harootunian AT, Adams SR, Wen W, Meinkoth JL, Taylor SS, Tsien RY.. Movement of the free catalytic subunit of cAMP-dependent protein kinase into and out of the nucleus can be explained by diffusion. MBoC 1993;4:993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Soto D, De Arcangelis V, Zhang J, Xiang Y.. Dynamic protein kinase a activities induced by beta-adrenoceptors dictate signaling propagation for substrate phosphorylation and myocyte contraction. Circ Res 2009;104:770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Richards M, Lomas O, Jalink K, Ford KL, Vaughan-Jones RD, Lefkimmiatis K, Swietach P.. Intracellular tortuosity underlies slow cAMP diffusion in adult ventricular myocytes. Cardiovasc Res 2016;110:395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Escobar M, Cardenas C, Colavita K, Petrenko NB, Franzini-Armstrong C.. Structural evidence for perinuclear calcium microdomains in cardiac myocytes. J Mol Cell Cardiol 2011;50:451–459. [DOI] [PubMed] [Google Scholar]
- 44. Ibarra C, Vicencio JM, Estrada M, Lin Y, Rocco P, Rebellato P, Munoz JP, Garcia-Prieto J, Quest AF, Chiong M, Davidson SM, Bulatovic I, Grinnemo KH, Larsson O, Szabadkai G, Uhlen P, Jaimovich E, Lavandero S.. Local control of nuclear calcium signaling in cardiac myocytes by perinuclear microdomains of sarcolemmal insulin-like growth factor 1 receptors. Circ Res 2013;112:236–245. [DOI] [PubMed] [Google Scholar]
- 45. Wang Y, De Arcangelis V, Gao X, Ramani B, Jung YS, Xiang Y.. Norepinephrine- and epinephrine-induced distinct beta2-adrenoceptor signaling is dictated by GRK2 phosphorylation in cardiomyocytes. J Biol Chem 2008;283:1799–1807. [DOI] [PubMed] [Google Scholar]
- 46. Zhu W, Petrashevskaya N, Ren S, Zhao A, Chakir K, Gao E, Chuprun JK, Wang Y, Talan M, Dorn GW, Lakatta EG, Koch WJ, Feldman AM, Xiao R-P.. Gi-biased beta2AR signaling links GRK2 upregulation to heart failure. Circ Res 2012;110:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD.. beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci U S A 2003;100:940–945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Salazar NC, Vallejos X, Siryk A, Rengo G, Cannavo A, Liccardo D, De Lucia C, Gao E, Leosco D, Koch WJ, Lymperopoulos A.. GRK2 blockade with betaARKct is essential for cardiac beta2-adrenergic receptor signaling towards increased contractility. Cell Commun Signal 2013;11:64.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fischmeister R, Castro LR, Abi-Gerges A, Rochais F, Jurevicius J, Leroy J, Vandecasteele G.. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res 2006;99:816–828. [DOI] [PubMed] [Google Scholar]
- 50. De Arcangelis V, Liu R, Soto D, Xiang Y.. Differential association of phosphodiesterase 4D isoforms with beta2-adrenoceptor in cardiac myocytes. J Biol Chem 2009;284:33824–33832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Terrin A, Di Benedetto G, Pertegato V, Cheung YF, Baillie G, Lynch MJ, Elvassore N, Prinz A, Herberg FW, Houslay MD, Zaccolo MPGE. ( 1) stimulation of HEK293 cells generates multiple contiguous domains with different cAMP: role of compartmentalized phosphodiesterases. J Cell Biol 2006;175:441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smith FD, Reichow SL, Esseltine JL, Shi D, Langeberg LK, Scott JD, Gonen T.. Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. Elife 2013;2:e01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smith FD, Esseltine JL, Nygren PJ, Veesler D, Byrne DP, Vonderach M, Strashnov I, Eyers CE, Eyers PA, Langeberg LK, Scott JD.. Local protein kinase A action proceeds through intact holoenzymes. Science 2017;356:1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kapiloff MS, Piggott LA, Sadana R, Li J, Heredia LA, Henson E, Efendiev R, Dessauer CW.. An adenylyl cyclase-mAKAPbeta signaling complex regulates cAMP levels in cardiac myocytes. J Biol Chem 2009;284:23540–23546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Carnegie GK, Soughayer J, Smith FD, Pedroja BS, Zhang F, Diviani D, Bristow MR, Kunkel MT, Newton AC, Langeberg LK, Scott JD.. AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol Cell 2008;32:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang L, Burmeister BT, Johnson KR, Baillie GS, Karginov AV, Skidgel RA, O'Bryan JP, Carnegie GK.. UCR1C is a novel activator of phosphodiesterase 4 (PDE4) long isoforms and attenuates cardiomyocyte hypertrophy. Cell Signal 2015;27:908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY, Chen X.. Cardiotoxic and cardioprotective features of chronic beta-adrenergic signaling. Circ Res 2013;112:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR.. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 1998;21:869–883. [DOI] [PubMed] [Google Scholar]
- 59. Boivin B, Lavoie C, Vaniotis G, Baragli A, Villeneuve LR, Ethier N, Trieu P, Allen BG, Hebert TE.. Functional beta-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc Res 2006;71:69–78. [DOI] [PubMed] [Google Scholar]
- 60. Vaniotis G, Del Duca D, Trieu P, Rohlicek CV, Hebert TE, Allen BG.. Nuclear beta-adrenergic receptors modulate gene expression in adult rat heart. Cell Signal 2011;23:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, von Zastrow M.. Conformational biosensors reveal GPCR signalling from endosomes. Nature 2013;495:534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Irannejad R, Pessino V, Mika D, Huang B, Wedegaertner PB, Conti M, von Zastrow M.. Functional selectivity of GPCR-directed drug action through location bias. Nat Chem Biol 2017;13:799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lohse MJ, Engelhardt S, Eschenhagen T.. What is the role of beta-adrenergic signaling in heart failure? Circ Res 2003;93:896–906. [DOI] [PubMed] [Google Scholar]
- 64. Sprenger JU, Perera RK, Steinbrecher JH, Lehnart SE, Maier LS, Hasenfuss G, Nikolaev VO.. In vivo model with targeted cAMP biosensor reveals changes in receptor-microdomain communication in cardiac disease. Nat Commun 2015;6:6965. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.