

Abstract
Objective
To report cross-sectional clinical findings in a large cohort of patients affected by type 1 spinal muscular atrophy.
Methods
We included 122 patients, of age ranging between 3 months and 22 years, 1 month. More than 70% (85/122) were older than 2 years and 25% (31/122) older than 10 years. Patients were classified according to the severity of phenotype and to the number of SMN2 copies.
Results
Patients with the more common and the most severe phenotype older than 2 years were, with few exceptions, on noninvasive ventilation and, with increasing age, more often had tracheostomy or >16-hour ventilation and a gastrostomy inserted. In contrast, 25 of the 28 patients with the mildest phenotype older than 2 years had no need for tracheostomy or other ventilatory or nutritional support. In patients older than 2 years, the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders scores were generally lower compared to those found in younger patients and showed distinct levels of functional abilities according to the severity of the phenotype. Similar findings were also observed on the Hammersmith Infant Neurological Examination.
Conclusions
Our findings confirm that, after the age of 2 years, patients with type 1 spinal muscular atrophy generally survive only if they have gastrostomy and tracheostomy or noninvasive ventilation >16 hours and have low scores on the functional scales. More variability, however, can be expected in those with the mildest phenotype, who achieve head control. These data provide important baseline information at the time treatments are becoming available.
Spinal muscular atrophy (SMA) is an autosomal recessive disorder caused by mutations in the survival of motor neuron 1 (SMN1) gene.1 Nusinersen, an antisense oligonucleotide designed to increase full-length SMN protein levels, has recently been used in phase 22,3 and phase 3 sham-controlled studies in infants with infantile-onset SMA showing increased survival and some improvement on motor functional scales.4 Similar results have been observed in a parallel phase 3 sham-controlled study in late-onset spinal SMA.5 Based on these results, the drug has recently been approved in several countries. While waiting for regulatory approval, nusinersen was made available via an early access program (EAP) to allow early access to all patients with type 1 SMA. In Italy, a national committee was created to identify all the patients potentially eligible for the EAP and facilitate the process.6 This allowed identification of patients who were not regularly followed in tertiary care centers and to systematically collect information in a large cohort of patients with type 1 SMA whose age ranged from neonatal to young adult.
Our aim was to report functional abilities and other clinical findings at baseline, before treatment with nusinersen was started, to provide a cross-sectional picture of a large cohort of patients with type 1 SMA in Italy with a wide age range and to retrospectively evaluate whether and how survival and functional abilities were influenced by onset and severity of the disease or different modalities of intervention.
Methods
A nationwide search was performed to identify all patients with type 1 SMA in Italy. The search started from the national registries of SMA, but all the tertiary neuromuscular centers, rehabilitation/counseling centers with known expertise in SMA, and genetic laboratories were also consulted and informed of the EAP. Details of this procedure have already been reported.6
Inclusion criteria included a genetically confirmed diagnosis of SMA with a homozygous deletion of exon 7 or other mutations in the SMN1 gene, and a clinically confirmed diagnosis of type 1 SMA (inability to achieve independent sitting). One hundred fifty-six patients with SMA1 were identified. Two of them, with onset of clinical signs before 6 months, were excluded because they had achieved the ability to sit unsupported. Another 3 died (ages between 1 and 5 months) before they could be seen at one of the participating centers. Of the remaining 151, 99 agreed to be part of the program while the other 52 did not reply or had no interest in joining the EAP (age range 1 month to 13 years). Twenty-three additional patients were diagnosed after the program started and were also included, thus resulting in a final cohort of 122 patients. None of these patients were part of other clinical trials.
The patients were followed in 5 centers (2 in Rome, 1 in Genova, 1 in Milan, and 1 in Messina) that had already been involved in previous nusinersen trials.
Standard protocol approvals, registrations, and patient consents
The study was approved by the ethics committee in each center. Written informed consent was obtained from all participants (or guardians of participants) in the study.
A structured pro forma was used to collect current and retrospective clinical data. This included age at onset, maximal motor function achieved (e.g., head control), acquisition of speech, and, when available, analysis of retrospective functional data. This allowed the investigators at each site to classify the patients according to the Dubowitz decimal classification7 that identifies as 1.5 the infants with the phenotype observed in the majority of type 1 SMA: inability to raise the legs against gravity or maintain the head posture either in the supine or prone position, but having, at diagnosis, no difficulty with feeding and swallowing, no accumulation of pharyngeal secretions, and no obvious distress. At the more severe end of the spectrum, the infants had severe paralysis at birth and early respiratory and bulbar difficulties and an overtly poor prognosis for survival (type 1.1), while at the other end of the spectrum, infants had some head control and less respiratory compromise (type 1.9). This classification partially overlaps with a more recently proposed classification in 3 subtypes8,9: type 1A, presentation at birth with joint contractures and need for respiratory support, or onset of motor and respiratory involvement in the first week; type 1B, symptom onset after the neonatal period, usually before age 3 months, and head control never achieved; and type 1C, onset after the neonatal period, usually between 3 and 6 months, and head control achieved. We applied both classifications in this analysis.
Need for ventilator or nutritional support and time when these were initiated were also collected. SMN2 copy number, if DNA samples were available, was also determined by the commercial MLPA kit (SALSA MLPA probemix P021-A2 SMA; MRC-Holland, Amsterdam, the Netherlands) or by quantitative PCR.10
Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders
The Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) is specifically designed to assess motor function in weak infants, and has been validated in type 1 SMA.11,12 It includes 16 items with a total score that can range from 0 to 64. The scale was performed by clinical evaluators trained by the senior physical therapist (R.d.S.) after training and reliability sessions.
Hammersmith Infant Neurological Examination
The developmental section of the Hammersmith Infant Neurological Examination (HINE)13 is a short assessment, easily performed even in fragile, weak type 1 SMA. It includes 8 selected motor items that document developmental progress (figure 1), allowing not only the opportunity to record whether the various milestones have been achieved but also to quantify intermediate steps leading to the full achievement of the milestone. Each item provides the opportunity to score the level of development on a 5-point scale with 0 as absence of the activity. These milestones were designed according to the gradient of normal maturation. The module was originally designed as a part of a general neurologic examination and was not specifically designed for patients with neuromuscular weakness. Nevertheless, 7 of the 8 items reflect the maturation of the milestone from absence of the response to the achievement of the milestone involving antigravity movements. The only item that does not follow a similar maturation and is more related to maturation of coordination in fine motor abilities is hand grasp and therefore was excluded from the analysis, as also suggested in a recent clinical trial using the same assessment.4
Figure 1. Details of the level of respiratory care by age and subtype.
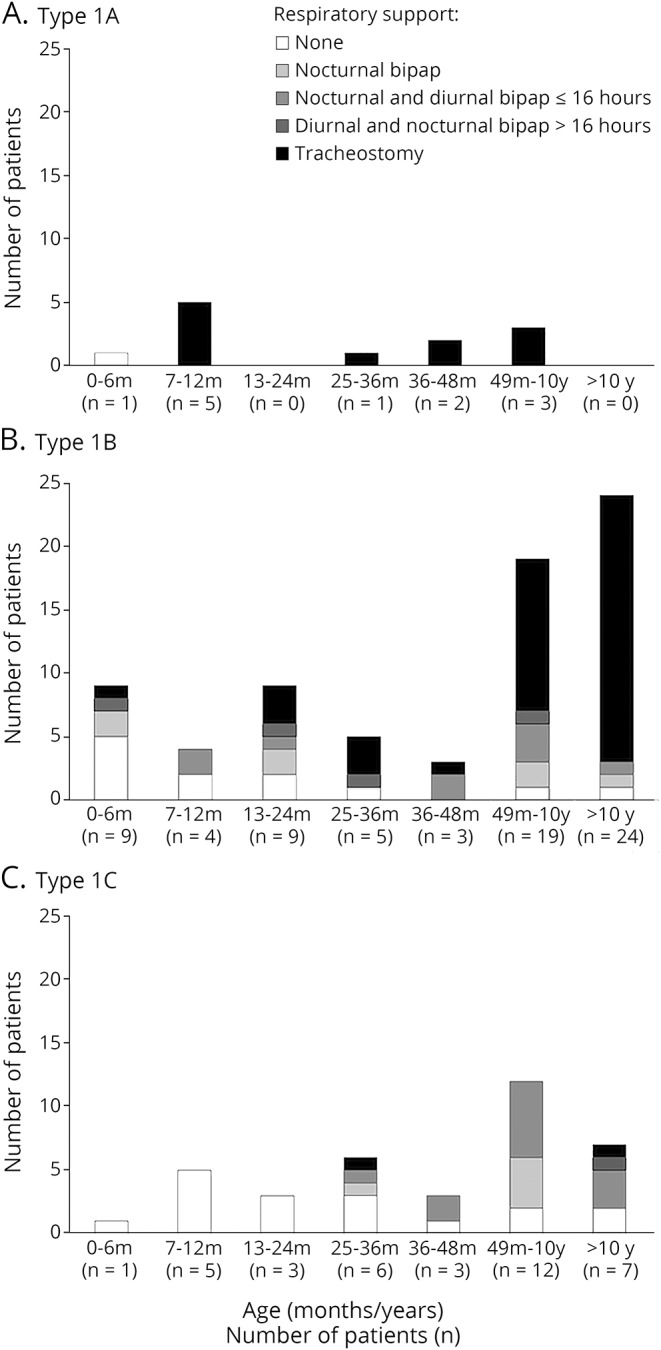
(A) Type 1A, (B) type 1B, and (C) type 1C. bipap = bilevel positive airway pressure.
Statistical analysis
Statistical analysis was performed with a dedicated software program, IBM SPSS Statistics release 22 (IBM, Armonk, NY). All data are presented as mean values and SD for continuous variables. One-way analysis of variance with Bonferroni post hoc test was used to compare continuous variables between groups. The level of significance was set at p = 0.05.
Results
One hundred twenty-two patients with type 1 SMA (66 female, 56 male) were included in the study. Their ages ranged between 3 and 266 months (mean 73, SD 64.7). The age at onset ranged between 0 and 8 months (mean 3.33, SD 1.9). Three of the 122 had 1 SMN2 copy, 75 had 2 copies, 25 had 3, and 2 had 4 copies. In 17 patients, a DNA sample was not available, and SMN2 copy number was not determined.
Classification
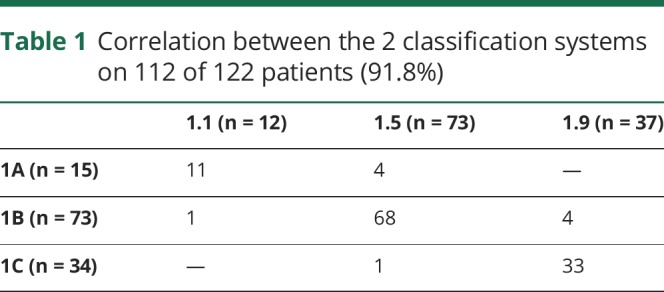
Twelve patients were classified as type 1.1, 73 as 1.5, and 37 as 1.9. Fifteen were classified as type 1A, 73 as 1B, and 34 as 1C. Table 1 shows the correlation between the 2 classification systems with 112 of the 122 patients (91.8%) having concordance between the 2 systems.
Table 1.
Correlation between the 2 classification systems on 112 of 122 patients (91.8%)
SMA subtype and ventilatory support
Figure 1 shows details of the level of respiratory care in the cohort according to the age when they were assessed and the subtype.
All the patients classified as 1.1 who survived after the age of 6 months had tracheostomy or >16 hours of ventilation.
Of the patients classified as 1.5, none of those younger than 1 year had a tracheostomy or >16 hours of ventilation. The number of tracheostomies progressively increased in the older patients.
In the patients classified as 1.9, only 3 of the 37 (1 seen at the age of 35 months, 1 at 12 years, and 1 at 13 years) had a tracheostomy or >16 hours of ventilation.
SMA subtype and nutritional difficulties
Figure 2 shows details of the level of nutritional difficulties in the cohort according to the age when they were assessed and the subtype.
Figure 2. Details of level nutritional difficulties by age and subtype.
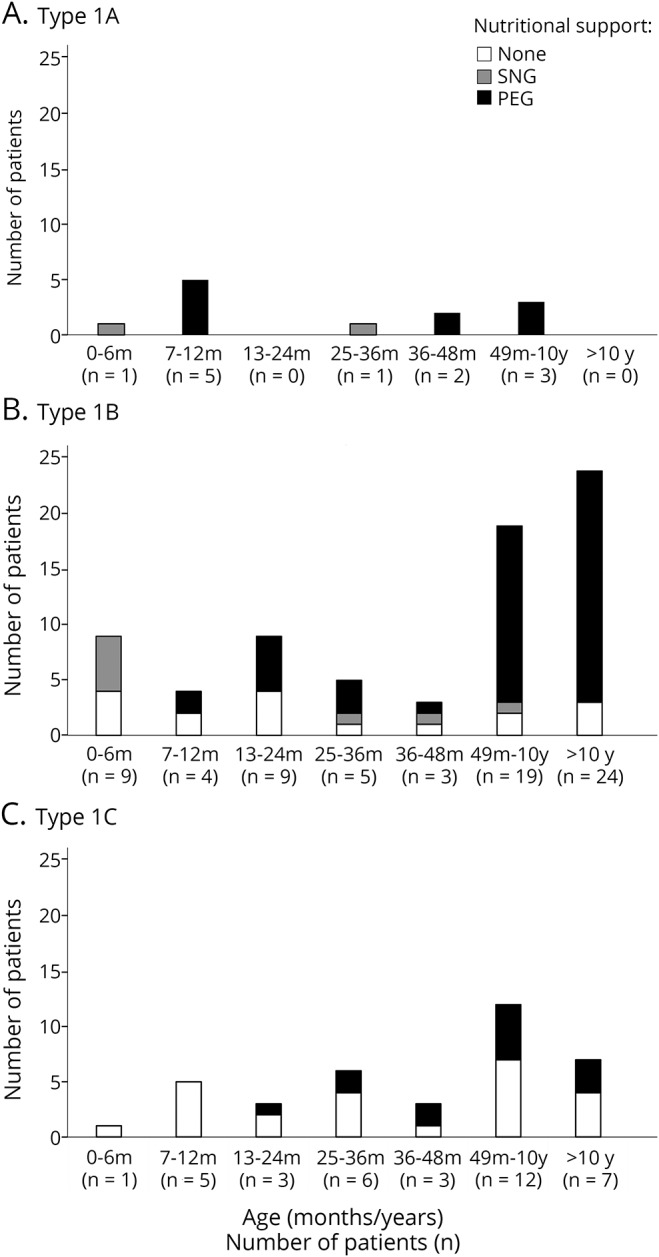
(A) Type 1A, (B) type 1B, and (C) type 1C. PEG = percutaneous endoscopic gastrostomy; SNG = nasogastric tube.
Of the patients classified as 1.1, with one exception, none survived after the age of 6 months without gastrostomy. In the patients classified as 1.5, 2 of the 4 assessed between 6 and 12 months of age had a percutaneous endoscopic gastrostomy (PEG). The number of PEGs progressively increased in the older patients. In the patients classified as 1.9, only one of the patients younger than 24 months had a PEG tube inserted. The number of PEGs progressively increased in the older patients, but even after the age of 4 years, less than 50% of the 1.9 patients had a PEG tube inserted.
Speech
Thirty-four patients (28%) acquired comprehensible speech, i.e., were able to produce at least short sentences that could be understood by the examiner and not just by the carers. Thirty were 1.9 patients (30/37 = 81% of the type 1.9 patients) and 4 were 1.5 (4/73 = 1.9% of the type 1.5 patients).
CHOP INTEND
The scores ranged between 0 and 52 (table 2). Patients with type 1.1, with one exception, never had scores above 8 with a mean score of 4.08 (SD 4.5); in patients with 1.5 SMA, the mean score was 9.42 (SD 10.13) and in patients with 1.9 was 25.67 (SD 12.5). As expected, with increasing age, there was a decrease in total scores in all the subgroups (figure 3).
Table 2.
CHOP INTEND and HINE scores according to the 2 classification systems and SMN2 copy number

Figure 3. Distribution of raw scores on the CHOP INTEND by age and SMA 1 subtype.

CHOP INTEND = Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders; SMA = spinal muscular atrophy.
There was a difference among patients with different subtypes of SMA according to the decimal classification (p < 0.001). Similarly, there was also a difference among patients with different subtypes of SMA according to the more recent classification (p < 0.001). The mean score in patients with 1A SMA was 5.4 (SD 7.2); in patients with 1B SMA, the mean score was 10 (SD 10.35) and in patients with 1C was 25.76 (SD 12.99). As expected, with increasing age, there was a decrease in total scores in all the subgroups.
Figure 4 shows the distribution of scores according to age and SMN2 copies, and that there are differences among patients with different SMN2 copy numbers (p < 0.001).
Figure 4. Distribution of raw scores on the CHOP INTEND by age and SMN2 copy number.

CHOP INTEND = Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders.
Hammersmith Infant Neurological Examination
All patients with 1.1 phenotype and 94.5% of the 1.5 had a total score of 0 (mean score 0.08, SD 0.36) (table 2), irrespective of their age. Scores of 1 or more were found in 74% of the 1.9 patients (mean score 1.57, SD 1.36) with higher scores in the younger group. There was a difference among patients with different subtypes of SMA, using both the decimal and the more recent classification (p < 0.001). Similarly, there was also a difference (p < 0.001) when patients were subdivided according to the number of SMN2 copies.
Discussion
In the last few years, there has been an effort to collect systematic prospective longitudinal studies in young infants with type 1 SMA.9,14–17 These studies have been important to provide natural history data for comparison to the results of the completed and ongoing clinical trials including young infants enrolled in the first months of life.4,18–20 The EAP has provided an opportunity to identify a wider group of type 1 patients, including older patients, and to assess their functional status. The involvement of genetic and rehabilitation centers, in addition to all the tertiary care centers, allowed us to identify a much higher number of type 1 patients than those registered in the Italian national registry for SMA. There were 152 type 1 patients identified, with an additional 21 newly diagnosed patients obtained during the 9 months after the program started. Because 122 of the 173 patients agreed to assessment and to discuss possible enrollment in the EAP, we were able to include more than 70% of the type 1 who could be identified in Italy.
The patients who refused intervention or to share their data were at the 2 extremes, either newly diagnosed for whom parents decided to follow a palliative approach, or some of the older patients with tracheostomy. For those patients, the families expressed concern that the risks associated with traveling and the time spent in hospital for the frequent procedures could outtake the possible benefits, because at that time, there was no evidence of the effect of nusinersen in patients older than those enrolled in the clinical trials.
While we cannot describe this as a population-based study, this is probably the closest attempt for a real-life prevalence study that, during the EAP, will prospectively collect functional data on a national level. The final cohort included 122 patients who were all assessed at baseline, before starting treatment. A number of patients were newly diagnosed or within 24 months of age, but more than 70% (85/122) were older than 2 years and 25% (31/122) were older than 10 years. We therefore included patients that are generally not reported in natural history studies.
Our data showed a marked difference between patients with the mildest phenotype compared to the others. As expected, in the more common and in the most severe phenotype, patients older than 2 years were, with few exceptions, on noninvasive ventilation, and with increasing age, more often had tracheostomy or >16 hours of ventilation. Similarly, more than 80% of the 1.1 and 1.5 patients older than 2 years had undergone gastrostomy, with increasing frequency with increasing age.
These results confirm previous observations that, after the age of 2 years, survival is generally only possible in the presence of tracheostomy/continuous ventilation and nutritional support.21,22 However, there were patients who had no need for tracheostomy or other ventilator or nutritional support after the age of 2. With few exceptions, these were patients with the mildest phenotype (1.9 or 1C). Because this was not a prospective longitudinal study, we cannot establish how these patients are representative of the whole cohort of 1.9, as we do not have the numbers of those who did not survive. All patients who survived after the age of 10 years without a tracheostomy or >16 hours of ventilation had relatively later onset, achieved head control, and had an overall milder disease course from onset. This was particularly true in 3 patients with 1.9 phenotype in whom the onset of clinical signs occurred soon after the age of 6 months. These patients had high scores on the CHOP and HINE, achieved head control, and, in 2 cases, also developed the ability to talk and had the best motor and respiratory outcomes. Because they never achieved the ability to sit unsupported, they were still strictly classified as type 1 and resemble the cases previously described by Dubowitz1 as borderline. This was not always related to a higher number of SMN2 copies; while 2 of the 3 had 3 SMN2 copies, the other had 2 copies.
The CHOP INTEND scores in patients older than 2 years were generally lower compared to those found in younger patients. As reported in a recent article describing CHOP INTEND scores in different type 1 subtypes in the first years after birth,17 we found that the CHOP INTEND can identify distinct levels of functional abilities in the 3 subgroups; patients with the mildest phenotype have relatively high scores compared to the others.
A similar trend was also observed on the HINE2 as patients with the mildest phenotypes were the only ones to have scores >2. In contrast, the HINE did not differentiate between 1.1 and 1.5, as the great majority had a score of 0, in keeping with our recent natural history data.16
Over the last few years, different classifications of type 1 SMA have become available and there are still some controversies on some aspects of classification, mainly related to the onset of the different subgroups. Using both the Dubowitz decimal classification23 and the more recently proposed classification,8 we found that the concordance between type 1.1 and 1A, type 1.5 and 1B, and 1.9 and 1C was overall very good (111/121 = 91%). The concordance was higher in the 1.5/1B and 1.9/1C subgroups (93% and 97%, respectively), while there was more discordance in the severe end of the spectrum. Onset at birth (n = 3) or within the first 2 weeks (n = 12), found in our 15 patients with type 1A, was not always associated with the most severe phenotype (1.1 in the decimal classification), as 4 of the 15 (1/3 at birth and 3/12 in the first 2 weeks) developed the milder, most typical phenotype (1.5 in the decimal classification). These patients, even though they had a very early onset of symptoms, had some distal movements and a less compromised respiratory function with less marked appearance of the involvement of the chest respiratory muscles, as observed in the more common type 1.5 phenotype. These findings suggest that caution should be used to classify severe patients only on the basis of onset and that a clinical assessment of severity of the motor and respiratory signs provides additional prognostic information.
Although patients with 3 copies had, as a group, better findings than those with 2 copies, as already reported in previous studies, this did not always hold true for individual cases. In our cohort, 2 patients had 4 copies and they did not show a milder phenotype. This information can be useful at the time there is the suggestion to use newborn screening algorithms focused on SMN2 copy number to identify patients with SMA eligible for treatment.
Our data provide a snapshot of functional abilities in a wide cohort of patients with type 1, subdivided according to subtypes and SMN2 copy number, including patients older than 10 years who are generally not included in type 1 natural history studies. Similar studies should be encouraged, because natural history studies in untreated patients will soon no longer be feasible as the prospectively collected natural history will henceforth reflect the increasing number of patients treated with nusinersen (Spinraza; Biogen, Cambridge, MA), and it is hoped that, in the near future, other treatments as well.
Another study reported systematic data on ventilatory and nutritional support in a large cohort of patients with type 1 SMA, but no detailed information on the subtypes or the SMN2 copy number was available.22
Our findings confirm that, after the age of 2 years, type 1 patients with early onset and those with the most common phenotype (1.1/A and 1.5/B) who survive, generally have tracheostomy and gastrostomy and have very low scores on the CHOP INTEND. More variability can be found in those with the mildest phenotype, who achieve head control. These data provide important baseline information to establish the possible efficacy of nusinersen in a real-life setting, in a wider population that will include patients who do not have the inclusion criteria used in the clinical trials.2,4
Glossary
- CHOP INTEND
Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders
- EAP
early access program
- HINE
Hammersmith Infant Neurological Examination
- PEG
percutaneous endoscopic gastrostomy
- SMA
spinal muscular atrophy
Footnotes
Editorial, page 337
Contributor Information
Collaborators: Italian EAP Working Group, Daniela Leone, Gloria Ferrantini, Beatrice Berti, Maria Carmela Pera, Nicola Forcina, Sara Carnicella, Giulia Norcia, Marco Piastra, Orazio Genovese, Alessandro Pedicelli, Paola Cimbolli, Antonio Versaci, Imma Rulli, Eloisa Gitto, Cristina Faraone, Stefania La Foresta, Maria Macrì, Giulia Colia, Anna Maria Bonetti, Adelina Carlesi, Renato Cutrera, Maria Beatrice Chiarini, Marta Ferretti, Alberto Garaventa, Giovanni Montobbio, Carlo Gandolfo, Valentina Iurilli, Paola Tacchetti, Emilia Bobeica, Valentina Lanzillotta, Alice Pirola, Sara Lupone, Elisa De Mattia, Elisa Falcier, Fabrizio Rao, Elisabetta Roma, Caterina Conti, Francesca Salmin, Cristina Grandi, Fausto Fedeli, Luca Mancini, Nicola Tovaglieri, Paolo Stoia, Maurizio Heinen, Valeria Cozzi, Beatrice Travaglia, Emma Mizzotti, Daniela Lauro, Luca Binetti, Anita Pallara, Simona Spinoglio, Maria Letizia Solinas, Grazia Zappa, Francesca Penno, Cristina Ponzanelli, and Jacopo Casiraghi
Author contributions
Design or conceptualization of the study: Marika Pane, Concetta Palermo, Sonia Messina, Valeria Sansone, Claudio Bruno, Enrico Bertini, Francesco Danilo Tiziano, Eugenio Mercuri. Analysis or interpretation of the data: Marika Pane, Concetta Palermo, Michela Catteruccia, Maria Sframeli, Emilio Albamonte, Marina Pedemonte, Adele D'Amico, Giorgia Brigati, Roberto de Sanctis, Giorgia Coratti, Simona Lucibello. Drafting or revising the manuscript for intellectual content: Marika Pane, Concetta Palermo, Sonia Messina, Valeria Sansone, Claudio Bruno, Michela Catteruccia, Maria Sframeli, Emilio Albamonte, Marina Pedemonte, Adele D'Amico, Giorgia Brigati, Roberto de Sanctis, Giorgia Coratti, Simona Lucibello, Enrico Bertini, Francesco Danilo Tiziano, Eugenio Mercuri.
Study funding
The study was supported by a Telethon UILDM grant (GSP 13002).
Disclosure
M. Pane has received funding from Biogen as a speaker in sponsored symposia. C. Palermo reports no disclosures relevant to the manuscript. S. Messina has received funding from Biogen as a speaker in sponsored symposia. V. Sansone has received funding from Biogen as a speaker in sponsored symposia. C. Bruno has received funding from Biogen as a speaker in sponsored symposia. M. Catteruccia has received funding from Biogen as a speaker in sponsored symposia. M. Sframeli reports no disclosures relevant to the manuscript. E. Albamonte has received funding from Biogen as a speaker in sponsored symposia. M. Pedemonte reports no disclosures relevant to the manuscript. A. D'Amico has received funding from Biogen as a speaker in sponsored symposia. G. Brigati reports no disclosures relevant to the manuscript. R. de Sanctis has received funding from Biogen as a speaker in sponsored symposia. G. Coratti has received funding from Biogen as a speaker in sponsored symposia. S. Lucibello reports no disclosures relevant to the manuscript. E. Bertini has received funding from Biogen as a speaker in sponsored symposia. Dr. Bertini has received funding as a member of advisory boards from Roche, Biogen, and AveXis. G. Vita has received funding from Biogen as a speaker in sponsored symposia. F. Danilo Tiziano has received funding from Biogen as a speaker in sponsored symposia. E. Mercuri has received funding from Roche, Biogen, and AveXis as a speaker in sponsored symposia and as a member of advisory boards. Dr. Mercuri has received funding as a member of advisory boards from Roche, Biogen, and AveXis. Go to Neurology.org/N for full disclosures.
References
- 1.Dubowitz V. Muscle Disorders in Childhood, 2nd ed. London: Baillière Tindall; 1995. [Google Scholar]
- 2.Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 2016;388:3017–3026. [DOI] [PubMed] [Google Scholar]
- 3.Chiriboga CA, Swoboda KJ, Darras BT, et al. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 2016;86:890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med 2017;377:1723–1732. [DOI] [PubMed] [Google Scholar]
- 5.Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med 2018;378:625–635. [DOI] [PubMed] [Google Scholar]
- 6.Messina S, Pane M, Sansone V, et al. Expanded access program with nusinersen in SMA type I in Italy: strengths and pitfalls of a successful experience. Neuromuscul Disord 2017;27:1084–1086. [DOI] [PubMed] [Google Scholar]
- 7.Dubowitz V. Chaos in classification of the spinal muscular atrophies of childhood. Neuromuscul Disord 1991;1:77–80. [DOI] [PubMed] [Google Scholar]
- 8.Finkel R, Bertini E, Muntoni F, Mercuri E; ENMC SMA Workshop Study Group. 209th ENMC International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7–9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord 2015;25:593–602. [DOI] [PubMed] [Google Scholar]
- 9.Finkel RS, McDermott MP, Kaufmann P, et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 2014;83:810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andreassi C, Angelozzi C, Tiziano FD, et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet 2004;12:59–65. [DOI] [PubMed] [Google Scholar]
- 11.Glanzman AM, Mazzone E, Main M, et al. The Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord 2010;20:155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glanzman AM, McDermott MP, Montes J, et al. Validation of the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND). Pediatr Phys Ther 2011;23:322–326. [DOI] [PubMed] [Google Scholar]
- 13.Haataja L, Mercuri E, Regev R, et al. Optimality score for the neurologic examination of the infant at 12 and 18 months of age. J Pediatr 1999;135:153–161. [DOI] [PubMed] [Google Scholar]
- 14.Kolb SJ, Coffey CS, Yankey JW, et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol 2017;82:883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolb SJ, Coffey CS, Yankey JW, et al. Baseline results of the NeuroNEXT Spinal Muscular Atrophy Infant Biomarker Study. Ann Clin Transl Neurol 2016;3:132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Sanctis R, Coratti G, Pasternak A, et al. Developmental milestones in type I spinal muscular atrophy. Neuromuscul Disord 2016;26:754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Sanctis R, Pane M, Coratti G, et al. Clinical phenotypes and trajectories of disease progression in type 1 spinal muscular atrophy. Neuromuscul Disord 2018;28:24–28. [DOI] [PubMed] [Google Scholar]
- 18.Aartsma-Rus A, Balabanov P, Binetti L, et al. Stakeholder collaboration for spinal muscular atrophy therapy development. Lancet Neurol 2017;16:264. [DOI] [PubMed] [Google Scholar]
- 19.Mendell JR, Goemans N, Lowes LP, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 2016;79:257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med 2017;377:1713–1722. [DOI] [PubMed] [Google Scholar]
- 21.Bach JR. Medical considerations of long-term survival of Werdnig-Hoffmann disease. Am J Phys Med Rehabil 2007;86:349–355. [DOI] [PubMed] [Google Scholar]
- 22.Bach JR, Saltstein K, Sinquee D, Weaver B, Komaroff E. Long-term survival in Werdnig-Hoffmann disease. Am J Phys Med Rehabil 2007;86:339–345. [DOI] [PubMed] [Google Scholar]
- 23.Dubowitz V. Chaos in the classification of SMA: a possible resolution. Neuromuscul Disord 1995;5:3–5. [DOI] [PubMed] [Google Scholar]