

Abstract
Purpose
MDM2 amplification can promote tumorigenesis directly or indirectly through p53 inhibition. MDM2 has increasing clinical relevance because inhibitors are under evaluation in clinical trials, and MDM2 amplification is a possible genomic correlate of accelerated progression, known as hyperprogression, after anti–PD-1/PD-L1 immunotherapy. We used next-generation sequencing (NGS) to ascertain MDM2 amplification status across a large number of diverse cancers.
Methods
We interrogated the molecular profiles of 102,878 patients with diverse malignancies for MDM2 amplification and co-altered genes using clinical-grade NGS (182 to 465 genes).
Results
MDM2 amplification occurred in 3.5% of patients (3,650 of 102,878). The majority of tumor types had a small subset of patients with MDM2 amplification. Most of these patients (99.0% [3,613/3,650]) had co-alterations that accompanied MDM2 amplification. Various pathways, including those related to tyrosine kinase (37.9% [1,385 of 3,650]), PI3K signaling (25.4% [926 of 3,650]), TP53 (24.9% [910 of 3,650]), and MAPK signaling (23.6% [863 of 3,650]), were involved. Although infrequent, mismatch repair genes and PD-L1 amplification also were co-altered (2.2% [79 of 3,650]). Most patients (97.6% [3,563 of 3,650]) had one or more co-alterations potentially targetable with either a Food and Drug Administration–approved or investigational agent. MDM2 amplifications were less frequently associated with high tumor mutation burden compared with the MDM2 wild-type population (2.9% v 6.5%; P < .001). An illustrative patient who harbored MDM2 amplification and experienced hyperprogression with an immune checkpoint inhibitor is presented.
Conclusion
MDM2 amplification was found in 3.5% of 102,878 patients, 97.6% of whom harbored genomic co-alterations that were potentially targetable. This study suggests that a small subset of most tumor types have MDM2 amplification as well as pharmacologically tractable co-alterations.
INTRODUCTION
The MDM2 proto-oncogene encodes a nuclear localized E3 ubiquitin ligase. The core function of MDM2 is to inhibit the tumor suppressor p53, which is critical for regulating genes involved in DNA repair, cell cycle, senescence, and apoptosis. When amplified, MDM2 facilitates proteasomal degradation of p53, which promotes tumorigenesis.1-3 MDM2 amplification has been reported in multiple tumor types4-6 and is a hallmark of tumorigenesis.3 In certain tumor types, such as glioblastoma and well-differentiated liposarcoma, MDM2 amplification and TP53 alterations are mutually exclusive,4,5 which is consistent with the inhibitory function of MDM2. However, in other tumors (eg, osteosarcoma, esophageal cancer), MDM2 amplification and TP53 alterations co-occur.5,6
Preclinical studies have suggested noncanonical p53-independent roles for MDM2. For instance, an in vitro study that used an MDM2-overexpressed/TP53 wild-type cancer cell line revealed a potential role for MDM2 in suppressing senescence in a TP53-independent fashion.7 Moreover, MDM2-amplified/TP53-null mice have a higher incidence of spontaneous tumorigenesis than TP53-null mice (without MDM2 amplification).8 Among several potential noncanonical roles for MDM2, a functional angiogenesis effect has been proposed.9 Indeed, a preclinical study showed that under hypoxic conditions, MDM2-overexpressed/TP53-null cancer cells produce vascular endothelial growth factor (VEGF) mRNA at higher levels than MDM2-negative/TP53-null cells. Hypoxia induces translocation of MDM2 from the nucleus to the cytoplasm, and subsequent binding of the MDM2 C-terminal domain to the 3′untranslated region of VEGF mRNA increases mRNA stability and translation.10 In addition, suppression of MDM2 activity with a small-molecule inhibitor leads to decreased hypoxia-inducible factor 1α and VEGF expression, which support a role for MDM2 in angiogenesis.11 TP53 mutations also lead to increased VEGF-A expression12,13 and have been associated with increased responsiveness to VEGF/VEGF receptor inhibitor therapy.14-16 Taken together, these data suggest that MDM2 promotes angiogenesis through either inhibition of p53 or mechanisms independent of p53.
An understanding of MDM2 amplification status has clinical relevance for patients with cancer because MDM2 inhibitors are in early-phase clinical development (Data Supplemental). Although preliminary results demonstrated no responses to MDM2 inhibitors among unselected patients,17-19 responses occurred in wild-type TP53 liposarcoma (MK-8242; response rate [RR], 11.1% [three of 27 patients]) or melanoma (AMG232; RR, 28.6% [six of 21 patients]).20,21
MDM2 amplification also has been implicated as a potential marker for accelerated tumor growth with receipt of immune checkpoint inhibitors.22 This phenomenon is called hyperprogression and affects approximately 9% of patients who receive PD-1/PD-L1 inhibitors.23 Hyperprogression has been defined as a time to treatment failure < 2 months from checkpoint inhibitor initiation, a > 50% increase in tumor burden compared with pre-immunotherapy imaging, and a more than two-fold increase in progression pace.22 To date, accelerated progression after anti–PD-1/PD-L1 agents has been reported by at least four groups.22-25 Although the mechanisms that mediate this phenomenon remain unclear, we and others have demonstrated that MDM2 family gene amplifications and EGFR alterations correlate with hyperprogression.22,25
Given the clinical importance of MDM2, we describe the landscape of cancer types that harbor MDM2 amplification and evaluate the comprehensive genomic profiles of 102,878 tumors from patients with malignancies. An illustrative patient with MDM2 amplification that demonstrated hyperprogression after checkpoint blockade is presented.
METHODS
Patients
We explored the MDM2 amplification status of patients with diverse malignances who were referred for comprehensive next-generation sequencing (NGS) from June 2012 through December 2016 (N = 102,878; Table 1; Data Supplement). A de-identified database with cancer diagnoses and molecular profiling results was available. This study was performed in accordance with the guidelines of the University of California, San Diego, institutional review board with regard to analysis and consent.
Table 1.
Cancer Characteristics and Their Association With MDM2 Amplification

Tissue Samples and Mutational Analysis
Tumors were provided as formalin-fixed, paraffin-embedded samples and evaluated by NGS in a Clinical Laboratory Improvement Amendments–certified laboratory (Foundation Medicine, Cambridge, MA). The methods used for NGS have been validated and previously reported.26-29 DNA was adaptor ligated, and hybrid capture was performed for all coding exons of 182 to 465 cancer-related genes plus select introns from 14 to 31 genes frequently rearranged in cancer (Data Supplement). For samples in which RNA was available, targeted RNA sequencing was performed for rearrangement analysis in 333 genes (Data Supplement). Sequencing was performed with an average sequencing depth of coverage > 250×, with > 100× at > 99% of exons. Somatic mutations are identified with 99% specificity and > 99% sensitivity for base substitutions at ≥ 5% mutant allele frequency and > 95% sensitivity for copy number alterations. Gene amplification is reported at eight or more copies above ploidy, with six or more copies considered equivocal. The exception is ERRB2 for which five or more copies are considered equivocal amplification.28,29 Only characterized genomic alterations (not variants of unknown significance) were curated for all analyses except those for tumor mutation burden (TMB).
TMB
TMB was calculated on the basis of the total number of mutations counted per megabase.30 Noncoding alterations, alterations reported as known somatic alterations in Catalog of Somatic Mutations in Cancer, truncations in tumor suppressor genes, and alterations predicted to be germline were not counted. High TMB was defined as ≥ 20 mutations/megabase.
Cancer Genomic Data Through Publicly Available Data Sets
MDM2 amplification status was also curated from the Genomics Evidence Neoplasia Information Exchange (GENIE) accessed in July 2017.31-33
End Points, Statistical Methods, and Case Study
Descriptive statistics were used to summarize the cancer diagnoses and genomic alterations identified in the data set. Statistical analyses were carried out using GraphPad Prism 7 software (GraphPad Software, La Jolla, CA). A patient with MDM2 amplification who experienced hyperprogression while receiving an immune checkpoint inhibitor is presented.
RESULTS
Evaluation of MDM2 Amplification Among Diverse Cancers
Among the 102,878 diverse cancers studied, MDM2 amplification was identified in 3,650 (3.5%). MDM2 amplification was most commonly seen among liposarcoma (63.6% [332 of 522]) followed by gallbladder, adenocarcinoma (11.1% [62 of 554]); sarcoma, not otherwise specified (10.7% [103 of 955]); and urothelial carcinoma (10.4% [198 of 1,898]; Table 1). MDM2 amplification was not found among anaplastic and papillary carcinoma of thyroid (zero of 166 and 376, respectively) and uncommonly among adenocarcinoma of the appendix, rectum, and colon (0.23% [one of 440], 0.28% [four of 1,448], and 0.33% [28 of 8,562], respectively; Data Supplement). In comparison, according to the GENIE database (total, 13,473 samples), MDM2 amplification has been reported in 5.5% (744 of 13,473) of diverse cancers, including liposarcoma (69.1% [47 of 68]); gallbladder, adenocarcinoma (17.5% [seven of 40]); sarcoma, not otherwise specified (25.0% [four of 16]); and urothelial carcinoma (10.7% [48 of 446]; Fig 1).
Fig 1.

Comparison of rate of MDM2 amplification in the current report (N = 102,878 samples) versus in Genomics Evidence Neoplasia Information Exchange (GENIE; N = 13,473 samples).31 Data from GENIE were obtained as previously described.31 The 10 most common diagnoses that harbor MDM2 amplification from the current report were selected for the comparison. MDM2 amplifications were seen in 3.5% (3,650 of 102,878) of patients in the current report versus 5.5% (744 of 13,473) from GENIE. MDM2 amplification was most commonly seen in patients with liposarcoma (63.6% [332 of 522] in the current report and 69.1% [47 of 68] from GENIE); gallbladder, adenocarcinoma (11.1% [62 of 554] in the current report and 17.5% [seven of 40] from GENIE); sarcoma, not otherwise specified (10.7% [103 of 955] in the current report and 25.0% [four of 16] from GENIE); urothelial carcinoma (10.4% [198 of 1,898] in the current report and 10.7% [48 of 446] from GENIE); and lung, adenosquamous carcinoma (9.8% [17 of 173] in the current report and 9.0% [one of 11] from GENIE).
Genomic Co-Alterations Associated With MDM2 Amplification
Among 3,650 patients with MDM2 amplification, 99.0% (3,613) were found to have genomic co-alterations (Data Supplement). Frequently co-altered genes were CDK4 (43.6% [1,591 of 3,650]), FRS2 (40.8% [1,491 of 3,650]), TP53 (20.1% [733 of 3,650]), and CDKN2A (18.2% [665 of 3,650]; Data Supplement). In contrast, among patients with wild-type MDM2, CDK4 and FRS2 alterations were rare compared with those with MDM2 amplification (1.2% and 0.20%, respectively; both P < .001). TP53 alterations were more common in patients with MDM2 wild type (53.6%; P < .001; Data Supplement). When co-alterations are grouped into specific pathways, cell cycle–associated genes were most commonly co-altered (68.5% [2,502 of 3,650]) followed by tyrosine kinase–associated genes (37.9% [1,385 of 3,650]), PI3K signaling–associated genes (25.4% [926 of 3,650]), TP53-associated genes (24.9% [910 of 3,650]), and MAPK signaling–associated genes (23.6% [863 of 3,650]). Although uncommon, mismatch repair genes and PD-L1 amplification were co-altered in 2.2% (79 of 3,650) of patients (Table 2).
Table 2.
Selected Co-Alterations That Accompany MDM2 Amplification and Potential Targeted Therapies

Potential Cognate-Targeted Therapies for Genomic Co-Alterations Associated With MDM2 Amplification
Among 3,650 patients with MDM2 amplification, the median number of alterations per patient was six (range, one to 25); 96.9% (3,536) had one or more co-alterations targetable with a Food and Drug Administration–approved agent (either on or off label). An additional 0.7% (27 of 3,650) of patients had one or more co-alterations targetable with investigational agents. Altogether, 97.6% (3,563 of 3,650) of patients had genomic co-alterations actionable with either a Food and Drug Administration–approved or investigational agent; the median number of potentially targetable genomic co-alterations was three (range, zero to 17; Data Supplement).
Association of MDM2 Amplification and Mutational Burden
Among diverse cancers (N = 102,878), high TMB status was significantly less frequent in patients with MDM2 amplification than in the MDM2 wild-type population (2.9% [105 of 3,650] v 6.5% [6,492 of 99,228], respectively; P < .001). A similar difference also was observed in the GENIE data set (frequency of high TMB among MDM2 amplification v MDM2 wild type, 3.4% [25 of 744] v 5.6% [714 of 12,729], respectively; P = .008).
In the current data set, subsets of cancer with MDM2 amplification also were associated with less-frequent high TMB status compared with MDM2 wild type (sarcoma, not otherwise specified, 0% [zero of 103] v 4.2% [36 of 852], respectively [P = .03]; urothelial carcinoma, 3.5% [seven of 198] v 11.8% [200 of 1,700], respectively [P < .001]; glioblastoma, 0.4% [one of 244] v 4.4% [120 of 2,725], respectively [P < .001]; Fig 2; Data Supplement). These differences in patient subsets were not seen in the GENIE data set perhaps because GENIE had considerably fewer patients (approximately 13% of the patients in our data set).
Fig 2.
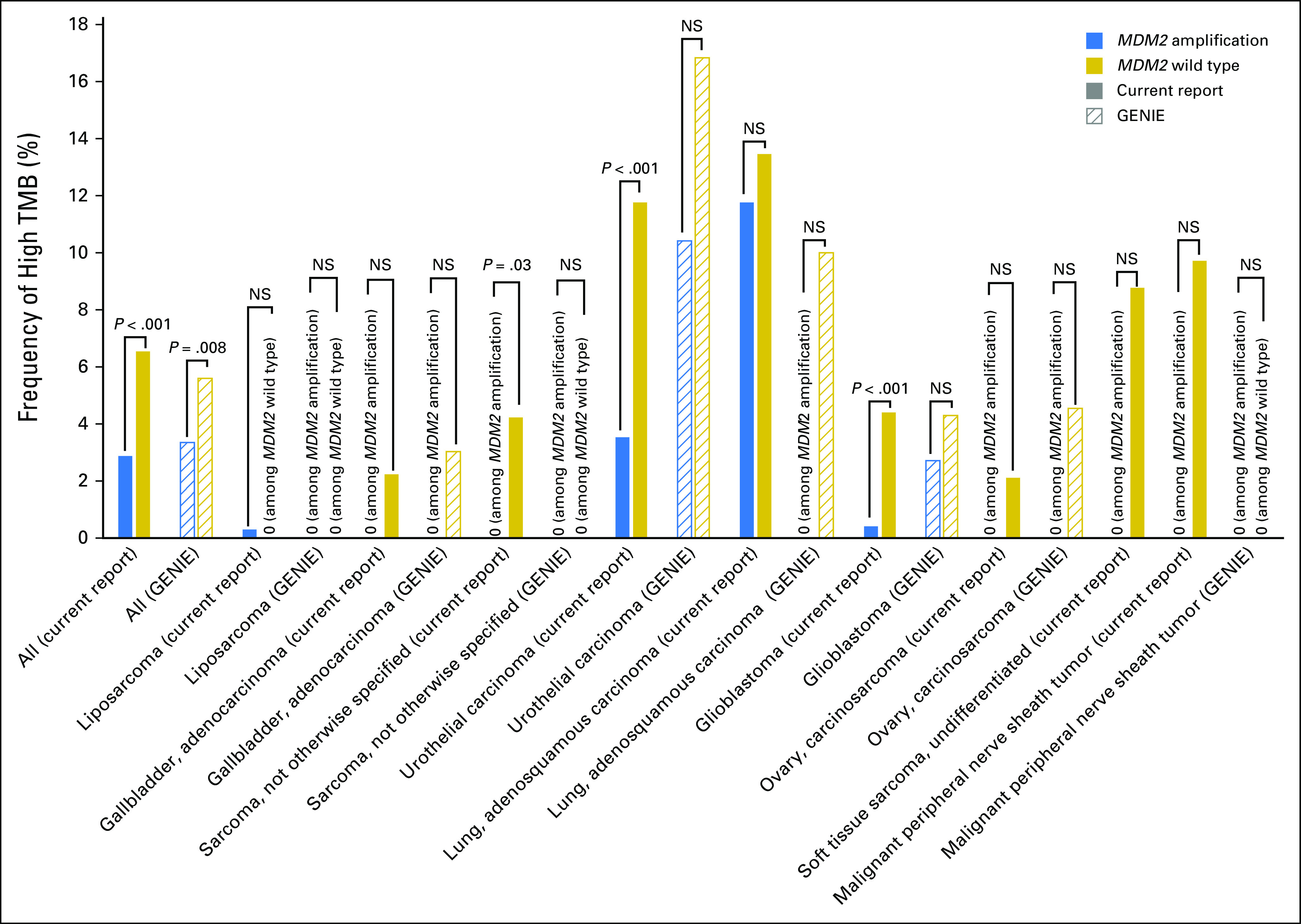
Association between MDM2 amplification and tumor mutation burden (TMB). Among patients with MDM2 amplification (n = 3,650), 2.9% (105) had high TMB, 23.3% (852) had intermediate TMB, and 73.8% (2,693) had low TMB. Among diverse cancers (N = 102,878), high TMB status was significantly less frequent in patients with MDM2 amplification than in those with MDM2 wild type (2.9% [105 of 3,650] v 6.5% [6,492 of 99,228]; P < .001). The Genomics Evidence Neoplasia Information Exchange (GENIE) data set also showed a similar observation (frequency of high TMB among MDM2 amplification v MDM2 wild type, 3.4% [25 of 744] v 5.6% [714 of 12,729], respectively; P = .008). In the current data set, certain cancers with MDM2 amplification were significantly less associated with high TMB than with MDM2 wild type (sarcoma, not otherwise specified, 0% [zero of 103] v 4.2% [36 of 852], respectively [P = .03]; urothelial carcinoma, 3.5% [seven of 198] v 11.8% [200 of 1,700], respectively [P < .001]; glioblastoma, 0.4% [one of 244] v 4.4% [120 of 2,725], respectively [P < .001]); these subsets did not show significant differences in GENIE, but the number of patient samples in GENIE was considerably smaller (Data Supplement). NS, not significant.
Among patients with MDM2 amplification (n = 3,650), TP53 was co-altered in 733. Most patients with MDM2 amplification and wild-type TP53 had low TMB (98.4% [2,817 of 2,917]); among patients who harbored both MDM2 amplification and TP53 alteration, 55.3% (405 of 733) had low TMB (P < .001).
MDM2 Amplification as a Potential Marker for Hyperprogression With Immune Checkpoint Inhibitors
We have previously reported that MDM2 amplification can be associated with hyperprogression after treatment with anti–PD-1/PD-L1 agents.22 We describe herein a 36-year-old woman (not previously reported) with adenocarcinoma of the gastro-esophageal junction who had stable disease (SD) while receiving second-line therapy with fluorouracil, oxaliplatin, and panitumumab (Fig 3, left and middle). For persistent, subcentimeter lymphadenopathy, the regimen was switched to nivolumab (anti–PD-1 inhibitor). The patient had rapid progression in the mediastinal and retroperitoneal lymph nodes as well as emergent massive ascites (time to treatment failure, 3 weeks; pace of progression increased by 6.4-fold compared with the 4 months before the start of checkpoint blockade, and tumor burden increased 460% compared with pre-immunotherapy imaging; Fig 3). The patient succumbed to disease 1.5 months after nivolumab administration. Molecular profiling of the primary tumor revealed alterations, including amplifications in MDM2, ERBB3, ARAF, CDK4, and EGFR and alterations in PIK3CA, FRS2, GLI1, and IKZF1. TMB was low and microsatellite stable. PD-1 and PD-L1 immunohistochemistry was not evaluated.
Fig 3.

Hyperprogression in a patient with MDM2 amplification treated with an anti–PD-1 checkpoint inhibitor.22 A 36-year-old woman presented with worsening dysphagia and anemia. Additional work-up revealed adenocarcinoma of the gastro-esophageal junction, stage IIIC. The patient was initially started on combination chemotherapy with epirubicin, oxaliplatin, and capecitabine with persistent lymphadenopathy. Therapy was switched to fluorouracil, oxaliplatin, and panitumumab with overall stable disease; however, the patient had persistent subcentimeter lymphadenopathy (left and middle). The regimen was then switched to nivolumab (anti–PD-1 inhibitor). Within 3 weeks, the patient showed marked clinical deterioration, and imaging showed rapid progression in mediastinal and retroperitoneal lymph nodes as well as emerging massive ascites (right). Pace of progression increased by 6.4-fold, tumor burden increased by 460% compared with pre-immunotherapy imaging, and time to treatment failure was 3 weeks (hyperprogression after immunotherapy previously defined as more than a two-fold increase in progression pace, a > 50% increase in tumor burden compared with pre-immunotherapy imaging, and a time to treatment failure < 2 months22). Therapy was then changed to fluorouracil, oxaliplatin, and trastuzumab, but the patient died 1.5 months after nivolumab was administered. Molecular profiling of the primary tumor revealed multiple alterations, including MDM2 amplification. Other alterations were ERBB3, ARAF, CDK4, and EGFR amplifications and alterations in PIK3CA, FRS2, GLI1, and IKZF1. Tumor mutation burden was low and microsatellite stable. PD-1 and PD-L1 status by immunohistochemistry were not evaluated.
DISCUSSION
We and others recently demonstrated that approximately 9% of patients treated with PD-1/PD-L1 checkpoint blockade exhibit a paradoxical acceleration in tumor progression (designated as hyperprogression). This phenomenon associates with MDM2 amplification.22-24 Therefore, caution is needed in treating patients who harbor MDM2 amplification with checkpoint inhibitors, and a thorough understanding of the MDM2 alteration landscape is clinically important. Therefore, we describe the genomic backdrop of MDM2 amplification among 102,878 patients with diverse malignancies. Overall, MDM2 amplification was found in 3.5% (3,650 of 102,878) of cancers (a number similar to that in the GENIE database [5.5% (744 of 13,473)]; Fig 1). MDM2 amplification most commonly has been seen in patients with liposarcoma (63.6% [332 of 522])5 but discerned in a subset of most tumor types, albeit at different frequencies (Table 1; Data Supplement). Certain diagnoses (eg, anaplastic and papillary thyroid cancer) were not associated with MDM2 amplification, and this anomaly was rare in acute myelocytic leukemia (one of 1,006 patients; Data Supplement).
An understanding of the comprehensive landscape of MDM2 amplification also is therapeutically relevant because MDM2 inhibitors are in clinical development (Data Supplement). Clinical activity of MDM2 inhibitors among unselected diverse cancers has been limited17-19; however, occasional responses have been observed in individuals selected for wild-type TP53.20,21 The low response rate with single-agent MDM2 inhibitors may be due to the lack of patient selection for MDM2 amplification or to co-altered genes (Data Supplement). Accumulating evidence has suggested that the matched targeted therapy approach can demonstrate better clinical outcomes than a nonmatched approach, but this implies the need to select patients for the relevant aberration.34-37 Wagner et al21 showed that responses with MK-8242 (MDM2 inhibitor) were exclusively observed in patients with liposarcoma (RR, 11.1% [three or 27]) whose molecular hallmark includes MDM2 amplification5 (nonliposarcoma; RR, 0% [zero of 14]). On the other hand, even in a disease such as liposarcoma where > 60% of patients have MDM2 amplification, the RR is relatively low, which may be due to, as mentioned previously, the presence of co-alterations. Indeed, the 12q13-15 amplicon on which MDM2 resides is large (but discontinuous); CDK4 and FRS2 reside on the amplicon and frequently are co-amplified with MDM238 but are rarely abnormal in patients without MDM2 amplification (Data Supplement).
In keeping with the notion that co-alterations are important, we also assessed the alterations that co-occurred with MDM2 amplification. The majority of MDM2-amplified tumors harbored co-alterations (99% [3,613 of 3,650]); the median number of alterations per patient was six (range, one to 25; Data Supplement). The most common co-alterations were indeed CDK4 and FRS2 amplification (Fig 4); therefore, the targeting of MDM2 amplification alone may be insufficient to achieve satisfactory antitumor activity (Data Supplement). Additional clinical trials that investigate the feasibility and efficacy of matched targeted combination strategies are required. Because FRS2 and CDK4 are on the MDM2 amplicon, the targeting of them may warrant specific study.
Fig 4.

Genomic co-alterations associated with MDM2 amplification (n = 3,650). The most common co-alterations associated with MDM2 amplification were CDK4 (43.6% [1,591 of 3,650]), FRS2 (40.8% [1,491 of 3,650]), TP53 (20.1% [733 of 3,650]), CDKN2A (18.2% [665 of 3,650]), and EGFR (12.7% [462 of 3,650]; Data Supplement).
Of note, we also observed that TP53 alterations were not mutually exclusive with MDM2 amplification, as previously reported.5,6 Although TP53 alterations were less commonly seen in patients with MDM2 amplification compared with wild type (Data Supplement), TP53 alterations were observed in 20.1% (733 of 3,650; Fig 2; Data Supplement). Because MDM2 amplification suppresses the function of p53, co-alteration of TP53 with MDM2 amplification suggests a noncanonical, p53-independent role for MDM2 in tumorigenesis. One of the proposed noncanonical roles of MDM2 is to facilitate angiogenesis.39 Zhou et al10 reported that MDM2-overexpressed/TP53-null cancer cells are associated with increased VEGF mRNA expression compared with MDM2-negative/TP53-null cells. Furthermore, Lakoma et al11 showed that pharmacologic inhibition of MDM2 is associated with a decrease in hypoxia-inducible factor 1α and VEGF expression in cancer cell lines. TP53 alterations also have been reported to be associated with increased VEGF expression in preclinical models as well as in patients with lung adenocarcinoma.12,13 Clinically, patients with cancer with TP53 alterations have been shown to experience longer progression-free survival (PFS) and a higher rate of SD of ≥ 6 months/partial and complete remission with bevacizumab (anti-VEGF antibody)–containing regimens compared with non-bevacizumab–containing regimens (median PFS, 11.0 v 4.0 months [P < .001]; SD ≥ 6 months/partial and complete remission, 31% v 7% [P ≤ 0.01]).15,16 TP53 alteration status also predicted longer PFS among patients with sarcoma treated with pazopanib (multikinase inhibitor that targets the VEGF receptor; hazard ratio, 0.38; P = .036).14 Thus, the harboring of either MDM2 amplification or TP53 alteration may lead to enhanced angiogenesis, which may be susceptible to anti-VEGF therapies. Additional investigation is warranted.
Mismatch repair genes and PD-L1 amplification were co-altered in 2.2% (79 of 3,650) of the patients with MDM2 amplification (Table 2). Tumors with mismatch repair deficiency or PD-L1 amplification have been associated with remarkable responses to immune checkpoint inhibitors.40-42 On the other hand, we have previously reported that MDM2 amplification and EGFR alterations (both of which were discerned in the current patient example) were significantly associated with hyperprogression when anti–PD-1/PD-L1 agents were used.22 In this prior report, all four patients with hyperprogression and available data had negative PD-L1 expression; the one patient with available data had high TMB. In the current study, we depict an individual with gastric cancer that harbored EGFR as well as MDM2 amplification who had indolent disease; the patient, however, showed explosive progression after being given the anti–PD-1 inhibitor nivolumab (Fig 4). Whether patients who have both PD-L1 amplification (a marker of sensitivity to checkpoint inhibitors in Hodgkin disease43) and MDM2 amplification would respond to checkpoint blockade is unclear. Of note, tumors that harbor MDM2 amplification had significantly lower rates of high TMB than MDM2 wild-type tumors (2.9% [105 of 3,650] v 6.5% [6,492 of 99,228]; P < .001). Because high TMB correlates with checkpoint blockade responsiveness,44,45 this observation may partially explain resistance to PD-1/PD-L1 inhibitors in MDM2-amplified tumors but does not clarify the mechanism that underlies the correlation between MDM2 amplification and hyperprogression. Furthermore, how patients whose tumors have high TMB as well as MDM2 amplification would fare on checkpoint inhibitors is unclear; however, one of our previously reported patients with MDM2 amplification who demonstrated hyperprogression with anti–PD-L1 immunotherapy had a high TMB.22 Finally, an issue that merits prospective exploration is how a combination of MDM2 and checkpoint inhibitors would affect the risk of hyperprogression in patients whose cancers bear an MDM2 amplification.
The current study has several limitations. First, the data set was de-identified, which limited the analyzable correlates; thus, clinical questions such as the frequencies of MDM2 amplification that depend on the disease state (early stage v metastatic and recurrent disease) could not be evaluated. Second, because the number of patients in each cancer type was based on the number of samples sent for NGS by the treating physicians, sample size bias is possible. Finally, the cancer diagnosis was annotated on the basis of the submitting physician’s description. Despite these limitations, the current study provides, to our knowledge, the largest and most comprehensive analysis of MDM2 amplification in diverse malignancies to date.
In summary, we have interrogated 102,878 patients with diverse cancers and demonstrated that amplification of MDM2 is found in 3.5% (3,650) of tumors. The majority of cancer types included a subgroup of patients, albeit small, with MDM2 amplification. Most patients (99.0% [3,613 of 3,650]) harbored co-alterations with MDM2 amplification (97.6% potentially targetable). Although infrequent, mismatch repair genes and PD-L1 amplification also were co-altered in 2.2% (79 of 3,650) of patients. In addition, high TMB was significantly less common among patients with MDM2 amplification. This study suggests that MDM2 amplification is found in a subset of most cancer diagnoses and that optimization of targeted therapy against MDM2 and immunotherapy might require relevant combinations of drugs.
Footnotes
Supported by the Joan and Irwin Jacobs fund and by National Cancer Institute Grant No. P30 CA016672.
AUTHOR CONTRIBUTIONS
Conception and design: Shumei Kato, Razelle Kurzrock
Financial support: Razelle Kurzrock
Administrative support: Razelle Kurzrock
Provision of study material or patients: Jeffrey S. Ross, Farshid Dayyani, Vivek Subbiah
Collection and assembly of data: Shumei Kato, Jeffrey S. Ross, Laurie Gay, Farshid Dayyani, Vivek Subbiah
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Shumei Kato
No relationship to disclose
Jeffrey S. Ross
Employment: Foundation Medicine
Leadership: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Research Funding: Foundation Medicine
Laurie Gay
Employment: Foundation Medicine
Stock and Other Ownership Interests: Gilead Sciences, Foundation Medicine
Farshid Dayyani
Employment: Roche (I)
Consulting or Advisory Role: Genentech, Roche
Speakers’ Bureau: Genentech, Roche, Amgen, Bayer AG
Jason Roszik
No relationship to disclose
Vivek Subbiah
Consulting or Advisory Role: MedImmune
Research Funding: Novartis (Inst), GlaxoSmithKline (Inst), NanoCarrier (Inst), Northwest Biotherapeutics (Inst), Genentech (Inst), Roche (Inst), BERG (Inst), Bayer AG (Inst), Incyte (Inst), Fujifilm (Inst), PharmaMar (Inst), D3 Oncology Solutions (Inst), Pfizer (Inst), Amgen (Inst), AbbVie (Inst), MultiVir (Inst), Blueprint Medicines (Inst), Loxo Oncology (Inst), Vegenics (Inst), Takeda Pharmaceuticals (Inst), Alpha Sigma (Inst), Agensys (Inst), Idera (Inst), Boston Biomedical (Inst)
Travel, Accommodations, Expenses: PharmaMar, Bayer AG
Razelle Kurzrock
Leadership: CureMatch
Stock and Other Ownership Interests: CureMatch
Honoraria: Cedars-Sinai, Jubilant Biosys, National Comprehensive Cancer Network, Janssen Pharmaceuticals, The Dedham Group, The Frankel Group, Segal Cancer Centre, Sequenom, American Association for Cancer Research, Global Biomarkers Consortium, UCSD Genomics Program Talk, Casdin Capital, NXT Health Strategies, Seminars in Oncology, Actuate Therapeutics, Yale Cancer Center, Northwestern University, XBiotech, Pancreatic Cancer Action Network, Sylvester Comprehensive Cancer Center, Mayo Clinic Cancer Center, Kaiser Permanente, Health Advances, Wiley, Scripps Translational Research Institute, Defined Health, CMEducation Resource, Avera Health, Roche, Loxo Oncology
Consulting or Advisory Role: Actuate Therapeutics, Loxo Oncology, Sequenom
Speakers’ Bureau: Roche
Research Funding: Incyte, Genentech, Merck Serono, Pfizer, Sequenom, Foundation Medicine, Guardant Health
Patents, Royalties, Other Intellectual Property: Patent
Travel, Accommodations, Expenses: EMD Serono, Gateway Research Advisory Committee, International Commission on Radiological Protection, Caris Life Sciences, American Association for Cancer Research, Quintiles, The Biomarkers Consortium, Association of American Cancer Institutes, Guardant Health, Global Source Ventures, Novena Therapeutics, American Society of Clinical Pharmacology & Therapeutics, Meyers Consulting, Food and Drug Administration Orange County Regulatory Affairs, Guardant Health, Genentech, OrbiMed, Sylvester Comprehensive Cancer Center, Journal of Precision Medicine, CureMatch, Lynx Group, Mayo Clinic Cancer Center, Kaiser Permanente, Pancreatic Cancer Action Network, Cedars-Sinai, MedImmune, JK Associates, ILANIT, Defined Health, Avera Health, Roche, XBiotech, Loxo Oncology, Southwest Oncology Group
REFERENCES
- 1.Chène P. Inhibiting the p53-MDM2 interaction: An important target for cancer therapy. Nat Rev Cancer. 2003;3:102–109. doi: 10.1038/nrc991. [DOI] [PubMed] [Google Scholar]
- 2.Li Q, Lozano G. Molecular pathways: Targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res. 2013;19:34–41. doi: 10.1158/1078-0432.CCR-12-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. Erratum: Nature 494:506, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ito M, Barys L, O’Reilly T, et al. Comprehensive mapping of p53 pathway alterations reveals an apparent role for both SNP309 and MDM2 amplification in sarcomagenesis. Clin Cancer Res. 2011;17:416–426. doi: 10.1158/1078-0432.CCR-10-2050. [DOI] [PubMed] [Google Scholar]
- 6.Shibagaki I, Tanaka H, Shimada Y, et al. p53 mutation, murine double minute 2 amplification, and human papillomavirus infection are frequently involved but not associated with each other in esophageal squamous cell carcinoma. Clin Cancer Res. 1995;1:769–773. [PubMed] [Google Scholar]
- 7.Kovatcheva M, Liu DD, Dickson MA, et al. MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget. 2015;6:8226–8243. doi: 10.18632/oncotarget.3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones SN, Hancock AR, Vogel H, et al. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc Natl Acad Sci U S A. 1998;95:15608–15612. doi: 10.1073/pnas.95.26.15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urso L, Calabrese F, Favaretto A, et al. Critical review about MDM2 in cancer: Possible role in malignant mesothelioma and implications for treatment. Crit Rev Oncol Hematol. 2016;97:220–230. doi: 10.1016/j.critrevonc.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 10.Zhou S, Gu L, He J, et al. MDM2 regulates vascular endothelial growth factor mRNA stabilization in hypoxia. Mol Cell Biol. 2011;31:4928–4937. doi: 10.1128/MCB.06085-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lakoma A, Barbieri E, Agarwal S, et al. The MDM2 small-molecule inhibitor RG7388 leads to potent tumor inhibition in p53 wild-type neuroblastoma. Cell Death Discov. 2015;1:15026. doi: 10.1038/cddiscovery.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ravi R, Mookerjee B, Bhujwalla ZM, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 13.Schwaederlé M, Lazar V, Validire P, et al. VEGF-A expression correlates with TP53 mutations in non-small cell lung cancer: Implications for antiangiogenesis therapy. Cancer Res. 2015;75:1187–1190. doi: 10.1158/0008-5472.CAN-14-2305. [DOI] [PubMed] [Google Scholar]
- 14.Koehler K, Liebner D, Chen JL. TP53 mutational status is predictive of pazopanib response in advanced sarcomas. Ann Oncol. 2016;27:539–543. doi: 10.1093/annonc/mdv598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Said R, Hong DS, Warneke CL, et al. P53 mutations in advanced cancers: Clinical characteristics, outcomes, and correlation between progression-free survival and bevacizumab-containing therapy. Oncotarget. 2013;4:705–714. doi: 10.18632/oncotarget.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wheler JJ, Janku F, Naing A, et al. TP53 alterations correlate with response to VEGF/VEGFR inhibitors: Implications for targeted therapeutics. Mol Cancer Ther. 2016;15:2475–2485. doi: 10.1158/1535-7163.MCT-16-0196. [DOI] [PubMed] [Google Scholar]
- 17.de Jonge M, de Weger VA, Dickson MA, et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur J Cancer. 2017;76:144–151. doi: 10.1016/j.ejca.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Gounder MM, Bauer TM, Schwartz GK, et al. A phase 1 study of the MDM2 inhibitor DS-3032b in patients (pts) with advanced solid tumors and lymphomas. J Clin Oncol. 2016;34(suppl; abstr 2581) [Google Scholar]
- 19.Kurzrock R, Blay J-Y, Bui Nguyen B, et al. A phase I study of MDM2 antagonist RG7112 in patients (pts) with relapsed/refractory solid tumors. J Clin Oncol. 2012;30(suppl; e13600) [Google Scholar]
- 20.Moschos SJ, Sandhu SK, Lewis KD, et al. Phase 1 study of the p53-MDM2 inhibitor AMG 232 combined with trametinib plus dabrafenib or trametinib in patients (Pts) with TP53 wild type (TP53WT) metastatic cutaneous melanoma (MCM) J Clin Oncol. 2017;35(suppl; abstr 2575) [Google Scholar]
- 21.Wagner AJ, Banerji U, Mahipal A, et al. Phase I trial of the human double minute 2 inhibitor MK-8242 in patients with advanced solid tumors. J Clin Oncol. 2017;35:1304–1311. doi: 10.1200/JCO.2016.70.7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato S, Goodman A, Walavalkar V, et al. Hyperprogressors after immunotherapy: Analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23:4242–4250. doi: 10.1158/1078-0432.CCR-16-3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Champiat S, Dercle L, Ammari S, et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res. 2017;23:1920–1928. doi: 10.1158/1078-0432.CCR-16-1741. [DOI] [PubMed] [Google Scholar]
- 24.Saâda-Bouzid E, Defaucheux C, Karabajakian A, et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol. 2017;28:1605–1611. doi: 10.1093/annonc/mdx178. [DOI] [PubMed] [Google Scholar]
- 25.Singavi AK, Menon S, Kilari D, et al. Predictive biomarkers for hyper-progression (HP) in response to immune checkpoint inhibitors (ICI) – Analysis of somatic alterations (SAs) Ann Oncol. 2017;28:v403–v427. [Google Scholar]
- 26.Wagle N, Berger MF, Davis MJ, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2:82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas RK, Nickerson E, Simons JF, et al. Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat Med. 2006;12:852–855. doi: 10.1038/nm1437. Erratum: Nat Med 12:1120, 2006. [DOI] [PubMed] [Google Scholar]
- 28.He J, Abdel-Wahab O, Nahas MK, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004–3014. doi: 10.1182/blood-2015-08-664649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.AACR Project GENIE Consortium AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017;7:818–831. doi: 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roszik J, Haydu LE, Hess KR, et al. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med. 2016;14:168. doi: 10.1186/s12916-016-0705-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.American Association for Cancer Research AACR Project GENIE. http://www.aacr.org/Research/Research/Pages/aacr-project-genie.aspx#.WZSaaD6GOUk
- 34.Jardim DL, Schwaederle M, Wei C, et al. Impact of a biomarker-based strategy on oncology drug development: A meta-analysis of clinical trials leading to FDA approval. J Natl Cancer Inst. 2015;107:djv253. doi: 10.1093/jnci/djv253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwaederle M, Zhao M, Lee JJ, et al. Impact of precision medicine in diverse cancers: A meta-analysis of phase II clinical trials. J Clin Oncol. 2015;33:3817–3825. doi: 10.1200/JCO.2015.61.5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwaederle M, Zhao M, Lee JJ, et al. Association of biomarker-based treatment strategies with response rates and progression-free survival in refractory malignant neoplasms: A meta-analysis. JAMA Oncol. 2016;2:1452–1459. doi: 10.1001/jamaoncol.2016.2129. [DOI] [PubMed] [Google Scholar]
- 37.Tsimberidou AM, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin Cancer Res. 2012;18:6373–6383. doi: 10.1158/1078-0432.CCR-12-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mejia-Guerrero S, Quejada M, Gokgoz N, et al. Characterization of the 12q15 MDM2 and 12q13-14 CDK4 amplicons and clinical correlations in osteosarcoma. Genes Chromosomes Cancer. 2010;49:518–525. doi: 10.1002/gcc.20761. [DOI] [PubMed] [Google Scholar]
- 39.Kim ES, Shohet JM. Reactivation of p53 via MDM2 inhibition. Cell Death Dis. 2015;6:e1936. doi: 10.1038/cddis.2015.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847–856. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 43.Armand P, Shipp MA, Ribrag V, et al. Programmed death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after brentuximab vedotin failure. J Clin Oncol. 2016;34:3733–3739. doi: 10.1200/JCO.2016.67.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598–2608. doi: 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mouw KW, Goldberg MS, Konstantinopoulos PA, et al. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7:675–693. doi: 10.1158/2159-8290.CD-17-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]