

Abstract
Ruthenium-based olefin metathesis catalysts, known for their functional group tolerance and broad applicability in organic synthesis and polymer science, continue to evolve as an enabling technology in these areas. A discussion of recent mechanistic investigations is followed by an overview of selected applications.
Introduction
Our laboratory has been engaged in the development and application of ruthenium-based olefin metathesis catalysts for over thirty years; the efforts of groups around the world and the chemistry of other metals, such as titanium, molybdenum, and tungsten, have also helped shape our thinking, both before and during this time period.1
The broad utility of ruthenium-based olefin metathesis catalysts rests upon their functional group, air, and moisture tolerance. In the mid-1980s, we overcame a synthetic challenge in polymer chemistry by demonstrating aqueous ring-opening metathesis polymerization (ROMP) using structurally-undefined polymeric Ru(II)-olefin complexes.2 This led to the discovery that strained olefins and Ru(II) provided a path to structurally well-defined ruthenium metathesis catalysts of the general structure shown in Scheme 1.3,4 Numerous variants (Figure 1) have emerged in the intervening years from our laboratory and others, with structural modification for enhancing catalyst initiation rate, turnover number, stereoselectivity, or lifetime. In our initial reports in the early 1990s, we used phosphine complex Ru-1-II to demonstrate ROMP2 and ring-closing metathesis (RCM) reactions in the presence of air and functional groups such as carboxylic acids, alcohols, aldehydes, and amine salts – a significant improvement when compared with known molybdenum catalysts.5 In that same timeframe, we developed procedures for the multi-kilogram-scale preparation of phosphine complex Ru-1-I,6–8 which accelerated its use in synthesis.
Scheme 1.
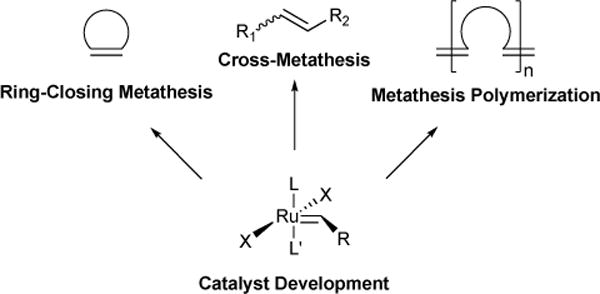
General Ru(II) catalyst structure and metathesis applications.
Figure 1.
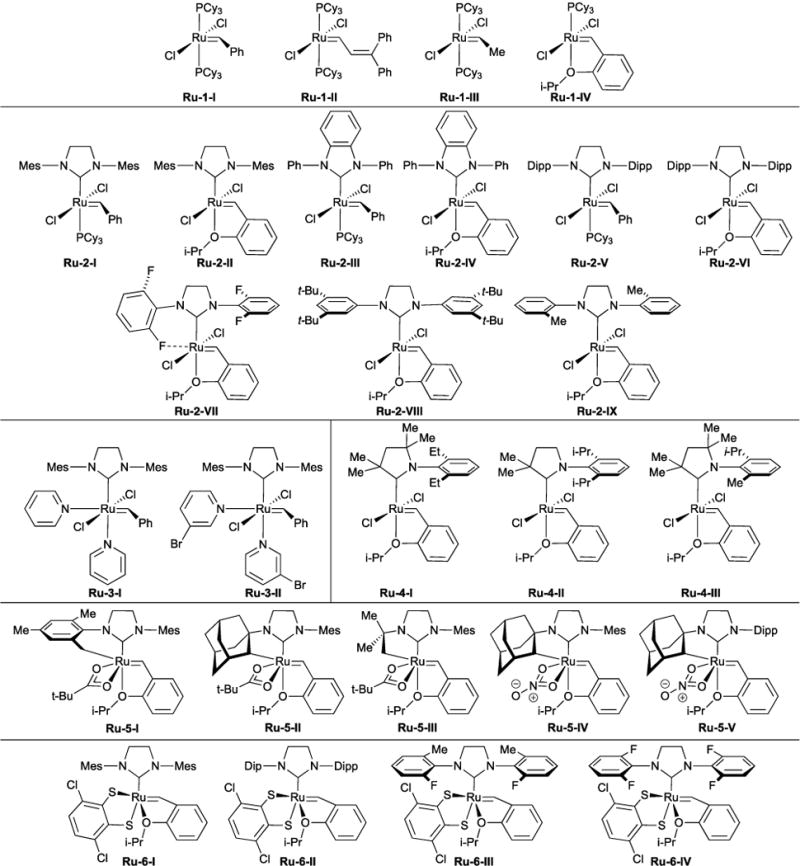
Select list of ruthenium olefin metathesis catalysts.
Ruthenium complexes with significantly improved properties were discovered by the early 2000s. Unsaturated N-heterocyclic carbene (NHC) complexes reported by Herrmann,9 Nolan,10 and Grubbs11 exhibited significantly greater metathesis activity and enhanced thermal stability. The saturated NHC complex Ru-2-I, disclosed by Grubbs and coworkers12 and Hoveyda’s phosphine-free variant Ru-2-II,13 have found widespread use in synthetic organic and polymer chemistry. Notably, Ru-2-I provided the first examples of selective cross-metathesis (CM) reactions.14 The NHC family of well-defined catalysts has also enabled several metathesis processes on an industrial scale. For example, Simeprevir, a hepatitis C treatment identified as an essential medicine by the World Health Organization, is prepared by RCM.15,16 In another example, a bio-refinery plant in Indonesia is currently using CM to process up to 180,000 metric tons of seed oil, providing access to olefins, oleochemicals, and specialty chemicals.17 This technology harkens back to the Phillips Triolefin Process of making ethylene and 2-butene from propylene or vice-versa – an early commercial cross metathesis process that paved the way for a broadly used and fundamentally new olefin transformation.1
Developments in metathesis chemistry are global in scope and have been exhaustively reviewed in 200318 and again in 2015.19 The aim of the present review is to provide a description of developments from our laboratory post-2005, including select applications from other laboratories. This review is divided into three sections: catalyst development and mechanistic studies, applications in organic synthesis, and in polymer chemistry.
2. Catalyst Development
2.1. Olefin Metathesis Catalytic Cycle
Extensive investigation into the olefin-metathesis mechanism have shown that the Chauvin mechanism20 is operative.21 The catalytic cycle (Figure 2) is initiated by dissociation of the phosphine in the ruthenium pre-catalyst to form an active 14-electron Ru-alkylidene intermediate (rate constant k1). The active Ru-alkylidene may either re-bind to the phosphine (rate constant k–1), inactivating the catalyst, or bind to an olefin in an η-2 fashion (rate constant k2), continuing the catalytic cycle. 2,2-cycloaddition between the η-2-bound olefin and the Ru-alkylidene (rate constant k3) results in the characteristic metallacyclobutane intermediate. Consequent 2,2-cycloreversion and olefin elimination produces the desired metathesis product and releases the active Ru-alkylidene. We have focused our mechanistic elucidation efforts on three areas: catalyst initiation kinetics (k1), olefin addition kinetics (k2) and stereochemistry, and metallacyclobutane intermediate stability, dynamics, and stereochemistry.
Figure 2.
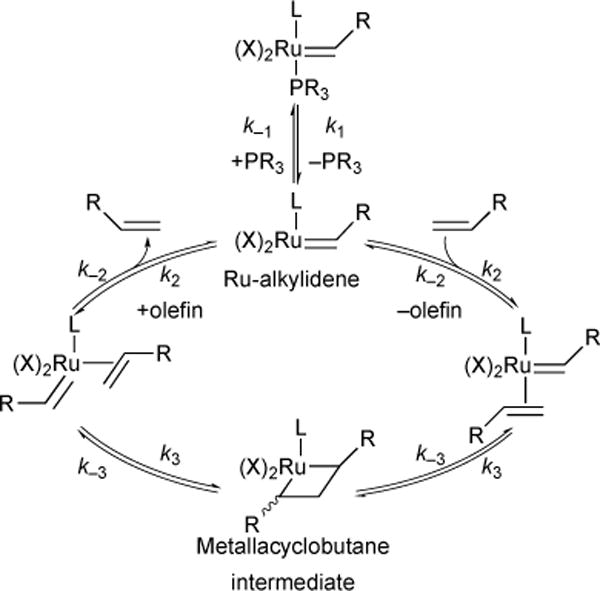
Generic ruthenium olefin metathesis catalytic cycle.
2.2. Perturbing Initiation Kinetics via Phosphine Substitution
Catalyst activity is affected by the rate of initiation/rebinding, olefin binding, cycloaddition, and catalyst decomposition. We sought to probe the effect of ligand substitution on initiation kinetics. Any modification to ancillary ligands affects all the above-mentioned elementary steps. However, close inspection of the catalytic cycle reveals that the active catalyst Ru-alkylidene is identical after the first cycle, regardless of the nature of the ruthenium pre-catalyst (Figure 3). Therefore, we reasoned that modifying the phosphine would only affect initiation kinetics without affecting the rate of the remaining steps in the catalytic cycle.
Figure 3.
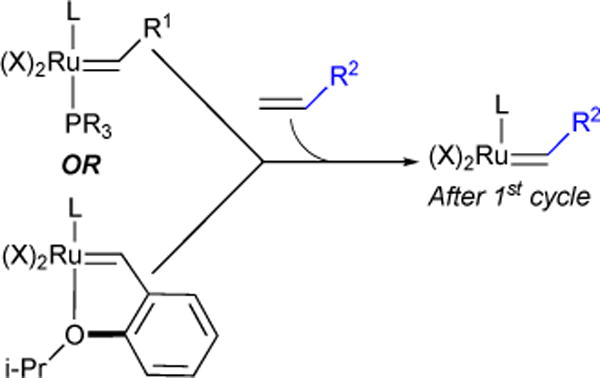
Active Ru-alkylidene is identical after first catalytic cycle, regardless of pre-catalyst structure.
Table 1 depicts the work our lab has done to probe the effects of phosphine substitution on the rate of catalyst initiation.22 We found that catalyst initiation is affected by both steric and electronic properties of the phosphine ligand. All other factors being equal, phosphine ligands with larger cone angles will dissociate more rapidly than those with small cone angles. Regardless of steric size, a weak donor ligand dissociates faster than a strong donor ligand. For example, a significant decrease in initiation rates is observed between Ru-2-I–PCy3 and Ru-2-I–P(n-Bu)3 (Entries I and II, respectively). The origin of this discrepancy arises from the difference in cone angles between the former (170°) and the latter (132°). In another example, Ru-2-I–P(Ph)2(OMe) has a ligand cone angle of 132°, but exhibits an initiation rate that is thirteen times larger than that of Ru-2-I–PCy3. In this case, the low donor strength of the P(Ph)2(OMe) ligand relative to PCy3 overrides the steric factor.
Table 1.
Effects of Ru-2-I phosphine substitution on initiation kinetics.

|
krel = k1 for each catalyst entry/k1 for catalyst in entry i
2.3. Side- vs Bottom-Bound Olefin Addition
Olefin binding may take two forms: one in which the olefin is bound cis to the L-type ligand (side-bound) and one in which the olefin is bound trans (bottom-bound, Figure 4). Both side- and bottom-bound complexes have been observed in the literature. Snapper and co-workers isolated bottom-bound complex 1 where the olefin is tethered via Ru-alkylidene moiety, suggesting bottom-bound mechanism.23 We isolated side-bound ruthenacyclobutane intermediate complex 2, suggesting olefin binding and 2,2-cycloadditon occurs via side-bound mechanism.24 In order to explore these modes of binding, we chose to use 1,2-divinylbenzene 3 due to its ability to chelate to the ruthenium center without undergoing ring-closing metathesis, as well as its expected slow homodimerization.25 We monitored via 1H NMR the reaction of diene 3 with pyridine-ligated ruthenium catalyst Ru-3-I and did not find evidence for the formation of the bottom-bound complex 4 (Figure 5). However, we observed two side-bound isomers 5a and 5b differentiated by the orientation of the η-2 bound chelated olefin at the ruthenium center. Although both were observed by 1H-NMR, 5b was isolated and characterized by X-ray crystallography.26
Figure 4.
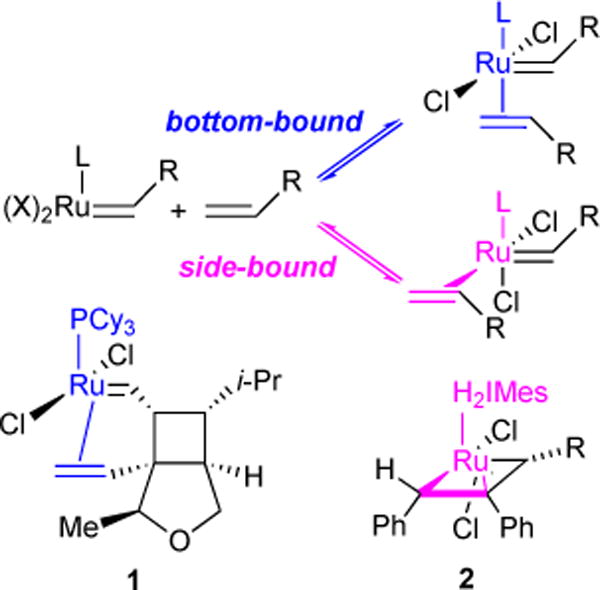
Side- vs. bottom-bound olefin addition.
Figure 5.
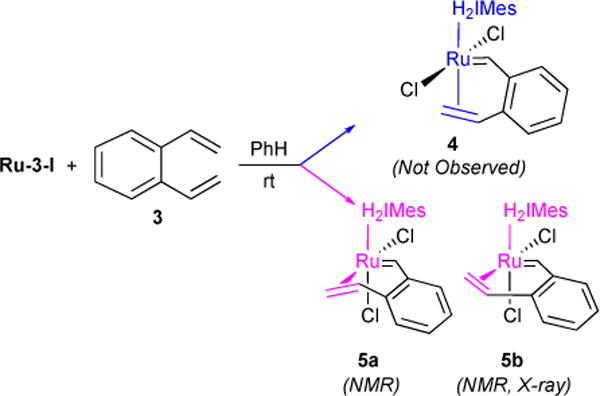
Evidence for side-bound olefin substrate binding.
2.4. Stability, Dynamics, and Stereochemistry of Metallacyclobutane Intermediate
While our previous work (vide supra) lent insights into the thermodynamic preference for olefin binding, the kinetic preference for subsequent metallacyclobutane formation was still largely elusive. Piers and co-workers reported the first direct observation of a ruthenium metallacycle.27 In this report, ruthenium-alkylidenium 6 was treated with 2.2 equivalents of ethylene at –50 °C, resulting in a C2v-symmetric ruthenacyclobutane 7 species observed by 1H-NMR (Figure 6). Their results suggested bottom-bound metallacyclobutane orientation.
Figure 6.
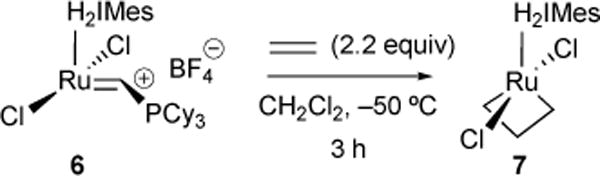
First direct observation of ruthenium metallacyle.
In our 2006 report, we prepared an unsymmetrical catalyst to investigate the preference for side- vs. bottom-bound metallacyclobutane formation, the dynamics of NHC ligand rotation and metallacycle formation/reformation, and the stereochemical orientation when substituted metallacycles are generated.28 We treated unsymmetrical ruthenium-alkylidenium 8 with ethylene (Figure 7A). The resulting ruthenacycle 9 exhibited a distinct two-proton signal at –2.66 ppm corresponding to the enantiotopic β-hydrogens. Furthermore, 2-D COSY and ROESY NMR data indicated that Ru-NHC rotation in 9 is sufficiently slow on an NMR-time scale at –40 °C. Furthermore, we reacted 6 with approximately 35 equivalents of propene. Three bottom-bound ruthenacyclobutane intermediates were observed (Figure 7B); 45% was ethylene-derived ruthenacycle 10, 29% propene-derived 11, and 2% butene-derived 12. Exchange cross-peaks at the α- and β-positions of 10 were indicative of non-productive metallacycle cycloreversion and re-formation occurring on an NMR time scale. In our more recent 2011 paper, we conducted similar studies with propene, 1-butene, and 1-hexene.29 We found that decreasing ethylene concentration favored an increase in populations of α-monosubstituted and α,α’-disubstituted ruthenacycles such as 11 and 12 (cis and trans), respectively. Moreover, trans-substituted metallacycles were favored over the cis counterpart by a factor of ca 2. Together with studies by Piers and coworkers, we showed that olefin cross metathesis reactions proceed via the formation of a bottom-bound, highly dynamic metallacyclobutane intermediate, with stereoisomeric preference for trans-1,3 substituted metallacycles.
Figure 7.
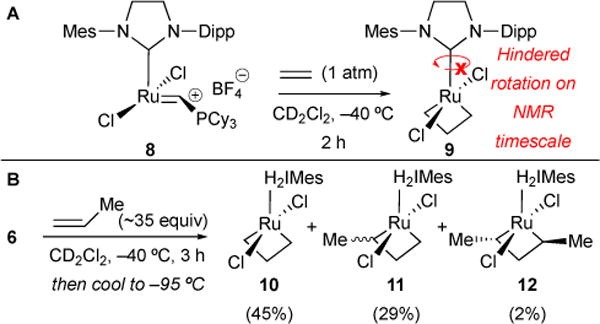
Bottom-bound, highly dynamic, ruthenacyclobutane intermediates observed.
2.5. Catalyst Decomposition
Despite advancements made toward developing functional-group-tolerant Ru-1 and Ru-2 catalysts for olefin metathesis, limitations such as high dilution, elevated temperatures, and extended reaction times were still required for closure of large rings. Thermal stability of the catalyst (i.e., the ability of a catalyst to avoid decomposition) plays a critical role in determining catalyst lifetime and turnover number.
We explored the first well-characterized decomposition products from the active Ru-methylidene intermediate of Ru-2-I in the presence and in the absence of ethylene (Figure 8A).30 In the absence of ethylene, we showed via 31P NMR spectroscopy and X-ray crystallography the formation of a dinuclear ruthenium complex 13. Three features are observed: (1) The ruthenium centers are bridged via a carbide, shown by a 13C chemical shift of 414.0 ppm and a carbide-hydride coupling constant of 10.4 Hz. (2) One ruthenium center uniquely consists of a hydride ligand (δ –8.6 ppm) and η6-binding to an N-heterocyclic carbene mesityl ring from the other ruthenium (δ mesityl proton 5.6 ppm). (3) A complete loss of a phosphine ligand from the starting complex Ru-2-I resulting in a byproduct with a 31P chemical shift of 34.5 ppm. Furthermore, complex 13 has been shown to promote olefin isomerization and migration, and we showed that p-benzoquinone additives can prevent this side reaction, if undesired (vide infra).31 In the presence of ethylene, we found that the major decomposition product of Ru-2-I is the C2-symmetric complex 14, isolated via X-ray crystallography.
Figure 8.
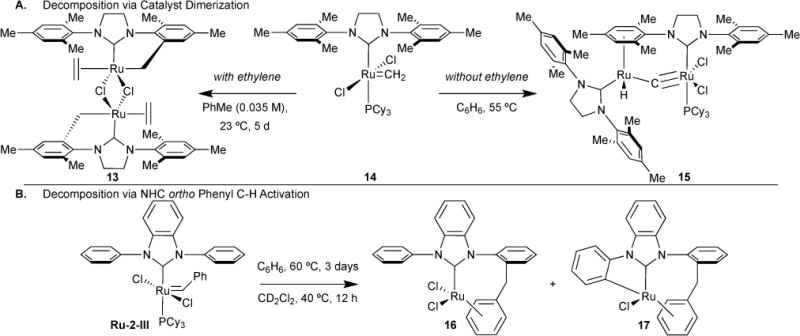
Ru-2 catalyst decomposition via dimerization into dinuclear complexes (A) and via C-H activation at the NHC ortho phenyl position (B).
Both Ru-2-III and Ru-2-IV are highly active for ring-closing metathesis.32 However, Ru-2-III decomposes faster than Ru-2-IV.33,34 We have shown that the Ru-2-III benzylidene carbon inserts into an ortho C–H on one of the N-phenyl rings (Figure 8B).35 Consequent η6-binding of ruthenium to the benzylidene phenyl group results in complex 16. We also showed that further ruthenium insertion into another ortho C-H can occur, resulting in complex 17. These studies indicated that substitutions at the ortho position may hinder unwanted C-H insertion reactions and hence reduce the propensity for catalyst decomposition.
2.6. Catalyst Development for Asymmetric Olefin Metathesis
There are three major classes of asymmetric olefin-metathesis: (1) asymmetric ring closing metathesis (ARCM), (2) asymmetric ring opening cross metathesis (AROCM), and (3) asymmetric cross metathesis (ACM). In ARCM, there is only one propagating alkylidene species. Consequently, to afford asymmetric catalysis, only the orientation of that propagating species, and enantiotopic olefin selection must be controlled. In AROCM, both the identity of the propagating species and its orientation must be controlled. Facial selectivity is however substrate-controlled when using norbornene as the substrate. In ACM, the identity of the propagating species, its orientation, and enantiotopic olefin selection all must be controlled, making this the most challenging class of asymmetric olefin metathesis. In this section, we summarize our work identifying ruthenium-based catalysts that can control for the factors described above.
2.6.1. Asymmetric Ring-Closing Metathesis (ARCM)
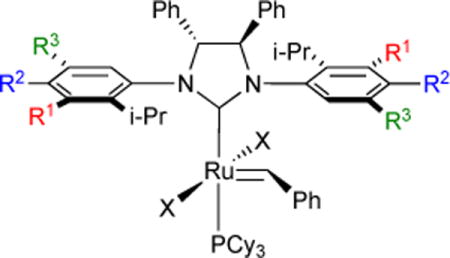
We initially showed that chiral ruthenium NHC catalyst Ru-2-X induced ARCM of prochiral trienes.36 Following this discovery, we explored a range of ruthenium olefin metathesis catalysts (Table 2), with varying substitution patterns at the NHC N-phenyl groups, for ARCM,37 using the conversion of 18 to 19 as a test case for catalyst screening (Table 3). High percent conversion is observed in all cases. However, Ru-2-XI and Ru-2-XVII afforded 90% ee; the highest reported for the reaction in this study.
Table 2.
Ru-2 asymmetric ring-closing metathesis catalysts.
| ||||
|---|---|---|---|---|
|
| ||||
| Catalyst | R1 | R2 | R3 | C |
| Ru-2-X | H | H | H | Cl |
| Ru-2-XI | H | H | H | I |
| Ru-2-XII | H | H | I-Pr | Cl |
| Ru-2-XIII | H | H | I-Pr | I |
| Ru-2-XIV | H | OMe | t-Bu | Cl |
| Ru-2-XV | H | OMe | t-Bu | I |
| Ru-2-XVI | I-Pr | H | H | Cl |
| Ru-2-XVII | I-Pr | H | H | I |
| Ru-2-XVIII | H | OMe | Me | Cl |
| Ru-2-XIX | H | OMe | Me | I |
Table 3.
Survey of some Ru-2 catalysts for asymmetric ring-closing metathesis.
| ||
|---|---|---|
|
| ||
| Catalyst | ee (%) | conv (%) |
| Ru-2-X | 35 | >98 |
| Ru-2-XII | 31 | >98 |
| Ru-2-XIV | 30 | >98 |
| Ru-2-XVI | 46 | >98 |
| Ru-2-XI | 90 | >98 |
| Ru-2-XIII | 84 | >98 |
| Ru-2-XV | 87 | >98 |
| Ru-2-XVII | 90 | >98 |
2.6.2. Asymmetric Ring-Opening Cross Metathesis (AROCM)
Following our initial exploration of Ru-2 catalysts for ARCM, we investigated the ability of the best ARCM catalysts for AROCM.38 Anhydride 20 was treated with 10 equivalents of styrene in DCM using 1 mol % of Ru-2-XVI, Ru-2-X, Ru-2-XII, or Ru-2-XIV (see Table 4). Out of the four catalysts, Ru-2-XIV was the most selective (76% ee). Additionally, no change in selectivity was observed when reaction was performed in various aprotic organic solvents.
Table 4.
Survey of some Ru-2 catalysts for asymmetric ring-opening cross metathesis.
| ||
|---|---|---|
|
| ||
| Catalyst | Product | ee (%) |
| Ru-2-XVI | ent-21 | 47 |
| Ru-2-X | ent-21 | 29 |
| Ru-2-XII | 21 | 62 |
| Ru-2-XIV | 21 | 76 |
We further explored the effectiveness of Ru-2-XIV and the diiodide variant Ru-2-XV for AROCM on a variety of substituted norbonene compounds. In all cases the E/Z ratio was close to 1:1, and ee values for the cis product were less than that for the trans. Within the trans product, enantioselectivities ranged from 57% to 81%. Aspects of the AROCM mechanism were elucidated from these studies: (1) E/Z ratios remained constant over 3 h, and secondary cross-metathesis is not observed, indicating that the E/Z ratio is a direct consequence of singular catalyst-substrate interactions. (2) Because the ee values of the E- and Z- products were significantly different, the nature of the important propagating species was inferred to be the ruthenium-benzylidene, not the ruthenium-methylidene species. (3) Further evidence supporting bottom-bound olefin binding was provided.
Given that catalyst Ru-2-XVI yielded the ent-21 and Ru-2-XIV yielded 21, and the propagating species is believed to be the ruthenium-benzylidene, we rationalized that a cis-coordinating species would lead to severe steric strain between the approaching norbonene and the Ru-NHC-phenyl substituents.
2.6.3. Asymmetric Cross Metathesis (ACM)
ACM reactions are the most challenging of the asymmetric metathesis reactions because the propagating species, its orientation, and enantiotopic olefin selection must be controlled. In our first report identifying an ACM catalyst, we employed substrate-control of both enantiotopic olefin selection and propagating species by using meso-diene substrates and an excess of the alkene metathesis reagent.38 Catalysts Ru-2-XVI and Ru-2-XII emerged as the most effective ACM catalysts, albeit in relatively moderate efficiency (ee up to 52 %).
2.7. Cyclic (Alkyl)(Amino) Carbene Ru-Catalysts
Bertrand and coworkers first reported the synthesis of olefin metathesis catalysts bearing cyclic (alkyl)(amino) carbenes (CAACs).39,40 The replacement of an sp2-hybridized nitrogen to an sp3-hybridized carbon results in: (i) greater σ-donor ability,41,42 (ii) a change in steric environment around the NHC, and (iii) a change in symmetry from C2v-symmetric in NHCs to CS or C1-symmetric in CAACs that possibly affects the microreversibility of the olefin-binding and cycloreversion steps in the metathesis catalytic cycle.43,44 Our investigations show that upon tuning of steric bulk of the CAAC ligand, CAAC-bearing Ru-4 catalysts are comparable to the NHC variants in ring-closing metathesis for the formation of di- and tri- substituted olefins.45 However, of note, we have shown that CAAC-bearing ruthenium olefin metathesis catalysts are remarkably effective for ethenolysis, achieving turnover numbers (TONs) of up to 340,000, at a catalyst loading of only 1 ppm (see Ethenolysis section below).46,47
2.8. Catalyst Development for Z-Selective Olefin Metathesis
The ability to selectively form the Z-olefin product in cross-metathesis reaction remains a significant challenge in metathesis research. In this section, we describe the recent progress made in our lab to design ruthenium-based Z-selective olefin metathesis catalysts. Hoveyda and Schrock reported the first example of a Z-selective olefin metathesis catalyst using tungsten and molybdenum. Z-selectivity was reported to be induced by a difference in size of the two apical ligands of the metallacyclobutane complex.48–53 From our lab, we reported a family of functional-group-tolerant Z-selective ruthenium based catalysts54,55 consisting of a chelating NHC ligand, derived from intramolecular carboxylate-driven C-H bond insertion of an N-bound substituent (Figure 1, Ru-5 catalysts).56,57 Of note, Ru-5-II and Ru-5-IV exhibit activity and Z-selectivity rivalling tungsten and molybdenum catalysts. Substituting the carboxylate ligand in Ru-5-II with a nitrato group such as in Ru-5-IV results in greater stability, Z-selectivity, and increase in turn-over number (TON) for homodimerization reactions. Altogether, the Ru-5 catalyst family has been used for olefin homodimerization,58 ring opening,59 ring-closing,60 cross61 metathesis, and ethenolysis.62 In this section, we highlight experimental and computational efforts by us and collaborators to elucidate the mechanism of action and catalyst decomposition.
We collaborated with Houk and coworkers to investigate the mechanism and origins of Z-selectivity in olefin metathesis using the chelated Ru-5-based catalysts.63 Structures were optimized using the B3LYP method and a mixed LANL2DZ/6-31G(d) basis set for Ru and other atoms, respectively. Both side- and bottom-bound mechanisms were computationally explored. In both mechanisms, four pathways, three involving a bidentate acetate and one involving a monodentate acetate, were computed, resulting in a total of eight possible pathways for the metathesis reaction. The minimum energy pathways for the side- and bottom- bound mechanism are shown in Figure 9. Computations revealed that the side-bound mechanism was energetically favored over the bottom-bound. The favored side-bound pathway proceeds via addition of ethylene to the ruthenium methylidene 22 to form the Ru-olefin π complex 23. Facile 2,2-cycloaddition (TS-24, ΔG‡ = 4.1 kcal/mol) leads to the exergonic metallacyclobutane intermediate 25 (ΔG = −4.9 kcal/mol). Ligand isomerization to intermediate 26 and consequent rate-determining 2,2-cycloreversion (TS-27, ΔG‡ = 11.4 kcal/mol) leads to ruthenium-alkyldene 28, where the resulting alkylidene is cis to the strong σ-donor adamantyl group.
Figure 9.
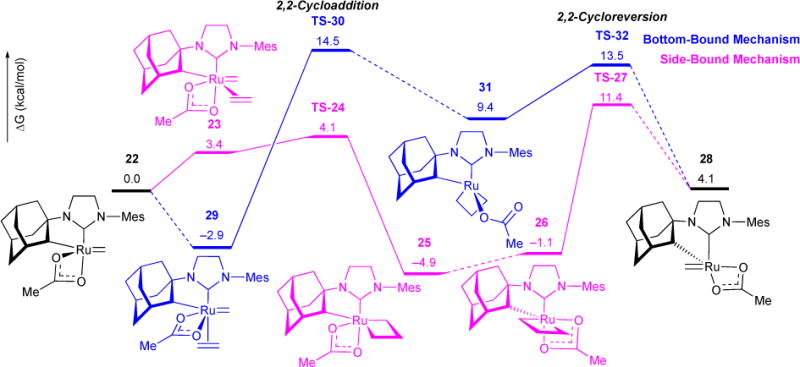
Minimum energy bottom-bound and side-bound mechanism for Z-selective olefin metathesis. Structures were optimized in B3LYP/LANL2DZ/6-31G(d) level of theory, and energies computed in M06/SDD/6-311+G(d,p) level of theory.
2.8.1. Side- vs. Bottom- Bound Selectivity
The bottom-bound Ru-olefin π complex 29 is more stable that the side-bound 23 by 6.3 kcal/mol. However, consequent bottom-bound 2,2-cycloaddition TS-30 is less stable than side-bound TS-24 by 10.4 kcal/mol. Moreover, all bottom-bound metallacyclobutane ground and transition state complexes proceeding TS-30 are higher in energy than the corresponding side-bound complexes due to two main factors: (i) Steric compression: Computations indicate that the strong preference for side-bound complexes can be readily explained via steric effects. The syn orientation of the metallacyclobutane and adamantyl groups in the bottom-bound complex TS-30 and TS-32 leads to unfavourable van der Waals interactions between adamantyl and metallacycle protons (Figure 10, Left). In the side-bound TS-24 and TS-27, the metallacyclobutane is positioned anti to the adamantyl group, and hence, no steric contact is observed between the two groups. (ii) d Orbital Backdonation: Both NHC and alkylidene ligands are strong σ-donors and π-acceptors. Consequently, the preferred conformation situates the Ru-NHC π* orbital in the same plane as the Ru-alkylidene bond (Figure 10, Right). In the side-bound mechanism, two different ruthenium d orbitals are involved in backdonation into the π* orbitals of the NHC and alkylidene. However, in the bottom-bound complexes, the same Ru d orbital is involved in backdonation between both the NHC and alkylidene π* orbitals (Figure 10).64–66 This weaker back-donation results in destabilization of the bottom-bound complexes.
Figure 10.
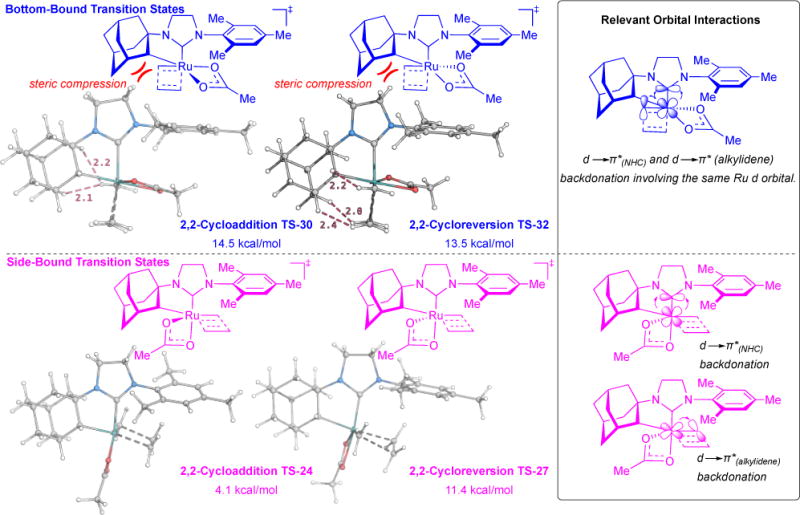
Computed 2,2-Cycloaddition and cycloreversion transition state structures for minimum energy side-bound and bottom-bound mechanisms (Left). Relevant orbital interactions depicting d orbital backdonation into π* of NHC and alkylidene moieties (Right).
2.8.2. Z- vs. E- Selectivity
The strong preference for the side-bound mechanism offers unique control via steric interactions for the Z- over the E- olefin product. Moreover, because affording Z-selectivity is necessarily a kinetically-controlled process, we investigated the origins of Z-selectivity by looking at the transition states for the Z/E-selectivity determining step, using propylene as our model substrate. Computations revealed that the TSs leading to E-olefin were less stable than those leading to the Z-olefin mainly because of unfavourable van der Waals interactions between one of the methyl groups in the (E)-2-butene product and the Ru-NHC mesityl group (Figure 11).
Figure 11.
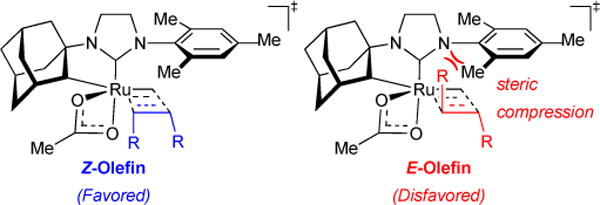
Z- vs E- selectivity model.
2.8.3. Decomposition of Z-Selective Catalysts
Catalyst decomposition studies shed light into improving catalyst stability, activity, and selectivity. Recently, we reported several unique catalyst decomposition products of Z-selective ruthenium-based catalyst Ru-5-II.67 Ru-5-II was exposed to carbon monoxide (CO) at –78 °C in an effort to afford a novel architecture characterized by a CO-ligated ruthenium catalyst complex with an intact alkylidene. Instead, complex 33, saturated with CO ligands and a covalent attachment of the N-adamantyl to the once-alkylidene carbon, was observed (Figure 12).68 We rationalized that the π-acidity of the CO ligands reduced the capacity for ruthenium π-backbonding with the alkylidene. To relieve this destabilization, the alkylidene inserts into the Ru-Cadamantyl bond to yield 33. This observation led us to propose that alkylidene insertion into the Ru-C bond of Ru-5 catalysts could still occur in the absence of CO ligands, and that consequent hydride elimination following alkylidene insertion would yield a complex incapable of productive metathesis.
Figure 12.
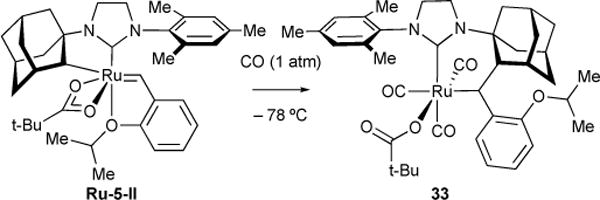
Decomposition of Ru-5-II via alkylidene insertion into Ru-C bond upon exposure to carbon monoxide.
In an effort to make a C-H activated analog of Ru-5-II, ruthenium complex 34 was treated with silver(I)pivalate. The expected formation of silver(I)chloride via ligand exchange with the pivalate was observed. However, instead of the desired catalyst architecture (i.e., C-H activated analog of Ru-5-II), X-ray crystal analysis revealed the formation of 35, formed from alkylidene insertion and subsequent β-hydride elimination (Figure 13). 1H NMR monitoring revealed that a monocarboxylate, mono chloride ruthenium alkylidene species was initially formed during the reaction. As this species disappeared, pivalic acid and a metastable C-H activated species was observed. This metastable species was slowly converted to a ruthenium hydride and subsequently, the η2-bound olefin ruthenium complex 35.
Figure 13.
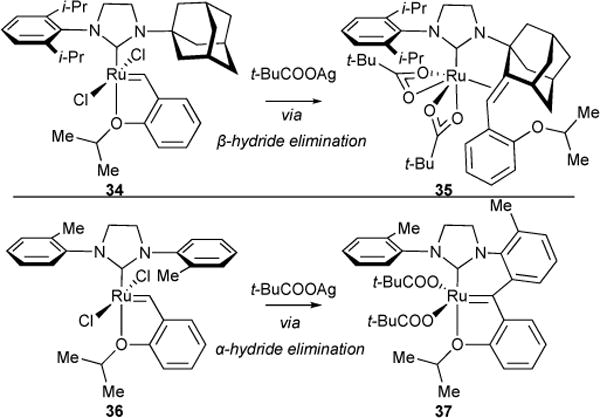
Treatment of 34 and 36 with silver(I) triflate in an effort to synthesize C-H activated ruthenium catalyst analogs of Ru-5-II yielded decomposition products 35 and 37, respectively.
In an effort to make another C-H activated ruthenium catalyst analog of 1 this time with a stronger, sp2-hybridized carbon-hydrogen bond,69 36 was treated with silver pivalate. Once again, the expected formation of silver(I)chloride via ligand exchange with pivalate was observed. However, in this case, a unique decomposition product 37 comprised of attachment of the benzylidene carbon to the ortho carbon of the N-aryl group. This suggests alkylidene insertion into a Ru-C bond followed by α-hydride elimination. Similar to the corresponding reaction with 34, 1H NMR monitoring showed the generation of pivalic acid as starting material alkylidene proton resonance disappeared, suggesting a C-H activation event had occurred prior to subsequent decomposition.
Ruthenium hydride formation via β-hydride elimination is further supported by the observed olefin migration in cross-metathesis reactions. Additives, such as p-benzoquinone, are typically used to prevent such migrations, albeit also leading to immediate catalyst decomposition.31 We showed that β-hydride elimination plays a role in the decomposition of C-H activated Ru-5 catalysts upon treatment with p-benzoquinone. Addition of this additive to Ru-5-II yielded disappearance of Ru-5-II, evidenced by disappearance of alkylidene resonance in the 1H NMR spectrum, and formation of ruthenium (0) dimer 44, observed by X-ray crystal analysis (Figure 14)
Figure 14.
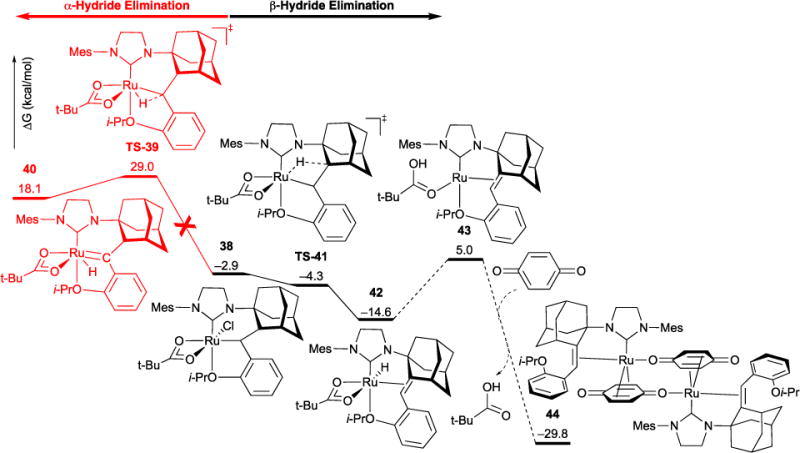
Computed α- vs β-hydride elimination pathways. The latter is favored over the former by 33.3 kcal/mol. Further decomposition of β-hydride elimination intermediate in the presence of p-benzoquinone leads to a dinuclear ruthenium complex. A Standard System for Catalyst Characterization
We collaborated with the Houk group to elucidate the two possible decomposition pathways (i.e., α- vs. β-hydride elimination) of Ru-5-II in the presence of p-benzoquinone (Figure 14). All electronic energies were calculated using M06/SDD/6-311+G(2d,p) on geometries optimized using B3LYP/LANL2DZ/6-31G(d) level of theory. Decomposition begins with the insertion of alkylidene into the ruthenium-carbon bond to form the thermodynamically favored alkyl ruthenium intermediate 38 (ΔG = –2.9 kcal/mol). From this intermediate, either α-hydride elimination product 40 or β-hydride elimination product 42 can form via the transition state structures TS-39 and TS-41, respectively. Computations reveal that free energy barrier leading to the former is 33.3 kcal/mol higher than that leading to the latter, indicating that β-hydride elimination is energetically preferred. Computations corroborate the experimentally-observed β-hydride elimination product. Following the formation of intermediate 42, reductive elimination of acetate leads to 43. Ligand exchange with p-benzoquinone via associative mechanism and consequent homodimerization leads to the highly exergonic decomposition product 44 (ΔG = –29.8 kcal/mol).
2.9. A Standard System for Catalyst Characterization
In 2006, we proposed a set of six reactions and conditions to establish a standard for comparing olefin metathesis catalysts.70 The reaction set included (i) three RCM reactions forming di-, tri-, and tetrasubstituted cyclopentenes, (ii) two CM reactions with coupling partners of variable reactivity, and (iii) a ROMP reaction using cyclooctene (Figure 15). Representative results for catalysts Ru-1-I, Ru-2-I, and Ru-2-II are shown in Table 5. This study established a means for benchmarking catalysts with respect to their activity, stability, and selectivity. The RCM results showed that the NHC-based catalysts are more efficient than the phosphine-based analogues, but also pointed out that no particular catalyst was superior for all reactions. Furthermore, closure of tetrasubstituted olefins was identified as a major challenge and one that was effectively dealt with in subsequent investigations. The CM reactions of allyl benzene and cis-1,4-diacetoxy-2-butene with NHC-based catalysts were rapid and, if given enough time for secondary isomerization, provided ca. 10:1 E/Z product selectivity. CM of electron-deficient methyl acrylate, known to be difficult with Ru-1-I, proceeded well with NHC-based systems Ru-2-I and Ru-2-II (each >80% conversion at 3 hours under the standard conditions). The standard ROMP reaction used 1,5-cyclooctadiene, whose polymerization could be conveniently followed by NMR spectroscopy when using a monomer to catalyst ratio of 1000:1. Here again, the NHC-derived catalysts were dramatically more reactive than the first-generation catalysts. In particular, the rapidly-initiating pyridine-derived Ru-3-II catalyzed a nearly instantaneous polymerization under the standard reaction conditions in less than 30 seconds.
Figure 15.
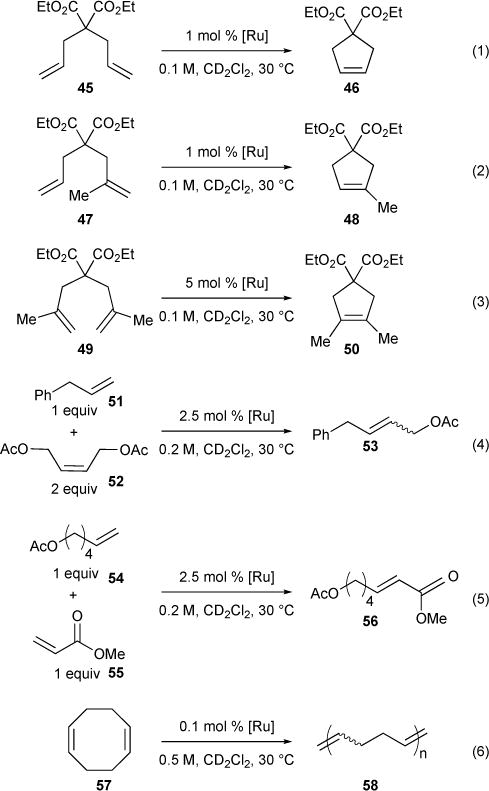
Set of six metathesis reactions for catalyst characterization.
Table 5.
Comparison of Ru Catalysts with Standard RCM, CM, and ROMP Reactions.
| Reaction (time) | Ru-1-I | Ru-2-I | Ru-2-II |
|---|---|---|---|
| 1 (30 min) | 66% | 97% | 99% |
| 2 (60 min) | 21% | 98% | 98% |
| 3 (4 d) | 0% | 17% | 6% |
| 4 (30 min) | 18 % | 79% | 72% |
| 5 (2 hr) | ~10% | 98% | ~90% |
| 6 (5 min) | ~5% | 98% | 99% |
3. Applications in Organic Synthesis
3.1. Ring-Closing Metathesis (RCM)
3.1.1. N-Boc-3-pyrroline Syntheses
We documented the facile RCM of N-Boc-diallylamine 59 (30 g) in a 2003 Organic Syntheses preparation using 0.5 mol% Ru-1-I to provide Boc-3-pyrroline 60 in 90-94% yield.71 Helmchen also reported an efficient large-scale (100 g) preparation, using lower Ru-1-III catalyst loading (0.1 mol%) and convenient product isolation after vacuum distillation of the crude reaction mixture.72 We have utilized this reaction for screening catalysts and found that reactions run in neat as-delivered substrate in the presence of as little as 0.05 mol% Ru-2-II proceeded in 87% yield (Figure 16).73
Figure 16.
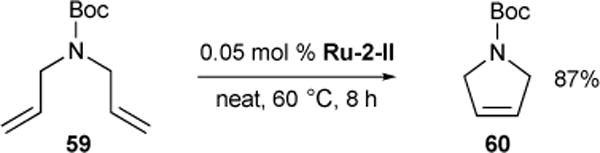
Synthesis of Boc-3-pyrroline.
3.1.2. Synthesis of Coumarins
Our group was interested in the synthesis of substituted coumarins both at the 3- and 4- positions (Figure 17). Synthesis of coumarin precursors 61 a-d is readily achieved via acylation of corresponding phenol. RCM of 61 using the highly active ruthenium catalyst Ru-2-I yielded coumarin 62a-d.74 Unsubstituted coumarin 62a was synthesized in high yields (89%). Comparatively high yields were maintained in the synthesis of mono-substituted coumarins 62c and 62d (88% and 74%, respectively). However, to achieve moderate yield (45%) of the di-substituted coumarin RCM product 62b, increased catalyst loading was required (10 mol%). At the time of publication of this report, this was one of the few successful cases of RCM to form tetra-substituted olefins, and was particularly remarkable given the electron-deficient nature of the olefins involved.
Figure 17.
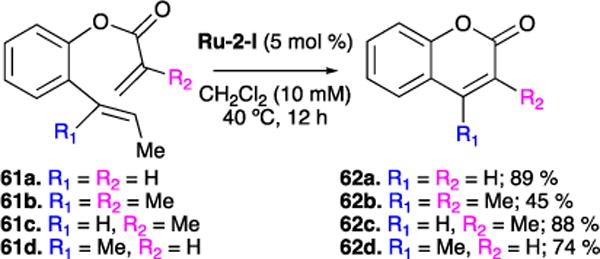
Synthesis of coumarins.
3.1.3. Synthesis of Rotaxanes
Rotaxanes75 are interlocked, dumbbell-shaped molecules in which one or more macrocycles are trapped. Much of the research involving rotaxanes focuses on the possibility of using these architectures in molecular machinery,76 such as molecular switches.77,78 Synthetic efforts have focused on kinetically-control synthesis,79 where the desired interlocking arises form an irreversible final transformation. Alternatively, our efforts have focused on taking advantage of the reversible nature of RCM reactions to impact thermodynamic control for rotaxane synthesis. We employed two strategies for synthesizing rotaxanes in our lab: (1) using the ruthenium alklylidene catalysts Ru-1-I and Ru-2-I for the synthesis of [2]-catenane80,81 and (2) using templating synthetic partners (or synthons) to facilitate ring-closing of macrocyclic structures.82–85
Our initial efforts were to capitalize on the significant ability of dibenzo[24]crown-8 to bind to R2NH2+ ions (Figure 18A).84 Treatment of macrocycle precursor 63a or 63b with ruthenium catalyst Ru-1-I resulted in the formation of macrocycle 64a or 64b in moderate yields (48% and 50%, respectively), as a mixture of E and Z isomers.86 In the presence of bis(3,5-dimethoxybenzyl)ammonium hexafluorophosphate 65, the corresponding [2]roxane 66a•PF6 and 66b•PF6 was formed in 73% and 30% yields, respectfully, also as a mixture of E and Z isomers. We attributed the increase in yield for 66a•PF6 over 64a to templating effects afforded by the ammonium ion. The product macrocycle complex was verified by 1H and 13C NMR spectroscopy, mass spectrometry, and single crystal X-ray analysis.
Figure 18.
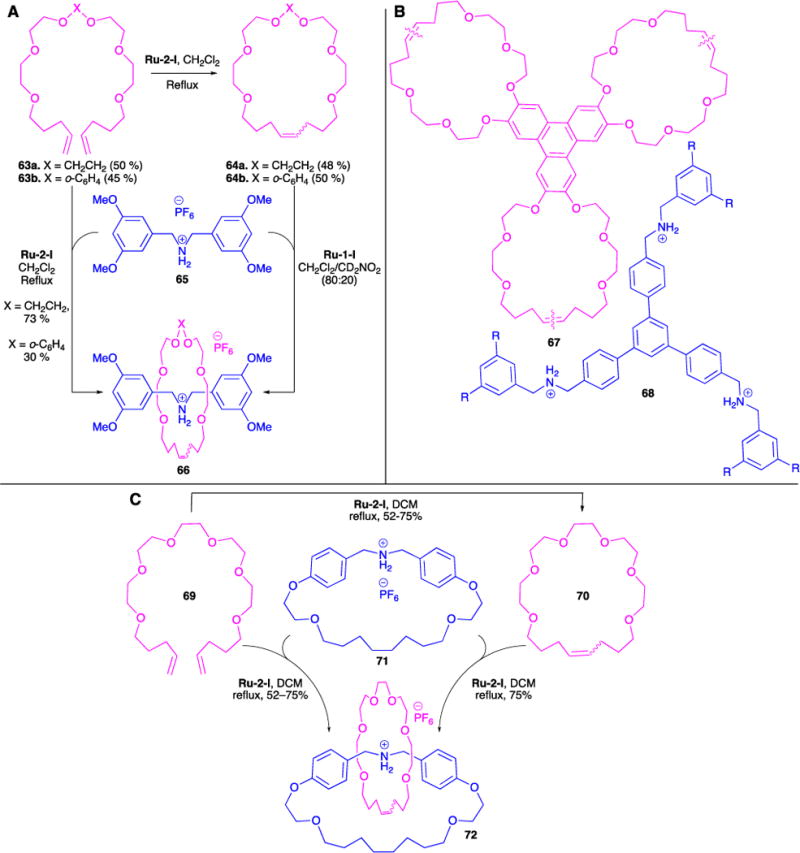
Synthesis of Magic Ring rotaxane macrocycles (A). Efficient preparation of a mechanically-interlocked bundle (B). Magic-ring catenation by olefin-metathesis (C).
In collaboration with Stoddart and coworkers, we extended this methodology to the RCM of a tritopic receptor (precursor of 67), in which three benzo[24]crown-8 rings are fused to a triphenyl core. This was templated by a trifurcated cation 68, where three dibenzoammonium ions are linked to a central benzenoid core (Figure 18B).87 We showed that by exploiting multivalency, achieved via non-bonding interactions between the three R2NH2+ and olefinic centers, as well as the dynamic covalent chemistry achieved via reversible ring-closing metathesis, a very stable (Ka = 107 mol L−1 in DCM) triply threaded, two-component superbundle 67 was formed.
The dynamic covalent nature of RCM was further employed in the clipping of the crown ether macrocycle to a cyclic secondary ammonium ion (Figure 18C).88 In this work, we were able to show that this clipped [2]catenane complex 72 can be formed from either a reaction between the macrocycle precursor 69 and the ammonium ion 71, or the closed macrocycle 70 and the ammonium ion 71. The latter of these further demonstrates the reversible nature of the olefin metathesis reaction in the synthesis of the dynamic [2]catenane.
3.1.4. Synthesis of peptide macrocycles
Intramolecular RCM is a useful method for altering the conformational and metabolic stability of α-helical peptides. The first examples of RCM-derived peptide turn and helical constraints were discovered in the mid- to late 1990s at Caltech.89–91 Later work by Verdine and co-workers showed that RCM of an olefin on the side-chain of a peptide residue position (i) with another side-chain olefin that is four (i + 4) or seven (i + 7) residues away serves to rigidify the peptide α-helix backbone by approximately one or two turns, respectively.92 By 2004, these ‘stapled peptides’ were shown to disrupt protein-protein interactions, have enhanced cell permeability and binding affinities, as well as enhanced bioavailability when compared with acyclic peptides.93 This work led to a commercialized venture (Aileron Therapeutics) seeking to develop metathesis-constrained helical peptide-based therapeutics targeting metabolic disorders and cancer.17,94
As mentioned earlier, RCM-based therapeutics have been demonstrated as a successful treatment for hepatitis C viral (HCV) infections.15,16 Ciluprevir 73 (BILN 2061) and Simeprevir 74 (Olysio®, TMC 345) are peptide-based macrocyclic protease inhibitors whose syntheses require efficient RCM reactions to form 15- and 14-membered rings, respectively (Figure 19). Boehringer-Ingelheim’s process development with Ciluprevir has been publically disclosed and has served as a case study for optimizing challenging RCM reactions in the large-scale (>400 kg) synthesis of pharmaceuticals.15 Details of the RCM process for making Simeprevir, which is presently being prescribed, remain in the patent literature.95
Figure 19.
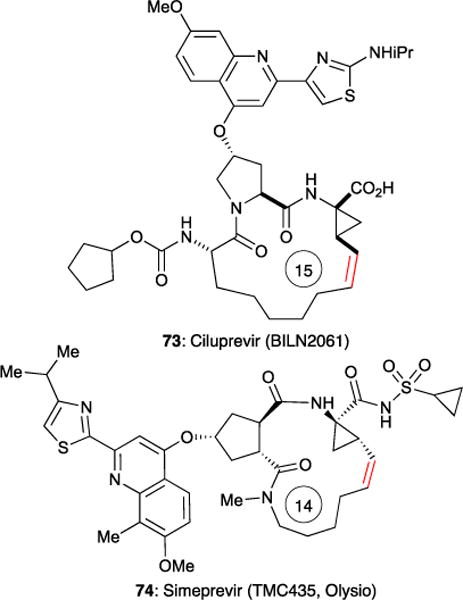
Structures of HCV protease inhibitors Ciluprevir and Simeprevir (trade name Olysio). Red olefin bond indicates site of RCM.
In collaboration with Toniolo and coworkers, we developed minimal RCM constraints for the 310-helix.96 An octapeptide with the sequence Boc-Aib-Aib-Aib-L-Ser(Al)-Aib-Aib-L-Ser(Al)-Aib-OMe was used in this study. We chose this sequence because the α-aminoisobutyric acid (Aib) residue has been shown to largely populate 310-helices.97–99 Reaction of the octapeptide using Ru-2-I yielded 18-membered cyclic peptide 75 (93% yield) with unusual high E-selectivity (>20:1 E/Z) (Figure 20). This 310-helical structure was validated by X-ray crystallography and circular dichroism (CD) spectra.
Figure 20.
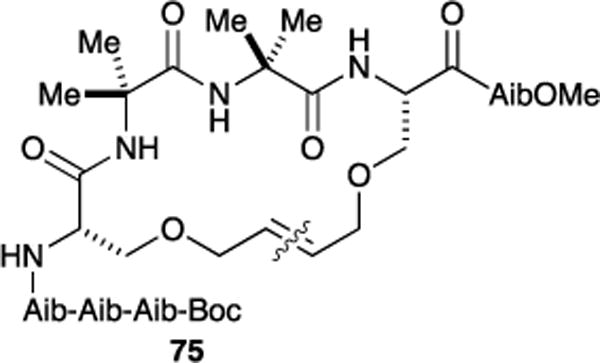
Synthesis of macrocyclized peptide constrained for the 310-helix. Wavy line across olefin bond indicate site of RCM.
In collaboration with Kwon and coworkers, we have further used this ring-closing metathesis protocol to synthesize a variety of cyclic amide N-alkylated peptoids of various ring sizes on a resin bead (Figure 21).100 These peptoids are more resistant to proteolytic cleavage101 and exhibit enhanced cell permeability102 compared to peptides, making this class of peptidomimetic compounds attractive for pharmacokinetic studies. We employed the ‘one-bead two-compound’ strategy103 with the RCM approach on a solid phase for the construction of cyclic peptoids. We screened ruthenium catalysts Ru-1-I, Ru-2-I, and Ru-2-II to determine which was most effective in the RCM to the cyclic peptoid 76. Under microwave conditions, Ru-1-I and Ru-2-I afforded 76 in low yields (10% and 20%, respectively). Notably, Hoveyda-chelate ruthenium catalyst Ru-2-II afforded the macrocycle in 80% yield. Encouraged by these results, Ru-2-II was employed in the solid-phase synthesis of tetramer to heptamer cyclic peptoids (77a–d in Figure 21). High yields, calculated as transformation efficiency as determined by HPLC, were observed (70-95% in DCM, 40 °C). However, small amounts of dimeric and metathesis products between cyclic and linear peptoids were also observed. Ring sizes of 16- to 25-membered cyclic peptoids were successfully synthesized, making this RCM-based approach a valuable addition to the toolbox of methods for cyclic peptoid synthesis.
Figure 21.
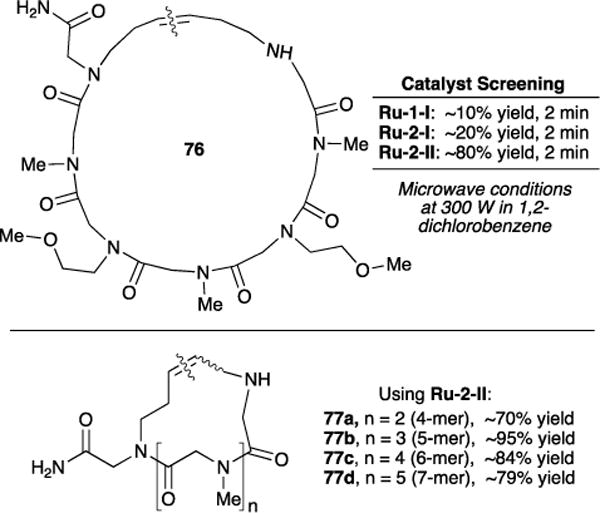
Solid-phase synthesis cyclic peptoids via RCM approach. Cyclic peptoids were cleaved from resin bead using trifluoroacetic acid (TFA). Wavy lines across olefin bond indicate site of RCM.
3.1.5. Z-Selective RCM
Using the newly developed Z-selective ruthenium-based catalysts (i.e., Ru-5 catalysts in Figure 1), we disclosed the Z-selective macrocyclic RCM (Table 6).60 RCM using Ru-5-V yielded a variety of macrolactones, macrolactams, macrocyclic ketones, acetals, and alcohols in yields ranging from 30–70%. Some general observations were gleaned from this study: (1) Dilute conditions and elevated temperatures were required to curb competing CM and oligomerization, and favor macrocyclic RCM. (2) Macrocyclic RCM to form large 16-, 17-, and 20- membered lactones gave the highest yields (see macrolactones 80 (72%), 82 (71%), and 81 (75%), respectively). (3) Z-selectivity was high for all macrocyclizations, although masking of ketones and alcohols were necessary to achieve higher Z-selectivity (i.e., 83 (68% Z) vs 84 (85% Z), and 85 (65% Z) vs 86 (75% Z), respectively). Furthermore, because the microscopic reverse of RCM is ethenolysis reaction, we successfully isolated E-macrocycles via selective ethenolysis of Z-macrocycle isomer from an E/Z mixture using the same catalyst.
Table 6.
Z-selective macrocyclization using Ru-5-V.
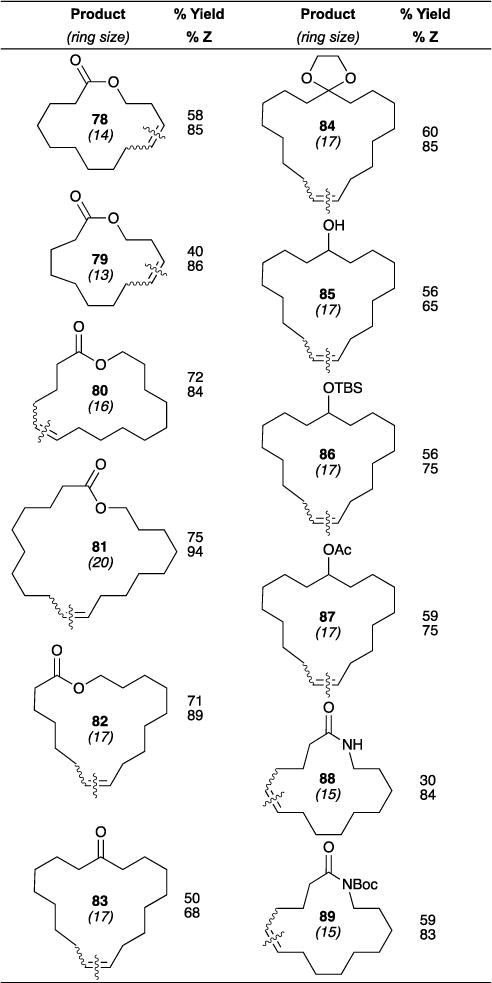
|
We have investigated Z-selective RCM of olefinic amino acids for the synthesis of “stapled” peptides of varying sizes.104,105 Prior to this report, the ability to control olefin geometry (i.e., E vs Z isomers) was a challenge during synthesis, and separation of the isomers were infeasible. Upon catalyst screening, we show that nitrato-ligated cyclometalated Ru-5-IV and Ru-5-V catalysts can be used to constrain resin-supported peptides from the i to the i + 2, i + 3, i + 4, and i + 7 residue positions. In search for an optimal resin, we found that resins bearing hydrophilic linkers, such as MBHA, led to increased conversions. Under optimized conditions, we synthesized the one α-helical turn-favored i to i + 4 peptide macrocycle 92 at 75% conversion and >90% Z-selective, after subjecting the resin-bound precursor to two cycles of catalyst addition. We investigated the synthesis of 93, a peptide constrained at the i, i + 7 positions favoring two α-helical turns. Under optimized conditions, 85% conversion and >90% Z selectivity was achieved. Efforts to synthesize the Z isomers of the more constrained i to i + 2, or i to i + 3 olefin crosslinks proved more challenging.105 Two example of such efforts – RCM to form 90 (41% yield, 93% Z) and 91 (30% yield, 90% Z) resulting in the desired product, albeit in low yields. Lastly, we also show that the ring-closing metathesis-ethenolysis approach to isolating E- and Z- macrocycles can also be used in the stereoselective synthesis of E- and Z- macrocyclic peptides.
3.1.6. Asymmetric RCM (ARCM)
A considerable amount of our efforts have gone towards ARCM catalyst development and synthetic applications. Products of ARCM are useful in target-oriented synthesis since two differentiated olefins are present in the final enriched product. We recently reported the ARCM of prochiral trienes (Table 7).106 Catalyst screening showed that nitrato-chelate cyclometalated catalyst Ru-5-IV (see Figure 1) was most effective in affording reactivity and selectivity. Some general observations were gleaned from the study: (1) Monosubstituted olefins cyclize readily, resulting in high yields. (2) Increasing bulk from dimethyl siloxy (94) to diphenyl siloxy (95) resulted in slower cyclization. (3) Trisubstituted olefins (96) did not undergo ring closure. (4) Saturated nitrogen containing heterocycles (98 and 100) were formed in high yields and enantioselectivities.
Table 7.
Asymmetric ring-closing metathesis with Ru-5-IV.
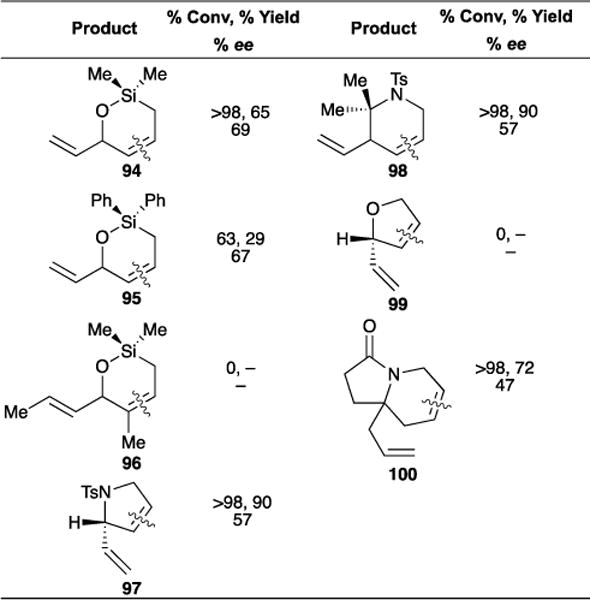
|
3.1.7. Tetrasubstituted Olefins
Ring-closing metathesis to form hindered tetrasubstituted olefins using ruthenium-based catalysts is a synthetic challenge. In the course of our work on Ru-catalyzed enantioselective metathesis, we unexpectedly isolated a five-membered ring containing a tetrasubstituted olefin when using a ruthenium catalyst lacking ortho substituents on the NHC N-phenyl rings. We then reported a series of ruthenium catalysts (Ru-2-VII, Ru-2-IV, Ru-2-VIII, and Ru-2-IX), with NHC phenyl rings with reduced bulk at the ortho position that were efficient in inducing ring-closing of tetrasubstituted olefins.32,107 Whereas ring-closing using Ru-2-I took 24 hours to achieve poor to moderate conversion of 1,1-disubstituted substrates, with Ru-2-IX 88-95% conversion was observed in <1 hour for many of the reactions. For example, the standardized reaction 3 in Table 5 proceeded in 100% isolated yield using Ru-2-IX (5 mol %) in benzene at 60 °C within an hour.107 We proposed that the remarkable increase in catalytic activity is a result of the reduced steric bulk around the NHC ligands, thus improving the ability of the catalyst to accommodate the steric bulk of tetra-substituted olefins.
In collaboration with Stoltz and coworkers, we reported an efficient and general strategy for the total synthesis of (+)-elatol 103, a member of the chamigrene family, which is a subclass of sesquiterpenes.108 Elatol has been shown to display antibiofueling, antibacterial, and antifungal activity, as well as cytotoxicity against HeLa and Hep-2 human carcinoma cell lines. We harnessed methodology developed in both of our labs for synthesizing elatol, namely enantioselective decarboxylative allylation of vinylogous ester derivatives and RCM of chlorinated, tetrasubstituted olefins using the newly discovered Ru-2-IX. When α,ω-diene 101 was subjected to RCM using Ru-2-Z, the desired fully-substituted chloroalkene 102 was formed in 97% yield. Subsequent acid-mediated elimination and hydrolysis, bis-bromination, and DIBAL reduction yielded (+)-elatol 103 (Figure 23).
Figure 23.
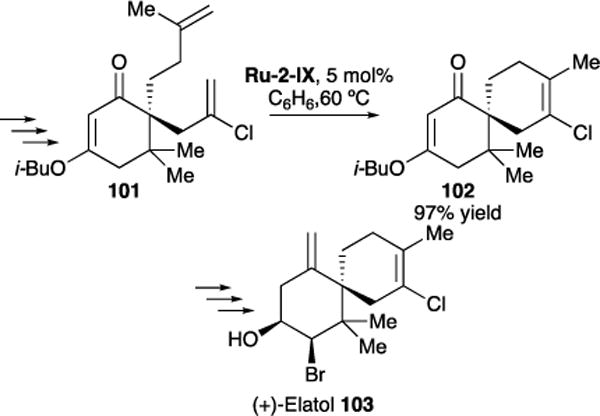
Using RCM for the synthesis of (+)-Elatol 103.
3.2. Cross-Metathesis (CM)
Over the years, olefin cross metathesis has become a powerful technique in organic chemistry for synthesizing complex, functionalized olefins from simple alkene precursors.109 However, in the early 2000’s, cross metathesis was underrepresented in the literature compared to ring-opening metathesis polymerization and ring-closing metathesis due to the difficulty in achieving product selectivity, stereoselectivity, and high catalyst activity in effecting productive metathesis. The enthalpic driving force, such as ring strain that drives ring-opening cross metathesis, or the entropic driving force such as the intramolecular process for ring-closing metathesis is notably absent in cross-metathesis reactions. Despite these challenges, cross metathesis is now regarded by synthetic organic chemists as a reliable method for functionalizing simple and complex olefins, as well as for complex fragment coupling reactions. For example, since the introduction of acrolein-based CM reactions in 2000110, α,β-unsaturated aldehydes and related derivatives have been utilized in these three applications (Figure 24). In their efforts to further develop efficient syn-1,3 diol syntheses, Evans and coworkers have reported a high-yielding coupling of acrolein with homoallylic alcohol 104, a reaction catalysed by Ru-2-II and proceeding to form olefin 105 in near-quantitative yield after in situ alcohol protection (Figure 24A).111 Krische and coworkers’ 20-step (+)-roxaticin total synthesis used acrolein and Ru-2-II to functionalize an advanced intermediate to form olefin 106 in 52% yield (Figure 24B).112 Finally, advanced intermediate 109 in Phillips and coworkers’ norhalichondrin B synthesis was prepared in 62% yield by Ru-2-IX coupling of enone 107 with allylic alcohol 108 (2 equiv) at elevated temperatures in 1,2-dichloroethane (Figure 24B).113
Figure 24.
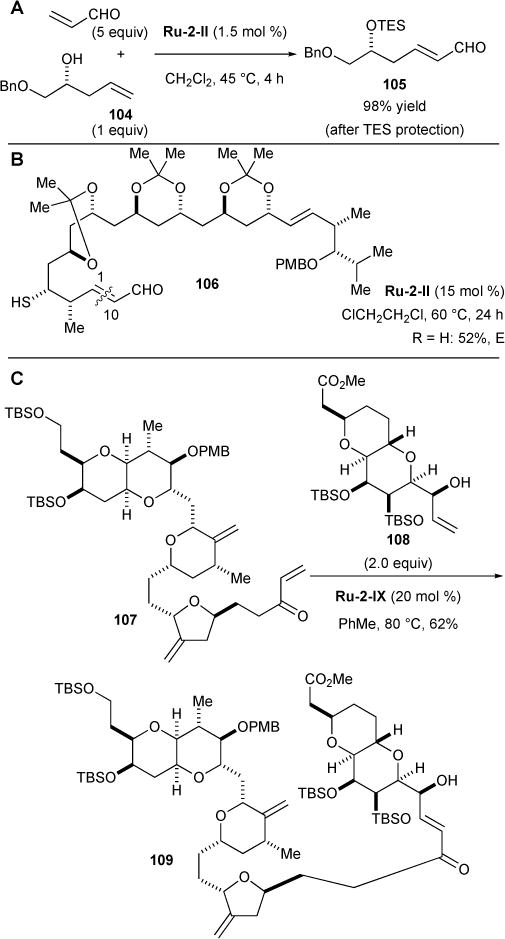
Examples of cross metathesis (CM) reactions used in natural product or natural product-inspired syntheses.
3.3. A General Model for Selectivity in Olefin Cross Metathesis
We developed rules for predicting product selectivity and stereoselectivity in olefin cross metathesis reactions.25 We investigated several classes of olefins, including styrenes, secondary and tertiary allylic alcohols, and generated four classifications of olefins based on their ability to undergo homodimerization (Figure 25): (1) “Type I” olefins are those that undergo rapid homodimerization, (2) “Type II” olefins undergo slow homodimerization, (3) “Type III” olefins do not undergo homodimerization, and (4) “Type IV” olefins are inert to cross metathesis and do not deactivate the ruthenium catalyst. Several olefin cross metathesis reactions performed with olefins within and between categories led to three rules for predicting product selectivity.
Figure 25.

Olefin categories and rules for selectivity in cross metathesis (CM) reactions.
3.3.1. Non-selective cross metathesis between two olefins of type I (Figure 26)
Figure 26.
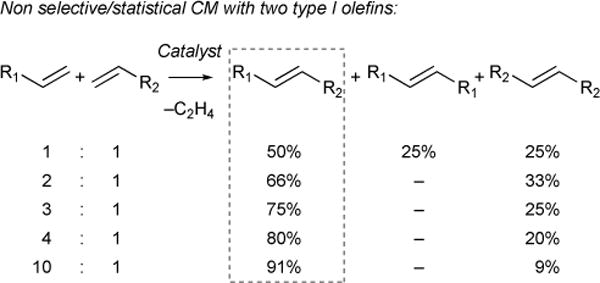
Statistical cross-metathesis with type I olefins as a function of relative stoichiometry.
With two type I olefins, the rates of homodimerization are similar and reactivity of resulting homodimers and cross products are high. In this scenario, equilibration of cross products with homodimers yields a statistical product mixture. To afford cross-metathesis product selectivity of over 90%, an excess of 10:1 for one olefin precursor over the other must be used.
An example of nonselective cross metathesis between type I olefins can be readily observed in the reaction between allylbenzene 110 and allylic alcohol 111 using ruthenium catalysts Ru-1-I or Ru-2-I (Figure 27). 2:1 equivalent of 111 to 110 yielded cross-metathesis product 112 in 80% yield.
Figure 27.
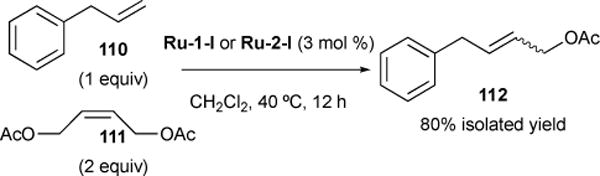
Nonselective CM with two type I olefins.
3.3.2. Selective Cross-Metathesis
In the same vein, any cross-metathesis reaction between olefins of the same type will yield non-selective products. Therefore, to avoid this inefficiency, one can design a cross-metathesis reaction with olefins of two different types to take advantage of the differing rates of homodimerization to afford product selectivity.
3.3.3. Selective cross metathesis between type I and type II olefins
In reactions between type I and type II olefins, the type I olefin may initially undergo rapid homodimerization, precluding direct CM with the type II olefin, and releasing ethylene as a byproduct. The product type I homodimer will also readily undergo rapid CM with the type II olefin, releasing one equivalent of type I olefin for further CM. This results in an equilibrated mixture of type I homodimer and cross products. Product distribution can however be driven toward cross product as ethylene is driven from the system. Furthermore, the inability of the catalyst to convert cross product to the type II homodimer in a secondary metathesis event prevents the erosion of product seclectivity. For example, in the reaction between type I 5-hexenyl acetate 113 and type II α,β-unsaturated ester 114,110 cross product 115 is formed with high product selectivity and stereoselectivity (Figure 28).
Figure 28.
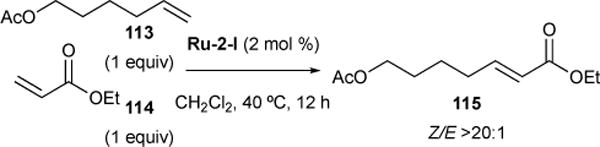
Selective CM with type I and type II olefins.
3.3.4. Selective cross metathesis between type I and type II vs type III olefins
Earlier studies showed that using first generation ruthenium catalyst Ru-1-I, fully substituted allylic centers/quaternary centers were insusceptible to cross metathesis, and did not disrupt catalyst activity (i.e., classified as Type IV with Ru-1-I).114 However, with second generation Ru-2-I, product selectivity and excellent stereoselectivity for the E-isomer is achieved (i.e., classified as Type II/III with Ru-2-I, see Figure 29). For example, reaction between 113 and unprotected tertiary alcohol 117 results in up to 93% yield of cross metathesis product 120, where only the trans isomer is observed by 1H NMR. Introduction of steric bulk protecting the tertiary alcohol (i.e., 118) results in almost exclusive formation of cross-product 121 (97% yield). We rationalized that the greater steric bulk results in initial trans product formation, and cross-product is completely hindered against secondary metathesis events that may erode product selectivity.
Figure 29.
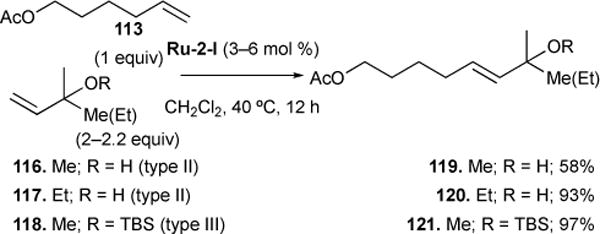
Selective CM of type I with type II vs type III olefins.
3.3.5. Synthesis of vinyl boronates
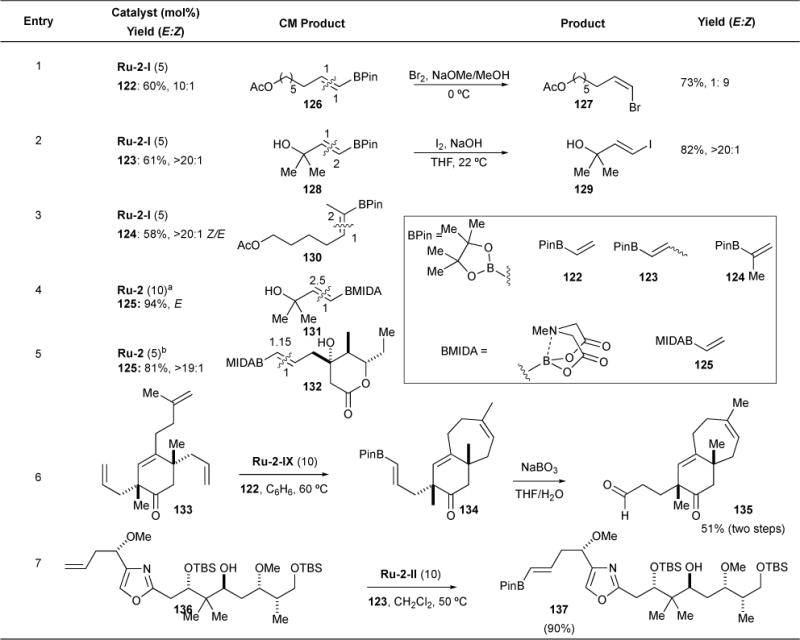
We reported a general strategy for the synthesis of a variety of vinyl boronates,115 which most notably are excellent cross-coupling partners.116–120 Importantly, this strategy was used in the preparation of β,β-disubstituted vinyl boronates, which cannot be synthesized by alkyne hydroboration, an alternate route for vinyl boronate synthesis. Upon realizing that 1-propenyl pinacol boronate (BPin) 123 was easier to prepare and isolate than vinyl boronate 122, we utilized it and the more hindered 2-isopropenyl pinacol boronate 124 in cross-metathesis with a variety of olefins, including styrenes, 1,1-disubstituted olefins, and tertiary allylic alcohols (Table 8). Yields and stereoselectivities ranged from good to excellent (60 – 91%, E/Z 10:1). We also showed that the E-boronates can be converted to Z-vinyl bromides via in situ bromination procedure, and into E-vinyl iodides via an analogous iodination procedure, thus affording the stereoselective formation of E and Z vinyl halides (Table 8, entries 1 and 2). Components of Suzuki and many other metal-catalyzed coupling reagents are thus formed simply from an alkene metathesis reaction.
Table 8.
Vinyl, 1-propenyl, and 2-propenyl pinacol and MIDA boronate (BPin, BMIDA) CM and halogenation processes.
|
Burke and coworkers introduced the CM-active vinyl N-methyliminodiacetic (MIDA) boronate 125 as a bench- and silica-gel stable boronic acid derivatives.121 This reagent was used by Fürstner and coworkers to prepare Suzuki coupling partner 130 using CM in 81% yield in their 2012 synthesis of leiodermatolide.122
Stoltz and coworkers’ (−)-cyanthiwigin F synthesis used a sequential one-pot RCM/CM process catalysed by Ru-2-IX to rapidly form a trisubstituted seven-membered ring (30 min), followed by vinyl BPin installation using 5 equivalents of vinyl boronate 122 (20 h). Treatment of silica-gel plug-purified material with aqueous sodium perborate provided the requisite aldehyde 135 in 51% isolated yield (Table 8, entry 6).123 Finally, boronate 137, an intermediate in Menche and coworkers’ rhizopodin total synthesis, was prepared in 90% yield by CM with vinyl boronate 123 and oxazole 136 using Ru-2-II in CH2Cl2 at 50 °C (Table 8, entry 7).124
3.3.6. CM with conjugated dienes
We extended this strategy to the chemoselective CM with conjugated dienes.125 We found that chemoselectivity can be achieved by installing either electron-withdrawing groups or steric bulk at one of the olefin sites, which deactivates the vicinal olefin and hinders metathesis at that site. In the course of optimizing electronic parameters, we found that the use of ethyl sorbate 139 and 5-hexenyl acetate 140, in the presence of Ru-1-I, yielded the homocoupling product 138. That is to say ethyl sorbate 139 is too deactivated to react, making it a type IV olefin in the presence of Ru-1-I. In the presence of Ru-2-I however, both olefins of the diene were subject to cross-metathesis, indicating that with Ru-2-I, the ester on 139 was sufficiently electron-withdrawing to selectively deactivate the vicinal olefin.
Using Ru-2-I, we found that tuning the electronics of the ester by installing a vinylic bromide yielded products 143, 144, 145, and 146 in good yields (48–70 %) and stereoselectivity (Z/E 6–10:1) (Table 9). We reasoned that bromide provided both electron-withdrawing functionality and steric bulk to the vicinal olefin, further deactivating the α,β-double bond and hence facilitating cross-metathesis chemoselectivity. Encouraged by these results, we investigated the possibility of replacing the ester functionality altogether with bromide. We found that reactions between 1,1-dibromo-1,3-pentadiene and methyl acrylate resulted in modest yield of product, albeit excellent stereoselectivity (147, 39% yield, >20:1); an increase in yield (60%) was observed between 1,1-dibromo-1,3-pentadiene and styrene to form 148.
Table 9.
Electronic control of chemoselective construction of conjugated dienes via olefin cross-metathesis.
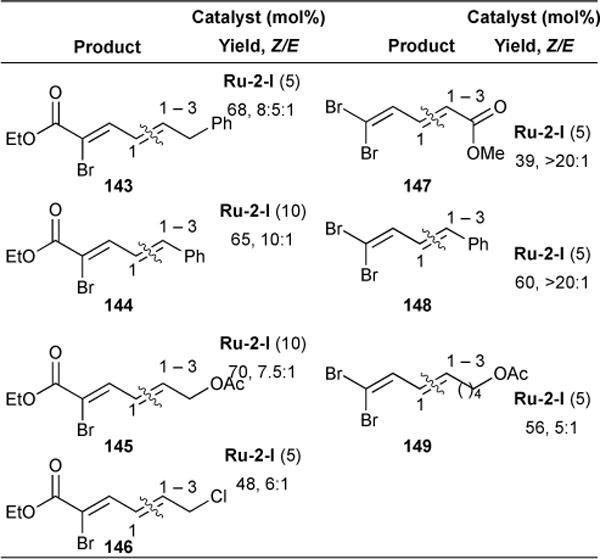
|
To probe further the effects of steric bulk on chemoselectivity, we reacted various 2-substituted 1,3-butadienes with functionalized olefins (Table 10). These transformations were performed in benzene solvent and at elevated temperatures to achieve conversion. Products formed in good yields and high chemoselectivity and diastereoselectivity. In fact, only the E product isomer was observed in solution.
Table 10.
Steric control of chemoselective construction of conjugated dienes via olefin cross-metathesis
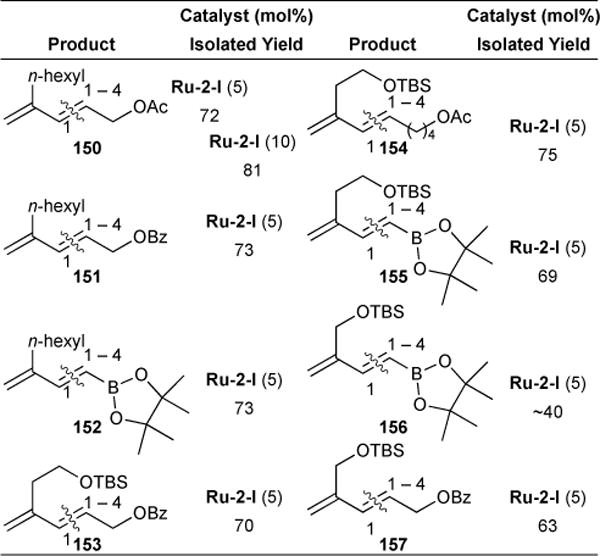
|
3.3.7. Z-Selective CM
We reported the first Z-selective homodimerization of terminal olefins using Ru-5-II (Figure 31A). Homodimerization of terminal olefins results in the release of ethylene, which serves to decompose the catalyst. However, through the efficient removal of ethylene from solution, homodimerization is robust – resulting in high conversions and Z-selectivity for a variety of terminal olefin substrates, including challenging alcohol substrates.58
Figure 31.
Scope of Z-selective cross-metathesis (CM) using Ru-5 catalysts.
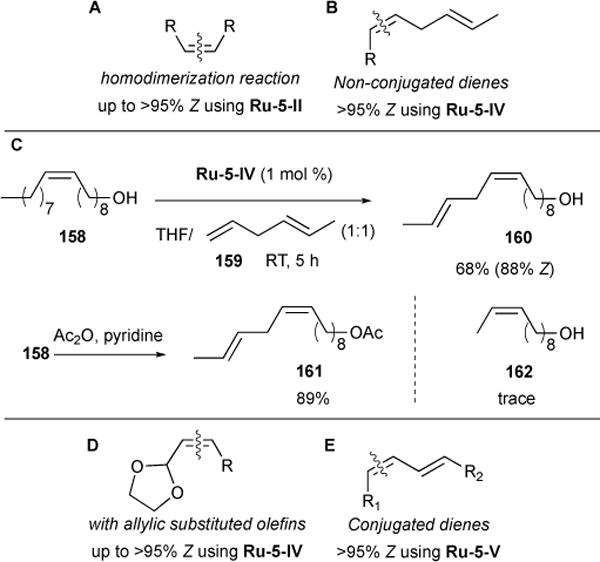
We further explored the general use of chelated ruthenium catalyst Ru-5-IV for complete Z-selective and chemoselective CM between terminal olefins and non-conjugated dienes (Figure 31B). A strong chemoselective preference for the terminal alkene in the cross-metathesis reactions, and high Z-selectivity of >95% were observed. In a series of CM with trans-1,4-hexadiene (159), a wide scope of functionalized terminal olefins carbonyls, aldehydes, ketones, esters, alkyl amines, and boronic acids – were all tolerated.126 In one application, we demonstrated concise syntheses of nine lepidopteran pheromones from renewable seed-oil derivatives using mild, economical, and safe processes.61 For example, Z-selective CM of seed-oil-derived oleyl alcohol (158) and hexadiene 159 provided unconjugated diene 160 and only trace quantities of olefin 162, formed from internal bond CM of hexadiene Y (Figure 31C). Diene alcohol 160 and acetylated derivative 161, made in two steps, are both sex pheromones capable of disrupting the mating behaviour of the olive pyralid moth, a pest native to areas of Europe and North Africa. Previous syntheses required at least six steps from commercial materials.127
Allylic substituted olefins create a significant challenge to Z-selectivity due to the increased steric bulk around the olefin, which further destabilizes the cis over the trans conformation of the carbon-carbon bond.128 We reported the use of nitrato ligated Ru-5-IV to afford Z-selective cross-metathesis of terminal olefins with a variety of allylic-substituted olefins (Figure 31D).129 Notably, in cases such as vinyl pinacol boronates, important cross-coupling reagents, where high E-selectivity have been afforded with Ru-1-I, Z-cross product was achieved with up to 92% Z-selectivity using Ru-5-V.
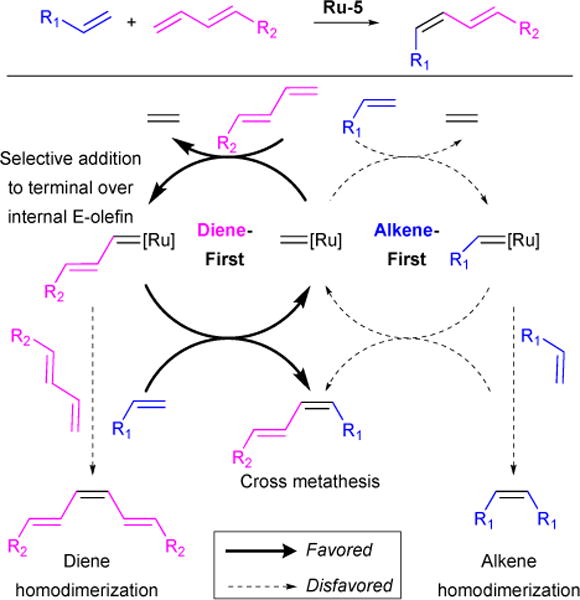
In collaboration with Houk and coworkers, we studied the highly chemoselective, Z-selective cross-metathesis of dienes to alkenes (Figure 31E).130 This reaction takes advantage of the pronounced Z-selectivity observed when cyclometalated ruthenium catalysts (Ru-5 catalysts, see Figure 1) are employed. Mechanistically, two pathways are envisioned where the active Ru-alkylidene adds to either the diene or alkene as the first propagation step (Figure 32). Computations using propene and E-1,3-pentadiene as a model showed that the diene-first pathway is favored over the alkene-first pathway. Following initial olefin addition to the ruthenium, either productive cross-metathesis with the substrate alkene or non-productive diene homodimerization may occur. However, the cross-metathesis pathway is favored by 3.5 kcal/mol over dimerization. These computations are consistent with the experimentally-observed product selectivity.
Figure 32.
Overall reaction and catalytic cycle of diene-alkene cross metathesis.
3.3.8. E-Selective CM
Trans-olefins are thermodynamically preferred in non-selective olefin metathesis reactions. Trans-olefins can be isolated in high purity via achieving equilibrium, followed by Z-selective ethenolysis/alkenolysis.50,131 However, the use of equilibrium mixture as starting material limits the overall yield of E-olefin product, and ethenolysis/alkenolysis adds an extra step in the purification process. We reported the first highly-controlled, highly trans-selective olefin cross-metathesis reaction using dithiolate-ligated ruthenium complexes (see Ru-6 catalysts in Figure 1).132 These Ru-6 catalysts were shown to achieve catalysis through stereoretention of the starting material. In one example shown below, all four catalysts were evaluated for efficiency in the demanding cross-metathesis of 1-decene 163 and cis- or trans- 4-octene 164 (Table 11). Using Ru-6-II, the reaction between 163 and cis-164 afforded cis-165 in high Z-selectivity; reaction with trans-164 afforded trans-165 in low yield and moderate E selectivity. We found that the slightly reduced steric bulk at the ruthenium NHC aryl ortho position (i.e., from an isopropyl in Ru-6-II to a methyl in Ru-6-I) resulted in improved stereoretention. Of note, replacing the ortho methyl groups with the much smaller fluoro group on the ruthenium catalyst (i.e., using Ru-6-III and Ru-6-IV) significantly increased E selectivity in the reaction. These findings support the proposed model whereby trans-olefinic substrates are increasingly compatible with catalysts as steric encumbrance is reduced.
Table 11.
Stereoretentive CM of 1-decene and 4-octene.
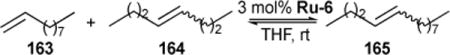
| ||||
|---|---|---|---|---|
|
| ||||
| Ru-6 | 159 | % conv | % yield | X/E |
| Ru-6-II | cis | 89 | 74 | 96/4 |
| Ru-6-II | trans | 92 | 4 | 13/88 |
| Ru-6-I | cis | 84 | 58 | >99/1 |
| Ru-6-I | trans | 37 | 7 | <1/99 |
| Ru-6-III | cis | 88 | 57 | 97/3 |
| Ru-6-III | trans | 53 | 29 | <1/99 |
| Ru-6-IV | cis | 79 | 54 | >99/1 |
| Ru-6-IV | trans | 55 | 31 | <1/99 |
3.3.9. Glycopeptide-based CM
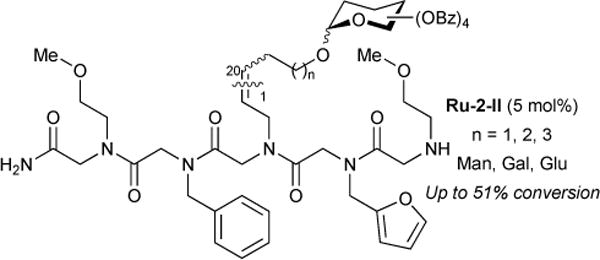
In collaboration with Kwon and co-workers, we reported, the solid-phase synthesis of glycopeptioids using CM chemistry.133 Peptoids with various side-chains were prepared on beads and alkenyl components were positioned at different chain lengths. Using Hoveyda chelate ruthenium catalyst Ru-2-II, cross metathesis of a peptoid with 20 equivalents of mannose, galactose, or glucose was achieved with up to 51% conversion (Figure 33).
Figure 33.
CM assisted solid-phase synthesis of glycopeptoids.
3.3.10. Template-Driven CM
We reported, in collaboration with the Stoddart and Tirrell groups, the template-driven dimerization of dibenzo[24]crown-8 derivative (Figure 34).134 The template consists of two dialkylammonium ion sites allowing for the recognition and preorganization of two crown ether molecules for efficient acyclic diene metathesis (ADMET). We expected that the two olefin sites on the crown ether derivative would afford polymerization. However, when a 1 mM solution of 167 in DCM was exposed to Ru-2-1 or Ru-2-2, the dimer was formed (Figure 34). Nonetheless, we observed an increase in both rate of dimerization and yield of dimerized product in the presence of dialkylammonium complex 166.
Figure 34.
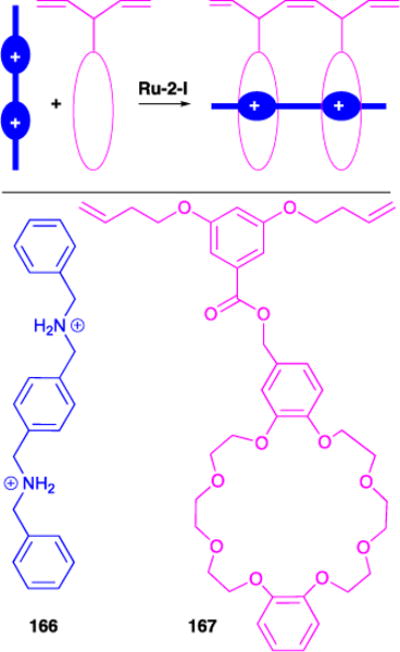
Template-driven olefin cross-metathesis.
3.3.11. Ethenolysis
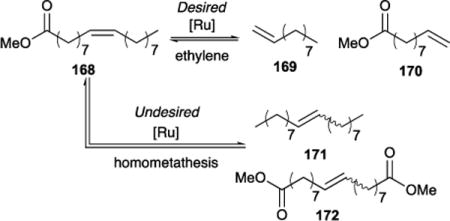
Ruthenium catalysts used in the CM reaction between internal olefins and ethylene (i.e., ethenolysis) are effective in producing desired low molecular weight products from renewable seed oils. Limited product selectivity and poor conversion have hampered the commercial use of this process. The use of microchemical systems provides effective strategies to overcome these challenges on an industrial scale. When reaction variables, such as surface area-to-volume are tightly controlled, waste is reduced, yields are maximized, and reaction times are reduced. Furthermore, the challenges of scale-up synthesis are avoided and replaced by parallelization of small-scale microfluidics. Consecutive reactions, such as separation, purification, and detections, can be integrated to complete the microchemical process.135–137 In collaboration with Kim and co-workers, we reported a facile and efficient microchemical ethenolysis, through a continuous segmented flow of ethylene gas and methyl oleate 168 in a capillary tube with a 0.5 mm inner diameter.138 At 60 psi of ethylene, the ethenolysis efficiencies were comparable to the results of batch reactions at 150 psi. Of the catalysts explored (see Table 12), CAAC-supported ruthenium metathesis catalyst Ru-4-I (50 ppm) yielded the largest turnover number (TON = 27200), and gave 80% conversion and 87% product selectivity.
Table 12.
Ethenolysis of methyl oleate
| ||||
|---|---|---|---|---|
|
| ||||
| [Ru] | % Conv | % Select | % Yield | TON |
| Ru-1-I | 65 | 96 | 62.4 | 5800 |
| Ru-1-IV | 39 | 94.8 | 37.0 | 7500 |
| Ru-2-II | 58 | 45 | 26.1 | 11000 |
| Ru-2-V | 54 | 52 | 28.1 | 13500 |
| Ru-2-VI | 61 | 58 | 35.4 | 22300 |
| Ru-4-I | 80 | 87 | 69.6 | 27200 |
More recently, in collaboration with Bertrand and coworkers, we reported several new CAAC-supported ruthenium catalysts for the ethenolysis of methyl oleate using.47 Remarkably, most of these catalysts supported a turnover of over 100,000 with catalyst loading of only 3 ppm. Of note, using just 1 ppm of Ru-4-III and 99.995% pure ethylene source, we were able to achieve catalyst turnover of up to 340,000 – the highest reported value for ethenolysis to date!
AROCM of norbonene derivatives
We reported a homochiral stereogenic-at-ruthenium complex Ru-5-II capable of asymmetric ring-opening-cross-metathesis of norbonene derivatives with high enantioselectivity.139 A variety of terminal olefins and norbonene derivatives were studied to determine their effect on efficiency, diastereoselectivity, and enantioselectivity (Table 13). Product yields ranged from 40 – 65% for all reaction entries. Z-selectivity and enantioselectivity were not appreciably affected by the steric or electronic nature of the terminal olefin substituents. Also, substitution at the norbonene did not significantly affect Z-selectivity or enantioselectivity. However, of note a significant erosion of Z-selectivity is seen in the formation of aryl ether 182, albeit with high enantioselectivity (95% ee). Both Z-182 and E-182 were formed with identical high ee, strongly supporting the rationale that the enantiodetermining step in this reaction precedes the olefin geometry-determining step.
Table 13.
Z-selective and Enantioselective Ring-Opening Cross Metathesis
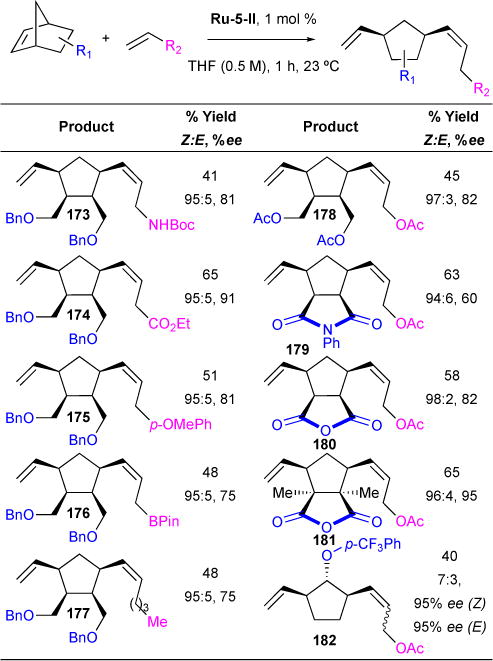
|
4. Ring Opening Metathesis Polymerization (ROMP)
The application of olefin metathesis to the synthesis of macromolecular materials has been most successfully and broadly demonstrated by way of ring-opening metathesis polymerization (ROMP). ROMP is a chain growth polymerization process by which cyclic olefins are converted to unsaturated polymeric material via the Chauvin mechanism. Among many others, our group has worked extensively to advance the utility of ROMP by developing well-defined, functional group tolerant catalysts. We reported the first titanacyclobutane system capable of mediating living ROMP, and the first well-defined, single-component ruthenium-based complex capable of catalyzing ROMP in the presence of heteronuclear and protic functionalities.140,141 These developments, together with those contributed by Yves Chauvin, Schrock and countless others, have established ROMP as a powerful tool for accessing polymeric materials of complex architecture, functionality, and size with unprecedented precision and ease. While this review focuses exclusively on those innovations set forth by our group since 2005, numerous more comprehensive reviews have been previously published on early ROMP catalyst development142–144 and emergent applications145,146 thereof.
4.1. Implications of Ruthenium ROMP Thermodynamics and Kinetics
The ROMP reaction is thermodynamically governed by the Gibbs Equation, ΔG = ΔH – TΔS, whereby enthalpic gains, associated with release of strain energy upon ring opening, drive the reaction forward, while entropic penalties curb the polymerization. For this reason, ideal ROMP candidates are typically widely-available cyclic olefins with excessive ring strain (e.g. norbornene (NBE), cyclobutene, dicyclopentadiene). In the case of low-strain cyclic olefins, the enthalpic reward from ring opening is often insufficient to offset the loss of entropy intrinsic to polymerization. Developing strategies for ROMP of low strain materials is thus not only an inherently challenging task, but a necessary one in order to establish ROMP’s versatility and utility in accessing polymeric materials with useful electronic, biological, and mechanical properties.
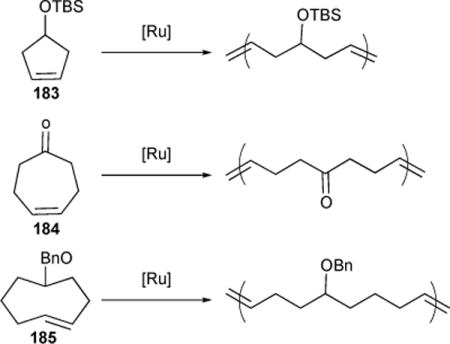
Traditionally, this challenge has been overcome by reducing the temperature of the reaction and employing a highly active, early transition metal-based catalyst in high concentrations to compensate for the resulting retardation of equilibrium kinetics. However, this approach is not viable for low strain starting materials bearing polar substituents, to which the highly active catalysts tend to be intolerant. Unlike catalysts based on early transition metals such as tungsten and molybdenum, fast initiating Ru-alkylidene Ru-3-II demonstrated both high ROMP activity and functional-group tolerance, affording the previously unrealized living ROMP of 5- and 7-membered cyclic olefins bearing polar symmetrical substituents (silyl ether 183, ketone 184, and ether 185).147 (Table 14)
Table 14.
Examples of the living ROMP of low strain or unhindered cyclic olefins with polar functionalities.
| ||||
|---|---|---|---|---|
|
| ||||
| Monomer | Limitation | [Ru] | Yield (%) | PDI |
| 183 | insufficient ring strain | Ru-3-II | 71 | 1.3 |
| 184 | insufficient ring strain | Ru-3-II | 88 | 1.2 |
| 185 | unhindered alkene | Ru-1-I + PPh3 | 97 | 1.06 |
Ru-mediated ROMP of low strain monomers has found high utility in enhancing the ease and precision with which commercially relevant materials can be produced to scale. Our group has demonstrated the production of well-defined poly(vinyl alcohol) (PVA) copolymer derivatives by ROMP of two functionalized, low strain moieties— 3-cyclopenten-1-ol and 5-cyclooctene-1,2-diol. In the former case, ROMP of unprotected 3-cyclopenten-1-ol followed by ADMET of non-protected 1,6-heptadiene-4-ol gave rise to well-defined poly(vinyl alcohol-alt-propenylene) copolymers with high polarity and precise distribution of the hydroxyl functionalities. Moreover, these copolymers could be accessed in a single-pot synthesis, affording a desirable commercial alternative to the multi-step procedures traditionally employed.148
We later reported the synthesis of a highly analogous ethylene vinyl alcohol (EVOH) copolymer by ROMP of 1,2-cis- and 1,2-trans-cyclooctenediol monomers. Protection of the monomer diols as acetates, carbonates, or acetonides conferred a temporary increase in the system strain, affording high yield polymers by Ru-1 and Ru-2 mediated ROMP. Post-polymerization hydrogenation and deprotection gave ethylene-vinyl alcohol (EVOH) copolymers with stereo- and regio- regularity.149
In other cases, however, the relatively low polymerizability of low-strain systems has proven advantageous. For example, the equilibrium ROMP of cyclopentene offers a clean route by which durable elastomeric polypentamers may be both synthesized and recycled via the same Ruthenium catalyst system. This potential application is possible on account of the extraordinarily low thermodynamic activation enthalpy and entropy unique to Ru-alkylidenes. For these polymerizations, the overall cyclopentene conversion is sensitive only to the temperature of the reaction, and independent of the catalyst loading or the catalyst activity. The polymerization/depolymerization equilibrium is thereby readily and cleanly manipulated by temperature control in the presence of a single well-defined catalyst and could find potential commercial application in the environmentally friendly synthesis/recycling of rubber analogs.150
More recently, in collaboration with the Choi group, we exploited the low ROM polymerizability of cyclopentene in the development of a novel method, multiple olefin metathesis polymerization (MOMP), by which all three olefin metathesis transformations— ring-opening, ring-closing, and cross metathesis— are combined in a one-pot reaction to afford an A,B-alternating copolymer.151 Well-defined reaction pathways were achieved by strategic design of dicyclopentene monomers capable of undergoing simultaneous ring-opening/ring-closing methathesis without significant thermodynamic bias towards one or the other. CM with diacrylate comonomers yielded A,B-alternating copolymers of uniform microstructure.
While low-strain cyclic olefins pose a challenge due to their low activity, unhindered alkenes have proven problematic on behalf of their unquenchable activity. In particular, the living ROMP of cyclooctene (COT) 185 to high MW polymeric materials has been historically frustrated by high rates of competing secondary metathesis on the flexible poly(COT) backbone. We showed that chain transfer associated with secondary metathesis of the unhindered backbone could be quenched nearly entirely by carrying out the Ru-1-I mediated polymerization in the presence of PPh3, a known suppressant of secondary metathesis, and a strongly coordinating solvent such as THF.152 (Table 14) Under these conditions, we demonstrated the first “living” ROMP of trans-COT as well as its protected hydroxy-functionalized derivatives and a variety of poly(NBE)-poly(COT) block copolymers.
4.2. Control of Polymer Microstructure in Ruthenium ROMP
The stereochemical and regiochemical makeup of a polymer, often referred to as the polymer ‘microstructure’, is widely known to have dramatic influence on its physical and mechanical properties (e.g. melting point, crystallinity, etc.). A tremendous amount of research has therefore been devoted to developing methods capable of producing polymers with highly controlled microstructure, and in turn, macroscopic properties. For ROMP polymers, this effort has focused mainly on controlling the double bond configuration of the unsaturated backbone, and to a lesser extent, on controlling the polymer tacticity (isotactic v. syndiotactic) and relative configuration of dyad substituents (head-head (H-H), head-tail (H-T), or tail-tail (T-T)) for polymers comprised of asymmetrically substituted monomers.
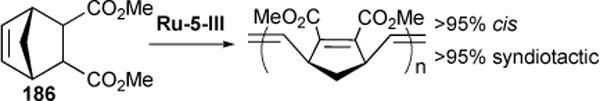
In 2012, we reported the use of Ru-5-IV to catalyze the ROMP of nine tested monomers to yield polymers of 48-96% (average: 80-95%) cis stereochemistry, essentially qualitatively reversing the stereoselectivity of Ru-1-I.153 Shortly thereafter, we reported the use of Ru-5-III, which unlike Ru-5-IV, demonstrated the capacity to mediate both polymer tacticity and stereochemistry, yielding polymers of high cis (62-95%) character and near total syndiotacticity (>95%).154 Notably, this system marked the first example of a ruthenium alkylidene complex capable of producing from olefin 186 a norbornene-derived polymer characterized by >95% of a single microstructure. (Figure 35) We attribute the unique capacity of Ru-5-III to control polymer tacticity to the reduced steric bulk of the cyclometalated N-t-butyl group as compared to the bulky N-adamantyl chelate in Ru-5-IV.
Figure 35.
Synthesis of a single microstructure polymer by ROMP with a stereoselective ruthenium-based catalyst. Conditions: [monomer]/[initiator] = 100:1 in THF (0.25 M in substrate) at room temperature.
We next explored the possibility of achieving enantiomerically pure ROMP products by kinetic resolution — a methodology that exploits the kinetic preference of a catalyst to selectively polymerize one enantiomer in a racemic monomer mixture, giving rise to both a chiral polymer and enantioenriched population of the unreacted monomer.155 Catalytic enantioselectivity was imparted by Ru-2-X (see Table 2), a chiral (4R, 5R)-diphenyl N-heterocyclic carbene that we had previously shown to give up to 92% ee in ARCM (vide supra).36 Despite displaying clear kinetic preference, the catalyst’s resolution selectivity (S) was found to change dramatically with the progression of polymerization, perhaps due to the increasingly dominant chiral secondary structure of the growing polymer. The trend of selectivity variance was also found to be solvent-dependent; resolution selectivity increased over the course of polymerizations performed in THF/DCM and decreased for those performed in toluene.
This recurrent, and thus far unaccounted for, inability to generalize methods of partially stereoselective ROMP across conditions and monomer types highlighted a lack of understanding in the mechanistic origins of cis selectivity and tacticity among Ru-alkylidenes. To this end, we initiated a detailed mechanistic investigation into the stereoselectivity of ROMP, relying heavily upon polymer microstructure as a sort of chronological “roadmap” of each catalytic cycle from which information on the propagation transition state could be gleaned.156
The ROMP of 2,3-dicarbomethoxynorbornadienes by eight stereoselective Ru-alkylidene catalysts produced polymers of predominantly cis, syndiotactic microstructure, with cis content varying from 68-99%. A combination of computational and experimental evidence suggested stereoselectivity derives from stereogenic metal control at the Ru-alkylidene center. The prevailing cis, syndiotactic motif is mechanistically explained by step-wise inversion of the metallic center followed by the monomer’s anti-approach (i.e. the approach in which the monomer bulk points away from the catalyst N-aryl group). As a result, the incoming monomer is added to alternating sides of the Ru-C double bond, giving a near-perfect cis, syndio-selectivity. Unlike cis-selectivity, HT bias among asymmetrically substituted monomers was found to be negligible in all tested cases. What little HT bias was observed, however, was highest for monomers polymerized at a faster rate — suggesting distinct propagating species with characteristic HT bias.
The origins of microstructural error were also investigated. The predominant erroneous motifs — trans, syndiotactic and cis, isotactic microstructures— were traced to anti or syn monomer addition to the higher energy syn alkylidene. Anti to syn isomerization of the alkylidene most likely occurs via rotation about the Ru-C bond, as computational methods reveal non-metathesis isomerization to be energetically infeasible under relevant reaction conditions. Therefore, the prevalence of error was deduced to be intrinsically related to the relative time scales of propagation and alkylidene isomerization, with error being more pronounced when these rates are more comparable.
4.3. Functionalized Polymers
Our group has fabricated polymers bearing heteronuclear functionalities for use in imaging, biomedicine, photonics, and energy. Among the earliest of these examples, we synthesized tumor-targeted fluorine-18 functionalized nanoparticles for use as in vivo PET imaging agents.157 (Figure 36)
Figure 36.
Synthesis of PET-active fluorinated nanoparticles. Blacklines of polymer backbone. Purple lines are pendant PEG groups. Red lines are cinnamoyl groups. Blue and green spheres are mesylate groups and fluorine atoms, respectively. (A) Ru-3 mediated sequential ROMP. (B) i) Dialysis against H2O. (ii) hv, 3 min. (iii) 1. K18F, kryptofix-222, K2CO3, BHT, MeCN, 120°C, 60 min. 2. K19F, kryptofix-222, MeCN, 80°C, 30 min.
The well-defined, radiotraceable nanoparticles were synthesized in a multistep procedure starting with the Ru-3-I mediated ROMP of cinnamoyl and PEG-mesylate functionalized norbornenes 187 and 188, respectively. The resulting polymers exhibited highly controlled MWs and in turn, hydrodynamic diameters following self-assembly into discrete micelles. Micelles were readily cross-linked by a UV-induced [2+2] dimerization reaction at the cinnamoyl trans-olefins and subsequently functionalized with 18F by nucleophilic displacement of the mesylate group for PET imaging.
In addition to proving highly useful for the probing of biological systems, ROMP has allowed for greater simplicity in the fabrication of complex, biologically-inspired materials. Elastin like peptides (ELPs) have been among the most attractive of these targets for synthetic chemists given the highly unique, dynamic physical properties of tropoelastin—the chief component of human blood vessels. In particular, chemists have sought to synthetically replicate the temperature-responsive behavior of tropoelastin. Non-crosslinked tropoelastin undergoes a conformational change marked by a phase transition at a precise temperature known as the lower critical solution temperature (LCST), below which the protein is insoluble. This property is essential to the protein’s characteristic elasticity and has proved challenging to reproduce synthetically. Specifically, the success of synthetic ELPs has been frustrated by the inability to access MW-independent LCSTs, a problem that is exaggerated in the case of polydisperse materials.
We thus investigated the potential of ROMP to afford a narrowly dispersed ELP with a MW-independent LCST.158 To this end, we synthesized a polynorbornene copolymer bearing PEG5 and ELP pendant sequences. (Figure 37) The LCST of the near-monodisperse polymers was independent of MW when the polymer was synthesized as a random copolymer with hydrophilic PEG5 co-monomers. Moreover, the LCST of the ELPs could be predictably tuned by altering the feed ratio of the monomers. As such, this ROMP-based approach affords a facile route to ELPs exhibiting controllable temperature-responsive behavior with previously unattainable reproducibility.
Figure 37.
Structure of an elastin-based random copoly(norbornene) with tunable temperature responsive behavior.
4.4. Surface-Functionalized Polymers
The ability to tolerate a wide range of heterofunctionalities lends itself well to the incorporation of interesting linkage chemistries, including those well-suited for the surface immobilization of polymers. Our group reported the first “universal method” for surface-initiated ROMP (SI-ROMP) in 1999.159 Since then, we and others have explored the utility of SI-ROMP for applications in fields such as energy, photonics, biology, and medicine.160,161 One of the most promising applications of SI-ROMP has been the fabrication of polymer dielectric layers for organic thin film transistors.162 (Figure 38) The integration of organic materials in electronic devices has driven their miniaturization, increased affordability, and streamlined production. Typically, organic polymers are incorporated into electronic devices such as FETs (field-effect transistors) as thin dielectric layers deposited on metallic surfaces by way of spin-coating, ink-jet printing, or screen printing. However, these methods—which require harsh conditions and lengthy reaction times—yield surfaces that are often dilute and non-conformal in topology, rendering the overlaying semiconductor ineffective in bridging the FET source and drain contacts. In contrast, SI-ROMP (surface-initiated ROMP) allows for the production of smooth, pinhole-free surfaces accessible under much more rapid, mild conditions. Moreover, film thickness, an essential parameter of the dielectric, is readily tuned by altering the identity of the surface tethered catalyst, the reaction time, or some combination thereof.
Figure 38.
FET fabricated from an SI-ROMP polymer dielectric layer.
4.5. Telechelic Polymers
Liquid Crystal (LC) elastomers and gels have attracted much attention on account of their high flexibility and responsiveness to stimuli such as heat, light, and electric/magnetic fields. However, the challenge of accessing LC elastomers and gels of precise, well-defined microscopic architecture has impeded fundamental understanding of the structure-property relationships characteristic of these materials. We envisaged a ROMP-based approach to the fabrication of LC gels with unprecedented control.163
ROMP-derived telechelic polymers were shown to give rise to well-ordered, controlled LC gels following click-chemistry mediated crosslinking. (Figure 39) The microscopic architecture of LC networks could be tuned with respect to cross-linker functionality, inter-cross-links strand length, and mesogen density. The reported LC gels accessed via ROMP also displayed desirable swelling behavior and rapid, reversible, low-threshold optic switching that could be predictably tuned by altering the length of the telechelic polymers. This novel route to well-defined, tunable LC gels serves both to elucidate the microscopic origins of their physical and mechanical properties, as well as to facilitate the facile production of LC gels with pre-selected properties.
Figure 39.
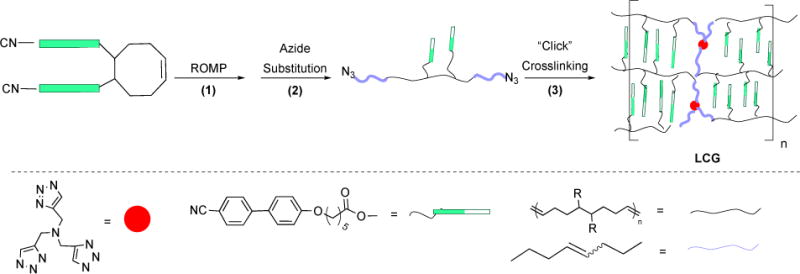
Preparation well-defined liquid crystal gels from telechelic ROMP polymers. (1) Ru-2-I, dichloroethane, 1,8-dibromo-4-octene, 55°C, 24 hr. (2) NaN3, DMF. (3) Triacetylene, tripropylargylamine, cat. CBr, PMDETA, DMF, 5 min (gelation) + 2 days (curing at 50°C).
4.6. Supramolecular Polymers
The extension and contraction of fibers is an intriguing natural motif with many potential applications in material science, particularly as molecular actuators. Fabrication of macromolecular networks coordinated by reversible, supramolecular interactions has been demonstrated as a viable approach to these systems, albeit one plagued by much synthetic challenge. One of the most promising synthetic routes to these switchable networks has been the incorporation of [c2]daisy-chain dimers (DCD) into polymers. The so-called ‘daisy-chain polymers’ are mechanically interlocked, rotaxane based macromolecules formed from the polymerization of macromers— bifunctional monomers possessing both host and guest functionality. Previous reports of [c2]daisy-chains relied upon irreversible bond formation to mechanical interlocking neighboring macromers. Unfortunately, this approach yields a product distribution favoring the kinetic, cyclic dimer product instead of the linear chain. In an effort to avoid this, we employed reversible ring closing metathesis (RCM) as the final stoppering event between macromers.164 This dynamic covalent chemistry— in this case, equilibirum-controlled metathesis— affords a new means by which the resulting product distribution can be biased toward the thermodynamically-favored linear chain. The high-yield [c2]daisy-chains are readily functionalized as bisolefins to undergo acyclic diene metathesis (ADMET) polymerization. (Figure 40)
Figure 40.
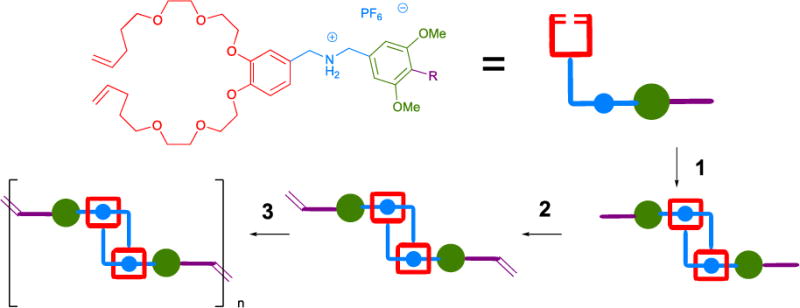
Dual-metathesis mediated preparation of mechanically-interlocked polymers based on a [c2]daisy-chain motif. R=H, O(CH2)6OH. (1) i. Ru-2-I, DCM. ii. H2\PtO2, EtOAc. (2) 4-Pentenoic acid, EDC, DMF. (3) DCM, 45°C.164
Following the synthesis of the mechanically-interlocked polymers in Figure 40, we sought to design a DCD characterized by enhanced stability in the contracted state— a requisite to engineering a ‘stronger’ molecular actuator. In an effort to do so, we envisaged a strongly coordinating biphenyl binding site between the macromer host and guest residues, expecting the functionality to promote dimer-dimer pre-assembly via π- π stacking interactions and facile threading afforded by the elongated, rigid linker. When incorporated into linear polymers, the obtained DCD exhibited excellent mechanical properties such as facile switching and particularly high stability in the contractile state, owing to strong coordination. Acyl protection of the ammonium guests increased the steric bulk of the residues, inducing slippage and thereby extension. Impressively, this extended conformation was concomitant with a 48% increase in size.165
Like DCD-polymers, polyrotaxanes represent yet another class of advanced supramolecular polymers. While the macromers of DCD-polymers bear host, guest, and stoppering functionality, each monomer unit of a polyrotaxanes is characterized by a single guest residue only; monomers are threaded, polymerized and the supramolecular polymers stoppered in a tedious succession of steps. Seeking an alternative, simplified approach, we developed a one-pot synthesis of polyrotaxanes that was both efficient and scalable.166 The reported approach employs a polymerizable ammonium bisolefin salt, dibenzo [24] crown-8 ether host, and end-capping chain transfer agent. The combination of efficient complexation and rapid equilibration of ADMET polymerization affords high yield polyrotaxanes (>80%) up to 19.3 kDa in MW in a single operation. Moreover, the polymers formed from simultaneous multicomponent threading, polymerization, and capping show no loss in threading efficiency (82%) compared to those polymerized from discretely pre-assembled supramolecular monomers.
4.7. ROMP of Cyclic Polymers (REMP)
For the larger part of ROMP’s history, cyclic polymers have been accessible via ROMP only as unstable byproducts produced under dilute conditions. This, however, changed with the advent of Ring Expansion Metathesis Polymerization (REMP) and the associated development of REMP specific catalysts. Fürstner and coworkers reported the first N-to-Ru tethered unsaturated NHC-based complexes167 and we subsequently demonstrated their use in polymerizations of cyclooctadiene to cyclic polybutadiene.168,169 We then prepared a series of saturated NHC-based cyclic Ru-alkylidene catalysts with enhanced stabilities and activities.170
The probing of four cyclic catalyst profiles revealed a high degree of kinetic tunability inherent to catalyst architecture. Catalysts bearing longer tethers were characterized by faster polymerization kinetics as compared to those with shorter tethers. The hindered kinetics of short tether catalysts could be attributed to high rates of catalyst release competing with propagation. Additionally, high intermolecular chain transfer among short tether catalysts indicated a step-growth polymerization mechanism, while longer tethered catalysts were shown to mediate chain growth polymerization.
We have used these catalysts to prepare cyclic dendronized polymers via REMP and have used atomic force microscopy to confirm their uniform cyclic topology (Figure 41).171
Figure 41.
REMP of dendronized macromonomer.
4.8. Photoinitiated ROMP
The ability to efficiently develop application-tailored derivatives of the parent Ru catalysts (Ru-1-I, Ru-2-I, Ru-3-I) has enabled technological advancements of ROMP across a diversity of fields. Among the highest impact and cross-sectional of these is photolithography— the process whereby UV light is used to cure a chemical pattern during the microfabrication of various materials, most notably integrated circuits. Despite the tremendous amount of work that has gone into developing this technique, the array of materials accessible by photolithography remains limited by the small functional diversity represented in commercially available photoresists, the light-sensitive chemicals from which a patterned surface coating is made.
Seeking a more versatile approach to patterned materials, we developed photolithographic olefin metathesis polymerization (PLOMP), a photolithography method employing a latent metathesis catalyst that reacts with olefins in the surrounding environment of the resist upon irradiation.172 The latent photocatalyst — a ruthenium vinyl ether complex 191 — is prepared by the ROMP of 1,5-cycloctadiene (COD, 189) with Ru-3-I, followed by quenching with ethyl vinyl ether 190 (Figure 42). 191 is widely recognized as being metathesis-inert However, we’ve shown that the catalyst activity can be regained upon exposure to UV light when under stabilizing conditions. With this in mind, we designed a viscous, olefin rich negative tone resist by dissolving 191 and poly(COD) in 5-ethylidene-2-norbonene 192; this stabilized photocatalyst 191 by way of dative bonding. Exposure to UV light triggered the cross-linking of difunctional ROMP monomer 192 within the matrix of linear poly(COD) polymer. The protypical example demonstrates the synergistic utility of ROMP photoinitiation and functional group tolerance in the facile, one-pot fabrication of resists from which a novel family of photocurable materials could be developed.
Figure 42.
Preparation of latent metathesis photocatalyst 189 via ROMP of 1,5-cyclooctadiene (COD) using Ru-3-I followed by quenching with ethyl vinyl ether 190. The resulting polyCOD is dissolved in 5-ethylidene-2-norbonene 192 to yield highly viscous resist material stabilizing 191.
4.9. Bottle-Brush Polymers
Bottle-brush polymers are novel class of macromolecules identified by their high MW, densely-packed side chains. This characteristic steric congestion forces the polymer backbone into an extended, wormlike conformation. The preparation of these unique structures is accomplished by one of three synthetic approaches. In the “grafting from” approach, monomers bearing initiation sites are polymerized and their macromolecular pendants grown directly from these sites. In the “grafting onto” approach, the polymer backbone and side chains are synthesized independently before grafting the pre-formed side chains onto the main chain polymer. In the “grafting-through”, macromonomers are polymerized to yield brush polymers of complete (per-monomer) grafting.
Prior study of the Ru-1-I mediated graft through ROMP of brush polymers showed that while narrowly-dispersed, the resulting graft polymers suffered from low DPs. Having already demonstrated the capacity of Ru-3-I to catalyze the ROMP of bulky monomers with high activity, we investigated its potential to mediate the ROMP of norbornene-poly(methacrylate), poly(t-butylacrylate) and polystyrene macromonomers.173 Fast-initiating living polymerization yielded brush polymers of high Mn, narrow dispersity and high DP with characteristic worm-like morphology. (Table 15)
Table 15.
Characteristics of bottle-brush homopolymers, block copolymers and random copolymers synthesized by ROMP with a high activity catalyst.
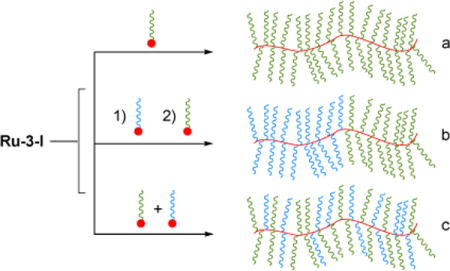
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| entry | motif | MM(s) | Mn theo | Mn GPC | PDI | DP | conv (%) |
| 1 | a | NB(PtBA)4700 | 921 | 1140 | 1.01 | 242 | 98 |
| 2 | a | NB(PMA)3700 | 348 | 420 | 1.02 | 114 | 94 |
| 3 | a | NB(PS)2200 | 210 | 231 | 1.02 | 105 | 93 |
| 4 | a | NB(PS)6600 | 607 | 701 | 1.01 | 106 | 92 |
| 5 | b | NB(PS)6600 NB(PnBA)4000 |
1060 | 1230 | 1.04 | — | 91 |
| 6 | b | NB(PtBA)4700 NB(PnBA)4000 |
1270 | 1260 | 1.04 | — | 98 |
| 7 | b | NB(PS)2200 NB(PtBA)4700 |
345 | 340 | 1.03 | — | 96 |
| 8 | b | NB(PLA)4700 NB(PnBA)4000 |
870 | 980 | 1.07 | — | 97 |
| 9 | c | NB(PLA)4700 NB(PnBA)4000 |
910 | 1030 | 1.04 | — | 98 |
Furthermore, we demonstrated the adaptability of our approach to random and block copolymers, employing norbornene-based macromonomers bearing polyacrylate, polystyrene, and polylactic acid pendants.174 Again, the obtained polymers exhibited excellent precision and conversion. (Table 15) Unlike the brush homopolymers previously reported, however, the brush copolymers exhibited nanostructures that could be highly controlled by altering the ratios and distribution of co-macromonomers. While symmetric block copolymers (i.e., those formed from a 1:1 macromonomer ratio) formed well-ordered morphologies, asymmetric ones did not. The side-chains of block copolymers segregated into large lamellar domains the size of which could be predicted by the backbone length. In contrast, the side-chains of random copolymers arranged into lamellar domains the spacing of which was independent of backbone length, likely due to segregation of proximal domains on alternating sides of the backbone. These predictable morphologies have established graft through ROMP as a viable platform for bottom-up fabrication of discrete nanostructures with diverse applications.
Inspired by the ease with which graft through ROMP could be employed for the bottom up fabrication of polymeric structures with well-ordered morphologies, we set forth to investigate its potential application to syntheses of more advanced nantostructures. Specifically, we showed that a hybrid graft through-REMP methodology could be employed to access cyclic brush polymers with MWs among the highest yet reported for any graft/brush polymers irrespective of structure.175 The ultrahigh MW polymers could be synthesized in a mild one-pot, single-step approach far simpler than those traditionally employed for the synthesis of cyclic dendrimers. Imaging techniques showed the cyclic nanostructures to have diameters ranging from ~100-180 nm— affording a novel class of cyclic structures roughly 14-40 times the size of standard cyclic dendronized polymers.
Undoubtedly, these novel, ordered nanoscale architectures afforded by graft polymers make these macromolecules excellent candidates for numerous applications, a sampling of which have been investigated by our group. First among these, we reported the use of bivalent-bottle-brush polymers as synthetically accessible substitutes to PEGylated dendrimers— nanoscale vehicles often employed for controlled delivery of chemotherapeutics with good success. Despite their efficacy, however, a facile synthetic route to accessing dendrimers with controlled size has yet to be reported. Alternatively, we showed that bivalent bottle-brush polymers— a sort of pseudo-alternating copolymer— loaded with an anticancer drug and PEG could be readily access by two approaches: graft through and “graft-through then click-to” ROMP. In the first of these, bivalent norbornene-PEG-drug macromonomers were prepared in advance of polymerization.176 In the latter, ROMP of bivalent norbornene-PEG-chloride was followed by halide-azide exchange and subsequent copper-catalyzed azide-alkyne cycloaddition (“click-to”) of a photocleavable drug derivative to the polymer.177 (Figure 43) Both methods afforded polymers with tunable size (6-50 nm) and DP, per-monomer drug loading, and stimuli-responsive (UV-triggered) drug release. Morphological characterization of the “graft-through then click-to” derived polymer revealed a unimolecular micelle morphology consisting of a hydrophobic drug core surrounded by a shielding corona of extended PEG moieties. EPR probing of these bivalent-brush polymers revealed greater structural homogeneity, increased rotational freedom of core bound hydrophobic pendants, and higher steric shielding from external reagents as compared to unbranched random copolymers of similar composition.178
Figure 43.
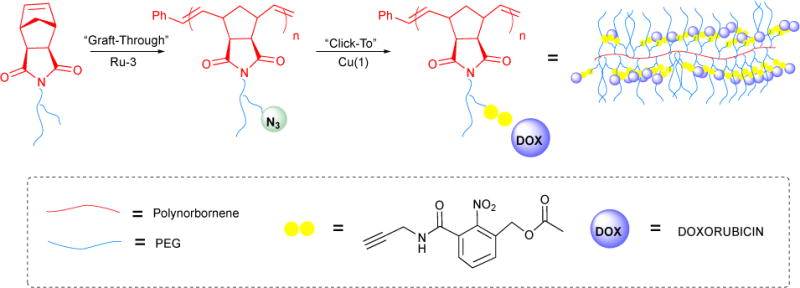
Adaptable “Graft-Though” then “Click-To” synthesis of bivalent bottle-brush polymers for triggered drug release.
In addition to their demonstrated suitability for biomedical applications, brush polymers have shown great promise in the fields of energy and photonics, most notably as photonic crystals (PCs). PCs are materials characterized by a periodic dielectric function whose photonic band gap elicits frequency-specific reflection. Fully realizing the potential of this unique property across a host of applications, however, requires an inexpensive, scalable method for the production of PCs with tunable frequencies of reflection.
Owing to the steric demand of their high MW side-chains, brush polymers are known to adopt elongated, linear conformations that reduce the incidence of multipolymer entanglement.179 In the absence of entanglement, the polymers readily self-assemble into ordered nanostructures with rapid equilibria. Self-assembly of brush block copolymers (BCPs) into ordered structures with domains (uniform regions of stacked lamellae) > 100 nm affords photonic crystals with reflection in the visible spectrum without the need for additives or complex swelling procedures required to achieve long wavelength reflectance in PCs fabricated from analogous linear BCPs.
In an effort to demonstrate the generalizability of this method, we reported the self-assembly of various brush BCPs into ordered nanostructures exhibiting tunable peak reflectances spanning the visible spectrum from ultraviolet (UV) into the near-infrared (NIR) region. For brush BCPs bearing more flexible macromonomers based on polylactides and polystyrene, peak reflective wavelengths as long as 540 nm could be achieved for PCs assembled under controlled evaporation and as long as 1311 nm for those assembled under thermal annealing compression. Impressively, substitution of these flexible macromonomers with ones based on rigid-rod isocyanate motifs facilitated the formation of large domains in the absence of thermal annealing, affording peak reflective wavelengths as long as 1120 nm under ambient conditions.180,181 In both cases, wavelengths of peak reflectance could be accessed by taking advantage of the linear correlation between polymer MW and the photonic bandgap of the crystal: a structure-property relationship unique to PCs formed from brush BCPs.
While this method affords a scalable route to PCs with application-tailored bandgaps, it necessitates the synthesis of a MW-specific block brush copolymer for each new wavelength of reflectance desired. To bypass this limitation, we developed a simple method of post-synthetically tuning the bandgap of brush copolymer PCs through blend ratios. By altering the relative compositions of high and low MW brush BCPs, we demonstrated the capacity to readily tune the peak wavelength of reflectance across the visible spectrum with high precision.182 (Figure 44)
Figure 44.

PCs tuned to have bandgaps across the visible spectrum by way of intervaled variance in brush BCP ratios. Brush BCP blends are composed of high MW (MW = 4167 × 103 g/mol) and low MW (MW = 1512 × 103 g/mol) otherwise equivalent brush BCPs varied in composition from 0 to 100% at 10% intervals.
This ability to fine tune the optoelectronic properties of brush polymer nanostructures makes them good candidates for a spectrum of electronic and optoelectronic applications. Most recently, our lab reported the synthesis of novel organic semiconductors formed from brush polymers whose pendant chains had been functionalized with conductive C60 fullerene units.183 The highly ordered, extended conformation of the C60-linked brush polymers enabled precise, high contact spatial organization of these conducting units. The resulting strong electronic coupling between neighboring fullerenes could be predictably tailored by altering the length of the pendant chain linking the C60 fullerene to the polymer backbone; shorter pendants were found to yield stronger couplings, while longer ones weakened the capacity for electron transfer.
Despite the intrigue of these ultrahigh MW polymers, their potential utility in future applications depends upon further development of methods for precisely tuning their physical and mechanical properties. Most recently, our group has reported methods for (1) modulating physical interactions in polymer networks and (2) controlling grafting density and distribution of brush polymer side chains. In the first of these examples, we reported the previously unrealized interdigitation of linear and brush macromolecules into densely grafted brush polymers. Despite significant steric congestion, interdigitation among linear and grafted poly(L-lactide) and poly(D-lactide) was driven by the enthalpic favorability of sterocomplexation, with quantitaive formation in instances of low MW linear/brush systems.184
Secondly, we demonstrated a method of tuning the grafting density of brush polymers and in turn, their physical properties, self-assembly, and stimuli responsiveness. Unlike previously reported protocols,185,186 this method was neither plagued by harsh conditions and long reaction times nor limited to the production of highly variable materials with capped grafting densities and high PDIs.187 Simultaneous control over grafting density and side chain distribution was achieved by rational design of a comonomer system composed of a macromonomer (norbornene-functionalized polystyrene, polylactide, or polydimethylsiloxane) and small molecule ‘diluent’ (an endo, exo-norbornene dialkylester). Variation of the macromonomer:diluent feed ratio yielded copolymers of predictably tuned grafting density, with lower diluent ratios resulting in more densely grafted polymers. Strategically selecting comonomers of evenly matched self-propagation kinetics favored the production of randomly distributed copolymers, while those with unevenly matched self-propagation kinetics lead to gradient distributions. (Figure 45)
Figure 45.
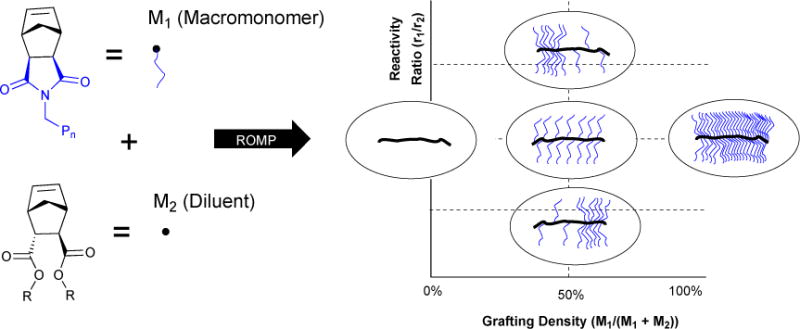
A method for simultaneously controlling the grafting density and side chain distribution in ROMP-derived brush copolymers by way of rational monomer design. PN is poly(styrene, lactide, or dimethyl siloxane). R is methyl, ethyl, or nBu.
4.10. Economical ROMP
The financial feasibility of ROMP is intrinsically linked to its scalability and in turn, its breadth of application. While efficient, traditional ROMP catalyst are expensive, required in high loadings (1/polymer chain), and often difficult to fully remove. Seeking a more cost-efficient alternative, we developed a method of catalyst recycling during ROMP.188 The so-called “pulsed addition” ROMP (PA-ROMP) affords homo- or block copolymers identical to those produced by traditional ROMP under conditions requiring seven-fold less catalyst. At its core, the method relies on a cyclic polymerization strategy; polymerization of an initial monomer population is followed by addition of a chain transfer agent (CTA). The CTA cleaves the initiator from the polymer end, simultaneously deactivating the growing chain and reforming the active initiator species. Addition of a second monomer population whose activity exceeds that of the CTA affords subsequent polymerization without the need to change pots. At this point, the cycle may be propagated or terminated. (Figure 46)
Figure 46.
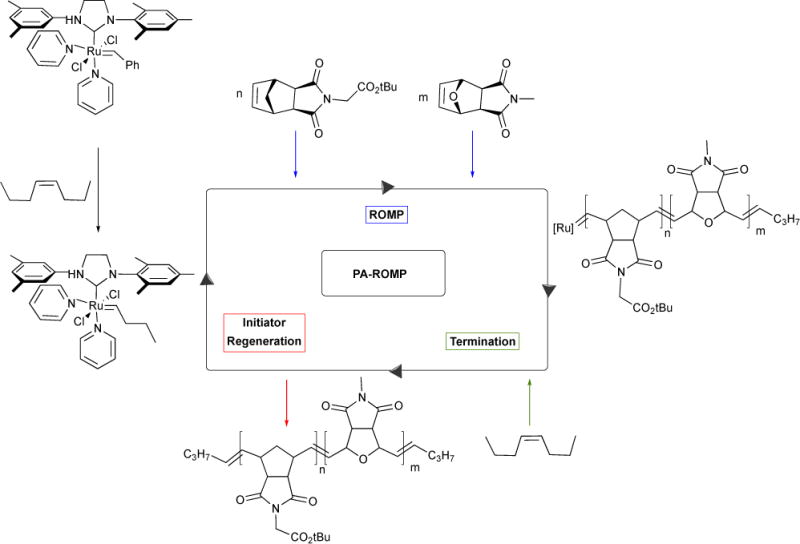
General scheme for the PA-ROMP of an exemplary poly(norbornene)-poly(oxanorbornene) block copolymer.
Ruthenium Removal from ROMP products
While PA-ROMP is unique in its cost efficiency, it comprises only one of the strategies reported for reducing Ru contamination in ROMP polymers, a challenge with ever-growing relevance as ROMP applications in the biomedical sphere continue to expand.189–191 Recently, our group reported a method for facile removal of heavy metal residues from a polymer chain by use a phase-Separable Polyisobutylene (PIB)-Supported Second-Generation Hoveyda-Grubbs Catalyst.192 The catalyst demonstrated activity analogous to its homogeneous counterpart in the polymerization of cyclopentene and a variety of its functionalized derivatives, affording commercially desirable polypentamer and poly(vinyl alcohol). The PIB Ru residue was readily removed from the concentrated crude product by a biphasic heptane-methanol wash, with the catalyst partitioning into the former and the polymer product into the latter phase. Polymer products synthesized with the PIB-bound Ru catalyst were considerably less contaminated than those synthesized by the non-supported analog even following multiple precipitations. As such, the PIB Ru catalyst holds the potential to improve purity standards and cut processing costs and time for large scale applications of ROMP-derived polymers.
The Future of Ru-ROMP
While lacking the precedent of the more established players in polymer chemistry, Ru-ROMP has quite rapidly gained recognition as a powerful, user-friendly tool for accessing well-defined materials with various applications— the breadth and significance of which can be evidenced by the medical, biological, electronic, and photonic examples discussed herein. Moreover, the highly tolerant, living nature of Ru-ROMP has shed new light on old materials, as functionalized polymers can now be accessed with unprecedented precision; undoubtedly this will elucidate the structure-property relationships characterizing many important materials, and in turn, facilitate rational design of their next generation derivatives. The importance of this contribution will only grow as Ru-ROMP continues to become both more precise and diverse, likely through the mechanism-driven development of new Ru catalysts. Industrially, the identity of Ru-ROMP is still very much in its infancy, although its potential is not difficult to appreciate given the ease of catalyst handling and their enhanced substrate scope. Several companies, such as Cymetech, LLC in Houston, TX, and Materia, a small company spun off from our lab in Pasadena, CA are currently working to expedite the translation of Ru-ROMP into the industrial setting with some promising early successes. For example, Ru-ROMP has been used industrially for the production of dicyclopentadiene-based thermoset resins for applications ranging from neat resins to syntactic foams to fiber-infused composites. These resins have high performance automotive applications, such as alternative fuel storage, body panels, and structural components, and are also used in electrical and electronic components. Other large-scale industrial applications include corrosive chemical storage, chlor-alkali electrolysis units, sanitary pipe and tank applications, heavy machinery, and high-pressure, high-temperature (HPHT) environments such as those encountered in resource extraction from deepwater or other high-pressure environments. These resins are also ideal for fabrication of longer, more durable, and more efficient wind turbine blades.193
5. Conclusions
In conclusion, we have reviewed recent advances from our laboratory in ruthenium-based olefin metathesis chemistry. Progress made by us and our collaborators toward experimental and computational studies on the mechanism of action, including modes of decomposition, has lent insights into developing novel catalyst architecture for enantioselective, Z-selective, and most recently E-selective olefin metathesis. We have employed these catalysts for RCM to form cyclic peptides, functionalized macrocyclic structures, as well as the challenging synthesis of tetrasubstituted olefins. We have also achieved chemoselective CM reactions using conjugated and non-conjugated dienes, as well as advancing industrial-scale ethenolysis reactions using CAAC-supported ruthenium catalysts with remarkable turnover of up to 340,000. Lastly, we reviewed advancements made in our laboratory to develop ruthenium-based ROMP catalysts, which has provided access to functionalized and well-defined polymers with a range of applications in polymer science and manufacturing.
Figure 22.
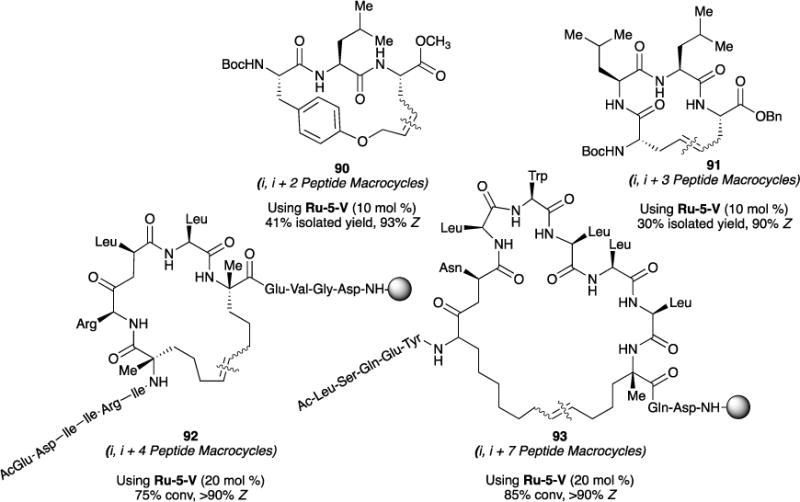
Z-selective RCM to form peptide macrocycles.
Figure 30.
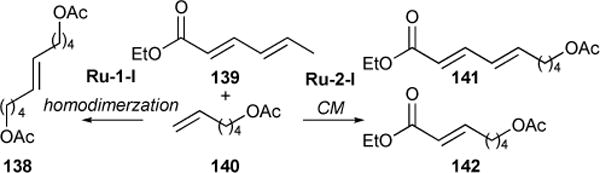
Achieving cross metathesis (CM) using more active Ru-2-I catalyst.
Acknowledgments
O.M.O. thanks Pomona College for provision of a Robbins Post-Doctoral Fellowship. N.C.W. thanks the Beckman Foundation for funding. D.J.O. thanks Pomona College, the National Science Foundation, and the Dreyfus Foundation for financial support. R.H.G. wishes to acknowledge the long-term support of the National Institute of Health and National Science Foundation that made this work possible. R.H.G. also acknowledges the many contributions both experimental and intellectual of the many students and postdoctoral fellows who have been associated with this research. We acknowledge the contributions of many groups who have helped push the area of ruthenium based metathesis to the present level. The commercial and practical developments in ruthenium metathesis technology resulting from the work at Materia, Inc. is gratefully acknowledged.
Notes and references
- 1.Grubbs RH. Angew Chem Int Ed. 2006;45:3760–3765. doi: 10.1002/anie.200600680. [DOI] [PubMed] [Google Scholar]
- 2.Novak BM, Grubbs RH. J Am Chem Soc. 1988;110:7542–7543. [Google Scholar]
- 3.Nguyen ST, Johnson LK, Grubbs RH, Ziller JW. J Am Chem Soc. 1992;114:3974–3975. [Google Scholar]
- 4.Nguyen ST, Grubbs RH, Ziller JW. J Am Chem Soc. 1993;115:9858–9859. [Google Scholar]
- 5.Fu GC, Nguyen ST, Grubbs RH. J Am Chem Soc. 1993;115:9856–9857. [Google Scholar]
- 6.Schwab P, France MB, Ziller JW, Grubbs RH. Angew Chem. 1995;107:2179–2181. [Google Scholar]
- 7.Schwab P, France MB, Ziller JW, Grubbs RH. Angew Chem Int Ed Engl. 1995;34:2039–2041. [Google Scholar]
- 8.Wilhelm TE, Belderrain TR, Brown SN, Grubbs RH. Organometallics. 1997;16:3867–3869. [Google Scholar]
- 9.Ackermann L, Fürstner A, Weskamp T, Kohl FJ, Herrmann WA. Tetrahedron Lett. 1999;40:4787–4790. [Google Scholar]
- 10.Huang J, Stevens ED, Nolan SP, Petersen JL. J Am Chem Soc. 1999;121:2674–2678. [Google Scholar]
- 11.Scholl M, Trnka TM, Morgan JP, Grubbs RH. Tetrahedron Lett. 1999;40:2247–2250. [Google Scholar]
- 12.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 13.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]
- 14.Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 15.Farina V, Horváth A. In: Handbook of Metathesis. Grubbs RH, Wenzel AG, O’Leary DJ, Khosravi E, editors. Wiley-VCH Verlag GmbH & Co KGaA; 2015. pp. 633–658. [Google Scholar]
- 16.WHO. WHO Model Lists of Essential Medicines. http://www.who.int/medicines/publications/essentialmedicines/en/, (accessed December 6, 2017)
- 17.Stoianova D, Johns A, Pederson R. Handbook of Metathesis. Wiley-VCH Verlag GmbH & Co KGaA; 2015. pp. 699–726. [Google Scholar]
- 18.Grubbs RH, editor. Handbook of Metathesis. 1. Wiley-VCH; Weinheim, Germany: 2003. [Google Scholar]
- 19.Grubbs RH, Wenzel AG, O’Leary DJ, Khosravi E, editors. Handbook of Metathesis, 3 Volume Set. 2. Wiley-VCH; Weinheim: 2015. [Google Scholar]
- 20.Jean-Louis Hérissonm P, Chauvin Y. Makromol Chem. 1971;141:161–176. [Google Scholar]
- 21.Adlhart C, Chen P. J Am Chem Soc. 2004;126:3496–3510. doi: 10.1021/ja0305757. [DOI] [PubMed] [Google Scholar]
- 22.Love JA, Sanford MS, Day MW, Grubbs RH. J Am Chem Soc. 2003;125:10103–10109. doi: 10.1021/ja027472t. [DOI] [PubMed] [Google Scholar]
- 23.Tallarico JA, Bonitatebus PJ, Snapper ML. J Am Chem Soc. 1997;119:7157–7158. [Google Scholar]
- 24.Trnka TM, Day MW, Grubbs RH. Organometallics. 2001;20:3845–3847. [Google Scholar]
- 25.Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 26.Anderson DR, Hickstein DD, O’Leary DJ, Grubbs RH. J Am Chem Soc. 2006;128:8386–8387. doi: 10.1021/ja0618090. [DOI] [PubMed] [Google Scholar]
- 27.Romero PE, Piers WE. J Am Chem Soc. 2005;127:5032–5033. doi: 10.1021/ja042259d. [DOI] [PubMed] [Google Scholar]
- 28.Wenzel AG, Grubbs RH. J Am Chem Soc. 2006;128:16048–16049. doi: 10.1021/ja0666598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wenzel AG, Blake G, VanderVelde DG, Grubbs RH. J Am Chem Soc. 2011;133:6429–6439. doi: 10.1021/ja2009746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong SH, Day MW, Grubbs RH. J Am Chem Soc. 2004;126:7414–7415. doi: 10.1021/ja0488380. [DOI] [PubMed] [Google Scholar]
- 31.Hong SH, Sanders DP, Lee CW, Grubbs RH. J Am Chem Soc. 2005;127:17160–17161. doi: 10.1021/ja052939w. [DOI] [PubMed] [Google Scholar]
- 32.Berlin JM, Campbell K, Ritter T, Funk TW, Chlenov A, Grubbs RH. Org Lett. 2007;9:1339–1342. doi: 10.1021/ol070194o. [DOI] [PubMed] [Google Scholar]
- 33.Ulman M, Grubbs RH. J Org Chem. 1999;64:7202–7207. [Google Scholar]
- 34.Hong SH, Wenzel AG, Salguero TT, Day MW, Grubbs RH. J Am Chem Soc. 2007;129:7961–7968. doi: 10.1021/ja0713577. [DOI] [PubMed] [Google Scholar]
- 35.Hong SH, Chlenov A, Day MW, Grubbs RH. Angew Chem. 2007;119:5240–5243. doi: 10.1002/anie.200701234. [DOI] [PubMed] [Google Scholar]
- 36.Seiders TJ, Ward DW, Grubbs RH. Org Lett. 2001;3:3225–3228. doi: 10.1021/ol0165692. [DOI] [PubMed] [Google Scholar]
- 37.Funk TW, Berlin JM, Grubbs RH. J Am Chem Soc. 2006;128:1840–1846. doi: 10.1021/ja055994d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berlin JM, Goldberg SD, Grubbs RH. Angew Chem Int Ed. 2006;45:7591–7595. doi: 10.1002/anie.200602469. [DOI] [PubMed] [Google Scholar]
- 39.Lavallo V, Canac Y, Präsang C, Donnadieu B, Bertrand G. Angew Chem Int Ed. 2005;44:5705–5709. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lavallo V, Canac Y, Präsang C, Donnadieu B, Bertrand G. Angew Chem. 2005;117:5851–5855. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lavallo V, Canac Y, DeHope A, Donnadieu B, Bertrand G. Angew Chem Int Ed. 2005;44:7236–7239. doi: 10.1002/anie.200502566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lavallo V, Canac Y, DeHope A, Donnadieu B, Bertrand G. Angew Chem. 2005;117:7402–7405. doi: 10.1002/anie.200502566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Romero PE, Piers WE. J Am Chem Soc. 2007;129:1698–1704. doi: 10.1021/ja0675245. [DOI] [PubMed] [Google Scholar]
- 44.Cavallo L. J Am Chem Soc. 2002;124:8965–8973. doi: 10.1021/ja016772s. [DOI] [PubMed] [Google Scholar]
- 45.Anderson DR, Lavallo V, O’Leary DJ, Bertrand G, Grubbs RH. Angew Chem Int Ed. 2007;46:7262–7265. doi: 10.1002/anie.200702085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anderson DR, Ung T, Mkrtumyan G, Bertrand G, Grubbs RH, Schrodi Y. Organometallics. 2008;27:563–566. doi: 10.1021/om7008028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marx VM, Sullivan AH, Melaimi M, Virgil SC, Keitz BK, Weinberger DS, Bertrand G, Grubbs RH. Angew Chem Int Ed. 2015;54:1919–1923. doi: 10.1002/anie.201410797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang AJ, Zhao Y, Schrock RR, Hoveyda AH. J Am Chem Soc. 2009;131:16630–16631. doi: 10.1021/ja908098t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flook MM, Jiang AJ, Schrock RR, Müller P, Hoveyda AH. J Am Chem Soc. 2009;131:7962–7963. doi: 10.1021/ja902738u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marinescu SC, Levine DS, Zhao Y, Schrock RR, Hoveyda AH. J Am Chem Soc. 2011;133:11512–11514. doi: 10.1021/ja205002v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Flook MM, Ng VWL, Schrock RR. J Am Chem Soc. 2011;133:1784–1786. doi: 10.1021/ja110949f. [DOI] [PubMed] [Google Scholar]
- 52.Meek SJ, O’Brien RV, Llaveria J, Schrock RR, Hoveyda AH. Nature. 2011;471:461–466. doi: 10.1038/nature09957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu M, Wang C, Kyle AF, Jakubec P, Dixon DJ, Schrock RR, Hoveyda AH. Nature. 2011;479:88–93. doi: 10.1038/nature10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Endo K, Grubbs RH. J Am Chem Soc. 2011;133:8525–8527. doi: 10.1021/ja202818v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keitz BK, Endo K, Patel PR, Herbert MB, Grubbs RH. J Am Chem Soc. 2012;134:693–699. doi: 10.1021/ja210225e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herbert MB, Suslick BA, Liu P, Zou L, Dornan PK, Houk KN, Grubbs RH. Organometallics. 2015;34:2858–2869. [Google Scholar]
- 57.Cannon JS, Zou L, Liu P, Lan Y, O’Leary DJ, Houk KN, Grubbs RH. J Am Chem Soc. 2014;136:6733–6743. doi: 10.1021/ja5021958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keitz BK, Endo K, Herbert MB, Grubbs RH. J Am Chem Soc. 2011;133:9686–9688. doi: 10.1021/ja203488e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hartung J, Grubbs RH. J Am Chem Soc. 2013;135:10183–10185. doi: 10.1021/ja4046422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marx VM, Herbert MB, Keitz BK, Grubbs RH. J Am Chem Soc. 2013;135:94–97. doi: 10.1021/ja311241q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Herbert MB, Marx VM, Pederson RL, Grubbs RH. Angew Chem Int Ed. 2013;52:310–314. doi: 10.1002/anie.201206079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miyazaki H, Herbert MB, Liu P, Dong X, Xu X, Keitz BK, Ung T, Mkrtumyan G, Houk KN, Grubbs RH. J Am Chem Soc. 2013;135:5848–5858. doi: 10.1021/ja4010267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu P, Xu X, Dong X, Keitz BK, Herbert MB, Grubbs RH, Houk KN. J Am Chem Soc. 2012;134:1464–1467. doi: 10.1021/ja2108728. [DOI] [PubMed] [Google Scholar]
- 64.Tsipis AC, Orpen AG, Harvey JN. Dalton Trans. 2005;0:2849–2858. doi: 10.1039/b506929g. [DOI] [PubMed] [Google Scholar]
- 65.Straub BF. Angew Chem Int Ed. 2005;44:5974–5978. doi: 10.1002/anie.200501114. [DOI] [PubMed] [Google Scholar]
- 66.Straub BF. Adv Synth Catal. 2007;349:204–214. [Google Scholar]
- 67.Herbert MB, Lan Y, Keitz BK, Liu P, Endo K, Day MW, Houk KN, Grubbs RH. J Am Chem Soc. 2012;134:7861–7866. doi: 10.1021/ja301108m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weng W, Parkin S, Ozerov OV. Organometallics. 2006;25:5345–5354. [Google Scholar]
- 69.Siegbahn PEM. J Phys Chem. 1995;99:12723–12729. [Google Scholar]
- 70.Ritter T, Hejl A, Wenzel AG, Funk TW, Grubbs RH. Organometallics. 2006;25:5740–5745. [Google Scholar]
- 71.Org Synth. 2003;80:85. [Google Scholar]
- 72.Stürmer R, Schäfer B, Wolfart V, Stahr H, Kazmaier U, Helmchen G. Synthesis. 2001;2001:0046–0048. [Google Scholar]
- 73.Kuhn KM, Champagne TM, Hong SH, Wei WH, Nickel A, Lee CW, Virgil SC, Grubbs RH, Pederson RL. Org Lett. 2010;12:984–987. doi: 10.1021/ol9029808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chatterjee AK, Toste FD, Goldberg SD, Grubbs RH. Pure Appl Chem. 2009;75:421–425. [Google Scholar]
- 75.Amabilino DB, Stoddart JF. Chem Rev. 1995;95:2725–2828. [Google Scholar]
- 76.Pease AR, Stoddart JF. Molecular Machines and Motors, Springer, Berlin, Heidelberg. 2001:189–236. [Google Scholar]
- 77.Bermudez V, Capron N, Gase T, Gatti FG, Kajzar F, Leigh DA, Zerbetto F, Zhang S. Nature. 2000;406:608–611. doi: 10.1038/35020531. [DOI] [PubMed] [Google Scholar]
- 78.Collin JP, Dietrich-Buchecker C, Gaviña P, Jimenez-Molero MC, Sauvage JP. Acc Chem Res. 2001;34:477–487. doi: 10.1021/ar0001766. [DOI] [PubMed] [Google Scholar]
- 79.Griffiths KE, Stoddart JF. Pure Appl Chem. 2008;80:485–506. [Google Scholar]
- 80.Leigh DA, Lusby PJ, Teat SJ, Wilson AJ, Wong JKY. Angew Chem Int Ed. 2001;40:1538–1543. doi: 10.1002/1521-3773(20010417)40:8<1538::AID-ANIE1538>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 81.Weck M, Mohr B, Sauvage JP, Grubbs RH. J Org Chem. 1999;64:5463–5471. doi: 10.1021/jo990268c. [DOI] [PubMed] [Google Scholar]
- 82.Wisner JA, Beer PD, Drew MGB, Sambrook MR. J Am Chem Soc. 2002;124:12469–12476. doi: 10.1021/ja027519a. [DOI] [PubMed] [Google Scholar]
- 83.Cantrill SJ, Pease AR, Stoddart JF. J Chem Soc Dalton Trans. 2000;0:3715–3734. [Google Scholar]
- 84.Cantrill SJ, Fulton DA, Heiss AM, Pease AR, Stoddart JF, White AJP, Williams DJ. Chem Weinh Bergstr Ger. 2000;6:2274–2287. doi: 10.1002/1521-3765(20000616)6:12<2274::aid-chem2274>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 85.Kolchinski AG, Busch DH, Alcock NW. J Chem Soc Chem Commun. 1995;0:1289–1291. [Google Scholar]
- 86.Kilbinger AFM, Cantrill SJ, Waltman AW, Day MW, Grubbs RH. Angew Chem Int Ed. 2003;42:3281–3285. doi: 10.1002/anie.200351167. [DOI] [PubMed] [Google Scholar]
- 87.Badjić JD, Cantrill SJ, Grubbs RH, Guidry EN, Orenes R, Stoddart JF. Angew Chem. 2004;116:3335–3340. doi: 10.1002/anie.200453963. [DOI] [PubMed] [Google Scholar]
- 88.Guidry EN, Cantrill SJ, Stoddart JF, Grubbs RH. Org Lett. 2005;7:2129–2132. doi: 10.1021/ol050463f. [DOI] [PubMed] [Google Scholar]
- 89.Miller SJ, Grubbs RH. J Am Chem Soc. 1995;117:5855–5856. [Google Scholar]
- 90.Blackwell HE, Grubbs RH. Angew Chem Int Ed. 1998;37:3281–3284. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3281::AID-ANIE3281>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 91.Blackwell HE, Sadowsky JD, Howard RJ, Sampson JN, Chao JA, Steinmetz WE, O’Leary DJ, Grubbs RH. J Org Chem. 2001;66:5291–5302. doi: 10.1021/jo015533k. [DOI] [PubMed] [Google Scholar]
- 92.Schafmeister CE, Po J, Verdine GL. J Am Chem Soc. 2000;122:5891–5892. [Google Scholar]
- 93.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, Filipovic Z, Qureshi FZ, Cai H, Berry P, Feyfant E, Shi XE, Horstick J, Annis DA, Manning AM, Fotouhi N, Nash H, Vassilev LT, Sawyer TK. Proc Natl Acad Sci. 2013;110:E3445–E3454. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Horvath A, Wuyts S, DEPRÉ DPM, COUCK WLJ, Cuypers JLJ, HARUTYUNYAN S, BINOT GFS. Improved process for preparing an intermediate of the macrocyclic protease inhibitor tmc 435. Google Patents. 2013 [Google Scholar]
- 96.Boal AK, Guryanov I, Moretto A, Crisma M, Lanni EL, Toniolo C, Grubbs RH, O’Leary DJ. J Am Chem Soc. 2007;129:6986–6987. doi: 10.1021/ja071148m. [DOI] [PubMed] [Google Scholar]
- 97.Toniolo C, Crisma M, Formaggio F, Peggion C. Pept Sci. 2001;60:396–419. doi: 10.1002/1097-0282(2001)60:6<396::AID-BIP10184>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 98.Marshall GR, Hodgkin EE, Langs DA, Smith GD, Zabrocki J, Leplawy MT. Proc Natl Acad Sci. 1990;87:487–491. doi: 10.1073/pnas.87.1.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Karle IL, Balaram P. Biochemistry (Mosc) 1990;29:6747–6756. doi: 10.1021/bi00481a001. [DOI] [PubMed] [Google Scholar]
- 100.Khan SN, Kim A, Grubbs RH, Kwon YU. Org Lett. 2011;13:1582–1585. doi: 10.1021/ol200226z. [DOI] [PubMed] [Google Scholar]
- 101.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Drug Dev Res. 1995;35:20–32. [Google Scholar]
- 102.Kwon YU, Kodadek T. J Am Chem Soc. 2007;129:1508–1509. doi: 10.1021/ja0668623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kwon YU, Kodadek T. Chem Commun. 2008;0:5704–5706. doi: 10.1039/b812735b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mangold SL, O’Leary DJ, Grubbs RH. J Am Chem Soc. 2014;136:12469–12478. doi: 10.1021/ja507166g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mangold SL, Grubbs RH. Chem Sci. 2015;6:4561–4569. doi: 10.1039/c5sc01507c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hartung J, Dornan PK, Grubbs RH. J Am Chem Soc. 2014;136:13029–13037. doi: 10.1021/ja506611k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stewart IC, Ung T, Pletnev AA, Berlin JM, Grubbs RH, Schrodi Y. Org Lett. 2007;9:1589–1592. doi: 10.1021/ol0705144. [DOI] [PubMed] [Google Scholar]
- 108.White DE, Stewart IC, Grubbs RH, Stoltz BM. J Am Chem Soc. 2008;130:810–811. doi: 10.1021/ja710294k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.O’Leary DJ, O’Neil GW. Handbook of Metathesis. Wiley-VCH Verlag GmbH & Co KGaA; 2015. pp. 171–294. [Google Scholar]
- 110.Chatterjee AK, Morgan JP, Scholl M, Grubbs RH. J Am Chem Soc. 2000;122:3783–3784. [Google Scholar]
- 111.Evans PA, Grisin A, Lawler MJ. J Am Chem Soc. 2012;134:2856–2859. doi: 10.1021/ja208668u. [DOI] [PubMed] [Google Scholar]
- 112.Han SB, Hassan A, Kim IS, Krische MJ. J Am Chem Soc. 2010;132:15559–15561. doi: 10.1021/ja1082798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jackson KL, Henderson JA, Motoyoshi H, Phillips AJ. Angew Chem Int Ed. 2009;48:2346–2350. doi: 10.1002/anie.200806111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Blackwell HE, O’Leary DJ, Chatterjee AK, Washenfelder RA, Bussmann DA, Grubbs RH. J Am Chem Soc. 2000;122:58–71. [Google Scholar]
- 115.Morrill C, Grubbs RH. J Org Chem. 2003;68:6031–6034. doi: 10.1021/jo0345345. [DOI] [PubMed] [Google Scholar]
- 116.Nicolaou KC, Li A, Edmonds DJ, Tria GS, Ellery SP. J Am Chem Soc. 2009;131:16905–16918. doi: 10.1021/ja9068003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nicolaou KC, Stepan AF, Lister T, Li A, Montero A, Tria GS, Turner CI, Tang Y, Wang J, Denton RM, Edmonds DJ. J Am Chem Soc. 2008;130:13110–13119. doi: 10.1021/ja8044376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nicolaou KC, Lister T, Denton RM, Montero A, Edmonds DJ. Angew Chem Int Ed. 2007;46:4712–4714. doi: 10.1002/anie.200701548. [DOI] [PubMed] [Google Scholar]
- 119.Nicolaou KC, Tang Y, Wang J, Stepan AF, Li A, Montero A. J Am Chem Soc. 2007;129:14850–14851. doi: 10.1021/ja076126e. [DOI] [PubMed] [Google Scholar]
- 120.Njardarson JT, Biswas K, Danishefsky SJ. Chem Commun. 2002;0:2759–2761. doi: 10.1039/b209941a. [DOI] [PubMed] [Google Scholar]
- 121.Uno BE, Gillis EP, Burke MD. Tetrahedron. 2009;65:3130–3138. [Google Scholar]
- 122.Willwacher J, Kausch-Busies N, Fürstner A. Angew Chem Int Ed. 2012;51:12041–12046. doi: 10.1002/anie.201206670. [DOI] [PubMed] [Google Scholar]
- 123.E JA, Jr, Stoltz BM. Nature. 2008;453:1228. doi: 10.1038/nature07046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dieckmann M, Kretschmer M, Li P, Rudolph S, Herkommer D, Menche D. Angew Chem Int Ed. 2012;51:5667–5670. doi: 10.1002/anie.201201946. [DOI] [PubMed] [Google Scholar]
- 125.Funk TW, Efskind J, Grubbs RH. Org Lett. 2005;7:187–190. doi: 10.1021/ol047929z. [DOI] [PubMed] [Google Scholar]
- 126.Cannon JS, Grubbs RH. Angew Chem Int Ed. 2013;52:9001–9004. doi: 10.1002/anie.201302724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ortiz A, Quesada A, Sanchez A. J Chem Ecol. 2004;30:991–1000. doi: 10.1023/b:joec.0000028463.43564.40. [DOI] [PubMed] [Google Scholar]
- 128.Turner RB, Jarrett AD, Goebel P, Mallon BJ. J Am Chem Soc. 1973;95:790–792. [Google Scholar]
- 129.Quigley BL, Grubbs RH. Chem Sci. 2014;5:501–506. doi: 10.1039/c3sc52806e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Luo SX, Cannon JS, Taylor BLH, Engle KM, Houk KN, Grubbs RH. J Am Chem Soc. 2016;138:14039–14046. doi: 10.1021/jacs.6b08387. [DOI] [PubMed] [Google Scholar]
- 131.Miyazaki H, Herbert MB, Liu P, Dong X, Xu X, Keitz BK, Ung T, Mkrtumyan G, Houk KN, Grubbs RH. J Am Chem Soc. 2013;135:5848–5858. doi: 10.1021/ja4010267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Johns AM, Ahmed TS, Jackson BW, Grubbs RH, Pederson RL. Org Lett. 2016;18:772–775. doi: 10.1021/acs.orglett.6b00031. [DOI] [PubMed] [Google Scholar]
- 133.Khan SN, Kim A, Grubbs RH, Kwon YU. Org Lett. 2012;14:2952–2955. doi: 10.1021/ol300808c. [DOI] [PubMed] [Google Scholar]
- 134.Cantrill SJ, Grubbs RH, Lanari D, Leung KCF, Nelson A, Poulin-Kerstien KG, Smidt SP, Stoddart JF, Tirrell DA. Org Lett. 2005;7:4213–4216. doi: 10.1021/ol051599g. [DOI] [PubMed] [Google Scholar]
- 135.Reyes DR, Iossifidis D, Auroux PA, Manz A. Anal Chem. 2002;74:2623–2636. doi: 10.1021/ac0202435. [DOI] [PubMed] [Google Scholar]
- 136.Auroux PA, Iossifidis D, Reyes DR, Manz A. Anal Chem. 2002;74:2637–2652. doi: 10.1021/ac020239t. [DOI] [PubMed] [Google Scholar]
- 137.Whitesides GM. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 138.Park CP, Van Wingerden MM, Han SY, Kim DP, Grubbs RH. Org Lett. 2011;13:2398–2401. doi: 10.1021/ol200634y. [DOI] [PubMed] [Google Scholar]
- 139.Hartung J, Grubbs RH. J Am Chem Soc. 2013;135:10183–10185. doi: 10.1021/ja4046422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gilliom LR, Grubbs RH. J Am Chem Soc. 1986;108:733–742. [Google Scholar]
- 141.Nguyen ST, Johnson LK, Grubbs RH, Ziller JW. J Am Chem Soc. 1992;114:3974–3975. [Google Scholar]
- 142.Grubbs RH, Tumas W. Science. 1989;243:907–915. doi: 10.1126/science.2645643. [DOI] [PubMed] [Google Scholar]
- 143.Novak BM, Risse W, Grubbs RH. Polymer Synthesis Oxidation Processes, Springer, Berlin, Heidelberg. 1992:47–72. [Google Scholar]
- 144.Grubbs RH. J Macromol Sci Part A. 1994;31:1829–1933. [Google Scholar]
- 145.Bielawski CW, Grubbs RH. Prog Polym Sci. 2007;32:1–29. [Google Scholar]
- 146.Leitgeb A, Wappel J, Slugovc C. Polymer. 2010;51:2927–2946. [Google Scholar]
- 147.Hejl A, Scherman OA, Grubbs RH. Macromolecules. 2005;38:7214–7218. [Google Scholar]
- 148.Tuba R, Al-Hashimi M, Bazzi HS, Grubbs RH. Macromolecules. 2014;47:8190–8195. [Google Scholar]
- 149.Scherman OA, Walker R, Grubbs RH. Macromolecules. 2005;38:9009–9014. [Google Scholar]
- 150.Tuba R, Grubbs RH. Polym Chem. 2013;4:3959–3962. [Google Scholar]
- 151.Lee HK, Bang KT, Hess A, Grubbs RH, Choi TL. J Am Chem Soc. 2015;137:9262–9265. doi: 10.1021/jacs.5b06033. [DOI] [PubMed] [Google Scholar]
- 152.Walker R, Conrad RM, Grubbs RH. Macromolecules. 2009;42:599–605. doi: 10.1021/ma801693q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Keitz BK, Fedorov A, Grubbs RH. J Am Chem Soc. 2012;134:2040–2043. doi: 10.1021/ja211676y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rosebrugh LE, Marx VM, Keitz BK, Grubbs RH. J Am Chem Soc. 2013;135:10032–10035. doi: 10.1021/ja405559y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Daeffler CS, Miyake GM, Li J, Grubbs RH. ACS Macro Lett. 2014;3:102–104. doi: 10.1021/mz4005953. [DOI] [PubMed] [Google Scholar]
- 156.Rosebrugh LE, Ahmed TS, Marx VM, Hartung J, Liu P, López JG, Houk KN, Grubbs RH. J Am Chem Soc. 2016;138:1394–1405. doi: 10.1021/jacs.5b12277. [DOI] [PubMed] [Google Scholar]
- 157.Matson JB, Grubbs RH. J Am Chem Soc. 2008;130:6731–6733. doi: 10.1021/ja802010d. [DOI] [PubMed] [Google Scholar]
- 158.Conrad RM, Grubbs RH. Angew Chem Int Ed. 2009;48:8328–8330. doi: 10.1002/anie.200903888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Weck M, Jackiw JJ, Rossi RR, Weiss PS, Grubbs RH. J Am Chem Soc. 1999;121:4088–4089. [Google Scholar]
- 160.Mendes PM. Chem Soc Rev. 2008;37:2512–2529. doi: 10.1039/b714635n. [DOI] [PubMed] [Google Scholar]
- 161.Vatansever F, Hamblin MR. J Nanoparticle Res. 2016;18:302. doi: 10.1007/s11051-016-3328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rutenberg IM, Scherman OA, Grubbs RH, Jiang W, Garfunkel E, Bao Z. J Am Chem Soc. 2004;126:4062–4063. doi: 10.1021/ja035773c. [DOI] [PubMed] [Google Scholar]
- 163.Xia Y, Verduzco R, Grubbs RH, Kornfield JA. J Am Chem Soc. 2008;130:1735–1740. doi: 10.1021/ja077192j. [DOI] [PubMed] [Google Scholar]
- 164.Guidry EN, Li J, Stoddart JF, Grubbs RH. J Am Chem Soc. 2007;129:8944–8945. doi: 10.1021/ja0725100. [DOI] [PubMed] [Google Scholar]
- 165.Clark PG, Day MW, Grubbs RH. J Am Chem Soc. 2009;131:13631–13633. doi: 10.1021/ja905924u. [DOI] [PubMed] [Google Scholar]
- 166.Momčilović N, Clark PG, Boydston AJ, Grubbs RH. J Am Chem Soc. 2011;133:19087–19089. doi: 10.1021/ja208515r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fürstner A, Ackermann L, Gabor B, Goddard R, Lehmann CW, Mynott R, Stelzer F, Thiel OR. Chem – Eur J. 2001;7:3236–3253. doi: 10.1002/1521-3765(20010803)7:15<3236::aid-chem3236>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 168.Bielawski CW, Benitez D, Grubbs RH. J Am Chem Soc. 2003;125:8424–8425. doi: 10.1021/ja034524l. [DOI] [PubMed] [Google Scholar]
- 169.Bielawski CW, Benitez D, Grubbs RH. Science. 2002;297:2041–2044. doi: 10.1126/science.1075401. [DOI] [PubMed] [Google Scholar]
- 170.Boydston AJ, Xia Y, Kornfield JA, Gorodetskaya IA, Grubbs RH. J Am Chem Soc. 2008;130:12775–12782. doi: 10.1021/ja8037849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Boydston AJ, Holcombe TW, Unruh DA, Fréchet JMJ, Grubbs RH. J Am Chem Soc. 2009;131:5388–5389. doi: 10.1021/ja901658c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Weitekamp RA, Atwater HA, Grubbs RH. J Am Chem Soc. 2013;135:16817–16820. doi: 10.1021/ja4093083. [DOI] [PubMed] [Google Scholar]
- 173.Xia Y, Kornfield JA, Grubbs RH. Macromolecules. 2009;42:3761–3766. [Google Scholar]
- 174.Xia Y, Olsen BD, Kornfield JA, Grubbs RH. J Am Chem Soc. 2009;131:18525–18532. doi: 10.1021/ja908379q. [DOI] [PubMed] [Google Scholar]
- 175.Xia Y, Boydston AJ, Grubbs RH. Angew Chem Int Ed. 2011;50:5882–5885. doi: 10.1002/anie.201101860. [DOI] [PubMed] [Google Scholar]
- 176.Johnson JA, Lu YY, Burts AO, Xia Y, Durrell AC, Tirrell DA, Grubbs RH. Macromolecules. 2010;43:10326–10335. doi: 10.1021/ma1021506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Johnson JA, Lu YY, Burts AO, Lim YH, Finn MG, Koberstein JT, Turro NJ, Tirrell DA, Grubbs RH. J Am Chem Soc. 2011;133:559–566. doi: 10.1021/ja108441d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Burts AO, Li Y, Zhukhovitskiy AV, Patel PR, Grubbs RH, Ottaviani MF, Turro NJ, Johnson JA. Macromolecules. 2012;45:8310–8318. [Google Scholar]
- 179.Hu M, Xia Y, McKenna GB, Kornfield JA, Grubbs RH. Macromolecules. 2011;44:6935–6943. [Google Scholar]
- 180.Miyake GM, Weitekamp RA, Piunova VA, Grubbs RH. J Am Chem Soc. 2012;134:14249–14254. doi: 10.1021/ja306430k. [DOI] [PubMed] [Google Scholar]
- 181.Sveinbjörnsson BR, Weitekamp RA, Miyake GM, Xia Y, Atwater HA, Grubbs RH. Proc Natl Acad Sci. 2012;109:14332–14336. doi: 10.1073/pnas.1213055109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Miyake GM, Piunova VA, Weitekamp RA, Grubbs RH. Angew Chem Int Ed. 2012;51:11246–11248. doi: 10.1002/anie.201205743. [DOI] [PubMed] [Google Scholar]
- 183.Fang L, Liu P, Sveinbjornsson BR, Atahan-Evrenk S, Vandewal K, Osuna S, Jiménez-Osés G, Shrestha S, Giri G, Wei P, Salleo A, Aspuru-Guzik A, Grubbs RH, Houk KN, Bao Z. J Mater Chem C. 2013;1:5747–5755. [Google Scholar]
- 184.Sveinbjörnsson BR, Miyake GM, El-Batta A, Grubbs RH. ACS Macro Lett. 2014;3:26–29. doi: 10.1021/mz400568j. [DOI] [PubMed] [Google Scholar]
- 185.Ohno S, Matyjaszewski K. J Polym Sci Part Polym Chem. 2006;44:5454–5467. [Google Scholar]
- 186.Matsuda M, Satoh K, Kamigaito M. Macromolecules. 2013;46:5473–5482. [Google Scholar]
- 187.Lin TP, Chang AB, Chen HY, Liberman-Martin AL, Bates CM, Voegtle MJ, Bauer CA, Grubbs RH. J Am Chem Soc. 2017;139:3896–3903. doi: 10.1021/jacs.7b00791. [DOI] [PubMed] [Google Scholar]
- 188.Matson JB, Virgil SC, Grubbs RH. J Am Chem Soc. 2009;131:3355–3362. doi: 10.1021/ja809081h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Vougioukalakis GC. Chem – Eur J. 2012;18:8868–8880. doi: 10.1002/chem.201200600. [DOI] [PubMed] [Google Scholar]
- 190.Sommer WJ, Weck M. Coord Chem Rev. 2007;251:860–873. [Google Scholar]
- 191.Buchmeiser MR. Chem Rev. 2009;109:303–321. doi: 10.1021/cr800207n. [DOI] [PubMed] [Google Scholar]
- 192.Al-Hashimi M, Tuba R, Bazzi HS, Grubbs RH. ChemCatChem. 2016;8:228–233. [Google Scholar]
- 193.Thermoset Resin Applications with Infinite Formulation Flexibility. Materia; http://www.materia-inc.com/products/thermoset-resins/applications, (accessed December 14, 2017) [Google Scholar]