

Clinically used antidepressants, such as selective serotonin reuptake inhibitors (SSRIs), aid only a fraction of patients. Furthermore, even successful use of SSRIs takes 2 to 6 weeks of maintained medication. Depressed patients need faster help. Since 2000, several clinical studies report that depressed patients given subanesthetic doses of ketamine showed improvement within 2 hours (1–4). Trials continue for various dosing regimens, formulations, and populations. It is not understood what causes the therapeutic action of the SSRIs, and it is also not clear how ketamine exerts its effects.
The best-known behavioral effect of ketamine is dissociative anesthesia. The drug retains Food and Drug Administration (FDA) approval for anesthesia in special populations as well as for veterinary use. The dissociative effects presumably arise from ketamine’s action to block N-methyl-D-aspartate (NMDA) receptor channels that have been opened by glutamate. The kinetics, equilibrium, and voltage sensitivity of open-channel blockers is a well-studied topic, and recent work (5) shows how ketamine becomes trapped within the channel pore of NMDA receptors at local concentrations of ~1 µM, which are expected to occur at the clinically effective antidepressant human doses (6).
How might blockade of NMDA receptors lead to the antidepressant effects? Most studies emphasize signal transduction pathways that could be modulated by the locally decreased Ca2+ flux through NMDA receptors, especially extrasynaptic GluN2B subunit-containing NMDA receptors (7–9). In one series of experiments, the decreased Ca2+ flux led to decreased activity of eukaryotic elongation factor 2 kinase, which in turn desuppressed eukaryotic elongation factor 2. This ribosome-binding protein then increased translation of brain-derived neurotrophic factor (BDNF) (10, 11). Many other experiments show that BDNF is released during antidepressant action.
WEAKNESSES OF THE NMDA RECEPTOR BLOCKADE HYPOTHESIS
It is not known whether a population of neurons has the appropriate combination of high firing rate and NMDA receptor expression to accumulate this “foot in the door” blockade. Recent data have diminished the likelihood that fast-spiking parvalbumin-sensitive interneurons, the most appropriate candidate, play a role (12).
Another argument in favor of NMDA receptor action, namely, that some other NMDA blockers also produce antidepressant effects (10, 13, 14), seems weaker. Anecdotally, most clinical researchers emphasize the uniquely rapid time course and effectiveness of ketamine. Furthermore, some open-channel blockers of NMDA receptors produce neither the intracellular signal transduction consequences nor the antidepressant effects (11).
A recent article showed that pyramidal neuron-restricted GluN2B-null animals do have activated mechanistic target of rapamycin (mTOR), less depression-like behavior, and activated protein synthesis and that these effects cannot be further enhanced by ketamine (9). Given that NMDA receptor knockouts have many defects, these data do not decisively prove the NMDA receptor hypothesis for ketamine action.
A more daunting challenge is to test whether ketamine’s entry into neurons is necessary for the drug’s rapid antidepressant action.
Ketamine blocks other ion channels. Researchers have proposed that α7 nicotinic receptor blockade partially underlies ketamine actions (15).
KETAMINE ACTIVATES THE mTOR PATHWAY
Regardless of the initial molecular interaction of ketamine, several research groups have shown that ketamine eventually activates the pathway involving mTOR (9, 13, 15). The best-known homeostatic action of this pathway is the adjustment of cell metabolism to match nutrient availability and to counteract autophagy, for instance during the first hours alter birth (16). The mTOR pathway can be activated by growth factors, presumably part of this pathway’s selective advantage. The Duman group produced evidence that mTOR activation lies downstream from BDNF action on TrkB receptors at the plasma membrane (17). In addition, mTOR activation has the major function of increasing protein synthesis, which itself could lead to increased BDNF secretion.
REASONS TO SUSPECT INTRACELLULAR ACTION OF KETAMINE
Low-molecular-weight neural drugs are usually assumed to act on the extracellular face of membrane proteins. This differs from the known action of most clinically used drugs, which act intracellularly. One can assess a molecule’s ability to enter cells via passive membrane permeation by the parameter logD(7.4), which equals logP, corrected for the fraction of permeant species at physiological pH. Based on published pKa and logP values (18), we calculate that logD for ketamine equals 1.85, similar to many FDA-approved drugs assumed to act intracellularly (see Figure 5.24 in reference 19). Thus, one should consider an intracellular action for ketamine. Innovative chemistry using copper to catalyze an azide-alkyne cycloaddition showed that some ketamine derivatives accumulate within cells (20). That ketamine readily permeates membranes underlies the several ongoing clinical trials using intranasal administration of ketamine and/or S-ketamine (also termed “esketamine”), because ketamine would permeate six plasma membranes to reach the CNS. One clinical trial of ketamine has achieved FDA fast-track status as a rapid antidepressant.
The mTOR Pathway Can Also Be Activated in Lysosomes
The mTORC1 branch of the mTOR pathway can also be activated by amino acids (21). The signaling pathway begins within the lumen of lysosomes. Lysosomal mTOR activation would enhance the selective advantage of the mTOR pathway.
Ketamine, like any weak base, is expected to accumulate in acidic compartments, such as lysosomes, via classical acid trapping (Figure 1) (24). As an α-amino ketone, ketamine bears some resemblance to an α-amino acid, and S-ketamine and L-amino acids have the same stereochemistry. Thus, it appears worthwhile to consider the hypothesis that ketamine, or one of its metabolites (25), can mimic amino acids within lysosomes. Activation of the mTOR pathway by amino acids is rather general, occurring in insect and mammalian cells. This action occurs when amino acids interact with at least two molecules residing in the lysosomal membrane: the V-ATPase (26) and the lysosomal amino acid transporter SLC38A9 (21, 27–29).
FIGURE 1. One Expects Ketamine to Accumulate in Acidic Vesicles of Any Living Cella.
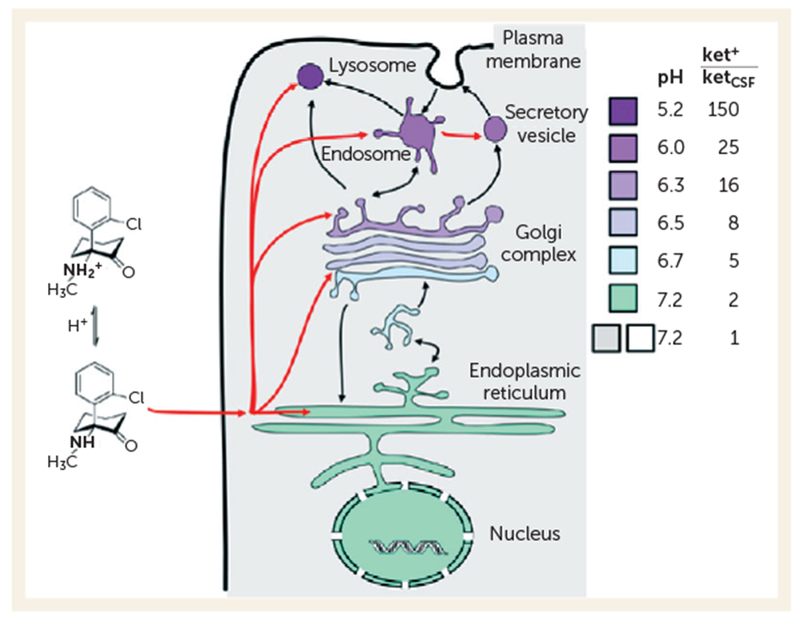
a ket+ = protonated ketamine concentration in organelles; ketCSF=ketamine concentration in CSF. The colors represent pH in cytoplasm and within organelles. The ratios are based on calculations by Trapp (22). That weakly basic psychiatric drugs accumulate in acidic vesicles, by a factor of 50–100, within neurons has been elegantly proven (23).
Note that mTOR pathways are complex in themselves and also interact with several other pathways. Also, mTOR participates in various CNS disorders, including tuberous sclerosis, some seizure disorders, and some autism spectrum disorders (30, 31). However, we do not imply that appreciable numbers of depressed patients have fundamental flaws in mTOR pathways (any more than appreciable numbers of depressed patients have low CSF serotonin).
The lysosome hypothesis, in its simplest form, predicts that any cell with lysosomes and the mTOR pathway—that is, almost any eukaryotic cell—would undergo mTOR activation by ketamine. This point has not been tested exhaustively, but it holds true for PC-12 cells despite their lack of NMDA receptors (25).
Pharmacological Chaperoning
A previous suggestion about the intracellular action of ketamine is that it acts within the endoplasmic reticulum and nearby regions of the Golgi as a pharmacological chaperone for NMDA receptors (6). A pharmacological chaperone is a small molecule that binds to and stabilizes a protein during early stages of synthesis, assembly, and processing. This suggestion arises from the emerging evidence that nicotine acts as a pharmacological chaperone for nicotinic receptors, underlying some aspects of both nicotine addiction (32) and the apparent neuroprotective effects of nicotine (33). Most studies on nicotine as a pharmacological chaperone have attempted to explain how days-to-weeks exposure can up-regulate nicotinic receptors and reduce endoplasmic reticulum stress (unfolded proteins within the endoplasmic reticulum lead to an intracellular homeostatic response, called the unfolded protein response, that rebalances protein and lipid processing). Of relevance to ketamine action, ~50% of up-regulation in response to nicotine occurs within the first 24 hours (34), and this action may well result from pharmacological chaperoning by nicotine within dendrites. Dendritic actions of ketamine are thought to underlie its antidepressant action (10, 13).
TESTING PROPOSED PATHWAYS ACTIVATED BY INTRACELLULAR KETAMINE
These two specific suggested intracellular mechanisms have guided our thinking, but other signaling pathways could be affected by intracellular ketamine. Whatever the target, one challenge would be to quantify the entry of ketamine, to study its dynamics, and to study its subcellular details.
A more daunting challenge is to test whether ketamine’s entry into neurons is necessary for the drug’s rapid antidepressant action. In the absence of a reductionist assay for antidepressant action, it would be appropriate to know whether cell-impermeant ketamine analogues—such as the quaternary amine—initiate mTOR activation, elongation factor 2 activation, or other signal transduction pathways. Such molecules would be in vitro probes, not drug candidates. Alternatively, selective delivery of ketamine to specific subcellular locales could also help tease out the effects of intracellular and extracellular ketamine (35).
Satisfying these challenges would provide the research community with a validated target, a way to define and localize target engagement, a relevant pharmacokinetic criterion at the subcellular level, and a set of additional molecules whose variation could explain the variability in patients’ response to ketamine. Such knowledge would provide a better rationale for developing antidepressants that act in less than a day.
Footnotes
The authors report no financial relationships with commercial interests.
REFERENCES
- 1.Berman RM, Cappiello A, Anand A, et al. : Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 2000; 47:351–354 [DOI] [PubMed] [Google Scholar]
- 2.Zarate CA Jr, Singh JB, Carlson PJ, et al. : A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 2006; 63:856–864 [DOI] [PubMed] [Google Scholar]
- 3.Zarate CA Jr, Mathews D, Ibrahim L, et al. : A randomized trial of a low-trapping nonselective N-methyl-D-aspartate channel blocker in major depression. Biol Psychiatry 2013; 74:257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murrough JW, Iosifescu DV, Chang LC, et al. : Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry 2013; 170:1134–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glasgow N, Johnson J: Voltage dependence of NMDA receptor inhibition by memantine and by ketamine depend on duration of glutamate application and on receptor subtype, in Neuroscience Abstracts (Program No 501.08). Washington, DC, Society for Neuroscience, 2014 [Google Scholar]
- 6.Lester HA, Miwa JM, Srinivasan R: Psychiatric drugs bind to classical targets within early exocytotic pathways: therapeutic effects. Biol Psychiatry 2012; 72:907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sutton MA, Taylor AM, Ito HT, et al. : Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron 2007; 55:648–661 [DOI] [PubMed] [Google Scholar]
- 8.Nosyreva E, Kavalali ET: Activity-dependent augmentation of spontaneous neurotransmission during endoplasmic reticulum stress. J Neurosci 2010; 30:7358–7368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller OH, Yang L, Wang CC, et al. : GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. eLife 2014; 3:e03581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Autry AE, Adachi M, Nosyreva E, et al. : NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011; 475:91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gideons ES, Kavalali ET, Monteggia LM: Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci USA 2014; 111: 8649–8654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pozzi L, Pollak Dorocic I,Wang X, et al. : Mice lacking NMDA receptors in parvalbumin neurons display normal depression-related behavior and response to antidepressant action of NMDAR antagonists. PLoS ONE 2014; 9:e83879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li N, Lee B, Liu RJ, et al. : mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010; 329:959–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Preskorn SH, Baker B, Kolluri S, et al. : An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol 2008; 28:631–637 [DOI] [PubMed] [Google Scholar]
- 15.Moaddel R, Abdrakhmanova G, Kozak J, et al. : Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholineevoked currents in α7 nicotinic acetylcholine receptors. Eur J Pharmacol 2013; 698:228–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuma A, Hatano M, Matsui M, et al. : The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032–1036 [DOI] [PubMed] [Google Scholar]
- 17.Lepack A, Fuchikami M, Dwyer J, et al. : Ketamine stimulates mTORC1 and BDNF release in primary cortical cultures, in Neuroscience Abstracts (Program No 808.09). Washington, DC, Society for Neuroscience, 2014 [Google Scholar]
- 18.Hansch C, Leo A, Hoekman D: Exploring QSAR: Hydrophobic, Electronic, and Steric Constants. Washington, DC, American Chemical Society, 1995 [Google Scholar]
- 19.Smith D, Allerton C, Kalgutkar A, et al. : Pharmacokinetics and Metabolism in Drug Design, 3rd ed. Edited by Mannhold R, Kubinyi H, Folkers G. Weinheim, Germany, Wiley, 2012 [Google Scholar]
- 20.Emnett C, Li H, Benz A, et al. : Novel reagents for cell biological and biochemical assessment of ketamine targets, in Neuroscience Abstracts (Program No 501.15). Washington, DC, Society for Neuroscience, 2014 [Google Scholar]
- 21.Bar-Peled L, Sabatini DM: Regulation of mTORC1 by amino acids. Trends Cell Biol 2014; 24:400–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapp S, Rosania GR, Horobin RW, et al. : Quantitative modeling of selective lysosomal targeting for drug design. Eur Biophys J 2008; 37: 1317–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tischbirek CH, Wenzel EM, Zheng F, et al. : Use-dependent inhibition of synaptic transmission by the secretion of intravesicularly accumulated antipsychotic drugs. Neuron 2012; 74:830–844 [DOI] [PubMed] [Google Scholar]
- 24.de Duve C, de Barsy T, Poole B, et al. : Commentary: lysosomotropic agents. Biochem Pharmacol 1974; 23:2495–2531 [DOI] [PubMed] [Google Scholar]
- 25.Paul RK, Singh NS, Khadeer M, et al. : (R,S)-Ketamine metabolites (R,S)-norketamine and (2S,6S)-hydroxynorketamine increase the mammalian target of rapamycin function. Anesthesiology 2014;121:149–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zoncu R, Bar-Peled L, Efeyan A, et al. : mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011; 334:678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jewell JL, Kim YC, Russell RC, et al. : Metabolism: differential regulation of mTORC1 by leucine and glutamine. Science 2015; 347:194–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang S, Tsun ZY, Wolfson RL, et al. : Metabolism: lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015; 347:188–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abraham RT: Cell biology: making sense of amino acid sensing. Science 2015; 347:128–129 [DOI] [PubMed] [Google Scholar]
- 30.Bourgeron T: A synaptic trek to autism. Curr Opin Neurobiol 2009; 19:231–234 [DOI] [PubMed] [Google Scholar]
- 31.Prather P, de Vries PJ: Behavioral and cognitive aspects of tuberous sclerosis complex. J Child Neurol 2004; 19:666–674 [DOI] [PubMed] [Google Scholar]
- 32.Henderson BJ, Lester HA: Inside-out neuropharmacology of nicotinic drugs. Neuropharmacology (Epub ahead of print, Feb 4,2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srinivasan R, Henderson BJ, Lester HA, et al. : Pharmacological chaperoning of nAChRs: a therapeutic target for Parkinson’s disease. Pharmacol Res 2014; 83:20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marks MJ, Stitzel JA, Collins AC: Time course study of the effects of chronic nicotine infusion on drug response and brain receptors. J Pharmacol Exp Ther 1985; 235:619–628 [PubMed] [Google Scholar]
- 35.Tian L, Yang Y, Wysocki LM, et al. : Selective esterase-ester pair for targeting small molecules with cellular specificity. Proc Natl Acad Sci USA 2012; 109:4756–4761 [DOI] [PMC free article] [PubMed] [Google Scholar]