

Abstract
The exciton binding energy (Eb) and the band gap energy (Eg) of poly(phenylene vinylene) are determined by high-resolution measurements of the photoconductivity excitation profile as a function of light polarization, applied electric field, and temperature. At high applied electric fields, a peak in the photoconductivity is observed when the sample is pumped at a photon energy just below the onset of the band-to-band π-π* absorption. This peak is interpreted as resulting from field ionization of a weakly bound exciton with Eb ≈ 60 meV. The binding energy is obtained from the energy of the exciton peak relative to the band edge and independently from analysis of the dependence of the exciton dissociation on field and temperature.
A central issue in the field of conjugated polymers is the strength of the electron-electron interaction relative to the bandwidth (1): Is the attraction of a geminate electron-hole pair so strong that the photoexcitations are localized and strongly correlated Frenkel excitons? Or rather, are the charge carriers sufficiently well screened that a band picture supplemented by the electron-phonon interaction (polaron formation) and the electron-electron interaction (weakly bound excitons) is justified? Determination of the exciton binding energy (Eb) is critically important to answering these questions and thereby to understanding the electronic structure of semiconducting polymers.
Because this issue has not been resolved, the extensive literature on the optical properties of semiconducting (conjugated) polymers contains two conflicting assignments for the lowest energy π-π* absorption (1).
The lowest energy π-π* absorption results from the creation of tightly bound neutral singlet excitons with the onset of the interband transition at a significantly higher energy, as for example, in molecular crystals such as anthracene (2).
The lowest energy π-π* absorption results from a direct band-to-band transition, as for example, in direct gap semiconductors such as GaAs.
These two different assignments imply very different results for the photogeneration of charged excitations. When the lowest energy π-π* absorption results from a direct band-to-band transition, one expects to observe a threshold for photogeneration of charge carriers close in energy to the onset of absorption (Eπ-π); i.e., at
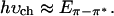 |
1a |
If, however, Eb is large, one expects to observe the threshold for photogeneration of charge carriers via the lowest band-to-band transition at energy greater than the onset of optical absorption by Eb; i.e., at
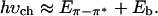 |
1b |
For poly(phenylene vinylene) (PPV) and several of its soluble derivatives, the quantum efficiency for photogeneration of charged excitations (polarons) has been measured, in zero external field, using ultrafast photo-induced absorption by infra-red active vibrational modes (3–5). The results demonstrated charge carrier photogeneration with a single threshold that is close in energy to the onset of absorption, in agreement with Eq. 1a (5). Although photoconductivity data have been reported with a second threshold well above the onset of absorption (6, 7), we have detected and characterized a contribution to the transient and steady-state photocurrent that originates from electron photoemission (8). After quenching the photoemission contribution, the true bulk photoconductivity data in PPV and all of the PPV derivatives show a threshold close to the onset of optical absorption, above which the photocurrent is nearly independent of excitation energy (up to 6.2 eV), again in agreement with Eq. 1a (8). These data demonstrate that the oscillator strength in the lowest energy optical absorption band of PPV (and its soluble derivatives) arises from the lowest energy band-to-band transition.
Theoretical models have yielded estimates for the Eb in PPV that range from values of order 0.1 eV to 1 eV (6, 7, 9). Moreover, the photoluminescnce is proportional to the light intensity (I) rather than I2, indicative of emission from a bound electron-hole pair. Thus, finding the exciton with spectroscopic accuracy and measuring the Eb remain as important goals for experimental studies of the photophysics of semiconducting polymers.
We have measured the Eb in chain-oriented PPV through high-resolution excitation profile spectroscopy of the steady-state photocurrent (Iphoto) at various external fields (F) and temperatures (T), and in samples with different defect concentrations. The spectral signature of the exciton is observed in the excitation profile as a narrow peak that emerges just below the band edge upon increasing the external electric field or the defect density. Because the exciton absorption and emission are polarized parallel to the chain axis, measuring the excitation profile of Iphoto with light polarized parallel and perpendicular to the PPV chain axis enables the identification (and separation) of carrier generation by means of exciton dissociation from carrier generation by means of the π-π* interband transition. From these studies, we have determined band gap energy (Eg) (2.42 eV) and Eb (≈60 meV), and we have clarified the role of the external field and defects in the carrier photogeneration process.
Details of the Experiment
Free-standing, oriented PPV films (draw ratio of 4 and thickness of 15 μm) were used. For Iphoto measurements, two gold electrodes, separated by a gap of 15 μm, were evaporated onto the surface. The steady-state photocurrent (normalized to constant incident photon flux) was measured by using the conventional modulation technique with a lock-in amplifier. The light incident on the sample (either from a tungsten or Xe source) was dispersed by a monochromator.
To demonstrate the sensitivity of the photoconductivty excitation profile as a method for detecting weakly bound neutral excitons, we carried out the experiment on single-crystal GaAs where the exciton and lowest energy band-to-band transition energies are known. Fig. 1 shows the Iphoto spectrum of GaAs obtained with the sample at T = 10 K; a peak is observed at a few meV below the step-like signature of the onset of the interband transition. This peak in the photoconductivity results from a combination of field ionization and defect/impurity dissociation of weakly bound excitons into mobile electrons and holes. We will return to the details of the exciton dissociation mechanism in the context of the discussion of similar results obtained from chain-oriented PPV. The point here is simply to demonstrate the opportunity for using high-resolution measurements of the photoconductivity excitation profile as a means of detecting the exciton and measuring the exciton energy with respect to the onset of the band-to-band transition. Note that photoconductivity measurements are particularly sensitive; because of the ability to measure extremely low currents with relative ease, one can typically detect a photocurrent even if the oscillator strength for exciton absorption is too weak to be easily measured directly by absorption.
Figure 1.

Photocurrent excitation spectrum in single crystal GaAs measured at T ∼10 K.
Results and Analysis of the Data
Fig. 2 shows Iphoto excitation profile spectra obtained from PPV at various external fields as measured with unpolarized light. The most striking observation is the appearance of a shoulder in the Iphoto spectrum near the absorption edge at relatively low fields that develops into a narrow peak at fields above ≈105 V/cm. The emergence of the narrow Iphoto peak just below the absorption edge at high applied fields suggests a contribution to the carrier generation process from field ionization of the exciton.
Figure 2.

Photocurrent excitation spectra in PPV at various external fields; the normalization factors of the curves are indicated on the right.
Carrier generation by means of direct band-to-band excitation and exciton dissociation can be separated by using Iphoto spectra measured with polarized light (10). Fig. 3 shows typical Iphoto spectra measured with light polarized parallel (I ) and perpendicular (I
) and perpendicular (I ) to the chain axis with F = 7 × 104 V/cm applied parallel to the chain axis. Two important features are evident:
) to the chain axis with F = 7 × 104 V/cm applied parallel to the chain axis. Two important features are evident:
Figure 3.

Photocurrent spectra in PPV as measured with light polarized parallel (∥, green) and perpendicular (⊥, blue) to the chain axis with F = 7 × 104 V/cm; the curve representing the sum (red) is shown.
The onset of Iphoto is significantly higher in energy for I than for I
than for I , in agreement with absorption data on similar samples (11).
, in agreement with absorption data on similar samples (11).
The peak at 2.365 eV appears only in I . The anisotropy of Iphoto with respect to light polarization indicates a relatively high degree of chain extension and alignment in the oriented PPV samples.
. The anisotropy of Iphoto with respect to light polarization indicates a relatively high degree of chain extension and alignment in the oriented PPV samples.
Considering that the oscillator strength for the 1Bu exciton is polarized along the chain axis, (as demonstrated by the polarized emission) (12), these data provide a clear spectroscopic signature for the 1Bu exciton. Moreover, the data imply that the onset of I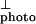 coincides with the onset of interband carrier generation. Assuming the onset energy for the interband transition is at the inflection point of I
coincides with the onset of interband carrier generation. Assuming the onset energy for the interband transition is at the inflection point of I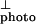 where d2I
where d2I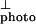 /d(ħω)2 = 0, we obtain the energy gap; Eg = 2.42 eV. Fig. 3 also shows the sum, I
/d(ħω)2 = 0, we obtain the energy gap; Eg = 2.42 eV. Fig. 3 also shows the sum, I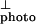 + I
+ I , which produces a spectrum essentially identical to that obtained with unpolarized light at the same applied field (see Fig. 2).
, which produces a spectrum essentially identical to that obtained with unpolarized light at the same applied field (see Fig. 2).
Because the oscillator strength for the exciton transition is exclusively along the chain axis, the exciton line can be seen more clearly by subtracting I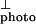 from I
from I . Note that I
. Note that I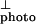 is larger than I
is larger than I at photon energies above 2.5 eV, most likely as a result of the larger absorption depth for ⊥-polarized light and thereby longer carrier lifetime (lower density of carriers and interchain recombination). Thus, to obtain the exciton line shape, the two Iphoto spectra (∥ and ⊥) were normalized to equal magnitude at 3.2 eV. As shown in Fig. 4, the exciton line is centered at 2.365 eV with full width at half-maximum of ≈100 meV. Therefore, in PPV, Eb ≈ 60 meV.
at photon energies above 2.5 eV, most likely as a result of the larger absorption depth for ⊥-polarized light and thereby longer carrier lifetime (lower density of carriers and interchain recombination). Thus, to obtain the exciton line shape, the two Iphoto spectra (∥ and ⊥) were normalized to equal magnitude at 3.2 eV. As shown in Fig. 4, the exciton line is centered at 2.365 eV with full width at half-maximum of ≈100 meV. Therefore, in PPV, Eb ≈ 60 meV.
Figure 4.

The exciton line shape, as obtained by subtracting I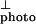 from I
from I (after normalization to equal magnitude at 3.2 eV).
(after normalization to equal magnitude at 3.2 eV).
The role of defects in the exciton dissociation was elucidated by measuring the Iphoto spectrum in a PPV sample with different defect concentrations. Defects were introduced by photo-oxidation using illumination by UV radiation while the sample was exposed to air. Fig. 5 compares the Iphoto spectra of a pristine PPV sample to that from the same sample after exposure to light from a Xe lamp (≈400 mW/cm2) for 2 h. After the initial irradiation, the 2.365-eV peak grew significantly relative to Iphoto at higher energies, consistent with defect-induced dissociation of the exciton into charged polaron pairs (7, 13). The more modest increase in Iphoto at higher energies is consistent with defect-induced dissociation of excitons formed after thermalization of charge carriers initially produced by direct π-π* absorption. After an additional 3 h of photo-oxidation of the same sample, the exciton peak remained distinct and narrow, but at higher energies the vibronic structure was absent and the overall magnitude of Iphoto was reduced, indicative of decreased carrier mobility. The efficiency of the defect-induced charge carrier generation is consistent with the relatively small Eb, a large Eb would suppress the tendency for exciton dissociation.
Figure 5.

Comparison of the excitation spectra of pristine PPV to that of the same sample after photo-oxidation by exposure in air to 400 mW/cm2 of UV light from a Xe lamp.
Fig. 6 shows I spectra at several different applied fields. As demonstrated in Fig. 2, with increasing field the 2.365-eV exciton peak increases in magnitude relative to the weaker and broader maxima at higher energies. Nevertheless, as shown in Figs. 3, 4, and 6, the equal energy separation (0.19 eV) between the secondary peaks is suggestive of vibronic replicas. Vibronic side-bands in the absorption and emission spectra are well known. However, because the final states of the transitions involving vibronic quanta (one- and two-phonon, etc.) are in the continuum above the onset of zero-phonon interband energy, an “exciton” associated with such transitions could not be a true bound state, but rather a resonance that would spontaneously decay into free carriers.
spectra at several different applied fields. As demonstrated in Fig. 2, with increasing field the 2.365-eV exciton peak increases in magnitude relative to the weaker and broader maxima at higher energies. Nevertheless, as shown in Figs. 3, 4, and 6, the equal energy separation (0.19 eV) between the secondary peaks is suggestive of vibronic replicas. Vibronic side-bands in the absorption and emission spectra are well known. However, because the final states of the transitions involving vibronic quanta (one- and two-phonon, etc.) are in the continuum above the onset of zero-phonon interband energy, an “exciton” associated with such transitions could not be a true bound state, but rather a resonance that would spontaneously decay into free carriers.
Figure 6.

I spectra as obtained at various applied fields; the Inset shows these curves normalized to similar magnitude at 3.2 eV.
spectra as obtained at various applied fields; the Inset shows these curves normalized to similar magnitude at 3.2 eV.
As noted earlier, we have found similar signatures of the exciton in the Iphoto spectra obtained from single-crystals of GaAs (see Fig. 1). Measurements on polydiacetylene-(toluene sulfonate) (PTS) also indicate a weak field-induced Iphoto response at the exciton absorption energy (≈2 eV), well below the onset of the interband transition. Thus, in GaAs, PTS, and PPV, field-induced ionization enables the observation of the exciton signature in the Iphoto spectrum.
Field Ionization of the Singlet Exciton
The 1Bu exciton binding energy and radius are, respectively,
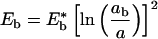 |
2a |
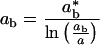 |
2b |
where
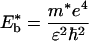 |
3a |
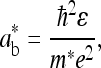 |
3b |
ɛ is the dielectric constant, ħ is Planck's constant, m* is the electron effective mass at the zone center in k-space (for PPV, m* ≈ 0.1 me), and e is the electron charge (14). For oriented PPV, the dielectric constant parallel to the chain axis is ≈8–10 (at 2.3 eV); whereas the dielectric constant in the perpendicular direction is ≈3 (11). Thus, depending on the value used for ɛ in Eqs. 2 and 3, estimated values for values for Eb range from 0.05 eV to 0.2 eV.
Calculations indicate that the higher energy states in the exciton series, Ebn ∼ Eb/n2, contain only about 20% of the total exciton oscillator strength. Although relatively weak, they might contribute to the band edge profile (N.K. and S.B., unpublished work).
Electric-field induced dissociation of a neutral electron-hole bound state is well known (7, 15, 16). At very high fields, F > F* = E*b/a*b, the bound state is destroyed. At intermediate fields, there is a barrier to field ionization, but the carriers can dissociate by tunneling. In this regime, the field-induced ionization rate, Γ, is given by (N.K. and S.B., unpublished work)
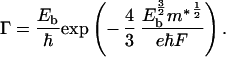 |
4 |
The densities of excitons (nex) and carriers (Nch) are determined by the following rate equations:
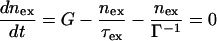 |
5a |
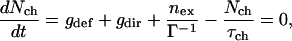 |
5b |
where G is the rate of exciton generation (proportional to the light intensity when pumped at the exciton line), τex is exciton lifetime (the decay time of the photoluminescence), τch is the carrier lifetime, gdef is the rate of carrier generation by exciton dissociation by defects, and gdir is the rate for direct carrier excitation. Note that gdir = 0 when light is absorbed directly into the exciton line. Under steady-state conditions Nch and Iphoto are given by:
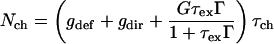 |
6a |
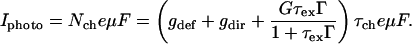 |
6b |
The importance of field-induced ionization is evident in Figs. 2 and 6. To determine Γ(F) more precisely, I vs. F was measured for two photon energies, at 2.365 and 2.6 eV (at the exciton peak and well above the onset of the interband transition). The dependence of the τchμ product on F was eliminated by dividing I
vs. F was measured for two photon energies, at 2.365 and 2.6 eV (at the exciton peak and well above the onset of the interband transition). The dependence of the τchμ product on F was eliminated by dividing I at 2.365 eV by that at 2.6 eV; this ratio (rphoto) is plotted in Fig. 7 The data were analyzed with the following expression:
at 2.365 eV by that at 2.6 eV; this ratio (rphoto) is plotted in Fig. 7 The data were analyzed with the following expression:
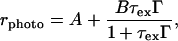 |
7 |
where A = gdef/gdir and B = G/gdir. In obtaining Eq. 7 from the expression for Iphoto (Eq. 6), we assumed that both the exciton generation rate through recombination of carriers following direct π-π* absorption at ħω > Eb and the rate of defect-induced carrier generation from excitons formed after absorption at ħω > Eb are small compared with G and gdef, respectively. In Fig. 7, A = gdef/gdir is determined by the F = 0 intercept, whereas the saturation at high fields determines B = G/gdir. The one-parameter (Eb) fit for rphoto is shown in Fig. 7. The fit is excellent in the low and high field limits, but the theoretical curve shows a sharper inflection. The “rounding” of the experimental results relative to the calculated curve might arise from a combination of disorder in the polymer and thermal broadening (the theoretical curve assumes zero temperature).
Figure 7.

The ratio, rphoto, of I measured at 2.365 and 2.6 eV vs. the applied field. The solid line is the theoretical fit to Eq. 1; the solid, dashed, and dotted theoretical curves represent A = 1, A = 0.5, and A = 0, respectively.
measured at 2.365 and 2.6 eV vs. the applied field. The solid line is the theoretical fit to Eq. 1; the solid, dashed, and dotted theoretical curves represent A = 1, A = 0.5, and A = 0, respectively.
The value obtained for Eb from the data in Fig. 7 depends weakly on the value chosen for the exciton lifetime. From the fits, we obtain Eb = 55 ± 15 meV for τex = 1 ns, and Eb = 65 ± 15 meV for τex = 300 ps, in excellent agreement with the value for Eb obtained independently from the Iphoto excitation spectrum. Fig. 7 includes theoretical curves for values of gdef/gdir other than those obtained from the intercept. As gdef is reduced, the low field value of rphoto decreases.
Because Eb is only a few times kBT, thermal excitation of charge carriers out of the neutral bound state is expected. When the exciton is pumped resonantly, measurements of the temperature dependence (220–300 K) indicate that rphoto = A + exp(−Ea/kBT) at F = 104 V/cm. The activation energy, Ea, was obtained from a fit of the above expression to the measured T dependence (see Fig. 8); Ea ≈ 70 ± 11 meV. Thus, the T dependence is quantitatively consistent with Eb as inferred above (more details on these experimental results will be published in a future publication).
Figure 8.

The dependence of rphoto on temperature, where the line represents the best fit of the data to thermally activated expression, which yields Eb = 70 ± 11 meV.
Reflectance data (11) from oriented PPV indicate a weak absorption feature on the leading edge of the π-π* transition corresponding to the field-induced peak in Iphoto. The weak oscillator strength in the exciton absorption relative to the π-π* transition is expected because the one-dimensional joint-density-of-states is divergent at the band edge while the interband matrix elements decease toward zero as ≈E1/2 for energies within Eb of the onset of the band-to-band transition (13).
One might attempt to interpret the peak in Iphoto at 2.365 eV as arising from field-induced ionization of defects rather than field-induced ionization of excitons. In that interpretation, however, the defect energy must be precisely defined at 55 meV below the band edge, implying that the oscillator strength above 2.42 eV is that of the interband transition and that the exciton must be even less strongly bound. Moreover, one would not expect such a narrow peak for charged defects in PPV. Finally, in the defect interpretation, the increase in Iphoto upon photo-oxidation must arise from increased defect absorption. Such a precisely defined defect-induced peak is not observed in the optical absorption after photo-oxidation; instead the absorption edge is simply broadened. Moreover, in the “optically thick” samples used in these experiments, the incident light is completely absorbed at 2.365 eV before any increase in the defect concentration by photo-oxidation. Thus, an increase in the defect density would not increase the peak in Iphoto. On the contrary, dissociation of excitons by defects is well known, it is not associated with a change in absorption and (at least at moderate defect levels) need not broaden nor shift the exciton peak.
Conclusions
In conclusion, measurements of the photocurrent excitation spectra at various external fields, temperatures, defect concentrations, and with light polarized parallel and perpendicular to the polymer chain axis reveal the basic properties of the electronic structure of PPV. The Eb is ≈60 meV, and the band gap energy is 2.42 eV. The relatively small Eb indicates that the charge carriers are well screened so that a band picture supplemented by the electron-phonon interaction and the electron-electron interaction is justified.
Acknowledgments
We thank Dr. G. Bahir for preparing the contacts on the GaAs sample and for important discussions and C. Soci for assistance with the activation energy measurements. The research was supported by the National Science Foundation under NSF-DMR 9812852.
Abbreviations
- Eb
exciton binding energy
- PPV
poly(phenylene vinylene)
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on May 1, 2001.
References
- 1.Sariciftci N S, editor. The Nature of Photoexcitations in Conjugated Polymers. Singapore: World Scientific; 1997. [Google Scholar]
- 2.Pope M, Swenberg C E. Electronic Processes in Organic Crystals. New York: Oxford Univ. Press; 1982. [Google Scholar]
- 3.Moses D, Dogariu A, Heeger A J. Chem Phys Lett. 2000;316:354–360. [Google Scholar]
- 4.Moses D, Dogariu A, Heeger A J. Phys Rev B. 2000;61:9373–9379. [Google Scholar]
- 5.Miranda, P., Moses, D. & Heeger, A. J. (2001) Phys. Rev. B, in press.
- 6.Chandross M, Mazumdar S, Jeglinski S, Wei X, Vardeny Z V, Kwock E W, Miller T M. Phys Rev B. 1994;50:14702–14705. doi: 10.1103/physrevb.50.14702. [DOI] [PubMed] [Google Scholar]
- 7.Harrison M G, Gruner J, Spencer G C W. Phys Rev B. 1997;55:7831–7849. [Google Scholar]
- 8.Moses, D., Soci, C., Miranda, P. & Heeger, A. J. (2001) Chem. Phys. Lett., in press.
- 9.Rohlfing M, Louie S. Phys Rev Lett. 1999;82:1959–1962. [Google Scholar]
- 10.Lee C H, Park J Y, Moses D, Heeger A J, Noguchi T, Ohnishi T. Synth Methods. 1999;101:444–445. [Google Scholar]
- 11.Comoretto D, Dellepiano G, Moses D, Cornil J, Santos D A, Bredas J L. Chem Phys Lett. 1998;289:1–7. [Google Scholar]
- 12.Hagler T W, Pakbaz K, Voss K F, Heeger A J. Phys Rev B. 1991;44:8652–8666. doi: 10.1103/physrevb.44.8652. [DOI] [PubMed] [Google Scholar]
- 13.Antoniadis H, Rothberg L J, Papadimitrakopoulos F, Yan M, Galvin M E, Abkowitz M A. Phys Rev B. 1994;50:14911–14915. doi: 10.1103/physrevb.50.14911. [DOI] [PubMed] [Google Scholar]
- 14.Kirova N, Brazovskii S, Bishop A R. Synth Methods. 1999;100:29–53. [Google Scholar]
- 15.Kersting R, Lemmer U, Deussen M, Bakker H J, Mahrt R F, Kurz H, Arkhipov V I, Bässler H, Göbel E O. Phys Rev Lett. 1994;73:1440–1443. doi: 10.1103/PhysRevLett.73.1440. [DOI] [PubMed] [Google Scholar]
- 16.Moses D, Okumoto H, Lee C H, Heeger A J. Phys Rev B. 1996;54:4748–4754. doi: 10.1103/physrevb.54.4748. [DOI] [PubMed] [Google Scholar]