

Abstract
Suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor (HDACI) approved by FDA for the treatment of cutaneous T cell lymphoma, is a promising anticancer drug for various cancers with a unique mode of action. However, it demonstrates limited clinical benefits in solid tumors as a single drug. In order to achieve enhanced and synergistic co-delivery of SAHA and Doxorubicin (DOX), a cleavable SAHA-based prodrug polymer (POEG-PSAHA), consisting of hydrophilic poly(oligo(ethylene glycol) methacrylate) (POEG) blocks and hydrophobic SAHA segments, has been developed. POEG-b-PSAHA prodrug polymer was able to form spherical micelles with a diameter around 60 nm, and well retained the pharmacological activity of SAHA in either inhibiting the proliferation of tumor cells or inducing histone acetylation. DOX formulated in POEG-b-PSAHA-based micelles showed a sustained release profile. DOX-loaded POEG-b-PSAHA exhibited more potent cytotoxicity towards tumor cells than free DOX and “inert” nanocarrier, POEG-b-POM. Consistently,DOX/POEG-b-PSAHA formulation resulted in an improved therapeutic effect in vivo compared to free DOX, Doxil, or DOX formulated in POEG-b-POM micelles. These results suggest that SAHA-based prodrug micelles may serve as a dual functional carrier for combination strategies in epigenetic-oriented anticancer therapy
Keywords: Vorinostat, Doxorubicin, Co-delivery, Prodrug micelles
1. Introduction:
Cancer development and progression is not only confined to genetic changes, but also involves epigenetic changes, leading to alterations in gene expression and cell phenotypes. Epigenetics is concerned with the heritable phenotypes based on changes in chromosome, as opposed to genetics, whose realm is on the basis of alterations in the primary DNA sequence (Avendaño and Menéndez, 2015, Berger et al., 2009). Alterations in the structure and modification status of chromatin represent powerful regulatory mechanisms for gene expression and genome stability (Avendaño and Menéndez, 2015). The major epigenetic modifications in mammals, and particularly in humans, are DNA methylation and post-translational histone modifications (acetylation, methylation, phosphorylation etc.) (Gibney and Nolan, 2010). In the context of histone modifications, the acetylation status of histones plays a crucial role in regulating gene expression by affecting the accessibility of DNA around them (Kouzarides, 2007). Acetylation levels are controlled by the balance of two enzymes families: histone acetyltransferase (HAT) and histone deacetylase (HDAC). HAT facilitates the acetylation of lysine in histone tails, which is associated with a more relaxed chromatin state, opening up access for transcription factors and polymerases, and therefore gene-transcription activation. In contrast, HDACs deacetylate the lysine residues thereby promoting a more condensed chromatin state and hence leading to transcriptional gene silencing (Grunstein, 1997, Gregory et al., 2001). Specially, the removal of the acetyl groups by HDACs also exposes the protonated amino groups of lysine units of histones, thereby increasing ionic interactions between the positively charged histones and negatively charged phosphate backbone of DNA, which yields a more compact chromatin structure and represses gene transcription by limiting the accessibility of the transcription machinery (Gregory et al.,2001).
A growing body of evidence indicates that the silence of tumor suppressor genes caused by HDAC overexpression could be a common phenomenon in tumor onset and progression (Zhu et al., 2004, Halkidou et al., 2004, Song et al., 2005). Therefore, there is growing interest in the development of HDAC inhibitors (HDACi) as anticancer agents and a large number of studies demonstrated the potential of HDACi in different cancer cell lines and in animal models of tumors (Marks et al., 2001, Johnstone, 2002, Falkenberg and Johnstone, 2014). The oral drug Vorinostat, also known as SAHA (from suberoylanilide hydroxamic acid) was the first HDAC inhibitor approved by the U.S. FDA in 2006 for the treatment of cutaneous T cell lymphoma in patients who have progressive, persistent or recurrent disease or following failure of two systemic therapies. It was also approved for multiple myeloma with slight advantage over Velcade®. SAHA is a pan-HDAC inhibitor and inhibits both classes I and II enzymes, but does not inhibit HDACs belonging to class III (Marks, 2007, Marks et al., 2001). SAHA acts as a micromolar inhibitor of HDACs by chelating the zinc atoms in HDAC catalytic sites via its hydroxamic end, with the phenyl ring protruding out of the binding pocket and lying on the hydrophobic surface of HDAC (Marks and Breslow, 2007). SAHA blocks the proliferation of cultured tumor cells and suppresses tumor growth in a variety of tumor models including prostate cancer (Butler et al., 2000), breast cancer (Cohen et al., 1999), leukemia (He et al., 2001), glioma (Eyupoglu et al., 2005) and lung cancer(Desai et al., 2003) with little or no toxicity.
Although SAHA has significant anticancer activity as a monotherapy in hematological malignancies, it demonstrates limited clinical benefit for patients with solid tumors, prompting the investigation of novel combination treatments with existing neoplastic interventions (Thurn et al., 2011). Besides, the acceptable toxicity profile of SAHA permits a broad integration into currently approved chemotherapy regimens (Munster et al., 2009,Marchion et al., 2005b, Marchion et al., 2005a). Based on a mechanistic rationale, SAHA has been shown to be additive or synergistic with a wide array of anticancer drugs, including conventional chemotherapeutics (doxorubicin, paclitaxel, cisplatin, gemcitabine, 5- fluorouracil and etoposide) (Arnold et al., 2007, Dowdy et al., 2006, Rikiishi et al., 2007, Kim et al., 2003), targeted agents (imatinib, bevacizumab and trastuzumab) (Nimmanapalli et al., 2003, Fuino et al., 2003) and transcriptional modulators (all-trans retinoic acid and the demethylating agent 5-aza-2’-deoxycytidine) (Cameron et al., 1999). One of the most significant combinations is with DNA damage-inducing therapies, which occurs, in part, through a SAHA-mediated increase in chromatin accessibility and downregulation of DNA repair (Thurn et al., 2011). Doxorubicin induces DNA strand breaks by binding to DNA, stabilizing the Topo II-DNA complex, and inhibiting the re-ligation of DNA strands during replication (Thom et al., 2011). SAHA potentiates DOX-induced Topo II-mediated DNA damage, growth inhibition and cell death through the chromatin decondensation, facilitating the access of topo II inhibitors to their DNA substrate (Marchion et al., 2005b, Marchion et al., 2005a). Moreover, the synergy is further intensified by the effect of SAHA on the expression, regulation and activation of a variety of DNA repair and DNA damage response genes, like Ku70 (Chen et al., 2007).
Although the combination of SAHA and other anticancer agents helps, to some extent, achieve better treatment of cancer, the triumph of simple physical mixture of multiple drugs is largely hindered as a result of poor solubility, minimal distribution to tumor tissue, and suboptimal dose ratio (Parhi et al., 2012). Carrier-mediated combination therapy could offer some advantages to overcome the above challenges, including improved solubility and bioavailability of each drug, increased accumulation at tumor sites through EPR effect, synchronized and controlled pharmacokinetics of each drug and ratiometric dosing, that is,the ability to tailor the relative ratios of each agent based on its pharmacological disposition (Lee and Nan, 2012).
Among the commonly used drug delivery systems, polymers with diverse structures have been widely selected as the nanocarriers for hydrophobic anticancer drugs, including SAHA, DOX, and others. Several nanoscopic formulations of SAHA have been developed for parenteral administrations to improve the solubility and overall disposition profile. For example, PEG-b-PLA block copolymer-based micelles have been employed as a carrier to encapsulate SAHA, and it was found that SAHA loaded in micelles provided sustained exposure and improved pharmacokinetics characterized by a significant increase in serum t1/2, AUC and mean residence time (Mohamed et al., 2012). However, the use of a large amount of pharmacologically inert carriers may not only add to the cost but also imposes safety issue (Croy and Kwon, 2006). Prodrug polymer nanocarriers represent an effective strategy to reduce the amount of inert materials. More import amphiphilic prodrug polymers self-assemble in aqueous solution and serve as dual-functional carriers to achieve the additive or synergistic effect with co-delivered anticancer drugs.
In this study, we designed synthesized SAHA-based prodrug polymer, denoted as POEG- b-PSAHA. These amphiphilic polymers could self-assemble into prodrug micelles and serve as nanocarriers for DOX delivery. The size and structure of POEG-b-PSAHA were characterized. Moreover, the in vitro pharmacological activity of POEG-b-PSAHA prodrug micelles was examined and compared with free SAHA. Finally, the antitumor activity of DOX-loaded POEG-b-PSAHA micelles was investigated in a syngeneic breast cancer model.
2. Methods
2.1. Materials
Aniline, Ν,Ν’-dicyclohexylcarbodiimide, 4-(dimethylamino)pyridine, hydroxylamine hydrochloride, 1-hydroxybenzotriazole hydrate, potassium carbonate, potassium hydroxide, vinylbenzyl chloride, 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid, oligo(ethylene glycol) methacrylate OEGMA (average Mn = 500), 2,2-Azobis(isobutyronitrile) (ΑIΒΝ), trypsin-EDTA solution, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and Dulbecco’s Modified Eagle’s Medium (DMEM) were all purchased from Sigma-Aldrich (MO, USA). ΑIΒΝ was purified by recrystallization in anhydrous ethanol. Monomethyl suberate was purchased from TCI (OR, USA). DOX.HC1 was purchased from LC Laboratories (MA, USA). Fetal bovine serum (FBS) and penicillin-streptomycin solution were purchased from Invitrogen (NY, USA.). Cell and were similarly handled as described before (Zhang et al., 2014b).
2.2. Methods
2.2.1. Synthesis of SAHA-monomer
SAHA was synthesized following a published method (Chung et al., 2013).Vinylbenzyl chloride (1.0 g, 6.6 mmol), succinic acid (7.7 g, 66.0 mmol, 10 equiv) and K2CO3 (4.5 g, 32.8 mmol, 5 equiv) were dissolved in 30 mL DMF and immersed into an oil bath at 90 °C. After 4 h, the mixture was filtered through cottons to remove excess amount of salt. The reaction was quenched by addition of excess amount of pure water and centrifuged to remove DMF. The precipitate was dissolved in DCM, and the crude product was purified by column chromatography on silica gel eluted with ethanol/DCM (v/v, 1: 9) to give the Compound 1 (1.4 mg, 91%).
For the synthesis of SAHA-monomer, CDI (69 mg, 0.43 mmol, 1.2 equiv) was added in portions to a stirred solution of Compound 1 (100 mg, 0.43 mmol, 1.2 equiv) in DCM. After stirring for 10 min, the reaction mixture turned into light yellow color. TLC showed that the intermediate was formed, and then SAHA (94 mg, 0.36 mmol) was added to the mixture in portions. After 2 h, the reaction was complete as confirmed by TLC, and the reaction was quenched by the addition of the saturated sodium bicarbonate. The aqueous layer was extracted with DCM, washed with brine, and dried over MgSO4. The crude product was chromatographed on silica gel (ethyl acetate/hexane =2:1) to give SAHA monomer 125 mg, 69%).
2.2.2. Synthesis of POEG macroCTA
POEG macroCTA was synthesized and purified following a published literature (Sun et al.,2016).
2.2.3. Synthesis of POEG-b-PSAHA
POEG macroCTA (150 mg, 0.02 mmol), SAHA-monomer (125 mg, 0.26 mmol, 13 equiv), AIBN (1 mg, 0.0061 mmol, 0.3 equiv) and 2 mL dried 1, 4-Dioxane were added into An Schlenk tube and deoxygenated by free-pump-thawing for three times. The mixture was then protected under N2 and immersed into an oil bath at 90 °C. After 24 h, the reaction was quenched by immersing the tube into liquid nitrogen. The mixture was purified through three cycles of dissolution/precipitation in DCM/hexane, and dried in vacuum to afford POEG-b-PSAHA (260mg, 96%).
2.2.4. Preparation and characterization of DOX-loaded POEG-b-PSAHA micelles
The blank or DOX-loaded POEG-b-PSAHA micelles were prepared through the film hydration method. First, DOX.HCl was neutralized with 3 equivalents of triethylamine in a mixture of DCM/methanol (1:1, v/v). DOX (5 mg/mL) was then mixed with POEG-b-PSAHA (50 mg/mL in DCM) at various carrier/drug weight ratios. After removing the solvent by nitrogen flow, the thin film was formed, which was further dried under vacuum for 2 h to remove any trace of remaining solvent. Then the thin film was hydrated with PBS followed by vortexing. The blank micelles were similarly prepared. The average diameter and the size distribution of micelles were examined via a Zetasizer (DLS). The morphology of blank or DOX-loaded POEG-b-PSAHA micelles was observed using transmission electron microscopy (TEM).
The drug loading efficiency of DOX was examined by Waters Alliance 2695 Separations Module combined with Waters 2475 Fluorescence Detector (excitation, 490 nm; emission, 590 nm; gain, 3; sensitivity (FUFS), 10 000) and high performance liquid chromatography (HPLC) respectively as described in previous publications (Zhang et al., 2014a). Drug loading capacity (DLC) and drug loading efficiency (DLE) were calculated according to the following equation: DLC %= [weight of drug loaded/(weight of polymer used + weight of drug used)]×100%, DLE %= (weight of loaded drug/weight of input drug)×100 %. The colloidal stability of drug-loaded micelles with various carrier/drug molar ratios at 4°C was evaluated by following the changes in sizes of the particles or visible precipitates every hour in the first 12 h and daily after 12 h following sample preparation.
The CMC of POEG-b-PSAHA was measured by fluorescence spectrometry using nile red as a fluorescence probe. Briefly, thirty microliters of nile red (0.05 mg/mL in DCM) were added to each tube and the solvent was removed by nitrogen flow. POEG-b-PSAHA micelles ranging from 6.lx 10 5 to 5 × 10−1 mg/mL were prepared with serial dilution as described above and then added into the tubes with nile red. The final concentration of nile red was kept at 6.0 × 10−7 M. The micelles were vortexed and kept overnight at room temperature to reach the equilibrium of solubilization. The samples were excited at an excitation wavelength of 550 nm and fluorescence intensities were recorded at an emission spectrum from 600 to 700 nm. The peak intensities at 647 nm were plotted versus polymer concentrations. The CMC value was calculated as the cross-point where a sharp increase in fluorescence intensity was observed.
The in vitro release of DOX from DOX-loaded POEG-b-PSAHA micelles was examined was observed.using a dialysis method. Briefly, two hundred and fifty microliters of DOX/POEG-b-PSAHA micelles or free DOX Cl (0.5 mg DOX/mL) were transferred to clamped dialysis bags (MWCO 3500 Da) in triplicate and incubated in 50 mL PBS as release medium, with gentle shaking at 100 rpm at 37 °C. One milliliter of medium was withdraw and same volume of fresh medium was supplemented at 10 min, 0.5, 1, 2, 4, 6, 8, 10, 12, 24, 48 and 72 h. DOX release from micelles was measured by fluorescence spectrometry at the excitation wavelength of 480 nm and emission wavelength of 590 nm by Synergy HI Hybrid Multi-Mode Microplate Reader (Winooski, VT).
2.2.5. In vitro cytotoxicity assay
Cytotoxicit;ty assay was performed with 4T1.2 mouse breast cancer cells, MCF-7 human breast cancer cells, and HCT-116 human colon cancer cells. 4T1.2 (1×103 cells/well), MCF-7 (5 ×103 cells/well), or HCT-116 (2x103 cells/well) cells were seeded in 96-well plates followed by 24 h of incubation in DMEM with 10% FBS and 1% streptomycin/penicillin. To evaluate the combinational effect of SAHA with other anticancer drugs, various concentrations of free SAHA, free DOXHCl, or the combination of both were added in triplicate to cells and incubated for 48 h. To study the cytotoxicity of blank POEG-b-PSAHA micelles, cells were challenged with various concentrations of POEG-b-PSAHA micelles or free SAHA (in DMSO) at equivalent SAHA concentrations for 48 h. The cytotoxicity of DOX-loaded POEG-b-PSAHA micelles was also examined, in comparison to DOX.HCl and DOX loaded in a biologically inert polymer POEG-b-POM, Cells were treated with different DOX formulations of varied concentrations. Blank POEG-b-PSAHA or POEG-b-POM at concentrations equivalent to those of carriers in the corresponding DOX formulation groups was also added into cells. Cells were incubated for 48 h and cell viability was determined by MTT assay. Briefly, ten microliters of 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) (5mg/mL in PBS) were added to cells. Following incubation for 4 h, the medium was removed and MTT formazan was solubilized by DMSO. The absorbance was measured by a microplate reader with wavelength at 590 nm and reference wavelength at 620 nm. Untreated cells were used as a control. Cell viability was calculated as [(ODtreat-ODblank )/(ODcontrol-ODblank)×100%]
2.2.6. Western blot
Western immunoblotting was conducted with 4T1.2 cells. Cells were treated with 0.1% DMSO (control) or indicated concentrations (1 μM or 5 μM) of SAHA or POEG-b-PSAHA, respectively. After 24 h treatment, cells were rinsed twice with ice-cold PBS and lysed by RIPA buffer. The lysates were centrifuged at 12,500 rpm for 10 min. Samples with equal amounts of total cellular proteins were subjected to sodium dodecylsulfate polyacryl amide gel electrophoresis (SDS-PAGE), followed by transferring to nitrocellulose membranes. The membranes were first blocked in 5% nonfat dry milk dissolved in DPBS with 0.1% Tween 20 (PBST) for 1 h at RT, and then incubated with primary antibody at a final dilution of 1:1,000 in 5% BSA in PBST overnight at 4 °C. After washing three times with PBST, the membranes were incubated with secondary antibody at a final dilution of 1:5,000 in PBST for 1 h at room temperature. After washing three times with PBST, bound antibodies were detected by chemiluminescence. Beta-actin was used as the loading control. Primary antibodies for AC- H3 and AC-H4 were from Cell Signaling Technology (MA, USA) and the antibody for beta-actin was from Sigma-Aldrich (MO, USA).
2.2.7. Plasma pharmacokinetics
For plasma pharmacokinetics, DOX/POEG-b-PSAHA micelles or free DOX.HClwas injected intravenously into tumor-free female CD1 mice at a DOX dose of 5 mg/kg, with five mice in each group. Blood samples were collected in heparinized tubes at designated time points (3min, 10 min, 30 min, 1 h, 2 h, 4 h, 8 h and 12 h) post injection. The samples were centrifuged at 12,500 rpm for 10 min at 4 °C and 20 The supernatant were collected, Then 200 μL methanol, containing 500 ng/mL daunorubicin as the internal standard, was added and vortexed for 3 min. The samples were centrifuged at 12,500 rpm for 10 min at 4 °C and 150 μL of the clear supernatant was collected and injected into HPLC for DOX analysis.
2.2.8. In vivo therapeutic study
The in vivo antitumor efficacy of the DOX loaded in POEG-b-PSAHA prodrug nanocarrier was tested in a syngeneic 4T1.2 mouse breast cancer model. Female BALB/c mice were s.c. inoculated with 4T1.2 cells at right mammary fat pad with 2 × 105 cells/mouse. When the tumor sizes reached around 50 mm3, mice were randomly divided into five groups (n=5), and treated via tail vein injection with PBS, DOX.HC1, Doxil, DOX/POEG-b-POM or DOX/POEG-b-PSAHA micelles at a DOX dose of 5 mg/kg on days 1, 4 and 7, respectively.Tumor sizes were measured with the digital caliper every three days following the initiation of the treatment and calculated by the formula: (L×W2)/2, in which L is the longest and W is the shortest in tumor diameters (mm). Data were plotted as the actual tumor volume. Body weights were also monitored for the indication of toxicity. On day 23 post injection, all mice were sacrificed and tumor tissues were collected for measurement of weights and photography.
2.2.8. Statistical analysis
In vitro or in vivo data are presented as mean ± standard deviation (SD) or mean ± standard error of mean (SEM), respectively. Statistical analysis was performed with two-tailed Student’s T test or one-way analysis of variance (ANOVA) for two groups or multiple groups, respectively, followed by Turkey simultaneous post hoc test. In all statistical analyses, P < 0.05 was considered statistically significant.
3. Results and Discussion:
3.1. Effect of combination of SAHA and DOX on tumor cell proliferation
The proliferation inhibitory effect of SAHA and DOX was examined in 4T1.2 and HCT-116 cell lines. As shown in Fig. 1A, SAHA or DOX inhibited the proliferation of 4T1.2 cells in a concentration-dependent manner. It was also apparent that combination of the two agents led to enhanced cytotoxicity. A similar combinational effect was found in HCT-116 cells (Fig. 1B). Combination index (Cl) was then calculated to assess a potential synergism between SAHA and DOX, by the equation CI=(dl/IC50l)+ (d2/IC502), with dl or d2 being the concentration of SAHA or DOX required to achieve 50% killing effect in co-treatment, while IC501 or IC502 being IC50 of SAHA or DOX in single treatment, respectively. The Cl of SAHA and DOX combination were 0.67 and 0.84 in 4T1.2 and HCT-116 cells respectively, suggesting a synergism between the two drugs.
Fig. 1.
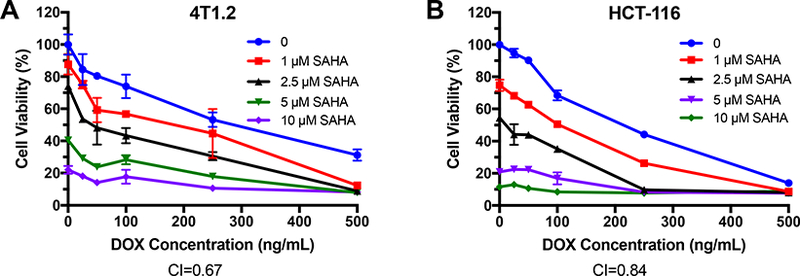
Synergistic effect between SAHA and DOX in inhibiting the proliferation of tumor cells. 4T1.2 (A) or HCT-116 (B) cells were treated with various concentrations of free SAHA, free DOX or the combination of SAHA and DOX. After 48 h, the cytotoxicity was determined by MTT assay. The experiments was performed in triplicate and repeated three times. Data are presented as means ± SD.
3.2. Synthesis and characterization of the POEG-b-PSAHA polymer
We initially designed and synthesized a SAHA-based monomer by directly conjugating SAHA with methacryloyl chloride. The macro-chain transfer agent POEG was then synthesized by RAFT polymerization of hydrophilic OEGMA monomer, which was further formed from this polymer had a size around 160 nm (data not shown), which is suboptimal for EPR effect. In order to decrease the hydrodynamic size, we re-designed a new SAHA monomer by introducing an additional 4 carbon hydrophobic chain and another benzyl ring to SAHA (Scheme. 1). Vinylbenzyl chloride was linked to succinic acid, followed by the condensation reaction with SAHA to form a hydrolysable diester bond. Then RAFT polymerization was similarly performed as described above, yielding the amphiphilic POEG-b-PSAHA block copolymer. The structure of POEG-b-PSAHA polymer was confirmed by 1H NMR (Fig. 2), and the average degree of polymerization of the SAHA monomer was calculated. Each POEG-b-PSAHA molecule contained 12 units of SAHA.
Scheme 1.
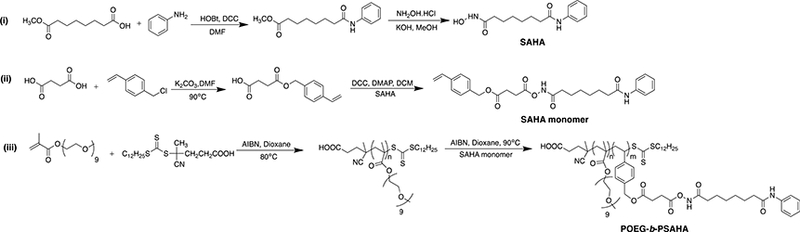
Synthesis of the SAHA-monomer and POEG-b-PSAHA polymers via RAFT polymerization.
Fig. 2.
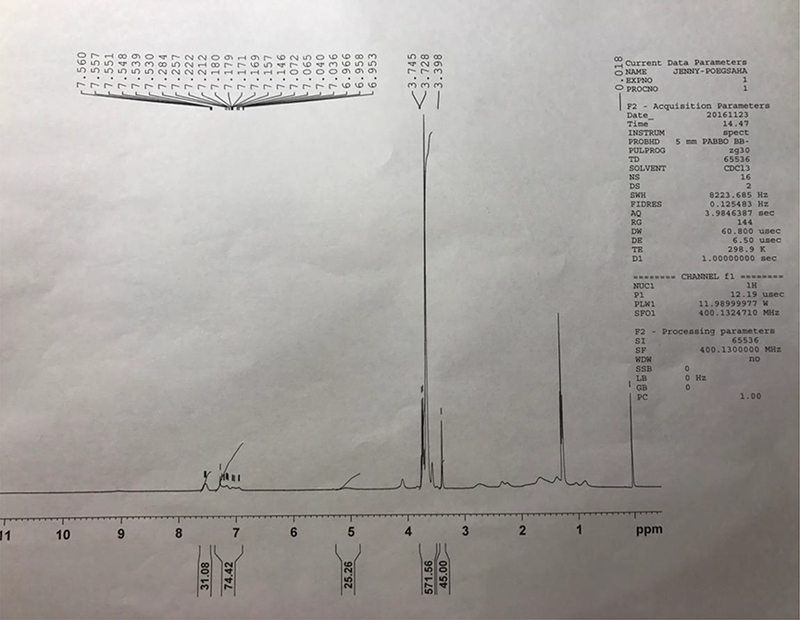
1H NMR spectra of POEG-b-PSAHA polymer in CDCl3.
3.3. Characterizations of DOX-loaded POEG-b-PSAHA micelles
Blank or DOX-loaded POEG-b-PSAHA micelles were prepared by a simple film hydration method. Unlike the initial SAHA polymer that formed relatively large-sized micelles (~160 nm), the size of drug-free POEG-b-PSAHA micelles was significantly smaller (63.7 nm). This is likely due to more effective carrier-carrier interaction as a result of incorporation of additional 4 carbon hydrophobic chain and another benzyl ring to SAHA monomer. Loading of DOX into POEG-b-PSAHA micelles at a carrier/drug ratio 20:1 only led to a used to initiate the polymerization of hydrophobic SAHA slight increase in the particle size (~70 nm) (Fig. 3B). TEM showed sphere shape with a uniform size distribution, which was consistent with DLS (Fig. 3C, D). The size, DLC, and formulation stability of drug-loaded POEG-b-PSAHA micelles were then examined and listed in Table 1. DOX could be loaded into POEG-b-PSAHA micelles at a carrier/drug ratio as low as 5/1 (mg/mg), at which ratio DOX loading capacity was at capacity was 14.9 % and DOX-loaded micelles were stable for 5 days at 4 °C. With an increase in the carrier/drug ratio, the drug encapsulation efficiency and colloidal stability were further improved. As shown in Table 1,at a carrier/drug ratio of 50:1, DOX-loaded micelles were stable for 50 days at 4 °C. A carrier/drug mass ratio of 20:1 and 5:1 was used for in vitro and in vivo study, respectively. Fig. 4 shows that the CMC of POEG-b-PSAHA micelles was around 0.004 mg/mL as determined by a rapid change of fluorescence intensity of loaded nile red probe during serial dilution. This low CMC of POEG-b-PSAHA shall provide a good stability for the micelles upon dilution in blood stream after i.v. injection.
Fig. 3.
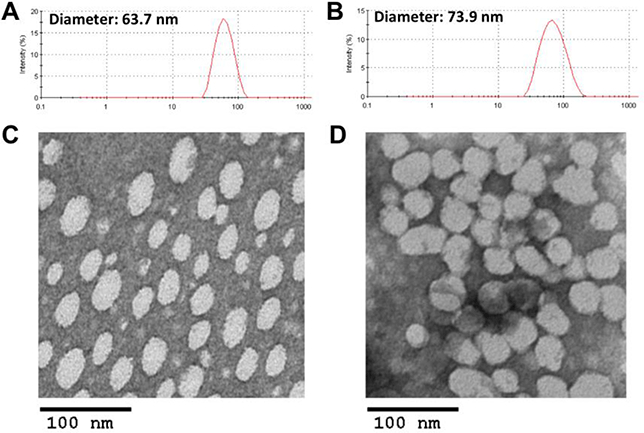
Particle size distribution of blank POEG-b-PSAHA micelles (A) and DOX-loaded POEG-b-PSAHA micelles (B). TEM images of blank POEG-b-PSAHA micelles (C) and DOX-loaded POEG-b-PSAHA micelles (D). Scale bar is 100 nm.
Table 1.
Physicochemical characterizations of DOX-loaded POEG-b-PSAHA micelles.
| Micelles | Mass ratio (mg: mg)a |
Size (nm)b | PDIC | DLC(%)d | DLE(%)e | Stabilityf |
|---|---|---|---|---|---|---|
| POEG-b-PSAHA: | 63.7 ± 0.7 | 0.14 ± 0.01 | ||||
| POEG-b-PSAHA: | 5:1 | 78.4 ± 0.4 | 0.22 ± 0.01 | 14.9 | 89.3 | 5 d |
| DOX | ||||||
| POEG-b-PSAHA: | 10: 1 | 75.9 ± 0.6 | 0.18 ± 0.01 | 8.2 | 90.6 | 9 d |
| DOX | ||||||
| POEG-b-PSAHA: | 20:1 | 73.9 ± 0.9 | 0.26 ± 0.01 | 4.5 | 93.5 | 17 d |
| DOX | ||||||
| POEG-b-PSAHA: | 30: 1 | 79.7 ± 0.3 | 0.23 ± 0.01 | 92.3 | 32 d | |
| DOX | 3.0 | |||||
| POEG-b-PSAHA: | 50:1 | 86.6 ± 0.2 | 0.20± 0.01 | 1.9 | 96.7 | 50 d |
| DOX |
DOX concentration in micelles was kept at 0.5 mg/mL.
Measured by dynamic light scattering particle sizer.
PDI = polydispersity index.
DLC = drug loading capacity.
DLE = drug loading efficiency.
Data mean there was no noticeable size change and visible precipitates during the follow-up period at 4 °C.
Fig. 4.
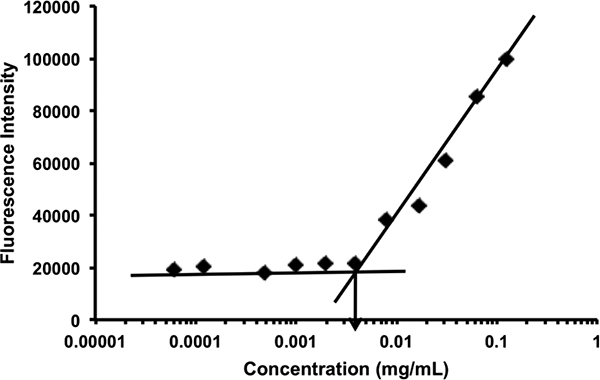
CMC of POEG-b-PSAHA micelles using nile red as a fluorescence probe. The fluorescence intensity of nile red was collected at the emission wavelength of 647 nm and the excitation wavelength of 550 nm. The fluorescence intensity was plotted versus concentrations of POEG-b-PSAHA copolymer. Values reported are the means ± SD for triplicate samples.
3.4. In vitro drug release
The release profile of DOX loaded in POEG-b-PSAHA micelles in PBS was investigated by a dialysis method and compared to that of free DOX. As shown in Fig. 5, around 50% of DOX was released from DOX.HC1 in the first two hours and 85% of total DOX was released less than 1% of DOX was released from the DOX-loaded POEG-b-first hour and a relatively sustained release continued for 72 hours, with with only around 30% of total drug released. The slow release kinetics might be due to strong carrier-drug interaction, including hydrophobic interaction and π-π stacking, which shall contrast provide a good stability of DOX/POEG-b-PSAHA in blood stream.
Fig. 5.
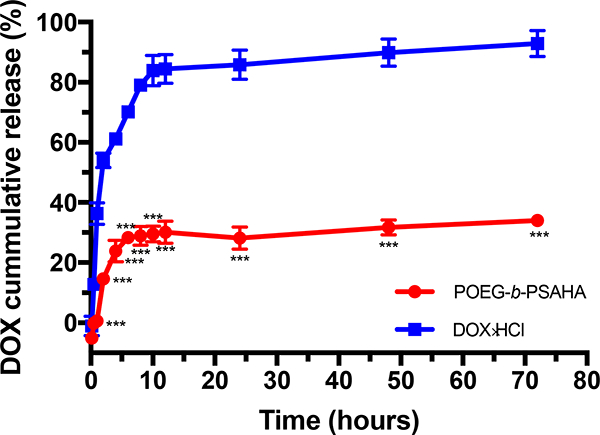
Cumulative DOX release profile from POEG-b-PSAHA micelles with free DOX as a control. PBS was used as the release medium. DOX concentration was fixed at 0.5 mg/mL. Values reported are the means ± SD for triplicate samples. *** P < 0.001 (POEG-b- PSAHA/DOX vs. free DOX).
3.5. In vitro cytotoxicity of drug-free or DOX-loaded POEG-b- PSAHA micelles
To evaluate whether POEG-b-PSAHA prodrug micelles maintained the ability of SAHA to inhibit tumor cell proliferation, 4T1.2, MCF-7 or HCT-116 cells were treated with POEG-b- PSAHA micelles or free SAHA (in DMSO) for 48 h. As shown in Fig. 6A, free SAHA inhibited the proliferation of 4T1.2 tumor cells in a dose-dependent manner, while POEG-b- PSAHA showed a comparable cytotoxicity. Similar results were shown in MCF-7 and HCT- 116 tumor cells (Fig. 6B & C), suggesting that SAHA could be effectively released from POEG-b-PSAHA via the cleavage of carbamide bond.
Fig. 6.
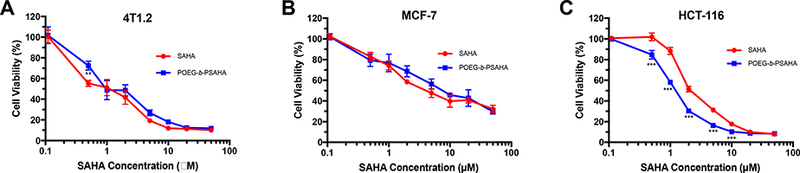
MTT cytotoxicity of POEG-b-PSAHA prodrug micelles in 4T1.2 mouse breast cancer cell line (A), MCF-7 human breast cancer cell line (B), and HCT-116 human colon cancer cell line (C) in comparison to free SAHA. Cells were treated for 48 h and values reported are the means ± SD for triplicate samples. **P < 0.01, ***P<0.01 (POEG-b-PSAHA vs. SAHA).
The in vitro cytotoxicity of DOX-loaded POEG-b-PSAHA micelles was also tested, in comparison with DOX.HC1 and DOXloaded in pharmacologically “inert” POEG-b-POM micelles. POEG-b-PSAHA and POEG-b-POM carrier alone were included as controls and were used at the same concentrations as those used in drug-loaded micelles. As shown in Fig. 7A, POEG-Λ-POM blank micelles showed minimal cytotoxicity even at the highest concentration (20 μg/mL), In contrast, blank POEG-b-PSAHA micelles exhibited significant cytotoxicity, which was comparable to that of DOX.HC1. DOX loaded in POEG-/1-PSAHA was more potent in killing tumor cells than DOX.HC1 and DOX/POEG-/1-POM formulation. Similar results were observed in MCF-7 and HCT-116 cells (Fig. 7B & C). It is likely that the improved cytotoxicity of DOX/POE G-b-PSAHA is attributed to a synergistic action between DOX and SAHA that was released from the carrier following intracellular delivery.
Fig. 7.
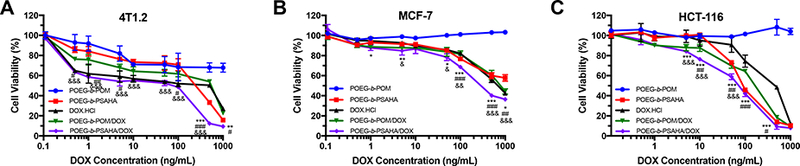
MTT cytotoxicity assay of DOX-loaded POEG-b-PSAHA micelles in 4T1.2 (A), MCF-7 (B) and HCT-116 (C) tumor cells after 48 h treatment. Data are presented as the means ± SD for triplicate samples. *P<0.05, **P<0.01, ***P<0.001 (POEG-b-PSAHA/DOX vs. DOX.HC1); #P< 0.05, ##P<0.01, ###P<0.001 (POEG-b-PSAHA/DOX vs. POEG-b- POM/DOX); &P < 0.05, &&P<0.01, &&&P<0.001 (POEG-b-PSAHA/DOX vs. POEG-b-PSAHA).
3.6. Effect of POEG-b-PSAHA on histone acetylation
To investigate if the POEG-b-PSAHA-mediated cytotoxicity is attributed to the specific action of released SAHA, we examined the efficiency of POEG-b-PSAHA in modulating the acetylation of histone in 4T1.2 breast cancer cells by Western blot. As shown in Fig. 8, the basal levels of acetylated H3 and H4 were very low. SAHA treatment at submicromolar concentrations for 24 h led to significant increases in their levels in a dose-dependent manner. Interestingly, POEG-b-PSAHA was comparable to or even more potent than free SAHA in inducing acetylation of H3 or H4 at the same concentrations of SAHA. These data indicated that POEG-b-PSAHA well retained the bioactivity of SAHA in inhibiting HDAC, resulting in the hyperacetylation of histone H3 and H4 and subsequently inhibition of cancer cell proliferation. The possible mechanism for the comparable or even further enhanced efficacy in histone acetylation is that POEG-b-PSAHA protected the hydroxamic acid of SAHA from hydrolysis and pharmacologically active SAHA was slowly released from the polymer over a prolonged period of time. On the other hand, free SAHA with unprotected hydroxamic acid may be subjected to rapid metabolic conversion to inactive metabolites, SAHA-glucuronide and 4-anilino-4-oxobutanoic acid (Hattori et al., 2011).
Fig. 8.
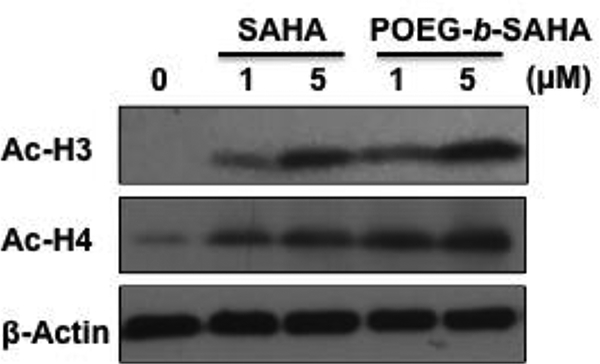
Effect of POEG-b-PSAHA prodrug micelles on expression of acetylated histone 3 (Ac-H3) and acetylated histone 4 (Ac-H4) compared to free SAHA. 4T1.2 cells were treated with various concentrations of SAHA or POEG-b-PSAHA at an equivalent concentration of SAHA for 24 h. Western blotting was performed with total protein extracts using antibodies against acetylated H3 and H4. Equal loading and transfer were verified by reproving the membranes for β-actin.
3.7. Plasma pharmacokinetics
The DOX/POEG-b-PSAHA or DOX.HCl was injected into tumor-free mice at a DOX dose of 5 mg/kg. The plasma concentrations of DOX were examined at different time points. The initial concentration of POEG-b-PSAHA was around 1 mg/mL, which was 250-fold higher than its CMC (0.004 mg/mL). The concentrations of DOX in the blood following i.v. injection of different treatments as a function of time were illustrated in Fig. 9. Compared to DOX.HCl group, the plasma concentrations of DOX from the group treated with POEG-b- PSAHA-b-DOX were significantly higher at early time points and maintained at relatively high levels until 12 h, which is likely attributed to the surface modification of PEG and stealth-shielding against RES system. These data demonstrated that DOX formulated in POEG-b- PSAHA prodrug micelles was able to circulate for a significantly longer period of time in the blood.
Fig. 9.
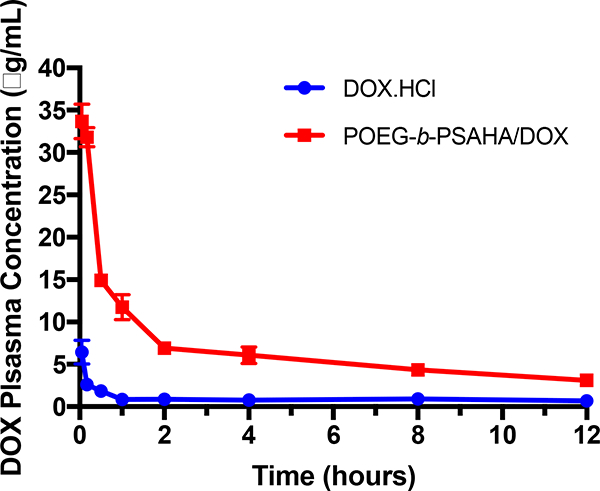
Plasma pharmacokinetics of DOX.HCl and DOX/POEG-b-PSAHA micelles in tumorfree female CD1 mice at the same dose of 5 mg DOX/kg. Values reported are the means ± SEM, n =5.
3.8. In vivo therapeutic study
Following the demonstration of a potential synergistic effect between POEG-b-PSAHA- based carrier and the co-delivered DOX in cultured cancer cells, we then evaluated the tumor growth inhibitory effect of DOX/POEG-b-PSAHA in a highly metastatic syngeneic murine breast cancer model (4T1.2, s.c.). As shown in Fig. 10A, free DOX exhibited a modest antitumor activity at a dose of 5 mg/kg. Doxil and DOX/POEG-b-POM formulations were more effective than free DOX. At the same dose of DOX, DOX/POEG-b-PSAHA was mosteffective in inhibiting the tumor growth (Fig. 10A). Fig. 10 B & D show images of tumor tissues collected at the end of the experiment, which were consistent with the result of tumor growth curves (Fig. 10A). The significantly improved anti-tumor activity of DOX/POEG-b-PSAHA is likely attributed to the effective accumulation of the nanocarrier at tumor sites due to its small size (~70 nm). The synergistic effect between the released SAHA and co-delivered DOX may also play an important role in tumor inhibition. More studies are needed in the future to further delineate the underlying mechanism. There were slight increases in body weights in all groups over the period of study, suggesting the negligible toxicity of DOX/POEG-b-PSAHA micelles in vivo.
Fig. 10.
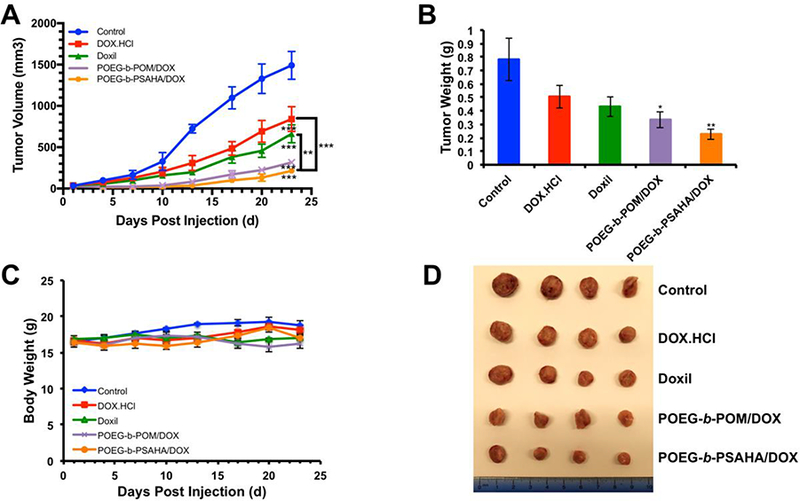
(A) Antitumor activity of DOX.HC1, Doxil, DOX-loaded POEG-b-POM and DOX- loaded POEG-b-PSAHA micelles in female BALB/c mice bearing 4T1.2 breast tumor. Three injections were given on days 1, 4, 7 and each point represents the mean of actual tumor volume. (B) Weights of tumors collected from different groups at the end of experiment. (C) Changes of body weight in mice receiving different treatments. (D) Photographs of tumors collected from different treatment groups at the end of experiment. Values reported are the means ± SEM, n = 5. *P < 0.05, **P < 0.01, ***P < 0.001 (vs. control).
4. Conclusion:
We have developed a well-characterized POEG-b-PSAHA prodrug micellar nanocarrier, which consists of a hydrophobic segment composed of 12 SAHA-based units, and a POEG hydrophilic segment for efficient entrapment of DOX. Our POEG-b-PSAHA prodrug micelles well retained the biological activity of SAHA in inhibiting the proliferation of tumor cells and promoting the acetylation of histone H3 and H4. Besides, POEG-b-PSAHA was effective in formulating DOX and demonstrated a slow kinetics of drug release. More importantly, in vivo delivery of DOX via POEG-b-PSAHA led to significant inhibition of 4T1.2 tumor, much more effectively than DOX.HCl, Doxil and DOX/POEG-b-POM.
Fig. 11.
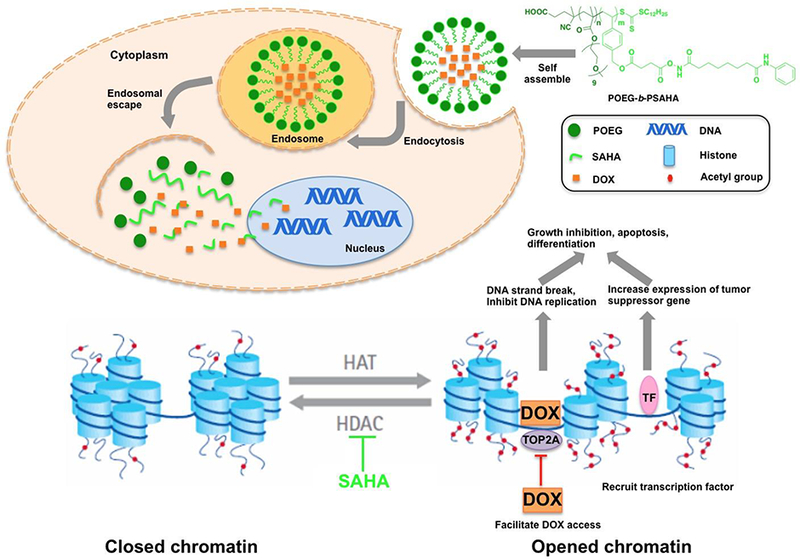
The potential mechanism for the synergistic action between DOX and SAHA in tumor cells following the intracellular delivery of DOX/POEG-b-PSAHA system.
Acknowledgments
We would like to thank Dr. Ming Sun for her help with negative EM study.
This work was supported by NIH grants no. R01CA174305 and no. R01CA219399.
Footnotes
Declaration of interest
Th authors declare no competing financial interest.
Reference:
- ARNOLD NB, ARKUS N, GUNN J & KORC M 2007. The Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Induces Growth Inhibition and Enhances Gemcitabine-Induced Cell Death in Pancreatic Cancer. Clinical Cancer Research, 13, 18–26. [DOI] [PubMed] [Google Scholar]
- AVENDANO C & MENENDEZ JC 2015. Chapter 8 - Epigenetic Therapy of Cancer. Medicinal Chemistry of Anticancer Drugs (Second Edition). Boston: Elsevier. [Google Scholar]
- BERGER SL, KOUZARIDES T, SHIEKHATTAR R & SHILATIFARD A 2009. An operational definition of epigenetics. Genes Dev, 23, 781–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUTLER LM, AGUS DB, SCHER HI, HIGGINS B, ROSE A, CORDON- CARDO C, THALER HT, RIFKIND RA, MARKS PA & RICHON VM 2000. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res, 60, 5165–70. [PubMed] [Google Scholar]
- CAMERON EE, BACHMAN KE, MYOHANEN S, HERMAN JG & BAYLIN SB 1999. Synergy of demethylation and histone deacetylase inhibition in the reexpression of genes silenced in cancer. Nat Genet, 21, 103–7. [DOI] [PubMed] [Google Scholar]
- CHEN CS, WANG YC, YANG HC, HUANG PH, KULP SK, YANG CC, LU YS, MATSUYAMA S, CHEN CY & CHEN CS 2007. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res, 67, 5318–27. [DOI] [PubMed] [Google Scholar]
- CHUNG Y-M, EL-SHAZLY M, CHUANG D-W, HWANG T-L, ASAI T, OSHIMA Y, ASHOUR ML, WU Y-C & CHANG F-R 2013. Suberoylanilide Hydroxamic Acid, a Histone Deacetylase Inhibitor, Induces the Production of Antiinflammatory Cyclodepsipeptides from Beauveria felina. Journal of Natural Products, 76, 1260–1266. [DOI] [PubMed] [Google Scholar]
- COHEN LA, AMIN S, MARKS PA, RIFKIND RA, DESAI D & RICHON VM 1999. Chemoprevention of carcinogen-induced mammary tumorigenesis by the hybrid polar cytodifferentiation agent, suberanilohydroxamic acid (SAHA). Anticancer Res, 19, 4999–5005. [PubMed] [Google Scholar]
- CROY SR & KWON GS 2006. Polymeric micelles for drug delivery. Curr Pharm Des, 12, 4669–84. [DOI] [PubMed] [Google Scholar]
- DESAI D, DAS A, COHEN L, EL-BAYOUMY K & AMIN S 2003. Chemopreventive efficacy of suberoylanilide hydroxamic acid (SAHA) against 4-(methylnitrosamino)- 1-(3-pyridyl)-1-butanone (NNK)-induced lung tumorigenesis in female A/J mice. Anticancer Res, 23, 499–503. [PubMed] [Google Scholar]
- DOWDY SC, JIANG S, ZHOU XC, HOU X, JIN F, PODRATZ KC & JIANG S-W 2006. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Molecular Cancer Therapeutics, 5, 2767–2776. [DOI] [PubMed] [Google Scholar]
- EYUPOGLU IY, HAHNEN E, BUSLEI R, SIEBZEHNRUBL FA, SAVASKAN NE, LUDERS M, TRANKLE C, WICK W, WELLER M, FAHLBUSCH R & BLUMCKE I 2005. Suberoylanilide hydroxamic acid (SAHA) has potent antiglioma properties in vitro, ex vivo and in vivo. J Neurochem, 93, 992–9. [DOI] [PubMed] [Google Scholar]
- FALKENBERG KJ & JOHNSTONE RW 2014. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov, 13, 673–91. [DOI] [PubMed] [Google Scholar]
- FUINO L, BALI P, WITTMANN S, DONAPATY S, GUO F, YAMAGUCHI H, WANG HG, ATADJA P & BHALLA K 2003. Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol Cancer Ther, 2, 971–84. [PubMed] [Google Scholar]
- GIBNEY ER & NOLAN CM 2010. Epigenetics and gene expression. Heredity, 105, 4–13. [DOI] [PubMed] [Google Scholar]
- GREGORY PD, WAGNER K & HORZ W 2001. Histone acetylation and chromatin remodeling. Exp Cell Res, 265, 195–202. [DOI] [PubMed] [Google Scholar]
- GRUNSTEIN M 1997. Histone acetylation in chromatin structure and transcription. Nature, 389, 349–52. [DOI] [PubMed] [Google Scholar]
- HALKIDOU K, GAUGHAN L, COOK S, LEUNG HY, NEAL DE & ROBSON CN 2004. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate, 59, 177–89. [DOI] [PubMed] [Google Scholar]
- HATTORI Y, NAGAOKA Y, KUBO M, YAMASAKU H, ISHII Y, OKITA H, NAKANO H, UESATO S & MAITANI Y 2011. Antitumor effect of liposomal histone deacetylase inhibitor-lipid conjugates in vitro Chem Pharm Bull (Tokyo), 59, 1386–92. [DOI] [PubMed] [Google Scholar]
- HE LZ, TOLENTINO T, GRAYSON P, ZHONG S, WARRELL RP JR., RIFKIND RA, MARKS PA, RICHON VM & PANDOLFI PP 2001. Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia. J Clin Invest, 108, 1321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSTONE RW 2002. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov, 1, 287–99. [DOI] [PubMed] [Google Scholar]
- KIM MS, BLAKE M, BAEK JH, KOHLHAGEN G, POMMIER Y & CARRIER F 2003. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res, 63, 7291–300. [PubMed] [Google Scholar]
- KOUZARIDES T 2007. Chromatin Modifications and Their Function. Cell, 128, 693–705. [DOI] [PubMed] [Google Scholar]
- LEE JH & NAN A 2012. Combination drug delivery approaches in metastatic breast cancer. Journal of drug delivery, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARCHION DC, BICAKU E, DAUD AI, SULLIVAN DM & MUNSTER PN 2005a. In vivo synergy between topoisomerase II and histone deacetylase inhibitors: predictive correlates. Mol Cancer Ther, 4, 1993–2000. [DOI] [PubMed] [Google Scholar]
- MARCHION DC, BICAKU E, TURNER JG, DAUD AI, SULLIVAN DM & MUNSTER PN 2005b. Synergistic interaction between histone deacetylase and topoisomerase II inhibitors is mediated through topoisomerase Ilbeta. Clin Cancer Res, 11, 8467–75. [DOI] [PubMed] [Google Scholar]
- MARKS P, RIFKIND RA, RICHON VM, BRESLOW R, MILLER T & KELLY WK 2001. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer, 1, 194–202. [DOI] [PubMed] [Google Scholar]
- MARKS PA 2007. Discovery and development of SAHA as an anticancer agent. Oncogene, 26, 1351–6. [DOI] [PubMed] [Google Scholar]
- MARKS PA & BRESLOW R 2007. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol, 25, 84–90. [DOI] [PubMed] [Google Scholar]
- MOHAMED EA, ZHAO Y, MESHALI ΜM, REMSBERG CM, BORG TM, FODA AΜ, TAKEMOTO JK, SAYRE CL, MARTINEZ SE, DAVIES NM & FORREST ML 2012. Vorinostat with sustained exposure and high solubility in poly(ethylene glycol)-b-poly(DL-lactic acid) micelle nanocarriers: characterization and effects on pharmacokinetics in rat serum and urine. J Pharm Sci, 101, 3787–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUNSTER PN, MARCHION D, THOMAS S, EGORIN M, MINTON S, SPRINGETT G, LEE JH, SIMON G, CHIAPPORI A, SULLIVAN D & DAUD A 2009. Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. British Journal of Cancer, 101, 1044–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIMMANAPALLI R, FUINO L, STOBAUGH C, RICHON V & BHALLA K 2003. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cells. Blood, 101, 3236–3239. [DOI] [PubMed] [Google Scholar]
- PARHI P, MOHANTY C & SAHOO SK 2012. Nanotechnology-based combinational drug delivery: an emerging approach for cancer therapy. Drug Discovery Today, 17, 1044–1052. [DOI] [PubMed] [Google Scholar]
- RIKIISHI H, SHINOHARA F, SATO T, SATO Y, SUZUKI M & ECHIGO S 2007. Chemosensitization of oral squamous cell carcinoma cells to cisplatin by histone deacetylase inhibitor, suberoylanilide hydroxamic acid. Int J Oncol, 30, 1181–8. [PubMed] [Google Scholar]
- SONG J, NOH JH, LEE JH, EUN JW, AHN YM, KIM SY, LEE SH, PARK WS, YOO NJ, LEE JY & NAM SW 2005. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS, 113, 264–8. [DOI] [PubMed] [Google Scholar]
- SUN J, CHEN Y, LI K, HUANG Y, FU X, ZHANG X, ZHAO W, WEI Y, XU L, ZHANG P, VENKATARAMANAN R & LI S 2016. A prodrug micellar carrier assembled from polymers with pendant farnesyl thiosalicylic acid moieties for improved delivery of paclitaxel. Acta Biomater, 43, 282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THORN CF, OSHIRO C, MARSH S, HERNANDEZ-BOUSSARD T, MCLEOD H, KLEIN TE & ALTMAN RB 2011. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics, 21, 440–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THURN KT, THOMAS S, MOORE A & MUNSTER PN 2011. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future oncology (London, England), 7, 263–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG X, HUANG Y, ZHAO W, LIU H, MARQUEZ R, LU J, ZHANG P, ZHANG Y, LI J, GAO X, VENKATARAMANAN R, XU L & LI S 2014a. Targeted delivery of anticancer agents via a dual function nanocarrier with an interfacial drug-interactive motif. Biomacromolecules, 15, 4326–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG X, LIU K, HUANG Y, XU I, LI I, ΜΑ X. & LI S. 2014b. Reduction- sensitive dual functional nanomicelles for improved delivery of paclitaxel. Bioconjug Chem, 25, 1689–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU P, MARTIN E, MENGWASSER I, SCHLAG P, JANS SEN KP & GOTTLICHER M 2004. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell, 5, 455–63. [DOI] [PubMed] [Google Scholar]