

Abstract
Recent years have seen a large increase in the discovery of allosteric ligands targeting muscarinic acetylcholine receptors (mAChRs). One of the challenges in screening such compounds is to understand their mechanisms of action and define appropriate parameter estimates for affinity, cooperativity and efficacy. Herein we describe the mechanisms of action and structure—activity relationships for a series of “pan-Gq-coupled” muscarinic acetylcholine (ACh) receptor (mAChR) positive allosteric modulators (PAMs). Using a combination of radioligand binding, functional inositol phosphate accumulation assays, receptor alkylation and operational data analysis, we show that most compounds in the series derive their variable potency and selectivity from differential cooperativity at the M1, M3 and M5 mAChRs. None of the PAMs showed greater than 10-fold subtype selectivity for the agonist-free receptor, but VU6007705, VU6007678, and VU6008555 displayed markedly increased cooperativity compared to the parent molecule and M5 mAChR-preferring PAM, ML380 (αβ > 100), in the presence of ACh. Most of the activity of these PAMs derives from their ability to potentiate ACh binding affinity at mAChRs, though VU6007678 was notable for also potentiating ACh signaling efficacy and robust allosteric agonist activity. These data provide key insights for the future design of more potent and subtype-selective mAChR PAMs.
Keywords: Muscarinic acetylcholine receptor, positive allosteric modulator, cooperativity, affinity, efficacy, operational model
Graphical abstract
INTRODUCTION
Muscarinic acetylcholine receptors (mAChRs) are Class A GPCRs that consist of five subtypes, denoted M1—M5. The M1, M3, and M5 mAChR subtypes primarily couple to Gq/11 proteins, which leads to the activation of phospholipase C and facilitates the release of calcium, while the M2 and M4 mAChR subtypes couple primarily to Gi/o proteins and inhibit adenylyl cyclase activity.1,2 The mAChRs are widely expressed within both the central nervous system (CNS) and peripheral tissues and have a key role in many physiological processes.3,4 Indeed, mAChRs have been implicated in cognition, motor control, attention, reward and motivation, appetite, muscle contractility, regulation of heart rate, thermoregulation and other functions.3–9 As a consequence, these receptors have emerged as interesting drug targets for treating a wide-range of CNS and peripheral disorders including schizophrenia, Alzheimer’s disease, drug addiction, asthma, and chronic obstructive pulmonary disease (COPD).2,4
Efforts to develop subtype selective mAChR therapeutics have historically focused on ligands that engage the highly conserved orthosteric (ACh) binding sites on these proteins.2,3,10,11 However, the high degree of sequence conservation makes identifying truly subtype selective ligands for individual mAChRs very challenging. Furthermore, directly targeting mAChRs with orthosteric agonists or antagonists increases the likelihood of on-target side effects by disruption of the spatiotemporal aspect of endogenous receptor signaling.4,6 In order to address these limitations, research has focused on developing small subtype-selective ligands that target allosteric sites on mAChRs, which are topographically distinct from the orthosteric binding site and less well conserved, offering opportunities for increased selectivity.12 Of note, the selectivity of an allosteric ligand for a given receptor subtype can be driven by two main factors. First, ligands can be developed to take advantage of structural differences at the level of the allosteric binding site to engender affinity-based selectivity between mAChRs.12 Second, allosteric ligands may bind to relatively well-conserved allosteric sites with similar affinity across all subtypes, but still only exert a positive, negative or neutral effect on an orthosteric ligand depending on the receptor subtype as a consequence of selective cooperativity between orthosteric and allosteric sites on the cobound receptor. Indeed, cooperativity driven selectivity is emerging as an alternative means of developing or understanding selective allosteric ligands,12,13 as evidenced by the proposed mechanisms of action of M4 mAChR-selective PAMs (such as thiochrome and LY203329813–15) and allosteric ligands targeting class C GPCRs.44
Medicinal chemistry efforts applied to allosteric modulator drug discovery programs are often considered challenging, with descriptions of structure—activity relationships (SAR) described as “flat” or “shallow”.16,17 However, this often reflects a focus of allosteric SAR on overall modulator potency in the presence of orthosteric agonist, which itself is composed of, at a minimum, three key molecular properties: allosteric modulator affinity for its binding site, its cooperativity with the orthosteric agonist, and the potential for direct activation of the receptor by the modulator itself, i.e., allosteric agonism.18 The delineation of the differential impact that chemical manipulation can have on each of these properties can often overcome common challenges associated with allosteric ligand SAR that focuses predominantly on potency measures.12
As an interesting case in point, VU0119498 was originally identified from a functional, cell-based, high throughput screen (HTS) as a PAM at M1, M3 and M5 mAChRs, while being devoid of appreciable activity at M2 and M4 mAChRs, thereby providing the first example of a “pan-Gq” mAChR PAM.19 Its discovery served as the impetus for subsequent studies focused on identifying more subtype-selective allosteric modulators for M1 and M5 mAChRs in particular,20–22 but despite improved mAChR subtype selectivity, next-generation allosteric modulators generated from these studies have limited rodent CNS penetration,20,21 thus hindering further translational studies.
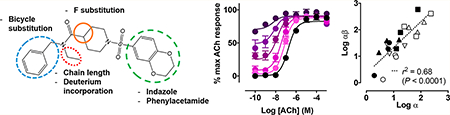
Further medicinal chemistry efforts led to the identification of ML380 as a moderately selective M5 mAChR PAM;23 subsequent studies yielded a range of newer analogues with improved CNS penetration,23,35 and these modifications are summarized in Figure 1. While compounds from this series have been characterized for their potency (i.e., their capacity to potentiate ACh-mediated responses) and mAChR subtype selectivity, there has been no mechanistic characterization of these modulators to determine the molecular allosteric parameters that engender activity and selectivity. Accordingly, the aim of the current study was to characterize the in vitro pharmacology of a cohort of pan-Gq mAChR PAMs based on a novel chemotype and predicted to have varying degrees of activity at M1 M3, and M5 mAChRs.23,35 Using both functional inositol phosphate (IP1) accumulation and [3H]-NMS binding studies we show that selectivity among these pan-Gq mAChR PAMs is driven by selective cooperativity, rather than binding affinity. Interestingly, in addition to a potentiation of ACh affinity, some of the PAMs also demonstrate modest efficacy modulation. An N-ethyl/propyl moiety appears to be important to the activity of all PAMs at the Gq-coupled mAChRs and the incorporation of a chiral indane motif yields higher cooperativity but relatively nonselective pan-Gq coupled PAM activity. Future studies with compounds of this series will be important to refine subtype selectivity while retaining increased activity.
Figure 1.

Modifications made to the novel chemotype that yielded the PAMs described in this study.23
RESULTS AND DISCUSSION
The discovery of a novel chemotype with M5 mAChR preferring PAM activity led to a series of allosteric modulators with improved mAChR subtype selectivity, but also modulators with diverse pan-Gq coupled mAChR PAM activity (Figure 1).23 Herein we have characterized the binding and functional properties of a selection of allosteric modulators at the M1, M3 and M5 mAChRs to understand which pharmacological parameters drive modulator activity and selectivity at Gq coupled mAChRs. Radioligand binding and functional assays were performed in whole CHO-hM1, -hM3, or -hM5 cells with a maximal binding capacities of 21.8 ± 0.6, 17.5 ± 0.3 or 7.3 ± 1.2 fmol/105 cells, respectively. In order to reduce the receptor reserve and visualize potential PAM-mediated effects on maximal agonist response(s), a subset of functional assays was performed after receptor alkylation with phenoxybenz-amine (PBZ), as previously described.24
ML380 (1-((1H-Indazol-5-yl)sulfonyl)-N-ethyl-N-(2-(trifluoromethyl)benzyl)piperidine-4-carboxamide).
For comparison, we built on existing pharmacological data available for the PAM ML380, which was noteworthy for its M5 mAChR potency and selectivity. We confirmed previous reports that ML380 is among the most potent M5 mAChR PAMs identified to date.23,24 In IP1 accumulation, ML380 demonstrated similar affinity for each Gq coupled mAChR and increased ACh potency at the M1 and M5 mAChR subtypes; the size of effect at the M5 mAChR being similar to previous reports (Figure 2A-C; Table 1).24 ML380 exhibited moderate positive cooperativity with ACh at the M1 and M5 mAChR subtypes and very weak positive cooperativity with ACh at the M3 mAChR (Figure 2A-C). ML380 also demonstrated intrinsic efficacy at Gq coupled mAChRs, the magnitude of which appeared to track with the PAM’s cooperativity at each receptor (Figure 2A-C). Indeed, ML380 appeared as almost a full agonist at the M5 mAChR, with minimal partial agonism at the M3 mAChR (Figure 2B, C). In [3H]-NMS equilibrium binding, the positive affinity cooperativity estimates were sufficient to explain the cooperativity estimates obtained from the functional data sets (Figure 2D-F, Tables 1 and 2), suggesting that ML380 exerts its effects mainly via changes in ACh affinity. This was confirmed with receptor alkylation studies, where ML380 (10 μM) caused a left-shift in the ACh curve with no effect on the ACh maximal response (Figure 2G-I). Given that ML380 displayed similar affinities for each of the Gq coupled mAChRs (Table 1), this suggests that any subtype selectivity for this PAM is derived from cooperativity.
Figure 2.

Functional and radioligand binding studies showing the effect of ML380 on ACh-stimulated IP1 accumulation in whole FlpIn-CHO cells stably expressing M1, M3, or M5 mAChRs (A-C; G—I) and on ACh-mediated inhibition of [3H]-NMS binding for the same cell lines (D-F), respectively. IP1 accumulation assays were performed with (A—C) or without (G—I) a 30 min pretreatment with PBZ (3 μM) to reduce the receptor reserve. ML380 (10 μM) did not change the maximal ACh response after PBZ treatment (extra sum-of-squares F-test: M1 mAChR: P = 0.14; M3 mAChR: P = 0.50; M5 mAChR: P = 0.35). Data are expressed as either a percentage of the maximal ACh response or as a percentage of specific [3H]-NMS binding and represent the mean ± SEM of at least three independent experiments performed in duplicate.
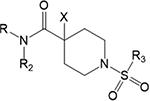
Table 1.
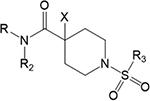
Operational Model Estimates for the Interaction between ACh and the Indicated Pan-Gq Coupled PAMs at M1, M3, and M5 mAChRs in IP1 Accumulation Assaysd
 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Structure | M1 mAChR | M3 mAChR | M5 mAChR | ||||||||||
| R | R2 | X | R3 | apKB | bLogβ | cLogτB | apKB | bLogβ | bLogτB | apKB | bLogβ | cLogτB | |
| ML380 | 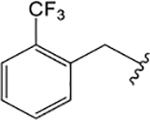 |
H | 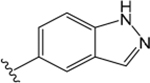 |
4.34 ± 0.37 | 0.95 ± 0.30 | 0.16 ± 0.26 | 4.65 ± 0.34 | 0.13 ± 0.13 | −0.25 ± 0.21 | 4.43 ± 0.30 | 0.97 ± 0.35 | 0.91 ± 0.26 | |
| VU0488129 | 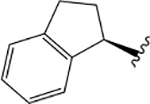 |
H | 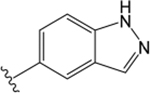 |
$5.28 ± 0.07 | $1.28 ± 0.04 | −0.14 ± 0.07 | = &4.5 | 1.51 ± 0.06 | −0.33 ± 0.14 | 6.43 ± 0.32 | 0.27 ± 0.09 | −0.74 ± 0.16 | |
| VU6007438 | 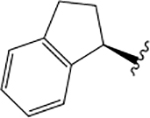 |
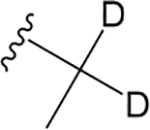 |
H |  |
$4.99 ± 0.19 | $1.59 ± 0.13 | −0.04 ± 0.06 | = &4.0 | 1.62 ± 0.06 | 0.01 ± 0.12 | 5.07 ± 0.25 | 1.14 ± 0.22 | 0.08 ± 0.12 |
| VU6007705 |  |
H |  |
4.87 ± 0.21 | 2.12 ± 0.21 | 0.42 ± 0.15 | 4.54 ± 0.24 | 1.75 ± 0.23 | 0.59 ± 0.20 | 4.59 ± 0.47 | 2.61 ± 0.48 | 1.03 ± 0.41 | |
| VU6007678 |  |
H | 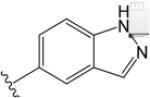 |
$4.94 ± 0.17 | $1.90 ± 0.13 | 0.34 ± 0.04 | $5.52 ± 0.04 | $1.28 ± 0.03 | −0.03 ± 0.04 | $4.82 ± 0.14 | $2.77 ± 0.13 | 0.84 ± 0.04 | |
| VU6008555 | 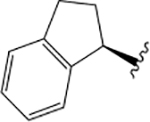 |
F | 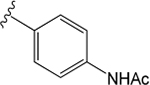 |
$4.74 ± 0.19 | $2.09 ± 0.15 | 0.42 ± 0.05 | = &4.0 | 1.91 ± 0.05 | 0.36 ± 0.07 | = &4.0 | 2.29 ± 0.08 | 0.95 ± 0.05 | |
| VU6007976 | 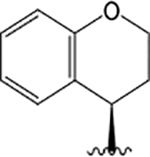 |
H | 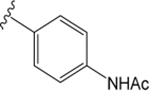 |
= &4.5 | 1.46 ± 0.08 | −0.003 ± 0.09 | = &4.0 | 1.39 ± 0.08 | −0.49 ± 0.26 | 5.15 ± 0.24 | 1.09 ± 0.25 | 0.32 ± 0.13 | |
Negative logarithm of the equilibrium dissociation constant for each pan Gq mAChR PAM for mAChRs. When the model did not converge, this parameter was constrained to a value derived from either an allosteric global pEC50 analysis (eq 3), as denoted by superscript $, or an absolute sum of squares analysis, as denoted by superscript &.
Logarithm of the efficacy scaling factor for the effect of the indicated pan Gq mAChR PAMs on ACh responses at the specified mAChRs; when the logarithm of the affinity cooperativity between ACh and the PAM is equal to zero (log a = 0), it is equivalent to the combined functional cooperativity between ligands (Log αβ). Combined cooperativity values with a corresponding $ or & were derived from applying an allosteric global pEC50 analysis (eq 3) or an absolute sum of squares analysis, respectively.
Logarithm of the operational efficacy parameter of the indicated pan Gq mAChR PAMs at the specified mAChRs.
Estimated parameters represent the mean ± SEM of at least three experiments performed in duplicate or triplicate. When the full operational model of allosterism (eq 1) was applied to data sets the negative logarithm of the equilibrium dissociation constant for ACh (pKA) was constrained to a predetermined value by applying the operational model of agonism to the ACh IP1 accumulation concentration—response curves (in the presence or absence of PBZ) from the alkylation experiments for the same mAChR. In the case of experiments at M1 mAChRs the pKA = 4.97 ± 0.21, at M3 mAChRs the pKA = 5.44 ± 0.21, and at M5 mAChRs the pKA = 5.84 ± 0.15.
Table 2.
Allosteric Ternary Complex Model Parameter Estimates for the Interaction between the Orthosteric Antagonist/Inverse Agonist [3H]-NMS and Agonist ACh with the Indicated Pan-Gq mAChR PAMs at M1, M3, and M5 mAChRsc
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Structure | M1 mAChR | M3 mAChR | M5 mAChR | ||||||||
| R | R2 | X | R3 | apKB | bLogαACh | apKB | bLogαACh | apKB | bLogαACh | ||
| ML380 | 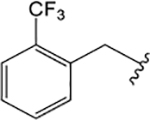 |
H | 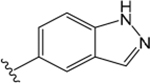 |
= 4.34 | 1.19 ± 0.09 | = 4.65 | 0.61 ± 0.11 | = 4.43 | 1.09 ± 0.12 | ||
| VU0488129 |  |
H |  |
= 5.28 | 0.80 ± 0.06 | = 4.50 | 0.84 ± 0.13 | = 6.43 | 0.29 ± 0.04 | ||
| VU6007438 |  |
 |
H | 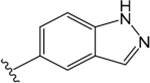 |
= 4.99 | 0.75 ± 0.08 | = 4.00 | 1.20 ± 0.11 | = 5.07 | 0.46 ± 0.11 | |
| VU6007705 | 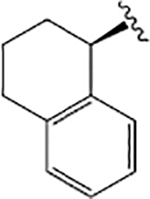 |
H | 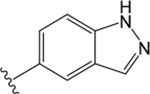 |
= 4.87 | 1.39 ± 0.07 | = 4.54 | 1.83 ± 0.04 | = 4.59 | 2.37 ± 0.08 | ||
| VU6007678 | 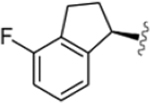 |
H | 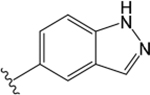 |
= 4.94 | 1.25 ± 0.08 | = 5.52 | 1.10 ± 0.07 | = 4.82 | 1.89 ± 0.08 | ||
| VU6008555 | 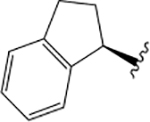 |
F | 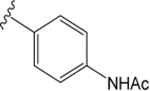 |
1.56 ± 0.07 | = 4.00 | 1.88 ± 0.06 | = 4.00 | 2.12 ± 0.07 | |||
| VU6007976 | 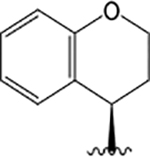 |
H | 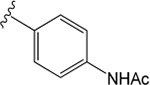 |
= 4.50 | 0.75 ± 0.11 | = 4.00 | 1.48 ± 0.09 | = 5.15 | 0.85 ± 0.10 | ||
Negative logarithm of the equilibrium dissociation constant of each pan Gq mAChR PAM (constrained to the estimate from the functional IP1 assays).
Logarithm of the affinity cooperativity between ACh and the indicated pan Gq mAChR PAM.
Estimated parameters represent the mean ± SEM of at least three experiments performed in duplicate.
VU0488129 ((R)-1-((1H-Indazol-5-yl)sulfonyl)-N-(2,3-dihydro-1H-inden-1-yl)-N-ethylpiperidine-4-carboxamide).
The introduction of a chiral center by replacement of the (trifluoromethyl)benzene with the 2,3-dihydro-1H-indene moiety reduced the number of possible conformations the compound is able to exhibit as compared to the more flexible ML380.
Unlike ML380, which has modest functional M5 mAChR selectivity, VU0488129 had very weak positive cooperativity with ACh at the M5 mAChR in IP1 assays, but moderate positive cooperativity at the M1 and M3 mAChR subtypes. These effects tracked broadly with its intrinsic agonist activity at the three receptor subtypes, consistent with a two-state model of receptor activation (Figure 3; Table 1).25,26 However, VU0488129 had 100-fold higher affinity at the M5 mAChR compared to ML380, suggesting that the reduced flexibility improved M5 mAChR binding, but reduced its cooperativity and functional subtype selectivity.
Figure 3.

Functional and radioligand binding studies showing the effect of VU0488129 on ACh-stimulated IP1 accumulation in whole FlpIn-CHO cells stably expressing M1, M3, or M5 mAChRs (A-C; G—I) and on ACh-mediated inhibition of [3H]-NMS binding for the same cell lines (D-F) as in Figure 2. VU0488129 (10 μM) did not change the maximal ACh response after PBZ treatment (M1 mAChR: P = 0.28; M3 mAChR: P = 0.12; M5 mAChR: P = 0.77; G—I). Data are expressed as either a percentage of specific [3H]-NMS binding or as a percentage of the maximal ACh response and represent the mean ± SEM of at least four independent experiments performed in duplicate.
Similar to the case of ML380, the affinity cooperativity estimates from [3H]-NMS binding assays tracked with (and were sufficient to explain) the combined cooperativity estimates for the same interactions in IP1 accumulation assays (Figure 3D—F; Table 2), a finding that was reflected in the receptor alkylation experiments where VU0488129 modestly potentiated ACh potency but did not significantly change the maximal ACh response (Figure 3G—I). Interestingly, as with ML380, the mechanism underlying the ability of VU0488129 to differentially potentiate ACh activity at mAChRs was driven by selective cooperativity between the modulator and ACh at the receptor.
VU6007438 ((R)-1-((1H-Indazol-5-yl)sulfonyl)-N-(2,3- dihydro-1H-inden-1-yl)-N-(ethyl-1,1-d2)piperidine-4-carboxamide).
The deuterated analogue of VU0488129, VU6007438, was synthesized to address any metabolic liability associated with the N-ethyl moiety.23 Unsurprisingly, given the very modest change, VU6007438 had very similar affinity and positive cooperativity with ACh at the M1 and M3 mAChRs compared to VU0488129 (Figure 4A, B; Table 1), and although it had >10-fold higher positive cooperativity at the M5 mAChR, this came at the cost of >10-fold reduced affinity for this subtype compared to VU0488129 (Figure 4C; Table 1). Notably, VU6007438 displayed higher intrinsic agonism at the M5 mAChR as compared to VU0488129. These observations imply that the incorporation of deuterium atoms into the N-ethyl moiety may subtly alter the binding affinity and transmission of positive cooperativity between VU6007438 and ACh at this mAChR subtype.
Figure 4.

Functional and radioligand binding studies showing the effect of VU6007438 on ACh-stimulated IP1 accumulation in whole FlpIn-CHO cells stably expressing M1, M3, or M5 mAChRs (A-C; G—I) and on ACh-mediated inhibition of [3H]-NMS binding for the same cell lines (D-F) as in Figure 2. VU6007438 (10 μM) modestly but significantly enhanced the maximal ACh response at all three Gq coupled mAChRs following PBZ treatment (M1 mAChR: P < 0.0001; M3 mAChR: P = 0.0049; M5 mAChR: P = 0.0052; G—I). Data are expressed as either a percentage of specific [3H]-NMS binding or as a percentage of the maximal ACh response, respectively, and represent the mean ± SEM of at least four independent experiments performed in duplicate.
VU6007705 ((R)-1-((1H-Indazol-5-yl)sulfonyl)-N-propyl-N-(1,2,3,4-tetrahydronaphthalen-1-yl)piperidine-4-carboxamide).
VU6007705 was the product of two modifications made to VU0488129: a change in the left-hand side from a 2,3-dihydro-1H-indene to a 1,2,3,4-tetrahydronaphthalene moiety and the replacement of the N-ethyl moiety with an N-propyl chain (Figures 3 and 5). These changes resulted in a dramatic change in ligand pharmacology, whereby VU6007705 had very high cooperativity with ACh in both [3H]-NMS binding and IP1 accumulation assays at all receptor subtypes (Figure 5A—C; Table 1). With such high positive cooperativity, it was unsurprising that the ligand displayed robust intrinsic allosteric agonism at each subtype (the magnitude of which tracked with the cooperativity between the modulator and ACh; Table 1) that was preserved even after receptor alkylation with PBZ (Figure 5 G-I). Despite the high positive cooperativity and robust allosteric agonism VU6007705 had no effect on the maximum ACh responses at the mAChRs, suggesting that the primary driver of cooperativity is the increase in ACh affinity (Tables 1 and 2). It is notable that despite markedly increased cooperativity across the receptor subtypes compared with ML380 and VU6007438, VU6007705 had similar affinities to these ligands, suggesting that the reduced flexibility of the structure may lock the ligand into a position that promotes contacts with binding site residues important to the transmission of cooperativity between the allosteric and orthosteric binding sites.
Figure 5.

Functional and radioligand binding studies showing the effect of VU6007705 on ACh-stimulated IP1 accumulation in whole FlpIn-CHO cells stably expressing M1, M3, or M5 mAChRs (A-C; G-I) and on ACh-mediated inhibition of [3H]-NMS binding for the same cell lines (D—F) as in Figure 2. Following PBZ treatment, VU6007705 (10 μM) did not change the maximal ACh response at M1 mAChRs (P = 0.39) but had a modest, significant, enhancement of the maximal ACh response at M3 (P = 0.022; 10 μM) and M5 mAChRs (P = 0.022; 1 μM), respectively (G—I). Data are expressed as either a percentage of specific [3H]-NMS binding or as a percentage of the maximal ACh response, respectively, and represent the mean ± SEM of at least four independent experiments performed in duplicate.
VU6007678 (R)-1-((1H-Indazol-5-yl)sulfonyl)-N-(4-fluoro-2,3-dihydro-1H-inden-1-yl)-N-propylpiperidine-4-carboxamide.
Fluorination of the indanyl core at the 4-position of VU0488129, and extension of the N-ethyl chain to propyl, yielded VU6007678, which has similar affinity for all Gq coupled mAChRs and high positive cooperativity with respect to both the signaling and binding affinity of ACh at all subtypes (Figure 6; Tables 1 and 2). These modifications seem to engender a broad increase in positive cooperativity with respect to ACh compared to that of VU0488129, with a modest (<10-fold) selectivity for the M5 mAChR, similar to that of ML380 (Figure 6; Tables 1 and 2).
Figure 6.

Functional and radioligand binding studies showing the effect of VU6007678 on ACh-stimulated IP1 accumulation in whole FlpIn-CHO cells stably expressing M1, M3, or M5 mAChRs (A-C; G—I) and on ACh-mediated inhibition of [3H]-NMS binding for the same cell lines (D-F) as in Figure 2. VU6007678 increased the maximal ACh response at mAChRs (M1 mAChR at 10 μM, P = 0.024; M3 mAChR at 10 μM, P = 0.0001; M5 mAChR at 300 nM, P < 0.0001). Data are expressed as either a percentage of specific [3H]-NMS binding or as a percentage of the maximal ACh response, respectively, and represent the mean ± SEM of at least three independent experiments performed in duplicate.
The affinity cooperativity estimates derived from the [3H]-NMS binding studies correlate with, but do not fully account for, the extent of the combined cooperativity estimates from the IP1 accumulation data. This suggests further effects on ACh signaling efficacy, a hypothesis validated after receptor alkylation, where VU6007678 increased maximal ACh responses and retained its allosteric agonism, even with reduced receptor reserve (Figure 6I—G). As with the other modulators, the degree of allosteric agonism tracked with the amount of cooperativity exhibited between ligands (Figure 6; Tables 1 and 2).
VU6008555 ((R)-1-((4-Acetamidophenyl)sulfonyl)-N-(2,3-dihydro-1H-inden-1-yl)-N-ethyl-4-fluoropiperidine-4-carboxamide).
Replacement of the indazole moiety of VU0488129 with the N-phenylacetamide moiety and the fluorination at the 4-position of the central piperidine ring yielded VU6008555. This change in the compound structure had little effect on the pharmacology of the ligand at the M1 and M3 mAChRs. Indeed, VU6008555 potentiated ACh signaling to a similar extent as VU0488129 and the affinity was largely unchanged at both the M1 and M3 mAChRs (Figures 3 and 7A, B, D, E; Tables 1 and 2). Interestingly, when compared to VU0488129, VU6008555 had >250-fold reduced affinity at the M5 mAChR, an effect largely offset by an ~ 100-fold increased positive cooperativity (Figures 3 and 7C, F; Tables 1 and 2). As with VU0488129 and ML380, the affinity cooperativity estimates in the binding assays were sufficient to account for the overall cooperativity estimates in the functional assays. This, combined with the lack of effect on ACh maximal responses after receptor alkylation, suggests that affinity cooperativity alone drives the observed functional effects of the PAM (Tables 1 and 2).
Figure 7.

Functional and radioligand binding studies showing the effect of VU6008555 on ACh-stimulated IP1 accumulation in whole FlpIn-CHO cells stably expressing M1, M3, or M5 mAChRs (A—C; G—I) and on ACh-mediated inhibition of [3H]-NMS binding for the same cell lines (D—F) as in Figure 2. Following PBZ pretreatment, 10 μM VU6008555 did not change the maximal ACh response at (G-I). VU6008555 (10 μM) had no effect on maximal ACh responses after PBZ treatment at M1 mAChRs (P = 0.59) or M5 mAChRs (P = 0.22), but did cause a small, but significant, increase in the response at M3 mAChRs (P = 0.03; H). Data are expressed as either a percentage of specific [3H]-NMS binding or as a percentage of the maximal ACh response, respectively, and represent the mean ± SEM of at least four independent experiments performed in duplicate.
VU6007976 ((R)-1-((4-Acetamidophenyl)sulfonyl)-N-(chroman-4-yl)-N-propylpiperidine-4-carboxamide).
Replacement of the left-hand side 2,3-dihydro-1H-indene moiety with a chromane (along with an N-ethyl chain to the N-propyl extension) yielded VU6007976. This compound has similar pharmacology to VU6007438, with relatively nonselective, moderately positive modulator activity in the IP1 assay across all the receptor subtypes, highest affinity for the M5 mAChR, and affinity cooperativity estimates that correlated with the functional assays (Figure 8A—F; Table 1). VU6007976 was without marked effects on the maximal ACh responses after PBZ treatment, suggesting that the cooperativity is driven through changes in ACh binding (Figure 8I—G).
Figure 8.

Functional and radioligand binding studies showing the effect of VU6007976 on ACh-stimulated IP1 accumulation in whole FlpIn-CHO cells stably expressing M1, M3, or M5 mAChRs (A-C; G-I) and on ACh-mediated inhibition of [3H]-NMS binding for the same cell lines (D-F) as in Figure 2. Following PBZ pretreatment, VU6007976 (10 μM) increased the maximal ACh response at M3 mAChRs (P = 0.0022) and M5 mAChRs (P < 0.0001), but not at M1 mAChRs (P = 0.11; G-I). Data are expressed as either a percentage of specific [3H]-NMS binding or as a percentage of the maximal ACh response, respectively, and represent the mean ± SEM of at least four independent experiments performed in duplicate.
Broadly speaking, our findings suggest that the compounds in this pan-Gq modulator series are relatively nonselective across the Gq coupled mAChRs with regard to affinity for the allosteric pocket on the unoccupied receptor; none of the compounds tested displayed any more than ~10-fold selectivity for any individual subtype. While highly subtype selective PAMs of the M1 mAChR have been extensively reported,18,27–33 identifying selective PAMs for the M3 and M5 receptor subtypes has met with less success, though efforts in this direction are fairly nascent. Indeed, any limited selectivity seems to derive from cooperativity; ML380 has similar binding affinity for all Gq coupled mAChRs but only shows PAM and allosteric agonist activity at the M1 and M5 mAChR subtypes, and only weakly potentiates ACh affinity at the M3 mAChR. This extends more generally to positive modulation across the series; VU6007678, VU6007705 and VU6008555 all have up to 60-fold higher cooperativity than ML380 at the Gq coupled mAChRs, though they have approximately the same affinity for the unoccupied receptor(s). With the exception of VU0488129 at the M5 mAChR, none of the compounds displayed submicromolar affinity for any of the receptor subtypes in the absence of agonist.
Given that the SAR depends heavily on changes on cooperativity between the modulators and ACh, the complementary use of [3H]-NMS binding and receptor alkylation studies to dissect any effects on affinity and efficacy was vital in providing additional mechanistic insights, suggesting that these complementary approaches should be more routinely incorporated into modulator SAR studies on key tool compounds. In general, there was a high degree of correlation between the overall cooperativity in the functional IP1 assays (log αβ) and the log α value estimated by the binding assay (Figure 9A), suggesting that changes in ACh binding affinity, rather than changes in signaling efficacy, drive the overall cooperativity. However, receptor alkylation studies provided some additional texture to these data, suggesting that VU6007678 can also modestly increase the maximal ACh responses after treatment of the receptors with PBZ (Figure 6). This implies that some compounds in the series can potentially modulate signaling efficacy, as an increase in the maximal agonist response cannot be reconciled in a model whereby the modulator increases only the affinity of the orthosteric agonist.
Figure 9.

Positive correlation between the composite functional cooperativity estimates (αβ) and (A) affinity cooperativity (α) and (B) relative efficacy (τB) estimates for allosteric modulators with ACh at the Gq coupled mAChRs. Each data point represents a parameter estimate for a single compound at an individual mAChR as determined by whole-cell [3H]-NMS competition binding and/or IP1 accumulation assays (Tables 1 and 2).
Given the high degrees of positive cooperativity displayed by some of the modulators, it was unsurprising that these same compounds displayed robust allosteric agonist activity, some of which was even preserved even after reduction of receptor reserve with PBZ. There was a significant correlation between the degree of allosteric agonism (as quantified by the parameter log τB) and the degree of positive cooperativity in the functional IP1 assay (log β; Figure 9B). This correlation implies that the modulators generally follow a two-state model of receptor activation and allostery, such that the binding of the PAMs shift the equilibrium toward a more active population of receptors, yielding both intrinsic allosteric agonism and increased sensitivity to ACh. Consistent with this mechanism, some of the PAMs also exerted weak effects to inhibit the binding of the inverse agonist radioligand, [3H]-NMS; however these effects were not always evident and most likely limited by the low affinity and/or solubility limit of the compounds under test.
Another general observation was that the incorporation of a chiral center into the structure correlated well with pan-Gq coupled PAM activity, and the N-substitution on the R2 moiety and N-ethyl/propyl moiety appeared important to PAM activity. More specifically, the incorporation of deuterium into VU6007488 favored M5 mAChR activity. Further investigation into analogues with these modifications may yield more selective M5 mAChR PAMs. It would be of interest to compare the in vivo efficacy of compounds with this mechanism of modulation as compared to pure affinity modulators, such as ML380, to determine which type of modulation, if any, is preferred for in vivo activity.
Since these PAMs exhibit different degrees of selectivity between Gq coupled mAChR subtypes, it would also be of interest to determine their binding sites on mAChRs. While structural studies have defined a prototypical allosteric modulator site in the extracellular vestibule of the M1-M4 mAChRs, currently there is minimal understanding as to the location and nature of the allosteric binding site on the M5 mAChR; it remains to be seen whether it is analogous to that described for other members of the receptor family.24,25,28,34
In summary, this study has provided the first detailed pharmacological characterization for the mechanistic basis of activity of a selection of pan-Gq coupled mAChR PAMs. We have shown that cooperativity, rather than binding affinity, appears to be the major mechanism driving the PAM activity and/or selectivity of these ligands. Moreover, this study has identified key structural modifications in the chemotype that increase the PAM activity of Gq coupled mAChR PAMs. Ideally, future studies will couple this increased cooperativity with greater selectivity for the M3 and M5 mAChRs, for which potent and selective modulators are still generally lacking relative to other mAChR subtypes.
METHODS
Materials.
Flp-In-Chinese hamster ovary (CHO) cells were obtained from Life Technologies (Mulgrave, Australia). Dulbecco’s modified Eagle’s medium was purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum was purchased from ThermoTrace (Melbourne, Australia). [3H]-N-Methylscopolamine ([3H]-NMS; specific activity, 70 Ci/mmol) and MicroScint scintillation liquid were purchased from PerkinElmer Life Sciences. The IP1 assay kit was purchased from Cisbio (Codolet, France). ACh and phenoxybenzamine (PBZ) were purchased from Sigma-Aldrich (St. Louis, MO). ML380, VU0488129, VU6007705, VU6007678, VU6008555, VU6007438, and VU6007976 were a generous gift of Craig Lindsley (Vanderbilt University, Nashville, TN3). All other chemicals, reagents and kits were from Sigma-Aldrich. For all procedures, purified water (18.2 MV cm) from a Milli-Q PF Plus system was used.
Cell Culture.
Human mAChR (hM1, hM3, and hM5 mAChR; Origene) constructs were isogenically integrated into Flp-In CHO cells (Invitrogen), and cells were selected in the presence of 600 mg/ mL hygromycin B at 37 °C, 5% CO2, as previously described for the hM1 mAChR.27 All cells were subcultured and seeded as previously described for the CHO-hM5 cells.24
[3H]-NMS Equilibrium Binding.
[3H]-NMS equilibrium binding assays were performed in CHO-hM1-hM3 and -hM5 mAChR cells as previously described.24
Inositol Phosphate Accumulation.
The IP1 assay kit (Cisbio) was used for direct quantitative measurement of inositol phosphate accumulation in CHO-hM1, -hM3, or -hM5 mAChR cells, as previously described in ref 24.
Receptor Alkylation Studies.
To determine functional affinity (KA) and efficacy (τA) estimates for acetylcholine at hM1, hM3, and hM5 mAChRs, in addition to determining the contributions made via affinity modulation (α) and efficacy modulation (β) to the overall PAM activity of allosteric ligands at each Gq coupled mAChR, additional IP1 accumulation assays were performed in CHO-hM1, -hM3, and -hM5 cells pretreated with phenoxybenzamine (PBZ) as per described.24
Data Analysis.
GraphPad Prism version 7.02 (San Diego, CA) was used for all curve fitting. For direct determination of the functional ACh equilibrium dissociation constants (KA) from the alkylation experiments, ACh concentration-response curves (in the presence or absence of PBZ) were globally fitted to the operational model of agonism as previously described in.24,36
For functional interaction studies between acetylcholine and PAMs in the IP1 accumulation assays, the following operational model of allosterism was applied:37
| (1) |
where basal is the response in the absence of ligand, Em is the maximal response of the system and n is the slope of the transducer function linking occupancy to response. [A] and [B] represent the concentrations of the orthosteric agonist (ACh) and allosteric ligand, respectively, and KA and KB represent their respective equilibrium dissociation constants. τΑ and tb denote the relative efficacies of the orthosteric and allosteric ligands, respectively. a denotes the affinity cooperativity between ACh and an allosteric modulator and β represents a scaling factor, which defines the magnitude and the direction of the effect of the allosteric modulator on ACh efficacy. For all data sets the Ka was constrained to the value derived from the alkylation experiments for the same mAChR (Mt mAChR pKA = 4.97 ± 0.21; M3 mAChR pKA = 5.44 ± 0.21; M5 mAChR pKA = 5.84 ± 0.15; n = 5-8), log a was constrained to 0 such that the resulting log β value was actually representative of the combined affinity and efficacy cooperativity (i.e., log αβ) between the interaction of a modulator with ACh for a particular mAChR. Derived cooperativity values greater than 1 indicate positive cooperativity; values < 1 (but >0) indicate negative cooperativity, and values of unity indicate neutral cooperativity.
In instances where application of eq 1 to interaction studies did not yield a reliable fit of the model or the model did not converge, the following two-step process was applied. First, the ACh concentration- response curves were fitted to the following four parameter logistic function:
| (2) |
where basal and Emax represent the shared minimum and maximal asymptotes, respectively, n is the shared Hill slope, and the log EC50 is the logarithm of the midpoint of the ACh stimulation curves. Second, agonist potencies for ACh in the presence and absence of modulator derived from eq 2 were then plotted against the corresponding concentration of the modulator, and eq 3 (below) was applied to the data to derive PAM affinity (KB) and combined affinity/efficacy cooperativity (αβ) estimates.38,39 This analysis was applied to interactions of ACh with VU0488129 at the M1 mAChR; VU6007678 at the M1, M3, and M5 mAChRs; VU6008555 at the M1 mAChR; and VU6007438 at the M1 mAChR (Supporting Information Figure 1).
| (3) |
where [B] is as previously described and d is the estimate of the ACh EC50 in the absence of the modulator. Interaction studies were then refitted to eq 1, where log a = 0, the log KB and log β were fixed to the values derived from application of eq 3 to data sets so that an estimate for the remaining parameter (intrinsic efficacy (τB) of the allosteric modulator) not yet quantified could be determined.
Finally, where eq 3 was applied to IP1 accumulation data sets and the model did not converge, a scanning of parameter space based on reduction in global absolute sum of squares was performed.40,41 Specifically, log α was constrained to 0 and the log KB was constrained to values ranging from −7.5 to −1.0, in 0.1 log unit increments while eq 1 was applied to each of the data sets under each of the aforementioned conditions until the global reduction in the sum of squares of the resulting fit approached an asymptotic value, suggesting that no further improvement in the fit could be obtained by changing the log KB. From this analysis, the reported functional log KB values were −4.5 for VU0488129 at the M3 mAChR, −4.0 for VU6008555 at the M3 and M5 mAChRs, −4.0 for VU6007438 at the M3 mAChR, and −4.5 and −4.0 for VU6007976 at the M1 and M3 mAChRs, respectively (Supporting Information Figure 2; Table 1); these values were used to fit the interaction data sets according to eq 1 to obtain an estimate of the combined cooperativity (log αβ) of the PAM with ACh and an estimate of the allosteric agonism (log τB) (Table 1).
For interaction [3H]-NMS equilibrium binding studies, data were analyzed according to an allosteric ternary complex model (ATCM) as previously described in refs 24 and 39.
| (4) |
where [A], [B], and [I] represent the concentrations of the radioligand ([3H]-NMS), allosteric ligand, and orthosteric inhibitor, respectively, KA, KB, and KI represent their respective equilibrium dissociation constants, and Bmax is the total number of receptors in a sample of tissue. The KA was fixed to 0.39 nM for interactions at the M1 mAChR, 0.34 nM for interaction at the M3 mAChR, or 0.3 nM for interactions at the M5 mAChR, as determined in separate saturation binding studies (data not shown). αA and αI denote the cooperativity values between the allosteric ligand and the radioligand or orthosteric inhibitor, respectively; values greater than 1 indicate positive cooperativity; values < 1 (but >0) indicate negative cooperativity, and values of unity indicate neutral cooperativity.
All estimates for potency, affinity, and cooperativity were reported as logarithms.42 Where appropriate, fitted parameters were compared by extra sum-of-squares F-test.43 Data are expressed as either a percentage of the maximal ACh response or as a percentage of specific [3H]-NMS binding and represent the mean ± SEM of at least three independent experiments performed in duplicate.
Supplementary Material
ACKNOWLEDGMENTS
A.C. is an NHMRC Senior Principal Research Fellow, P.M.S. is an NHMRC Principal Research Fellow, and C.W.L. is the William K. Warren, Jr. Chair in Medicine.
Funding
This work was funded by an NHMRC Program Grant (1055134), NIH (MH082867), NIMH (MH106839), and NIDA (DA037207).
ABBREVIATIONS
- CHO
Chinese hamster ovary
- mAChR
muscarinic acetylcholine receptor
- NMS
N-methyl scopolamine
- PAM
positive allosteric modulator
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.8b00136.
Graphical representation of global EC50 shift analysis to ACh potencies; graphical representation of absolute sum-of-squares analyses for global fits; allosteric ternary complex parameters for PAMs and [3H]-NMS binding (PDF)
REFERENCES
- (1).Matsui M, Yamada S, Oki T, Manabe T, Taketo MM, and Ehlert FJ (2004) Functional analysis of muscarinic acetylcholine receptors using knockout mice. Life Sci. 75, 2971–2981. [DOI] [PubMed] [Google Scholar]
- (2).Langmead CJ, Watson J, and Reavill C (2008) Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther 117, 232–243. [DOI] [PubMed] [Google Scholar]
- (3).Wess J (2004) Muscarinic acetylcholine receptor knockout mice: novel phenotypes and clinical implications. Annu. Rev. Pharmacol. Toxicol 44, 423–450. [DOI] [PubMed] [Google Scholar]
- (4).Wess J, Eglen RM, and Gautam D (2007) Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat. Rev. Drug Discovery 6, 721–733. [DOI] [PubMed] [Google Scholar]
- (5).Weiner DM, Levey AI, and Brann MR (1990) Expression of muscarinic acetylcholine and dopamine receptor mRNAs in rat basal ganglia. Proc. Natl. Acad. Sci. U. S. A 87, 7050–7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bymaster FP, McKinzie DL, Felder CC, and Wess J (2003) Use of M1-M5 muscarinic receptor knockout mice as novel tools to delineate the physiological roles of the muscarinic cholinergic system. Neurochem. Res 28, 437–442. [DOI] [PubMed] [Google Scholar]
- (7).Porter AC, Bymaster FP, DeLapp NW, Yamada M, Wess J, Hamilton SE, Nathanson NM, and Felder CC (2002) M1 muscarinic receptor signaling in mouse hippocampus and cortex. Brain Res. 944, 82–89. [DOI] [PubMed] [Google Scholar]
- (8).Vilaro MT, Palacios JM, and Mengod G (1990) Localization of M5 muscarinic receptor mRNA in rat brain examined by in situ hybridization histochemistry. Neurosci. Lett 114, 154–159. [DOI] [PubMed] [Google Scholar]
- (9).Ito Y, Oyunzul L, Seki M, Fujino Oki T, Matsui M, and Yamada S (2009) Quantitative analysis of the loss of muscarinic receptors in various peripheral tissues in M1-M5 receptor single knockout mice. Br. J. Pharmacol 156, 1147–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymaster FP, Hurley DJ, Potter WZ, and Paul SM (1997) Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch. Neurol 54, 465–473. [DOI] [PubMed] [Google Scholar]
- (11).Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, and Felder CC (2008) Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am. J. Psychiatry 165, 1033–1039. [DOI] [PubMed] [Google Scholar]
- (12).Christopoulos A (2014) Advances in G protein-coupled receptor allostery: from function to structure. Mol. Pharmacol 86, 463–478. [DOI] [PubMed] [Google Scholar]
- (13).Lazareno S, Dolezal V, Popham A, and Birdsall NJ (2004) Thiochrome enhances acetylcholine affinity at muscarinic M4 receptors: receptor subtype selectivity via cooperativity rather than affinity. Mol. Pharmacol 65, 257–266. [DOI] [PubMed] [Google Scholar]
- (14).Chan WY, McKinzie DL, Bose S, Mitchell SN, Witkin JM, Thompson RC, Christopoulos A, Lazareno S, Birdsall NJ, Bymaster FP, and Felder CC (2008) Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc. Natl. Acad. Sci. U. S. A 105, 10978–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Suratman S, Leach K, Sexton P, Felder C, Loiacono R, and Christopoulos A (2011) Impact of species variability and ‘probe-dependence’ on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. Br. J. Pharmacol 162, 1659–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, and Lindsley CW (2012) Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J. Med. Chem 55, 1445–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wood MR, Hopkins CR, Brogan JT, Conn PJ, and Lindsley CW (2011) “Molecular switches” on mGluR allosteric ligands that modulate modes of pharmacology. Biochemistry 50, 2403–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mistry SN, Valant C, Sexton PM, Capuano B, Christopoulos A, and Scammells PJ (2013) Synthesis and pharmacological profiling of analogues of benzyl quinolone carboxylic acid (BQCA) as allosteric modulators of the M1 muscarinic receptor. J. Med. Chem 56, 5151–5172. [DOI] [PubMed] [Google Scholar]
- (19).Marlo JE, Niswender CM, Days EL, Bridges TM, Xiang Y, Rodriguez AL, Shirey JK, Brady AE, Nalywajko T, Luo Q, Austin CA, Williams MB, Kim K, Williams R, Orton D, Brown HA, Lindsley CW, Weaver CD, and Conn PJ (2009) Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol. Pharmacol 75, 577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bridges TM, Kennedy JP, Cho HP, Breininger ML, Gentry PR, Hopkins CR, Conn PJ, and Lindsley CW (2010) Chemical lead optimization of a pan G(q) mAChR M(1), M(3), M(5) positive allosteric modulator (PAM) lead. Part I: Development of the first highly selective M(5) PAM. Bioorg. Med. Chem. Lett 20, 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bridges TM, Kennedy JP, Noetzel MJ, Breininger ML, Gentry PR, Conn PJ, and Lindsley CW (2010) Chemical lead optimization of a pan Gq mAChR M1, M3, M5 positive allosteric modulator (PAM) lead. Part II: development of a potent and highly selective M1 PAM. Bioorg. Med. Chem. Lett 20, 1972–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bridges TM, Marlo JE, Niswender CM, Jones CK, Jadhav SB, Gentry PR, Plumley HC, Weaver CD, Conn PJ, and Lindsley CW (2009) Discovery of the first highly M5-preferring muscarinic acetylcholine receptor ligand, an M5 positive allosteric modulator derived from a series of 5-trifluoromethoxy N-benzyl isatins. J. Med. Chem 52, 3445–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gentry PR, Kokubo M, Bridges TM, Noetzel MJ, Cho HP, Lamsal A, Smith E, Chase P, Hodder PS, Niswender CM, Daniels JS, Conn PJ, Lindsley CW, and Wood MR (2014) Development of a highly potent, novel M5 positive allosteric modulator (PAM) demonstrating CNS exposure: 1-((1H-indazol-5-yl)sulfoneyl)-N-ethyl-N-(2-(trifluoromethyl)benzyl)piperidine-4-car-boxamide (ML380). J. Med. Chem 57, 7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Berizzi AE, Gentry PR, Rueda P, Den Hoedt S, Sexton PM, Langmead CJ, and Christopoulos A (2016) Molecular mechanisms of action of M5 muscarinic acetylcholine receptor allosteric modulators. Mol. Pharmacol 90, 427–436. [DOI] [PubMed] [Google Scholar]
- (25).Canals M, Lane JR, Wen A, Scammells PJ, Sexton PM, and Christopoulos A (2012) A Monod-Wyman-Changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation. J. Biol. Chem 287, 650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Monod J, Wyman J, and Changeux JP (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol 12, 88–118. [DOI] [PubMed] [Google Scholar]
- (27).Abdul-Ridha A, Lane JR, Mistry SN, Lopez L, Sexton PM, Scammells PJ, Christopoulos A, and Canals M (2014) Mechanistic insights into allosteric structure-function relationships at the M1 muscarinic acetylcholine receptor. J. Biol. Chem 289, 33701–33711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Abdul-Ridha A, Lopez L, Keov P, Thal DM, Mistry SN, Sexton PM, Lane JR, Canals M, and Christopoulos A (2014) Molecular determinants of allosteric modulation at the M1 muscarinic acetylcholine receptor. J. Biol. Chem 289, 6067–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danziger A, Regan C, Flick R, Pascarella D, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Sur C, Kinney G, Seabrook GR, and Ray WJ (2009) Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. U. S. A 106, 15950–15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Kennedy JP, Jadhav SB, Menon UN, Xiang Z, Watson ML, Christian EP, Doherty JJ, Quirk MC, Snyder DH, Lah JJ, Levey AI, Nicolle MM, Lindsley CW, and Conn PJ (2009) A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J. Neurosci 29, 14271–14286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Davie BJ, Valant C, White JM, Sexton PM, Capuano B, Christopoulos A, and Scammells PJ (2014) Synthesis and pharmacological evaluation of analogues of benzyl quinolone carboxylic acid (BQCA) designed to bind irreversibly to an allosteric site of the M1 muscarinic acetylcholine receptor. J. Med. Chem 57, 5405–5418. [DOI] [PubMed] [Google Scholar]
- (32).Melancon BJ, Poslusney MS, Gentry PR, Tarr JC, Sheffler DJ, Mattmann ME, Bridges TM, Utley TJ, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, and Wood MR (2013) Isatin replacements applied to the highly selective, muscarinic M1 PAM ML137: continued optimization of an MLPCN probe molecule. Bioorg. Med. Chem. Lett 23, 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Poslusney MS, Melancon BJ, Gentry PR, Sheffler DJ, Bridges TM, Utley TJ, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, and Wood MR (2013) Spirocyclic replacements for the isatin in the highly selective, muscarinic M1 PAM ML137: the continued optimization of an MLPCN probe molecule. Bioorg. Med. Chem. Lett 23, 1860–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Valant C, Felder CC, Sexton PM, and Christopoulos A (2012) Probe dependence in the allosteric modulation of a G proteincoupled receptor: implications for detection and validation of allosteric ligand effects. Mol. Pharmacol 81, 41–52. [DOI] [PubMed] [Google Scholar]
- (35).Preparation, m. i. DOI: 10.1021/acschemneuro.8b00126. [DOI]
- (36).Black JW, and Leff P (1983) Operational models of pharmacological agonism. Proc. R. Soc. London, Ser. B 220, 141–162. [DOI] [PubMed] [Google Scholar]
- (37).Leach K, Sexton PM, and Christopoulos A (2007) Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci 28, 382–389. [DOI] [PubMed] [Google Scholar]
- (38).Davey AE, Leach K, Valant C, Conigrave AD, Sexton PM, and Christopoulos A (2012) Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor. Endocrinology 153, 1232–1241. [DOI] [PubMed] [Google Scholar]
- (39).Christopoulos A, and Mitchelson F (1997) Application of an allosteric ternary complex model to the technique of pharmacological resultant analysis. J. Pharm. Pharmacol 49, 781–786. [DOI] [PubMed] [Google Scholar]
- (40).Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, Baell JB, Sexton PM, Christopoulos A, and Shaw DE (2013) Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–299. [DOI] [PubMed] [Google Scholar]
- (41).Ehlert FJ (1985) The relationship between muscarinic receptor occupancy and adenylate cyclase inhibition in the rabbit myocardium. Mol. Pharmacol 28, 410–421. [PubMed] [Google Scholar]
- (42).Christopoulos A (1998) Assessing the distribution of parameters in models of ligand-receptor interaction: to log or not to log. Trends Pharmacol. Sci 19, 351–357. [DOI] [PubMed] [Google Scholar]
- (43).Motulsky H, and Christopoulos A (2004) Fitting models to biological data using linear and nonlinear regression A practical guide to curve fitting, Oxford University Press. [Google Scholar]
- (44).Hellyer SD, Albold S, Wang T, Chen ANY, May LT, Leach K, and Gregory KJ (2018) “Selective” Class C G proteincoupled receptor modulators are neutral or biased mGlu5 allosteric ligands. Mol. Pharmacol 93, 504–514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.