

SUMMARY
Genetic instability of the mitochondrial genome (mtDNA) plays an important role in human aging and disease. Thus far, it has proven difficult to develop successful treatment strategies for diseases that are caused by mtDNA instability. To address this issue, we developed a model of mtDNA disease in the nematode C. elegans, an animal model that can rapidly be screened for genes and biological pathways that reduce mitochondrial pathology. These worms recapitulate all the major hallmarks of mtDNA disease in humans, including increased mtDNA instability, loss of respiration, reduced neuromuscular function, and a shortened lifespan. We found that these phenotypes could be rescued by intervening in numerous biological pathways, including IGF-1/insulin signaling, mitophagy, and the mitochondrial unfolded protein response, suggesting that it may be possible to ameliorate mtDNA disease through multiple molecular mechanisms.
In Brief
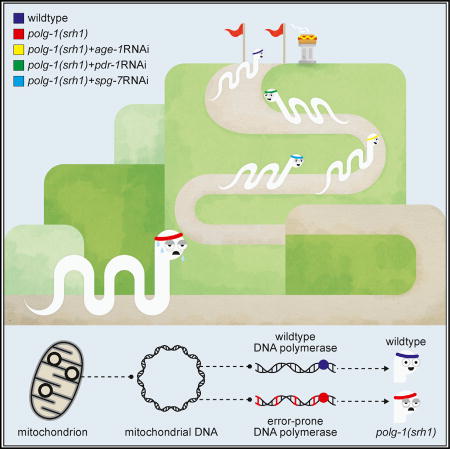
Haroon et al. describe a genetically engineered C. elegans that carries an error-prone copy of DNA polymerase γ, the enzyme that replicates the mitochondrial genome. This worm recapitulates the major hallmarks of mitochondrial disease in humans. The authors identify multiple biological pathways that could potentially delay disease progression.
INTRODUCTION
Energy animates life. Whether it’s protein synthesis, autophagy, or signal transduction, almost every biological process is driven by the consumption of energy. Most of this energy is generated by mitochondria, small tubular organelles that are frequently called the powerhouses of our cells. To optimize energy production, mitochondria carry their own genome called “mtDNA,” a small, circular molecule that encodes numerous protein products that are essential to ATP synthesis (Anderson et al., 1981). As a result, loss or mutation of mtDNA invariably affects energy production, which is particularly harmful to cells with high energy demands such as neurons and muscle fibers (Wallace, 2005). Most diseases caused by mtDNA instability are therefore characterized by some form of neuromuscular dysfunction. For example, point mutations, deletions, or overt loss of mtDNA molecules can result in premature deafness, myopathy, or severe encephalomyopathy in children (Saneto and Sedensky, 2013) and contributes to neurodegeneration (Kraytsberg et al., 2006) and muscle wasting (Wanagat et al., 2001) in aging adults. In most cases, mtDNA instability arises spontaneously, although it also can be caused by mutations in DNA polymerase γ, the enzyme that replicates the mitochondrial genome. Thus, the impact of mtDNA instability on human aging and disease is well documented. To this day though, no child has ever been cured of an mtDNA disease, nor does a treatment exist for the mtDNA component of age-related diseases. Although it is now possible to manage the symptoms of a number of pediatric mtDNA diseases, two bottlenecks have traditionally kept the scientific community from developing a more robust treatment strategy: a shortage of animal models and a lack of promising molecules to target. Here, we address these issues by establishing a model of mtDNA disease in worms that can give rise to countless additional models with the proper breeding scheme. Moreover, because worms can be easily manipulated by genetic and pharmaceutical means, we were able to use RNAi, drugs, and genetic mutants to identify multiple cellular pathways that can ameliorate the pathological consequences of mtDNA instability on organismal health, thus providing potential targets for therapeutic intervention. Together, these results suggest that multiple pathways may have evolved to prevent mtDNA disease, underlining the central role that energy plays in cellular life.
RESULTS
A Model of mtDNA Disease in C. elegans
To successfully design a therapeutic strategy for mtDNA disease, it will be essential to identify cellular mechanisms that can either increase or decrease the pathology that is caused by mtDNA instability. These experiments will reveal promising targets that can be exploited for therapeutic intervention, and may provide valuable insight into the basic biology that underlies mitochondrial function. Identifying these mechanisms requires a flexible animal model that is well suited for “discovery experiments.” The most promising model available today is the mitochondrial mutator mouse (Kujoth et al., 2005; Trifunovic et al., 2004). This mouse model carries an error-prone copy of DNA polymerase γ (PolgAD257A), the enzyme that replicates the mitochondrial genome. Because of error-prone DNA replication, the PolgAD257 mice exhibit a >100-fold increase in mtDNA mutations, which results in extensive mtDNA disease (Kujoth et al., 2005; Trifunovic et al., 2004; Vermulst et al., 2007, 2008). However, logistical considerations prohibit these mice from being screened en masse to identify genes or molecular pathways that modulate their pathology. Therefore, we recreated the PolgAD257A mutation in C. elegans, a genetically tractable model organism that is amenable to high-throughput screens. Using CRISPR/Cas9 technology, we directly edited the polg-1 gene (the C. elegans homolog of PolgA) and introduced the analogous D207A mutation (polg-1(srh1)) (Figure 1A). Similar to the mutator mice, we found that the polg-1(srh1) worms display a large increase in mtDNA mutations (Vermulst et al., 2007, 2008) (Figure 1B), which was primarily driven by C:G > T:A transitions (Figure 1C). These transitions were not equally distributed over the TaqI restriction site, despite the fact that it is a palindrome, suggesting that polg-1-mediated mutagenesis has a substantial strand bias (Figure 1C). Moreover, homozygous carriers of the polg-1(srh1) allele displayed a ~50% loss in mtDNA copy number (Figure 1D), mirroring the mtDNA depletion of homozygous mutator mice. Thus, even though mtDNA is replicated differently in worms compared to mice (Lewis et al., 2015), the polg-1(srh1) worms closely mimic the genetic instability of the mutator mice and display an increased rate of mtDNA mutation and depletion, two forms of mtDNA instability that cause mtDNA disease in humans (Copeland, 2008; El-Hattab and Scaglia, 2013).
Figure 1. Characterization of the polg-1(srh1) Worms.

(A) The exonuclease (green) domain of DNA polymerase γ contains three highly conserved regions (red) that control the fidelity of DNA synthesis, including an aspartic acid in exonuclease domain II that is essential for the proofreading activity of polymerase γ across the tree of life. We used CRISPR/Cas9 technology to mutate this residue to alanine in the error-prone allele polg-1(srh1).
(B) The mtDNA mutation frequency of the polg-1(srh1) worms is >70-fold higher compared to WT worms.
(C) The mutation spectrum of WT and polg-1(srh1) depicted as peaks above and below the WT nucleotide according to standard electrophoretogram color coding (red, T; blue, C; black, G; green, A). The percentage of each type of mutation is listed by the peaks.
(D) The mtDNA copy number is reduced by 56% in the polg-1(srh1) worms.
(E) The basal respiration of polg-1(srh1) worms worsens with age compared to WT worms.
(F) The mitochondria of polg-1(srh1) worms display reduced reserve capacity upon FCCP treatment at 5 days of age.
(G and H) Confocal images of day 10 polg-1(srh1) and WT worms (G) do not reveal a significant difference in oxidation stress (H).
(I–K) Neuromuscular function assessed by chemotaxis (I), thrashing (J), and a gentle touch assay (K) reveals increased dysfunction in polg-1(srh1) worms compared to WT animals.
(L) The polg-1(srh1) worms have a median lifespan of 13 days compared to 16 days for WT worms (log-rank test p < 0.01).
Data for WT and polg-1(srh1) worms are in blue and pink, respectively (in B, D–F, and H–L).
Bar graphs represent the mean ± SEM of at least three biological replicates, and the lifespan assay was performed using at least 100 worms per genotype. Unpaired t tests were performed to determine significance (*p < 0.05, **p < 0.01; ns, no significant difference).
Mutator Worms Replicate Major Hallmarks of mtDNA Disease in Humans
In human patients, mtDNA instability invariably results in mitochondrial dysfunction. To determine whether the mutator worms display mitochondrial dysfunction as well, we analyzed intact worms by respirometry. We found that at day 1 of adulthood, the mutator worms respire at the same rate as wild-type (WT) worms; however, at 11 days of age, they display a ~50% decrease in oxygen consumption (Figure 1E), indicating that they suffer from a progressive decline in mitochondrial function. Closer examination showed that this decline starts at 5 days of age, when basal respiration is optimal, but the reserve capacity of mitochondria has been reduced to 50% of WT levels (Figures 1F, S1A, and S1B). However, similar to the mutator mice, reduced respiration did not result in overt production of reactive oxidative species (Figures 1G, 1H, and S1C–S1F) (Kujoth et al., 2005; Trifunovic et al., 2005). The most prevalent symptom of inherited, as well as age-related mtDNA disease is neuromuscular dysfunction (Wallace, 2010). To determine whether the polg-1(srh1) worms display neuromuscular dysfunction as well, we tested their locomotion toward a chemo-attractant. This assay tests the ability of the worms to sense the chemo-attractant (Wes and Bargmann, 2001) (neuronal function), as well as their ability to crawl toward it (muscle function) (Kashyap et al., 2012). At day 1 of adulthood, the polg-1(srh1) worms were able to reach the chemo-attractant with the same efficiency as WT worms; however, as the polg-1(srh1) worms grew older, they became increasingly incapable of accomplishing this task (Figure 1I). On average, this phenotype emerged at 5 days of adulthood, coinciding with the onset of mitochondrial dysfunction, and worsened as the worms grew older (Figure 1I). To better understand the physiological underpinnings of this neuromuscular dysfunction, we used a “gentle touch” and “thrashing” assay. The gentle touch assay documents the ability of worms to detect a gentle tap on their head and their tail, which primarily tests the function of highly specialized mechanosensory neurons (Hobert et al., 1999). This assay revealed that the mutator worms are increasingly unresponsive to gentle touches, which became significantly different from WT worms at day 7 of adulthood and lasted until measurements concluded at day 10 (Figure 1J). The thrashing assay documents the number of times worms bend their bodies over in rapid succession in liquid media (Bansal et al., 2015; Han and Beckerle, 2009). Similar to the gentle touch assay, we found that the polg-1(srh1) worms display an age-related decrease in thrashing (Figure 1K; Movies S1 and S2). However, in contrast to the gentle touch assay, this phenotype was more severe and emerged concurrently with the chemotaxis defect at day 5 of adulthood. This observation suggests that the dysfunction of the polg-1(srh1) worms highlighted by the chemotaxis assay is most likely due to an inability to move, and not an inability to sense the chemoattractant. Further experimentation will be required to determine whether their inability to move is caused by dysfunction of the muscle cells themselves, or the motor neurons that innervate them. The neuromuscular dysfunction associated with mtDNA disease in humans is typically incompatible with normal lifespan (Wallace, 2005, 2010). To determine whether the polg-1(srh1) worms display a shortened lifespan as well, we compared the lifespan of WT worms to polg-1(srh1) worms and found that the polg-1(srh1) worms are indeed shorter lived than WT animals (Figure 1l), further confirming the extent of their disease, and their similarity to the short lived mutator mice (Trifunovic et al., 2004). Taken together, these experiments indicate that the polg-1(srh1) worms recapitulate several hallmarks of mtDNA disease in humans including increased mtDNA instability, loss of mitochondrial respiration, extensive neuromuscular dysfunction, and a shortened lifespan.
Mutation Load and Tissue Dysfunction of polg-1(srh1) Worms Worsen over Generations
The number of mutated mtDNA molecules that are transmitted from mother to child controls the severity and the type of pathology that emerges in children (Wallace, 2010). The higher this number is, the more complex the disease tends to be; however, it has proven difficult to study germline transmission of mtDNA mutations in detail, because it is not possible to transform the mitochondrial genome at will. As a result, researchers are forced to rely on natural mtDNA variants to decipher the parameters that regulate mutation inheritance. We reasoned that the increased mutation rate of the polg-1(srh1) allele could greatly accelerate the process of isolating organisms with specific mtDNA mutations of interest, so that novel models of inherited mtDNA mutations can rapidly be generated. To test this hypothesis, we tracked the offspring of individual WT and polg-1(srh1) worms over 35 generations and sequenced the mtDNA of 3 siblings at generations 5, 15, 25, and 35. Over these generations, the mutator worms accrued an increasing number of detectable heteroplasmic mutations (Figures 2A, 2B, and S2), while the mtDNA of the WT worms remained pristine. Consistent with the idea that these mutations contribute to the dysfunction of the polg-1(srh1) worms, we found that the performance of the polg-1(srh1) worms in the chemotaxis (Figure 2C), gentle touch (Figure 2D) and thrashing (Figure 2E) assay decreased with increasing generation number. To prevent these generational effects from confounding our results, all studies were performed on generation-matched worms (generations 27–33). However, in some cases, the rationale of our experiments required a change in generation number. If so, this change has been clearly denoted. We further found that these germline mutations both increased and decreased over successive generations, indicating substantial genetic drift. To formally demonstrate that these mutations can be isolated to study their inheritance, we decided to remove the error-prone polg-1(srh) allele from the nuclear genome. We reasoned that this strategy would prevent additional mutations from accumulating in the mitochondrial genome, and allow desirable mtDNA mutations to be fixed in a WT nuclear background. To this end we crossed the mutagenic polg-1(srh) allele out of a worm that carried a single base-pair deletion in the ND1 gene (Figures 2A, 2B, and S2E). After the removal of the polg-1(srh) allele, we selectively picked individual progeny from parents with ~70% heteroplasmy over several generations (Figure 2F), and were able to successfully retain this mtDNA mutation. Interestingly, we found that, three generations after breeding out the mutator allele, 33% of the worms laid inviable embryos, 33% of the worms laid progeny that became larval phase 2 dauers, and 33% of the worms laid normal embryos that developed fully. This experiment demonstrates that it is possible to exploit the polg-1(srh) worms to generate novel animal models that display gross phenotypic abnormalities, and carry highly desirable mtDNA mutations in their germline, which could fill an important void in the mitochondrial research community.
Figure 2. The Phenotype of the polg-1(srh1) Worms Worsens from Generation to Generation.

(A) The outer circle represents the worm mitochondrial genome highlighting the protein-coding genes (green), rDNA (blue), AT-rich region (yellow), and all the mutations (red) that arose over 35 generations of maintaining the polg-1(srh1) mutator allele. The inner circle represents the regions that were sequenced (blue).
(B) mtDNA mutations were tracked over 35 generations in WT and polg-1(srh1) worms. At generations 5, 15, 25, and 35, ~11 kb of mtDNA of 3 progenies of a single WT and polg-1(srh1) worm were sequenced. The mutations depicted here were present in all 3 progenies, indicating that they were successfully transmitted through the germline. Three mutations, here denoted in purple, brown, and orange polg-1(srh1) were present across multiple generations and showed substantial genetic drift. No new mutations arose in the WT strain.
(C–E) Neuromuscular dysfunction was assessed by chemotaxis (C), thrashing (D), and gentle touch (E) revealed progressive dysfunction with increasing generation in the polg-1(srh1) worms.
(F) Representative electrophoretograms tracking the T2085 deletion that results in an early stop codon in the ND1 gene in both the parent with polg-1(srh1) allele and its resultant progeny where the mutator allele has been mated out. Three individuals without the polg-1(srh1) allele in the F1 generation, 10 individuals in the F2 generations, and 21 individuals in the F3 generation were isolated and tested for the transmission of T2085 heteroplasmy in a background WT for polg-1.
Bar graphs represent the mean ± SEM of at least three biological replicates. Unpaired t tests were performed to determine significance (*p < 0.05, **p < 0.01; ns, no significant difference).
Also see Figure S2.
Multiple Pathways Modulate mtDNA Disease in Worms
The chemotaxis assay described above is highly quantitative and reproducible, and tests the most important clinical aspects of mtDNA disease. Therefore, we used this assay as a screening tool to identify genes that can ameliorate the pathology associated with mtDNA disease (Figure 3). To this end, we performed a candidate RNAi screen using >130 RNAi constructs that control either C. elegans lifespan, overall health, or various aspects of mitochondrial function, parameters that are likely to intersect with mtDNA disease. We considered a gene a positive hit if RNAi against that gene improved the performance of the mutator worms by >30%. This cutoff is based on the observation that the inherent variation between randomly selected polg-1(srh1) worms rarely exceeded this threshold (0 out of 10 trials), indicating a <10% chance of discovering a false positive (Figure S3A). Second, RNAi against age-1, which we found to be a potent modulator of mtDNA disease (35% on average) (Figures 3A–3C), improved the performance of the mutator worms by >30% in 6 out of 9 trials (Figure S3B), indicating a > 60% chance of detecting a gene that increases the performance of the polg-1(srh1) worms by 35% or more. Excitingly, RNAi against numerous genes decreased the pathology associated with mtDNA disease in worms. For example, manipulation of the insulin growth factor (IGF)-1/insulin signaling (IIS) pathway, mitophagy, autophagy, apoptosis, and the mitochondrial unfolded protein response (UPRmt) all ameliorated the neuromuscular defect of the polg-1(srh1) worms (Figure 3D). Interestingly, reduced autophagy was previously shown to ameliorate mitochondrial dysfunction in a model of mitochondrial disease that is caused by a nuclear mutation (Peng et al., 2015), while suppression of apoptosis activates the highly beneficial stress response in worms (Judy et al., 2013). Similarly, mitophagy and UPRmt are implicated in various diseases that contain a mitochondrial component (Haynes et al., 2013; Youle and Narendra, 2011). We frequently found that RNAi against multiple components of the same pathway rescued the mutator worms, strongly implicating that pathway in the progression of mtDNA disease (Figure 3D). Furthermore, we found that pathways that do not impact mitochondrial biology (such as the unfolded protein response of the ER) did not alter the performance of the mutator worms (Table S1). To validate the results of our screen and investigate the molecular mechanisms that underlie our observations, we picked three pathways that emerged from our screen and used drugs and genetic mutations to knockout or activate the molecular pathways they control.
Figure 3. An RNAi Screen Identified Multiple Genes That Control mtDNA Disease in the polg-1(srh1) Worms.

(A and B) 20–35 WT (A) and polg-1(srh1) (B) worms were aged for 5 days and spotted inside the red circle. A chemo-attractant was spotted 5 cm away from the worms (green circle), and the worms were allowed to crawl toward the chemo-attractant for 1 hr. Over this time span, most WT worms reached the chemo-attractant, while the polg-1(srh1) worms performed significantly worse.
(C) Twenty 5-day-old polg-1(srh1) adults on RNAi against age-1 perform similar to WT worms.
(D) A list of genes that rescued the polg-1(srh1) neuromuscular defect by >30% when suppressed by RNAi.
The IGF-1/Insulin signaling pathway emerged as the strongest modulator of mtDNA disease, as RNAi against age-1, par-5, and akt-1 all rescued the mobility defect of the polg-1(srh1) worms. In contrast, RNAi against daf-18, which directly opposes AGE-1 (Murphy and Hu, 2013), exacerbated the phenotype of the polg-1(srh1) worms (Table S1). At a mechanistic level, age-1, par-5, and akt-1 share a common goal as well: they all increase the phosphorylation of the transcription factor DAF-16, trapping it in the cytoplasm and suppressing its activity (Murphy and Hu, 2013). RNAi against age-1, par-5, and akt-1 releases this suppression and allows DAF-16 to translocate to the nucleus where it initiates a transcriptional program that promotes organismal health (Kenyon, 2010). Consistent with this idea, we found that RNAi against daf-16 greatly exacerbated the neuromuscular defect of the polg-1(srh1) worms (Table S1). To verify our findings further, we introduced a hypomorph of the IGF-1/insulin receptor (daf-2(e1370)) into the polg-1(srh1) worms. This allele displays reduced IIS activity (Kenyon et al., 1993) and consistent with our RNAi results, we found that it rescued the neuromuscular defect of the mutator worms (Figure 4A). Conversely, a partial deletion of daf-16 (daf-16(mu86)) exacerbated their phenotype (Figure 4B). Thus, these genetic mutations recapitulated the results of our RNAi screen. We further found that the daf-2(e1370) allele also improved the basal respiration rate of the mutator worms (Figure 4C), indicating that reduced IIS activity improves the mobility of the worms by correcting their underlying mitochondrial dysfunction. Interestingly, the daf-2(e1370) allele also improved mtDNA copy number in the mutator worms (Figure 4D), suggesting that it can partially rescue the etiology of the disease itself, although the mutation rate of the polg-1(srh1) worms was unaffected (Figure 4E).
Figure 4. Verification of the RNAi Screen with Genetic Mutants and Small Molecules.

(A) The daf-2(e1370) allele rescues the neuromuscular defect of 5-day-old polg-1(srh1) worms.
(B) The daf-16(mu86) allele worsens the neuromuscular defect of 5-day-old polg-1(srh1) worms. This set of experiments was performed at generation 7 instead of generation 30, when the phenotype of the mutator worms is not significantly different from the WT worms until 7 days of age. Accordingly, the detrimental effect of the daf-16(mu86) allele is better illustrated.
(C) The daf-2(e1370) allele rescues the basal respiration rate of 10-day-old polg-1(srh1) worms.
(D and E) The daf-2(e1370) allele (D) increases mtDNA copy number by 118%, but (E) has no effect on the mutation frequency of polg-1(srh1) worms.
(F) The pdr-1(gk448) allele rescues the chemotaxis defect of 5-day-old polg-1(srh1) worms.
(G) Induction of mitopagy with 50 µM urolithin A (UA) worsens the chemotaxis phenotype of the 5-day-old polg-1(srh1) worms.
(H) The pdr-1(gk448) allele rescues the basal respiration rate of 10-day-old polg-1(srh1) worms.
(I and J) The pdr-1(gk448) allele (I) increases mtDNA copy number by 25% and (J) results in a 5.5-fold increase in the mutation frequency of polg-1(srh1) worms.
(K and L) The atfs-1(et15) allele (K) rescues the chemotaxis defect of 5-day-old polg-1(srh1) worms and (L) the reserve capacity of 5-day-old polg-1(srh1) worms.
(M and N) The atfs-1(et15) allele (M) increases mtDNA copy number by 63%, but (N) has no effect on the mutation frequency of polg-1(srh1) worms.
Bar graphs represent the mean ± SEM of at least three biological replicates. Unpaired t tests were performed to determine significance (*p < 0.05, **p < 0.01; ns, no significant difference).
Our screen further indicated that knockdown of dct-1 and pdr-1 could rescue the mobility defect of the mutator worms. Because these genes play a pivotal role in mitophagy (Palikaras et al., 2015), these results suggest that reduced mitophagy may be beneficial for the polg-1(srh1) worms. To verify these observations with a genetic mutant, we introduced the pdr-1(gk448) allele into the mutator worms. Pdr-1 encodes the C. elegans homolog of the parkin ubiquitin ligase PARK2, and the pdr-1(gk448) allele carries a partial deletion that greatly reduces mitophagy in worms (Springer et al., 2005). Consistent with our RNAi screen, we found that the pdr-1(gk448) allele rescued the neuromuscular defect of the polg-1(srh1) worms (Figure 4F). Conversely, exposure of the mutator worms to urolithin A, a drug that induces mitophagy (Ryu et al., 2016), worsened their phenotype (Figure 4G). Finally, we found that the pdr-1(gk448) allele rescued the basal respiration rate of the mutator worms (Figure 4H), similar to the daf-2(e1370) allele. In contrast to the daf-2(e1370) allele though, we found that the pdr-1(gk448) allele did not alter mtDNA copy number (Figure 4I) and paradoxically increased the mitochondrial mutation frequency by 5.5-fold (Figure 4J).
Finally, our screen indicated that RNAi against the mitochondrial protease spg-7 and several components of the electron transport chain (atp-3, nuo-2, D2030.4, and T02H6.11) rescued the neuromuscular defect of the mutator worms. Knockdown of spg-7 or certain components of the electron transport chain are known to induce the unfolded protein response in mitochondria (Jovaisaite et al., 2014), indicating a possible involvement of this adaptive stress response in the rescue mechanism. Indeed, suppression of UPRmt by RNAi against haf-1, atfs-1, and ubl-5, exacerbated the phenotype of the mutator worms (Table S1). To further verify the protective role of UPRmt in mtDNA disease progression, we introduced the deletion allele atfs-1(tm4919) (Pellegrino and Haynes, 2015) into the polg-1(srh1) background. Atfs-1 encodes a transcription factor required for UPRmt activation. Interestingly, we found that homozygous carriers of the atfs-1(tm4919) allele rendered heterozygous polg-1(srh1) worms infertile, indicating that loss of UPRmt greatly exacerbates the phenotype of the polg-1(srh1) worms. Accordingly, we were unable to generate homozygous polg-1(srh1); atfs-1(tm4919) worms for experiments. To answer the question of whether activation of this adaptive response is able to rescue mtDNA disease, we used a constitutively active atfs-1 allele (atfs-1(et15)) and found that it indeed partially rescued the neuromuscular defect of the mutator worms (Figure 4K). In addition, this allele rescued the decreased reserve capacity of mitochondria (Figure 4L) and slightly increased mtDNA copy number (Figure 4M), although it did not alter the mtDNA mutation frequency (Figure 4N).
DISCUSSION
MtDNA instability is associated with a remarkable number of human diseases. Although it is now possible to manage, and even improve some of the symptoms of mtDNA disease, there is currently no cure for either inherited mtDNA diseases or the mtDNA component of age-related diseases. To address this problem, we generated a model of mtDNA disease in C. elegans, which displays two forms of mtDNA instability known to cause mtDNA disease in humans: mtDNA depletion and mtDNA mutation. Both of these phenotypes are driven by a mutant DNA polymerase γ, which is a major source of mtDNA instability in patients as well. As a result, the polg-1(srh1) worms do not model any specific mtDNA disease, but serve as a general model of mtDNA disease caused by mtDNA instability. The polg-1(srh1) worms are especially useful because they can rapidly be screened for large numbers of genes, drugs, and small molecules that modulate mtDNA disease, which allows them to serve as a motor for discovery. Moreover, any findings can immediately be tested in the mitochondrial mutator mice to determine whether they are translatable to mammalian biology. Another useful feature of the mutator worms is that they rapidly accumulate mtDNA mutations in their germline, which allows for countless additional models of mtDNA disease to be generated. For example, after crossing the error-prone polg-1(srh1) allele out of the nuclear genome to prevent further mutation accumulation, worms with desirable germline mutations could be used to determine which parameters control the inheritance of mutated molecules. A similar approach was recently proposed for the mitochondrial mutator mice (Kauppila et al., 2016). Because mitochondrial research is greatly handicapped by a lack of inherited mutant mtDNA models, these worms would complement the mtDNA models that can be generated by restriction site targeting (Xu et al., 2008) to fill an important void in the research community.
A second problem that prevents the research community from developing a treatment for mtDNA disease is the lack of promising molecules to target. Here, we used the polg-1(srh1) worms to show that numerous molecular pathways control the severity of mtDNA disease. The most promising candidate we have identified thus far is the IGF-1/insulin signaling pathway. It has long been known that reduced IGF-1/insulin signaling has beneficial effects on the overall health of organisms (Kenyon, 2010); however, our results now suggest that for a discrete set of diseases, reduced IIS activity may also have a direct therapeutic application. Since reduced IIS activity increased mtDNA copy number, this approach may be especially useful for diseases caused by mtDNA depletion. Second, it is well known that reduced IGF-1 signaling is particularly beneficial to aging organisms (Kenyon, 2010). Since mtDNA mutations and mitochondrial dysfunction are associated with aging, these findings also provide insight into the molecular mechanisms by which reduced IGF-1 signaling prevents age-related dysfunction. We further found that reduced mitophagy could rescue the polg-1(srh1) worms from mtDNA disease, while increased mitophagy exacerbated their phenotype. This counter-intuitive observation suggests that while increased mitophagy is beneficial under normal circumstances, it can result in an adverse outcome in the context of disease. One potential explanation for this paradox could be that in the mutator worms recycled mitochondria are replaced with equally dysfunctional organelles, leading to futile cycles of mitophagy and mitochondrial biogenesis that results in further energy depletion. Interestingly, loss of PARK2 has both beneficial (reduced splenomegaly) and detrimental (substantia nigra) effects on the mutator mice (Pickrell and Youle, 2015), suggesting that the effect of reduced mitophagy on mammalian biology is highly cell-type and tissue specific. Another surprising observation was that reduced mitophagy increased the mutation burden of the mutator worms. These results suggest that mitophagy can potentially cull mutated mtDNA molecules from cells, which is consistent with the idea that mitophagy recycles dysfunctional organelles. Surprisingly though, knocking out PARK2 in the mitochondrial mutator mice did not seem to result in an increased mutation frequency. The reason for this discrepancy is currently unclear; however, an important difference between these organisms is that the mutator mice carry 10-fold more mutations than the mutator worms, which may limit the ability of mitophagy to significantly impact the mutation burden. Finally, we found that activation of UPRmt could rescue the neuromuscular defect of the polg-1(srh1) worms. Interestingly, it was previously shown that a reduction in mtDNA copy number causes an imbalance in the number of proteins derived from the nuclear and mitochondrial genome. This imbalance causes proteotoxic stress inside mitochondria by preventing proteins from finding their natural binding partner, and UPRmt ameliorates this stress. Since the polg-1(srh1) worms suffer from mtDNA depletion, we hypothesize that a reduction in proteotoxic stress partially underlies the improved performance of the polg-1(srh1); atfs-1(et15) worms.
Taken together, these observations demonstrate that the polg-1(srh1) worms are a useful model to identify modulators of mtDNA disease. We further note that all of the modifiers we analyzed here seem to ameliorate mtDNA disease by improving mitochondrial function, suggesting that they could be beneficial for abroad range of mtDNA diseases. Moreover, since the genetic instability of the mutator worms is substantial, these modifiers are likely to be fairly powerful. It is further interesting to note that despite the fact that few targets are available today, a substantial number of genes can modulate the pathological consequences of mtDNA instability, suggesting that numerous targets may exist for therapeutic treatment of mtDNA disease. One potential conclusion from these observations is that precisely because energy is required for every biological process in our cells, multiple pathways may have evolved to manage or prevent mtDNA disease. Using the polg-1(srh1) worms, the mutator mice, and numerous other models that are still on the horizon, it now finally may be possible to identify these pathways in a reasonable period of time, which holds enormous promise for our ability to understand the molecular basis of mtDNA disease and to develop comprehensive treatment plans for patients.
EXPERIMENTAL PROCEDURES
Strains and Growth Conditions
The polg-1(srh1) worms were created using CRISPR/Cas9 technology in accordance with published protocols (Waaijers et al., 2013) and using the guide RNA sequence UGAUCGAGCCAGAUGUCGGG and the donor DNA sequence atcggagagattggaatggaaaaagtgattattggacataatgttggatttgCccgGgccCgTtgCcgTgaggcttatcaatcgataaatgggtcaaagattcgatttatggatacaatgtct. Capitalized bases differ from the WT polg-1 sequence. These changes were required for changing the aspartic acid at residue 207 to alanine, as well as inhibiting CRISPR/Cas9 cleavage after integration of the donor DNA. Additionally, these changes introduced a SmaI restriction site for genotyping purposes. Like in many other models of mitochondrial dysfunction, we found that the polg-1(srh1) worms displayed pronounced fertility problems when in a homozygous state. Therefore, we maintained the polg-1(srh1) allele as heterozygote animals with the balancer chromosome mnC1 that carries a pharyngeal fluorescent GFP reporter (CGC strain MT20110). Only the non-fluorescent homozygous worms were used for experiments. The daf-2(e1370), daf-16(mu86), pdr-1(gk448), and atfs-1(tm4919) alleles were obtained from CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). The atfs-1(et15) strain was created by Dr. Marc Pilon (University of Gothenburg, Sweden) and was obtained from Dr. Cole Haynes. All strains were backcrossed into the laboratory N2 WT strain at least four times before comparative experiments were carried out. Strains were maintained without starvation at 20°C for at least two generations prior to experimental use. The L4 stage of worms was counted as day 0 of adulthood for all experiments.
mtDNA Copy Number
mtDNA copy number was determined by qPCR using at least three replicates per genotype by adapting a published protocol (Polyak et al., 2012). For each replicate, 10 L4 worms were collected in 10 µL of a buffer containing 10 mM Tris-HCl, 50 mM KCl, 1.5 mM MgCl2, 0.001% gelatin, and 5 mg/mL Proteinase K. The worms were then incubated at 65°C for 75 min, followed by a 20-min incubation at 95°C, and diluted with 10 µL of H2O. Subsequently, 5.5 µL of each sample was used to run a TaqMan assay with a Universal Master Mix (cat. no. 444040, Thermo Fisher Scientific), where ND4 (cat. no. 4440043, assay ID: AIFAT8G) and ACT-4 (assay ID: Ce02508047_s1, Thermo Fisher Scientific) assays quantified mtDNA and nDNA content, respectively. mtDNA copy numbers were then normalized to nDNA content.
Oxygen Consumption
To measure oxygen consumption, we adapted a published protocol (Dancy et al., 2016) and used approximately 300 L4 worms grown on plates to the appropriate age and collected them in M9 media (22 mM KH2PO4, 42 mM Na2HPO4, 86 mM NaCl, and 1 mM MgSO4). The worms were washed 3 times and aliquoted over 3–5 wells of a 24-well plate from Seahorse Biosciences containing 500 µL of M9 media. Images were made of the wells to account for the exact number of worms deposited in each well, and the plates were analyzed with a Seahorse Biosciences XF24 Extracellular Flux Analyzer in accordance with the following program: 10 cycles of 2 min mixing, 2 min resting, and 2 min reading. This method demonstrates a linear increase in oxygen consumption rate (OCR) reading with increasing number of N2 and polg-1(srh1) worms (Figures S1A and S1B). To assess maximal respiration, we injected carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) to a final concentration 25 µM, and ten additional readings were made.
Oxidative State
For each data point, five age-matched worms with the roGFP::orp1 construct were anesthetized on a 3% agarose pad with 10 mM levamisole-HCl, and their heads were imaged by excitation at 488 and 405 nm and emission at 500–600 nm. Confocal images were analyzed using ZEN Black software where the total fluorescence emitted by the excitation of each wavelength was calculated in arbitrary units (Figure S2C). Heads of WT and generation-matched polg-1(srh1) worms without the roGFP::orp1 construct were imaged using the same experimental parameters to ascertain autofluorescence and correct the data (Figure S2D). The 405-nm fluorescence was divided by the fluorescence produced by 488 nm to ascertain the oxidative state of the animal. The average of the oxidative state of all animals in a group is reported. This method was adapted from a published protocol (De Henau et al., 2015). WT animals with the construct were treated with paraquat to ascertain the sensitivity of the construct (Figures S2E and S2F).
Chemotaxis
This protocol has been previously published (Kashyap et al., 2012; Wes and Bargmann, 2001). Briefly, 30 to 50 age-matched worms were collected and washed in M9 buffer. These worms were placed 5 cm away from a spot with 2 µL of 10% isoamyl alcohol in 100% ethanol and 1 µL 10% sodium azide. The worms were then allowed to crawl toward the chemo-attractant for 1 hr, after which the distance between the chemo-attractant and individual worms was noted and measured using ImageJ software. Studies involving urolithin A were performed by placing parents on plates containing 50 µM urolithin A. L4 progeny were collected on the third day and placed onto fresh plates with urolithin A every 2 days prior to chemotaxis on day 7.
Gentle Touch
Appropriately aged worms were gently touched with an eyelash attached to the end of a pick on their head and their tail. These touches were repeated 5 times for a total of ten touches, and any movement in the opposite direction was counted as a response. At least 20 worms were tracked longitudinally over a time span of 10 days, with measurements taken every 2 days (adapted from Hobert et al., 1999).
Thrashing
Thrashing assays were performed by placing 5–6 appropriately aged worms in a 25-µL drop of M9 buffer on a glass slide per movie. They were allowed to acclimate for 1 min, and then a 1 min long time-lapse movie was obtained at 16.7 frames per second. At least five movies using different sets of five individuals were made for each data point. Each movie was processed using the ImageJ plugin “wrmtrck” (Nussbaum-Krammer et al., 2015). The average body bends per second for each movie pertaining to a single genotype and age were averaged. Six to ten replicate movies were assayed per genotype and age.
Lifespan
For each strain, three replicates of at least 30 L4 worms were grown at 20°C and transferred onto fresh a plate every other day. Each day, the worms were observed to identify live and dead animals. Worms that were not visibly moving were gently prodded, observed for a response, and counted as dead if there was no response, after which they were removed from the plate. Worms that escaped or died due to bagging or bursting were omitted from the final analysis.
Mitochondrial Genome Sequencing
Single worms were boiled at 65°C for 75 min and 20 min at 95°C in 10 µL of buffer containing 10 mM Tris-HCl, 50 mM KCl, 1.5 mM MgCl2, 0.001% gelatin, and 5 mg/mL Proteinase K. They were diluted with 10 µL of water, and four 100 µL PCR reactions were carried out per worms using Kapa HiFi polymerase (cat. no. KK2102, Kapa Biosystems) to amplify 11 kb of mitochondrial DNA (Figure S4A). Primer sequences and details for amplification cycling are denoted in Figure S4. The resultant electrophoretograms were scanned for double peaks to indicate heteroplasmic mutations arising in the mtDNA and aligned against WT mtDNA to identify homoplasmic mtDNA mutations.
Random Mutation Capture and Mutation Spectrum
The original protocol (Vermulst et al., 2007) was modified from using DNA isolated from mitochondrial preps to using whole-genome preps. Briefly, we sorted 2,000 homozygous polg-1(srh1) worms and performed whole-genome preps. 10–20 µg of total DNA was digested with TaqαI restriction enzyme (cat. no. R0149T, New England Biolabs) for 10 hr, with 1 µL of fresh enzyme added every hour. Digested DNA was amplified by qPCR or digital droplet PCR using two primer sets to measure total mtDNA copy number and mutant mtDNA copy number. PCR reactions across TaqαI sites that record mutant mtDNA molecules were digested with TaqαI post-PCR to determine that digestion was complete, and mtDNA mutation rates were calculated based on copy number measurements and qPCR efficiency.
Control Primers
Forward: gagcgtcatttattgggaaga
Reverse: aataaagcttgtgctaatcccat
TaqI Primers
Forward: acaccggtgaggtctttggttcat
Reverse: aacctaagccctaggcccaaagtaac
To separate the individual mutations, amplicons were diluted to a single-molecule level and PCR amplified separately and then sequenced.
RNAi
We used RNAi constructs from the Arhinger library and adapted the protocol from the Ahringer laboratory (Kamath and Ahringer, 2003). For RNAi treatments, WT or polg-1(srh1) heterozygote parents were placed on plates spotted with bacteria that had the L4440 (no RNAi) or RNAi construct of interest. On the third day, L4 progeny were placed on fresh L4440 or RNAi plates and transferred to fresh plates on days 2 and 4. If the RNAi caused any developmental defects, then L4 progeny from the L4440 plates were placed on the RNAi plates. On the fifth day of the adulthood, the chemotaxis assay was performed as described above.
Supplementary Material
Highlights.
A genetically engineered worm recapitulates the hallmarks of mtDNA disease in humans
This worm can be exploited to generate countless additional models of mtDNA disease
An RNAi screen identifies 25 genes that can prevent or delay mtDNA disease in worms
IGF-1/insulin signaling, mitophagy, and UPRmt strongly affect disease progression
Acknowledgments
We would like to thank Marni J. Falk, Douglas C. Wallace, and Robert B. Wilson for comments on the manuscript and the project design. This work was supported by grants from the NIH (R01-GM124532 to M.V.; CA204894 and ES026222 to J.H.B.), a grant from NIEHS (T32-ES019851 to C.F.), and a pilot award from UPenn (FP20457 to M.V.). The contents of this publication do not necessarily represent the official views of the NIH.
Footnotes
Supplemental Information includes four figures, one table, and two movies and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.099.
AUTHOR CONTRIBUTIONS
S.H. and M.V. conceived the project. S.H., N.G.E., and J.H.B. performed the mutation frequency analyses. S.H. and J.L.W. determined the mutation spectrum. A.L. performed the oxidative state studies. S.H., A.L., and C.F. carried out the mobility assays. S.H. generated the strains, performed the gentle touch, thrashing, and lifespan studies, and analyzed worm mtDNA over multiple generations. C.M.H., B.P.B., J.A.-F., and T.G. provided worm strains, technical expertise, and analysis tools. S.H. and M.V. wrote the manuscript. All authors contributed to and commented on this manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Bansal A, Zhu LJ, Yen K, Tissenbaum HA. Uncoupling lifespan and healthspan in Caenorhabditis elegans longevity mutants. Proc. Natl. Acad. Sci. USA. 2015;112:E277–E286. doi: 10.1073/pnas.1412192112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland WC. Inherited mitochondrial diseases of DNA replication. Annu. Rev. Med. 2008;59:131–146. doi: 10.1146/annurev.med.59.053006.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancy BM, Brockway N, Ramadasan-Nair R, Yang Y, Sedensky MM, Morgan PG. Glutathione S-transferase mediates an ageing response to mitochondrial dysfunction. Mech. Ageing Dev. 2016;153:14–21. doi: 10.1016/j.mad.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Henau S, Tilleman L, Vangheel M, Luyckx E, Trashin S, Pauwels M, Germani F, Vlaeminck C, Vanfleteren JR, Bert W, et al. A redox signalling globin is essential for reproduction in Caenorhabditis elegans. Nat. Commun. 2015;6:8782. doi: 10.1038/ncomms9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics. 2013;10:186–198. doi: 10.1007/s13311-013-0177-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HF, Beckerle MC. The ALP-enigma protein ALP-1 functions in actin filament organization to promote muscle structural integrity in Caenorhabditis elegans. Mol. Biol. Cell. 2009;20:2361–2370. doi: 10.1091/mbc.E08-06-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Fiorese CJ, Lin YF. Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol. 2013;23:311–318. doi: 10.1016/j.tcb.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O, Moerman DG, Clark KA, Beckerle MC, Ruvkun G. A conserved LIM protein that affects muscular adherens junction integrity and mechanosensory function in Caenorhabditis elegans. J. Cell Biol. 1999;144:45–57. doi: 10.1083/jcb.144.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovaisaite V, Mouchiroud L, Auwerx J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014;217:137–143. doi: 10.1242/jeb.090738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judy ME, Nakamura A, Huang A, Grant H, McCurdy H, Weiberth KF, Gao F, Coppola G, Kenyon C, Kao AW. A shift to organismal stress resistance in programmed cell death mutants. PLoS Genet. 2013;9:e1003714. doi: 10.1371/journal.pgen.1003714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Kashyap L, Perera S, Fisher AL. Identification of novel genes involved in sarcopenia through RNAi screening in Caenorhabditis elegans. J. Gerontol. A Biol. Sci. Med. Sci. 2012;67:56–65. doi: 10.1093/gerona/glr072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppila JHK, Baines HL, Bratic A, Simard ML, Freyer C, Mourier A, Stamp C, Filograna R, Larsson NG, Greaves LC, Stewart JB. A Phenotype-driven approach to generate mouse models with pathogenic mtDNA mutations causing mitochondrial disease. Cell Rep. 2016;16:2980–2990. doi: 10.1016/j.celrep.2016.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lewis SC, Joers P, Willcox S, Griffith JD, Jacobs HT, Hyman BC. A rolling circle replication mechanism produces multimeric lariats of mitochondrial DNA in Caenorhabditis elegans. PLoS Genet. 2015;11:e1004985. doi: 10.1371/journal.pgen.1004985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT, Hu PJ. Insulin/insulin-like growth factor signalling in C. elegans. Eisenmann DM, editor. The C. elegans Research Community. WormBook: The Online Review of C. elegans Biology. 2013 doi: 10.1895/wormbook.1.7.1. https://www.ncbi.nlm.nih.gov/books/NBK179230/ https://doi.org/10.1895/wormbook.1.7.1 . . https://www.ncbi.nlm.nih.gov/books/NBK179230/ [DOI] [PMC free article] [PubMed]
- Nussbaum-Krammer CI, Neto MF, Brielmann RM, Pedersen JS, Morimoto RI. Investigating the spreading and toxicity of prion-like proteins using the metazoan model organism C. elegans. J. Vis. Exp. 2015;95:52321. doi: 10.3791/52321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015;22:1399–1401. doi: 10.1038/cdd.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MW, Haynes CM. Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 2015;13:22. doi: 10.1186/s12915-015-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Ostrovsky J, Kwon YJ, Polyak E, Licata J, Tsukikawa M, Marty E, Thomas J, Felix CA, Xiao R, et al. Inhibiting cytosolic translation and autophagy improves health in mitochondrial disease. Hum. Mol. Genet. 2015;24:4829–4847. doi: 10.1093/hmg/ddv207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak E, Zhang Z, Falk MJ. Molecular profiling of mitochondrial dysfunction in Caenorhabditis elegans. Methods Mol. Biol. 2012;837:241–255. doi: 10.1007/978-1-61779-504-6_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Félix AA, Williams EG, Jha P, Lo Sasso G, Huzard D, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016;22:879–888. doi: 10.1038/nm.4132. [DOI] [PubMed] [Google Scholar]
- Saneto RP, Sedensky MM. Mitochondrial disease in childhood: mtDNA encoded. Neurotherapeutics. 2013;10:199–211. doi: 10.1007/s13311-012-0167-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer W, Hoppe T, Schmidt E, Baumeister R. A Caenorhabditis elegans Parkin mutant with altered solubility couples alpha-synuclein aggregation to proteotoxic stress. Hum. Mol. Genet. 2005;14:3407–3423. doi: 10.1093/hmg/ddi371. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, Loeb LA. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- Waaijers S, Portegijs V, Kerver J, Lemmens BB, Tijsterman M, van den Heuvel S, Boxem M. CRISPR/Cas9-targeted mutagenesis in Caenorhabditis elegans. Genetics. 2013;195:1187–1191. doi: 10.1534/genetics.113.156299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 2010;51:440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- Wanagat J, Cao Z, Pathare P, Aiken JM. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 2001;15:322–332. doi: 10.1096/fj.00-0320com. [DOI] [PubMed] [Google Scholar]
- Wes PD, Bargmann CI. C. elegans odour discrimination requires asymmetric diversity in olfactory neurons. Nature. 2001;410:698–701. doi: 10.1038/35070581. [DOI] [PubMed] [Google Scholar]
- Xu H, DeLuca SZ, O’Farrell PH. Manipulating the metazoan mitochondrial genome with targeted restriction enzymes. Science. 2008;321:575–577. doi: 10.1126/science.1160226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.