

Abstract
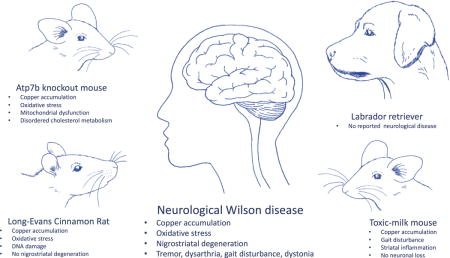
Wilson disease (WD) is an autosomal recessive disorder of copper metabolism manifesting with hepatic, neurological and psychiatric symptoms. The limitations of the currently available therapy for WD (particularly in the management of neuropsychiatric disease), together with our limited understanding of key aspects of this illness (e.g. neurological vs hepatic presentation) justify the ongoing need to study WD in suitable animal models. Four animal models of WD have been established: the Long-Evans Cinnamon rat, the toxic-milk mouse, the Atp7b knockout mouse and the Labrador retriever. The existing models of WD all show good similarity to human hepatic WD and have been helpful in developing an improved understanding of the human disease. As mammals, the mouse, rat and canine models also benefit from high homology to the human genome. However, important differences exist between these mammalian models and human disease, particularly the absence of a convincing neurological phenotype. This review will first provide an overview of our current knowledge of the orthologous genes encoding ATP7B and the closely related ATP7A protein in C. elegans, Drosophila and zebrafish (Danio rerio) and then summarise key characteristics of rodent and larger mammalian models of ATP7B-deficiency.
Graphical abstract
Wilson disease is an autosomal recessively inherited copper storage disease. Neurological impairment is common. In this review, we describe the different animal models available for this condition with particular focus on rodent models. Many of them closely resemble the hepatic involvement observed in human WD patients, but do not develop a neurological deficit.
First described in 1912 by S.A.K. Wilson, MD, (Kinnier Wilson 1912), Wilson disease (WD) is an autosomal recessive disorder of copper metabolism manifesting predominantly with hepatic, neurological and psychiatric symptoms (Bandmann et al. 2015). The gene mutated in WD is ATP7B, which encodes a P1B-type copper-transporting adenosine triphosphatase (ATPase) (Bull et al. 1993; Tanzi et al. 1993). ATP7B is expressed most highly in the liver and to a lesser degree also in the brain, kidney, placenta, mammary glands and testis (Bull et al. 1993; Tanzi et al. 1993; Michalczyk et al. 2008).
ATP7B has two functions in hepatic tissue: to supply copper to the trans-Golgi network for incorporation into ceruloplasmin and to facilitate the biliary excretion of excess copper (Terada et al. 1999; Terada et al. 1998).
At normal copper concentrations, ATP7B is situated at the trans-Golgi network where it transfers copper into the Golgi lumen for incorporation into ceruloplasmin (Hung et al., 1997; Lutsenko and Cooper, 1998; Yang et al., 1997). In polarised hepatocytes in response to physiologically-relevant copper concentrations, ATP7B migrates from the trans-Golgi network to apical vesicles (Roelofsen et al. 2000; Guo et al. 2005; Braiterman et al. 2009; Lalioti et al. 2016), whereas under conditions of copper overload, ATP7B moves to lysosomes, facilitating lysosome-mediated exocytosis and release of copper in bile (Polishchuk et al. 2014). The targeting of ATP7B to late endosomes have also been reported (Harada et al. 2005).
The specific roles of ATP7B in the brain, kidneys, lungs, and eye are much less understood and may involve buffering of Cu levels in the cytosol and regulating the amount of copper available for other copper-containing enzymes. Mutations in ATP7B impair biliary copper excretion and copper incorporation in ceruloplasmin. This leads to hepatic copper overload, with subsequent accumulation of copper in other organs including the brain, kidney and eye (Ala et al. 2007).
In the liver, copper toxicity can manifest either as acute fulminant hepatic failure or as chronic hepatitis with progressive fibrosis and cirrhosis (Huster 2010; Ostapowicz et al. 2002). Copper accumulation in the liver is associated with deteriorating liver function (as evidenced by elevated serum transaminases), inflammation, ballooning of hepatocytes, enlarged nuclei, fibrosis and frequent steatosis. We refer to excellent recent reviews focussing on the hepatic aspects of WD for more detail (Harada 2014; Rosencrantz and Schilsky 2011). The presentation of neurological WD is variable, with symptoms including dysarthria, tremor, dystonia, gait disturbance and rigidity (Machado et al. 2006; Bandmann et al. 2015). The most commonly reported psychiatric disorders in WD include depression, anxiety, irritability, cognitive impairment and personality change with disinhibition (Dening and Berrios 1990; Svetel et al. 2009). Neurological and psychiatric presentations are particularly difficult to diagnose, leading to sometimes very significant delays of many months or even years in the initiation of treatment (Merle et al. 2007; Zimbrean and Schilsky 2014).
The diagnosis is typically first prompted by clinical observations, including the presence of characteristic Kayser-Fleischer rings (commonly observed in patients with neurological symptoms, but present in less than half of hepatic patients). Reduced serum ceruloplasmin and increased 24-hour urinary copper levels can then confirm the clinical diagnosis. MRI imaging and direct genetic testing are also increasingly used in the diagnosis of WD. Direct analysis of hepatic copper content following biopsy should typically be reserved for patients with the hepatic presentation (Weiss and Stremmel 2012)
Without treatment, WD is fatal (Weiss and Stremmel 2012). The mainstay of management is lifelong copper chelation (trientine, penicillamine and tetrathiomolybdate) or zinc salt therapy (Bandmann et al. 2015). Zinc therapy is of limited use as initial treatment but can be of use as follow-on treatment to copper chelation or in individuals with preclinical WD (typically clinically-unaffected siblings). Importantly, neurological worsening can be seen with all currently available treatments. Outcomes for neuropsychiatric manifestations are least favourable, with over one-third of patients with neurological disease failing to improve with treatment and up to 70% of patients experiencing mental illness after the initiation of treatment (Merle et al. 2007; Zimbrean and Schilsky 2014; Weiss et al. 2013).
The limitations of the currently available therapy for WD, together with our limited understanding of key aspects of this illness (e.g. neurological vs hepatic presentation) justify the ongoing need to study WD in suitable animal models. This review will first provide an overview of our current knowledge of ATP7B and the closely-related ATP7A in Caenorhabditis. elegans (C. elegans), Drosophila and zebrafish (Danio rerio) and then summarise key characteristics of rodent and larger mammal models of ATP7B deficiency. Wherever possible, we have tried to emphasize clinically relevant aspects of each animal model for WD with particular emphasis on hepatic vs neurological manifestation and treatment response.
C. elegans, Drosophila and Danio rerio
Small animal models are relatively inexpensive and can provide a wealth of information about key physiological processes and suggest disease mechanisms (Lenartowicz et al. 2015). Figure 1 demonstrates the evolutionary relationship of the copper-transporting P-type ATPases of commonly-used animal models for human disease. Round worms (Caenorhabditis elegans) and fruit flies (Drosophila melanogaster) both have just one copper-transporting P-type ATPase, orthologous to both ATP7A, the Menkes disease protein, and ATP7B (Norgate et al. 2006; Sambongi et al. 1997). Xenopus and Danio rerio (zebrafish) have orthologues of both ATP7A and ATP7B (Mendelsohn et al. 2006).
Fig. 1.
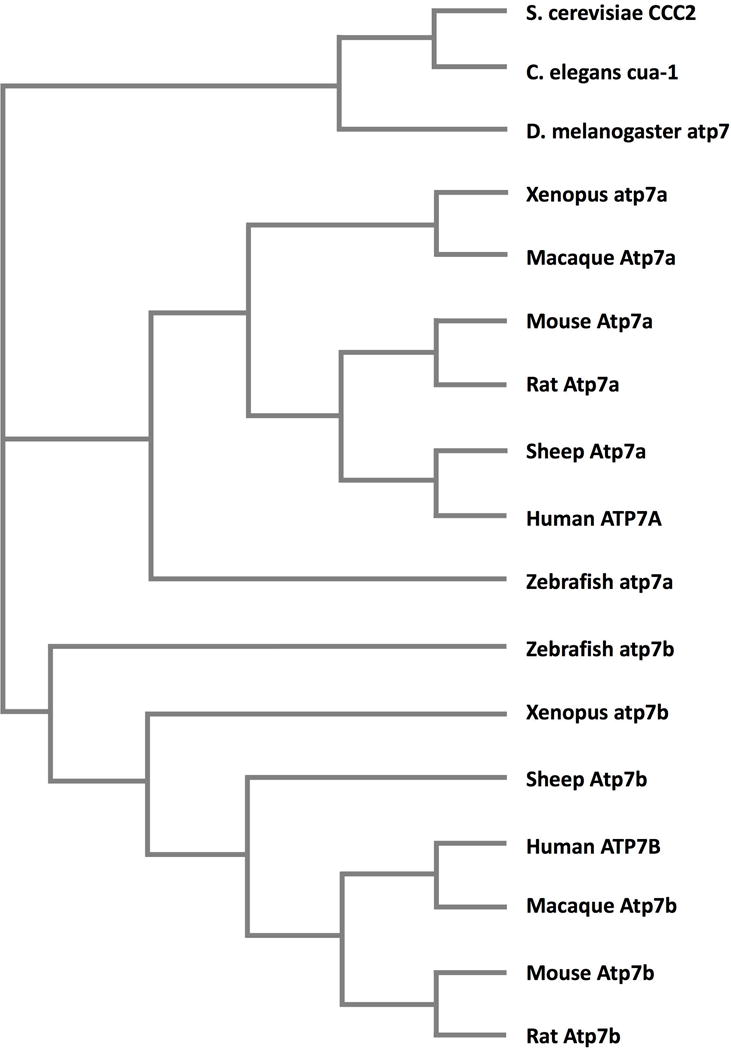
A dendrogram demonstrating the evolutionary relationships of the P1B- type ATPases from human, zebrafish, rat, mouse, macaque, sheep, Xenopus (frog), Drosophila (fruit fly), C. elegans (roundworm) and S. cerevisiae (yeast). Orthologues were identified on Blast and the protein sequences for each were identified on Ensembl and the dendrogram generated using Clustal Omega.
Drosophila has one copper-transporting ATPase, DmATP7. DmATP7A has 49.9% sequence identity to both ATP7A and ATP7B, with conservation of key motifs including the catalytic site, metal-binding sites and transmembrane regions (Southon et al. 2004). DmATP7 is expressed in the midgut, Malpighian tubules (analogous to the kidneys) and in neuronal tissue (Burke et al. 2008). Inactivation of DmATP7 causes copper to accumulate in cells, whereas overexpression reduces copper toxicity, suggesting that DmATP7 plays a role in efflux of excess copper (Southon et al. 2004; Balamurugan et al. 2007). DmATP7 does not appear to traffic under altered copper conditions and remains localised to the basolateral membrane (Burke et al. 2008). DmATP7 is also required for the dietary uptake of copper from the midgut (Norgate et al. 2006). Bahadorani et al used RNA interference to silence DmATP7 selectively in the digestive tract. This procedure produced a copper-deficiency phenotype with resultant neurodevelopmental defects (Bahadorani et al. 2010), including impaired larval growth and survival. Lethargy and reduced pigmentation in adults suggests that DmATP7 is also required for the delivery of copper to cuproenzymes, including those required for neuronal function and pigmentation. Altogether the phenotype resembles the loss of ATP7A function in human patients with Menkes disease (MD). MD is an X-linked recessive disorder characterised by copper deficiency, neurodegeneration, connective tissue disorder, “kinky” hair and hypopigmentation (Tumer and Moller 2010). Thus, using Drosophila to model human WD is unlikely to be informative. However, this model may yield useful insights into the evolution of the copper-transporting ATPases and their role in copper metabolism.
C.elegans express one copper-transporting P-type ATPase, CUA-1. CUA-1 shares 46.1% identity with human ATP7A protein and 42.6% with ATP7B. When expressed in yeast, CUA localises to a vacuolar membrane and corrects the growth defects of the mutant that lack endogenous Cu-ATPase CCC2 (Sambongi et al. 1997), In worms, CUA1 exists as two variants generated from alternatively spliced mRNA:CUA1.1, which protects worms from copper overload by sequestering copper in lysosome-like granules, and CUA1.2, which is constitutively targeted to the basolateral membrane and responsible for exporting copper from the gut to peripheral tissues (Chun et al. 2017). The CUA1.2 spliced variant in the gut is functionally equivalent to human ATP7A, whereas CUA1.1 may functionally resemble ATP7B. Characterization of the ATP7B role in human intestine would help to determine whether C. elegans is a suitable model for WD.
Zebrafish (Danio rerio), like humans, have two distinct genes for copper-transporting ATPase. The zebrafish orthologue of ATP7B, atp7b, is expressed in the liver, while atp7a is expressed in the notochord in developing embryos (Mendelsohn et al. 2006). Our own work has expanded this observation to demonstrate, via in-situ hybridisation, the expression of atp7b in the brain as wells as the liver up to 5 days post-fertilisation (Figure 2). Morpholino oligonucleotide-mediated knockdown of atp7b expression results in small-sized livers and brains with evidence of oxidative stress in the hindbrain, as demonstrated by increased heat shock protein 70 expression (Figure 3). These initial results studies suggest that zebrafish may be a promising candidate for a small vertebrate model of WD and warrant further, more detailed characterization in an atp7b stable mutant zebrafish line.
Fig. 2.
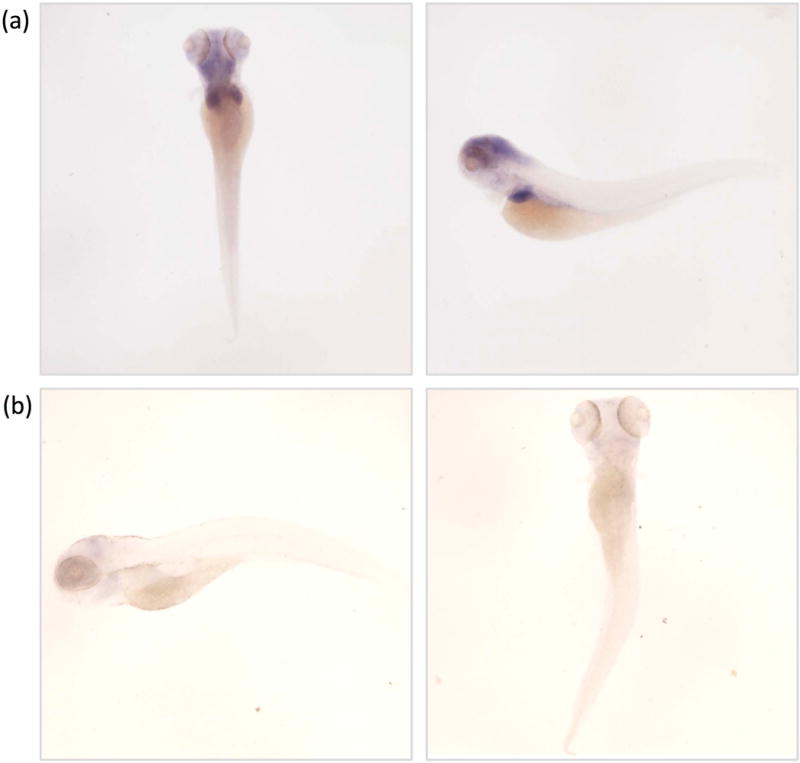
Whole mount atp7b in situ hybridisation of zebrafish embryos. (a) Dorsal and left lateral views zebrafish embryos at 5 days post fertilization, stained with the antisense atp7b probe demonstrating expression of atp7b in the head, and liver at this age. (b) Dorsal and lateral view of embryo stained with sense atp7b control/sense probe, demonstrating absence of staining.
Fig. 3.
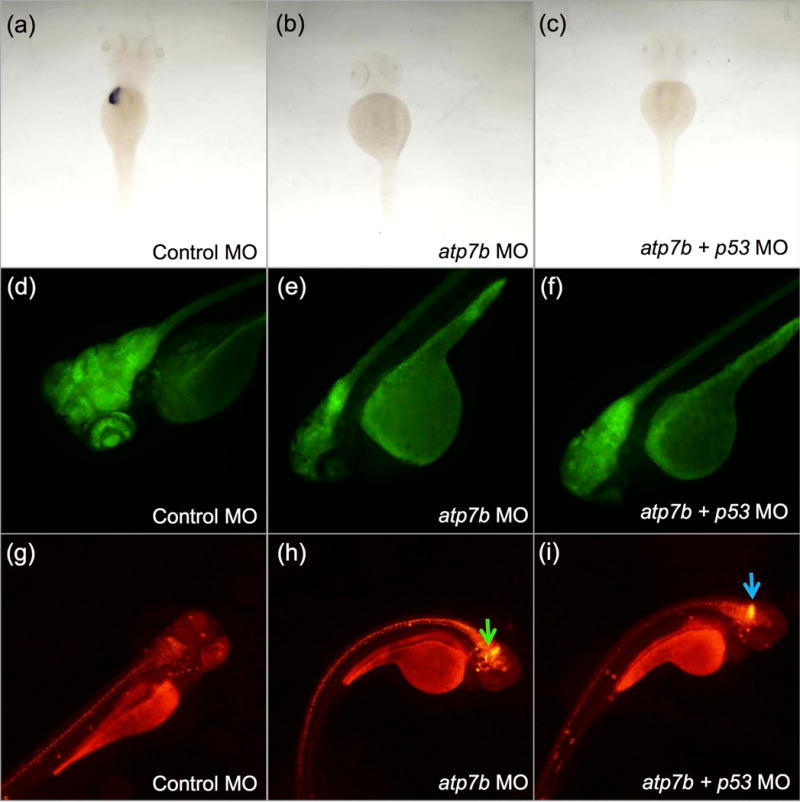
Atp7b deficiency results in marked reduction of the liver marker expression fabp10a and a severe neurological disease phenotype in zebrafish embryos at 4 days post fertilization (dpf). (a, b, c) Whole mount in-situ hybridisation using a probe for liver marker fabp10a was performed on control morpholino-injected, atp7b morpholino-injected and atp7b and p53 morpholino co-injected embryos. p53 morpholino co-injection was used to demonstrate that the phenotype was unlikely to be due to p53-mediated off-target effects of the morpholino. A decrease in expression of fabp10a, presumably reflecting a decrease in size or absence of the liver in was seen in atp7b morphants. (a) control morpholino-injected. (b) atp7b morphants. (c) atp7b morphants co-injected with p53 morpholino. (d, e, f) A transgenic HuC reporter line was used to study the effect of Atp7b deficiency on brain size. (d) control morpholino-injected. (e) atp7b morpholino-injected. (f) atp7b and p53 morpholino co-injected. Atp7b deficiency was associated with a decreased in the size of the brain. (g, h, i) WTos4 embryos which express DsRed on the hsp70 promoter show increased fluorescence in the zebrafish hind brain (green and blue arrows) in response to Atp7b deficiency. (g) control morpholino-injected. (h) atp7b morpholino-injected. (i) atp7b and p53 morpholino co-injected.
Rodent models
The Toxic Milk Mouse
“Toxic milk” (tx) is a mouse strain on DL background, bearing a naturally occurring point mutation in Atb7b, at position 2135 in exon 8 of the mouse orthologue of human ATP7B, located on chromosome 8 (Rauch and Wells 1995; Reed et al. 1995; Theophilos et al. 1996; Voskoboinik et al. 2001). The mutation changes methionine at position 1356 to valine, in the eighth transmembrane domain. This methionine is conserved in all copper ATPases including those from bacteria to yeast. This substitution causes loss of copper transport function and disrupts copper-dependent trafficking of Atp7b from the trans-Golgi network, a step essential for excretion of excess Cu (La Fontaine et al. 2001; Theophilos et al. 1996; Voskoboinik et al. 2001). Of note, a pathogenic mutation at the same amino acid position (Met1359Ile) has been reported in a human WD patient (Davies et al. 2008).
Original studies reported that pups of mutant females are copper deficient and show poor growth, hypopigmentation, tremors, abnormal locomotor behaviour and die within the first two weeks of life (Rauch 1983). This severe phenotype was predominantly attributed to the mutant mother producing copper-deficient milk (Michalczyk et al. 2000) and foster-nursing by a normal female rescued these effects (Rauch 1983). In rodents, copper content in tissues depends on age, which is especially apparent in the liver and the brain (Tarohda et al. 2004; Schaefer et al. 1999). In newborns, the tissue copper content may also be dependent on the copper content in the milk. Atp7b is expressed in the in the mammary gland, where if facilitates transport of copper to the milk in response to hormonal stimuli (Michalczyk et al. 2000; Michalczyk et al. 2008). Impairment of Atp7b-mediated copper incorporation into secretory vesicles and into ceruloplasmin contributes to the low copper content in the milk of Tx mice (Lutsenko et al. 2007).
However, Tx mice have low hepatic copper at birth, suggesting a contributing disturbance in the prenatal environment (Rauch 1983). The loss of ATP7B in placental cells and copper misbalance may also contribute to copper dis-homeostasis in fetal and new-born rodents (Hardman et al. 2004; Muramatsu et al. 1998).
More recent studies have nevertheless suggested that foster nursing is not required if female breeders are kept within certain age (over 6-8 weeks) and the overall phenotype appears milder than originally reported (Czachor et al. 2002). These mice still have reduced life-expectancy, with just 60% surviving to 1 year of age. The adult mice exhibit alopecia and severe tremors not seen in healthy controls (Czachor et al. 2002). In older mice, the liver demonstrates pathological changes and copper levels rise in the liver, brain, spleen, kidney and eyes (Rauch 1983; Howell and Mercer 1994).
A second tx mouse on a C3H/FeJ background (Atp7btx-j or “txj”) was characterized by Cox and co-workers (Coronado et al. 2001). Toxic milk Jackson (Atp7btx-J) arose spontaneously in strain C3H/HeJ in 1988. One outcross was made to C3HeB/FeJ and then sibling matings were used to maintain the stock” (Jackson Laboratory). This strain is commercially available through the Jackson laboratory, and it has now been actively employed for studies of Atp7b-dependent copper misbalance. The txj mice have a single nucleotide mutation, which replaces glycine 712 with aspartate (Coronado et al. 2001). In humans, the equivalent residue, Gly710, is a hot spot for mutations with replacement by Arg, Ser, Ala and Val reported in numerous patients (see Wilson disease mutation database (http://www.wilsondisease.med.ualberta.ca/database.asp).
Hepatic disease
TxJ adult mice have high hepatic copper and reduced ceruloplasmin activity (Rauch 1983; Howell and Mercer 1994). Homozygous txJ mice liver copper levels are elevated from as early as 3 weeks, reaching levels up to 100 times that of controls at 3 to 5 months (Czachor et al. 2002; Rauch and Wells 1995; Rauch 1983; Koropatnick and Cherian 1993). Inflammation is seen at 6 months of age (Czachor et al. 2002). At 3 months, prior to the development of overt liver disease, mitochondrial abnormalities became detectable. At this age, mitochondrial cristae have cystic dilatation at their tips, as seen in human patients with WD (Roberts et al. 2008; Sternlieb 1968). Pleiomorphic changes, increased matrix density and electron-dense inclusions are seen in the mitochondria of 6 month-old txJ mice, while mitochondrial complex assays showed a 25% reduction in complex IV activity and elevated citrate synthase activity at 5 months (Roberts et al. 2008). Liver histology is markedly abnormal with enlarged nuclei and intranuclear inclusions (Howell and Mercer 1994). Nodules develop in the liver at 8–12 months with otherwise relatively normal gross morphology, high copper and low metallothionein levels (Czachor et al. 2002). Mice heterozygous for the txJ mutation show impaired copper excretion. Following copper loading, they have increased hepatic copper concentrations compared to normal controls, though no histological changes are seen in the liver. These findings may be of relevance to human heterozygote carriers of ATP7B mutations (Cheah et al. 2007).
Neurological disease
TxJ mutant mice show defects in spatial memory and, to a lesser degree, in motor behaviour (Terwel et al. 2011a). Motor changes include a wide-based gait and shorter stride-length, resembling the neurological abnormalities seen in some WD patients. It remains unclear whether this apparent neurological phenotype is caused directly by the loss of Atp7b function in the CNS or results from metabolic abnormalities in the liver and general poor health of txJ mice at 12 months (Przybyłkowski et al. 2013a). Foster-nursed tx mice have increased copper levels in the brain, particularly in the cerebral cortex, striatum, thalamus, hypothalamus, cerebellum and brain stem, from as early as 3 months (Allen et al. 2006; Ono et al. 1997; Czachor et al. 2002; Terwel et al. 2011a). Zinc levels are also significantly increased in the striatum, hippocampus and cerebellum (Czachor et al. 2002). While copper-induced apoptosis is evident in the livers of 10 month old mice, neuronal apoptosis is absent in tx mouse brains (Chan et al. 2008). However, there is a slight reduction in dopamine levels and raised serotonin (Przybyłkowski et al. 2013b; Terwel et al. 2011a). TxJ mice also show striatal inflammation without neuronal loss (Terwel et al. 2011b).
Treatment studies
Following penicillamine treatment at a dose of 200mg/kg twice daily for 3 days, 16-week-old tx mice show increased serum and brain concentrations of free copper and decreased concentrations of protein-bound copper in the brain. With continued administration, for 10 and 14 days, total copper levels consequently decreased. The increase in free copper is associated with increased oxidative stress in the cortex and basal ganglia, suggesting a possible mechanism of the frequently observed neurological worsening in WD patients after initiation of chelation therapy (Chen et al. 2012). Tetrathiomolybdate treatment for 2, 8 or 14 days significantly reduced hepatic copper and metallothionein. A transient increase of serum and, later, renal copper subsequent to initiation of tetrathiomolybdate therapy reflects the effect of tetrathiomolybdate treatment on urinary copper excretion (Czachor et al. 2002).
The Atp7b Knockout Mouse
In 1999, the Atp7b knockout (Atp7b−/−) mouse was generated on a hybrid C57Blx129S6/SvEv background via insertion of early termination codons into exon 2 of mouse Atp7b gene. This process produced a truncated Atp7b mRNA, resulting in a complete knockout of Atp7b protein, and loss of Atp7b function (Buiakova et al. 1999). Atp7b−/− mice are born with low hepatic copper, which rises to 27 times that of healthy controls at 1 month and 50 times that of controls at 2 months. After two months, no further rise in hepatic copper is seen (Buiakova et al. 1999). High copper is also found in the kidney, brain, mammary gland and placenta of these animals. Contrary to hepatic copper, brain copper levels continue to rise throughout life. Like txJ mice, copper levels in the milk of mutant females are low. When breeding program involves heterozygous Atp7b+/− females and homozygous males, the Atp7b−/− pups do not show obvious neurologic abnormalities, despite biochemical changes in the brain (SL, own data). If both parents lack Atp7b, pups may have neurological symptoms including tremor and ataxia. When such pups are born, the pathology appears early and the majority of these pups die within 3 weeks of birth unless they are foster-nursed by normal mothers (Buiakova et al. 1999). Consistent with human WD, Atp7b−/− mice show impaired biliary excretion of copper, defective copper incorporation into ceruloplasmin, and increased urinary copper excretion (Buiakova et al. 1999; Gray et al. 2012; Huster et al. 2006).
Hepatic disease
Liver pathology in the Atp7b−/− mice develops with time and depends on the length of copper exposure and intracellular copper distribution (Buiakova et al. 1999; Huster et al. 2006; Ralle et al. 2010). Consequently, liver disease in Atp7b−/− mice can be broadly divided into three stages based on age (Huster et al. 2006; Ralle et al. 2010). Stage I disease (up to 8 weeks), is characterised by mild or absent liver histopathology, with nuclear and mitochondrial pleomorphisms evident on light microscopy (Huster et al. 2006). Serum transaminases and total bilirubin are already elevated at this stage. Copper accumulates in nuclei and cytosol (Ralle et al. 2010) and hepatic iron can also be elevated (Sauer et al. 2011). The apparent lack of oxidative stress at this stage is thought to be due to the incorporation of copper into metallothioneins, which are markedly upregulated (Huster et al. 2007). Instead, elevated copper cause significant changes in mRNA profiles by affecting components of mRNA splicing and trafficking machinery, as well as dysregulating activity of nuclear receptors (Burkhead et al. 2011; Wilmarth et al. 2012; Hamilton et al. 2016). At this stage, genes involved cell cycle machinery and DNA repair are prominently upregulated (Ralle et al. 2010; Wilmarth et al. 2012), whereas lipid metabolism is downregulated (see details below).
Between 13 and 20 weeks (stage II disease), widespread inflammation and necrosis is observed alongside further nuclear enlargement and bile duct proliferation (Huster et al. 2006). Intracellular deposits appear at this stage and cytoskeleton and cell adhesion genes are upregulated (Ralle et al. 2010). Merle and colleagues reported that hepatic iron continues to be elevated but we (SL) were unable to confirm this. In addition, serum iron and haemoglobin levels are low, suggesting that the low serum ceruloplasmin oxidase activity leads to defective iron homeostasis and anaemia in WD (Merle et al. 2010). However, the status of iron homeostasis in Atp7b−/− mice and WD patients’ needs further investigation because the results in either Atp7b−/− mice of human WD livers are inconsistent (Hachmoller et al. 2016; Boaru et al. 2015). Levels of iron and zinc were reported as high in hepatic tissue of Atp7b−/− mice between 10-24 months, in concordance with samples from hepatic tissue of human WD patients (Boaru et al. 2015); however no significant change in hepatic iron levels was seen in the US Atp7b−/− mouse colony (own data, SL). Presently, we cannot explain the reasons for this discrepancy. Atp7b−/− mice have disturbances of iron metabolism in the intestine, which may contribute (Pierson et al. 2018). Also, age may be a contributing factor. Further studies are needed.
Stage III disease (beyond 28-30 weeks), is characterised either by regeneration of liver parenchyma along with extensive bile duct proliferation consistent with cholangiocarcinoma (Huster et al. 2006). This stage is associated with transcriptional upregulation of genes involved in the inflammatory response and fibrosis (Ralle et al. 2010; Hamilton et al. 2016). Macroscopic changes in the liver of Atp7b−/− mice can be seen at seven months, with irregular shapes and regeneration nodes apparent (Buiakova et al. 1999; Huster et al. 2006). Oxidative stress is evident at this stage through changes in GSH levels and an increased GSSG:GSH ratio (Sauer et al. 2011). Structural mitochondrial changes in stage III disease include condensation and increased matrix density, loss of cristae structure, progressive matrix loss and vacuolisation. Compensatory giant mitochondria with increased cristae are also observed (Yurkova et al. 2011). Furthermore, there is altered respiratory chain function, with increased complex I and reduced complex II and III activity in the liver at 47 weeks (Sauer et al. 2011).
Increasingly coming to light is the role of altered lipid metabolism in WD. Deranged lipid metabolism and lipoprotein levels were first documented in the LEC rat (Levy et al. 2007). However, most research in this area has been performed using the Atp7b−/− mice. Lipid and cholesterol metabolism genes are downregulated during all stages of the disease in Atp7b−/− hepatic tissue (Ralle et al. 2010; Huster et al. 2007). Concomitantly, serum very low density lipoproteins and liver cholesterol are reduced (Huster et al. 2007). Transcriptional analysis has implicated the reduced function of nuclear receptors, particularly the liver X receptor (LXR)/retinoid X receptor (RXR) heterodimer (involved in the regulation of lipid metabolism and the inflammatory response) in the pathogenesis of liver disease at the Stage I; the farnesoid X receptor (FXR, activation of which usually inhibits production of excessive concentrations of bile acids in the liver) becomes increasingly involved as disease progresses (Wooton-Kee et al. 2015; Hamilton et al. 2015; Wilmarth et al. 2012). These abnormalities are also present in the liver of human WD patients (Wooton-Kee et al. 2015; Hamilton et al. 2015). Furthermore, the abnormality in FXR function can be reversed in Atp7b−/− mice by the administration of zinc (Wooton-Kee et al. 2015), while administration of an LXR agonist can normalise lipid metabolism, decrease liver inflammation and fibrosis and improve liver function in Atp7b−/− mice (Hamilton et al. 2015).
It should be noted that the relationship between copper and lipid metabolism in WD is far from being fully understood. Hepatic steatosis (fat accumulation) is a common clinical manifestation of human WD, observed in about 50% of patients (Liggi et al. 2013). These observations raised concerns that mice (who have down-regulated lipid metabolism at the early stage of the disease) may not adequately model human hepatic WD (Sintusek and Dhawan 2017). The discrepancy is likely not so much due to marked differences between the species with respect to Cu-lipid interactions (a recently developed mouse strain with Atp7b inactivated only in hepatocytes develops steatosis) but rather the inter-species and individual variations of modifying factors, which remain poorly understood (Muchenditsi et al. 2017). One such modifying factor has recently been described by Ferenci and colleagues, who identified allelic variation in triacylglycerol lipase PNPLA3 and pediatric age as independent variables associated with steatosis (Stattermayer et al. 2015). None of the published study has so far carefully compared copper levels/phenotypes in males and females, which could be an important variable as suggested by Muchenditsi et al and Medici et al (Medici et al. 2016; Muchenditsi et al. 2017).
Neurological disease
Why some WD patients develop neurological symptoms and others don’t remains unclear. In Atp7b−/− mice, cerebral copper rises throughout life and is double that of controls by 11 months of age without developing a neurological/behavioural phenotype (Lutsenko 2008; Buiakova et al. 1999; Boaru et al. 2014). Atp7b−/− brains also show signs of oxidative stress as well as respiratory chain dysfunction (Sauer et al. 2011). Further studies failed to demonstrate increased cerebral copper at 10 months of age, but found altered mitochondrial structure and deformed axons of basal ganglia neurons at this age. This suggests that copper mislocalization rather than just copper toxicity may also be implicated in neurological WD (Dong et al. 2015). Interestingly, the altered cholesterol metabolism seen in hepatic tissue is also evident in the brain (Sauer et al. 2011).
The compensatory role of Atp7a
Copper misbalance caused by Atp7b inactivation in tissues where it is normally expressed (such as the liver and the brain) also affects organs and tissues where Atp7b is not normally present. For example, although Atp7b is not expressed in the mouse adrenal, activity of the adrenal cuproenzyme dopamine-beta-hydroxylase (DBH) is low in Atp7b−/− mice, resulting in reduced noradrenaline and adrenaline levels. The DBH mRNA levels and copper levels in the adrenal glands do not differ significantly from controls; however, DBH protein levels are lower (Gerbasi et al. 2003). The mechanism behind these effects of Atp7b inactivation remains unclear. Copper elevation may cause ATP7A to move out of the trans-Golgi network towards the plasma membrane to export excess copper instead of supplying copper to copper-dependent enzymes. Such an Atp7a response to the loss of Atp7b function is observed in the Atp7b−/− mouse kidneys and cerebellum. Renal copper concentrations in 6-8-week-old Atp7b−/− mice are similar to that of controls. This is attributed to the re-localisation of Atp7a from intracellular vesicles to the basolateral membrane, suggesting that Atp7a attempts to compensate for the loss of copper export/sequestration function of Atp7b (Linz et al. 2008). In the mouse cerebellum, ceruloplasmin is expressed in Purkinje neurons alongside Atp7b and Atp7a. Holoceruloplasmin, containing copper, is synthesised in the Atp7b−/− mice cerebellum, unlike in the liver, suggesting that Atp7a in these cells facilitates copper incorporation into ceruloplasmin (Barnes et al. 2005).
Treatment studies
Using a lentivirus vector-mediated early gestational gene transfer of Atp7b in Atp7b−/− mice, Roybal et al demonstrated that prenatal gene therapy could potentially be used as a treatment of WD. In this proof-of-principle study, the vector was introduced into mouse embryos at an age corresponding to approximately 8-9 weeks of gestation in humans. Characterization of Atp7b−/− pups at 14 weeks postnatally (when liver disease is typically prominent) illustrated that liver morphology and copper incorporation into ceruloplasmin was restored as a result of Atp7b expression. However, the effect on hepatic copper levels was highly variable and the procedure of intracardiac injection was associated with a high mortality of 70% (Roybal et al. 2012). More recently, Murillo et al achieved human ATP7B cDNA transfer using an adeno-associated vector serotype 8 under the control of the liver-specific alpha-1 antitrypsin promotor (Murillo et al. 2016). The virus was administered via intravenous injection in 6-week old mice. Improvement in liver function was seen at all treatment doses and restoration of ceruloplasmin function at the highest dose of treatment. Gene transfer was also associated with normalisation of liver function up to 6 months of age, paralleled by normalised copper excretion in the bile. This careful and very promising study, nevertheless, highlights the need for better understanding of WD-related mechanisms. Either low or high dose of the virus resulted in expression of recombinant Atp7b at protein levels that exceeds endogenous Atp7b normally present in the liver, yet only high dose of virus provided a fairly complete and sustained beneficial outcome. This observation may suggest that limiting expression of Atp7b only to hepatocytes (using the low dose of the viral vector) may not be sufficient to ameliorate the disease and that restoration of Atp7b function in both parenchymal and nonparenchymal liver cells is required for complete normalization of liver function. This hypothesis is supported by recent findings in mice with selective deletion of Atp7b in hepatocytes (Muchenditsi et al. 2017). These animals accumulate copper in the liver, lack active ceruloplasmin, have enlarged nuclei and develop steatosis, but show markedly diminished inflammatory response compared to Atp7b−/− mice with global inactivation of Atp7b.
PET/CT imaging
Atp7b−/− mice have been used in the development of PET/CT using radioactive 64Cu as a diagnostic tool for WD. Compared to controls, Atp7b−/− mice had prolonged accumulation of 64Cu in the liver and consistently lower 64Cu-linked radioactivity in the brain (Peng et al. 2012a). Administered orally, the radioactive copper showed prolonged trapping in the gut and higher radioactivity in the kidneys (suggesting increased urinary excretion) in Atp7b−/− mice relative to controls (Peng et al. 2012b). A longitudinal study using the orally-administered tracer demonstrated reduced hepatic 64Cu uptake (suggesting a decrease in copper absorption in the diet) and associated increased urinary excretion of copper with age in Atp7b−/− mice (Gray et al. 2012).
Long-Evans Cinnamon Rat
The Long-Evan’s Cinnamon (LEC) rat is the most extensively researched rodent model of WD.
History and characterisation
In 1987, an inbred strain of the Long-Evans rat with cinnamon colouring was initially thought to have hereditary hepatitis (Yoshida et al. 1987; Sawaki et al. 1990). Several features of the LEC rat then strongly suggested it as a suitable animal model for WD. This included the autosomal recessive nature of the pathological trait (Yoshida et al. 1987; Masuda et al. 1988) and the identification of high hepatic copper levels causing the hepatitis (Sugawara et al. 1991; Okayasu et al. 1992). Following the identification of ATP7B as the causative gene for WD in human patients in 1993 (Bull et al. 1993; Tanzi et al. 1993; Petrukhin et al. 1994), a 900bp deletion within the 3’ end of the Atp7b gene was identified as the causative mutation in this rat strain, resulting in loss of protein expression (Wu et al. 1994).
Experiments in the LEC rat were instrumental in developing early understanding of ATP7B function and copper metabolism. Copper accumulates in the livers of LEC rats in concentrations up to 40 times that of controls (Sugawara et al. 1991). Through detection of low serum and biliary copper and low serum ceruloplasmin, it was evident that the LEC mutation impaired biliary excretion of copper and copper incorporation into ceruloplasmin, resulting in hepatic copper accumulation (Abe et al. 1994; Murata et al. 1995; Yamada et al. 1993; Sugawara et al. 1991). By partially ameliorating these defects with recombinant adenovirus mediated delivery of human ATP7B into the LEC rat, Terada et al demonstrated the dual functions of ATP7B in the biliary excretion of copper and in copper incorporation into ceruloplasmin (Terada et al. 1999; Terada et al. 1998).
Hepatic disease
Hepatic copper in the LEC rat increases from 4 weeks to the onset of hepatitis and declines thereafter (Hayashi et al. 2004; Klein et al. 1998). While serum copper is usually low, the decrease in hepatic copper seen following hepatitis is associated with an increase in copper in the serum (Sugawara et al. 1992; Komatsu et al. 2002). At 12-16 weeks the LEC rats develop fulminant hepatitis with jaundice, subcutaneous bleeding, oliguria, weight loss and convulsions (Yoshida et al. 1987). Those LEC rats that survive the hepatitis then develop hepatocellular carcinoma between 31 and 81 weeks of age (Yoshida et al. 1987; Sawaki et al. 1990). Liver histology demonstrates inflammation with focal necrosis, enlarged hepatocytes, steatosis, nuclear enlargement, oval cell proliferation and cholangiofibrosis, and additional hepatocellular carcinoma and cholangiocarcinoma in older rats (Irani et al. 2001; Li et al. 1991; Siaj et al. 2012; Yoshida et al. 1987; Lee et al. 2011).
Experiments in LEC rats helped delineate the role of metallothioneins in the pathophysiology of WD. The increase in metallothionein expression in hepatic tissue of LEC rats correlates with the extent of copper accumulation (Yamada et al. 1992; Klein et al. 1997). They function in an antioxidant capacity by binding excess copper to prevent the formation of hydroxyl radicles through Haber-Weiss reactions (Shishido et al. 2001; Klein et al. 1997; Nakamura et al. 1997). Copper-loaded metallothioneins are subsequently incompletely degraded in lysosomes (Nakamura et al. 1997; Klein et al. 1998).
Considerable evidence from LEC rat livers supports the role of oxidative stress in the pathophysiology of WD hepatitis. Copper accumulation is associated with membrane lipid peroxidation and DNA damage, which increases with age (Hayashi et al. 2000; Wang et al. 2011; Levy et al. 2007; Samuele et al. 2005; Klein et al. 1998; Nair et al. 2005; Yamashita et al. 1996). Furthermore, levels of hydrogen peroxide and lipid peroxide in the serum increase during acute hepatitis (Yasuda et al. 2006). Oxidative stress is associated with the upregulation of genes of protective proteins including Mn-superoxide dismutase (Sod-2), heat shock protein 70 (Hsp70) and Akr1b7 (Jia et al. 2006; Suzuki et al. 1993; Klein et al. 2003).
Mitochondrial damage occurs in correlation with increased mitochondrial copper resulting in altered mitochondrial morphology with matrix condensation, enlarged cristae and widened intramembrane spaces evident from 7 weeks of age. These changes appear to precede oxidative stress response. In LEC hepatocytes, oxidative stress (as measured by reduction in mitochondrial aconitase activity, depletion of unsaturated mitochondrial fatty acids) and defects of oxidative phosphorylation, occurs at a late stage (around 16 weeks of age) and not before the disease is clinically apparent (Zischka et al. 2011).
Inflammation and steatosis are also observed with an increase in the number of Kupffer cells and polymorphs as well as an upregulation of inflammation-related genes (Klein et al. 2003; Fong et al. 2004; Levy et al. 2007). Apoptosis follows, potentially mediated by tumour necrosis factor alpha (TNF-α) (Fong et al. 2004; Klein et al. 2003; Lee et al. 2011). Iron accumulates in liver of LEC rats in an age-dependent manner (Kato et al. 1996; Sugawara et al. 1992). Free iron in particular increases during hepatitis and is believed to contribute to the tissue damage via further generation of free radicals (Kato et al. 1996; Sugawara et al. 1999b). Iron accumulation alone, however, is not sufficient to cause DNA damage, which is a consequence of the copper accumulation (Hayashi et al. 2000).
Neurological disease
Over time, copper also accumulates in the brains of LEC rats (Li et al. 1991). Initially, at 4 weeks of age, copper concentrations in the cerebellum, medulla oblongata, pons, hypothalamus, mid-brain, thalamus, striatum, hippocampus and cerebral cortex are lower than those in controls (Hayashi et al. 2006; Saito et al. 1995). At this early stage, neurological copper deficiency is associated with altered monoamine metabolism: global noradrenaline deficiency and dopamine excess in the cerebral cortex but deficiency in the striatum (Saito et al. 1996; Samuele et al. 2005). By 20 weeks of age, copper levels in the cerebellum, medulla oblongata, pons and hypothalamus are elevated at levels up to 6 times higher than in healthy controls (Hayashi et al. 2006; Saito et al. 1995). The rise in copper levels in the brain coincides with the decrease in hepatic copper, suggesting copper is released from damaged hepatocytes and is deposited in the brain (Hayashi et al. 2006). At this age, there is no abnormal accumulation of copper in the striatum or substantia nigra, the areas of most interest in modelling human WD. Accordingly, no nigrostriatal degeneration is seen at this stage (Ahn et al. 2005; Kawano et al. 2001). Striatal copper continues to accumulate over time and is significantly higher than controls by 30 weeks of age (Kim et al. 2005b). The observation of increased Cu/Zn-SOD1 levels at 11 weeks and of lipid peroxidation in the striatum at 14 weeks suggests the presence of oxidative stress but overt nigrostriatal degeneration does not occur. Cu/Zn-SOD levels are also raised at 11 and 14 weeks in the cortex, while Mn-SOD levels are increased in the dopaminergic neurons of the substantia nigra at 14 weeks (Kim et al. 2005a; Samuele et al. 2005). This 14 week time-point typically marks the onset of chronic hepatitis in the LEC rats (Yoshida et al. 1987). Elsewhere in the brain, catecholaminergic (TH-positive) cell densities are reduced in the cingulate cortex, hippocampus and cerebellum of LEC rats at 4 and 10 weeks of age, reflecting reduced density of noradrenergic fibres in these areas associated with deficiency of dopamine-beta-hydroxylase (Saito et al. 1996; Kawano et al. 2001). Oxidative DNA damage is detectable in the brain as a whole from 12 weeks (Wang et al. 2011). Evaluation of DNA damage in the cerebrum and cerebellum of LEC rats via detection of DNA single strand breaks (SSBs) demonstrated a decrease in the number of healthy nuclei, with severe DNA damage occurring from 24 weeks (Hayashi et al. 2006). Ferrous and ferric iron levels are noticeably elevated in the substantia nigra and striatum (Kim et al. 2005b). Furthermore, transcription levels of genes for iron metabolism-related proteins, ceruloplasmin and hepcidin are increased in the LEC brain (Lee et al. 2013). Lee and co-workers unlocked many potential molecular processes involved in neurologic WD through gene expression studies in LEC rat brains. The affected pathways included mitochondrial respiration, S-adenosylhomocysteine metabolism and calcineurin-related processes. Of particular note was the upregulation of alpha-synuclein (involved in the pathogenesis of Parkinson’s disease) and genes related to amyloid precursor protein (involved in the pathogenesis of Alzheimer’s disease) (Lee et al. 2013).
Renal disease
Copper also accumulates in the kidneys of LEC rats from 12 to 18 weeks and reaches levels 10-20 times that of healthy rat controls (Hayashi et al. 2005). However, prior to development of hepatitis, renal copper is significantly lower than in controls (Sugawara et al. 1991). DNA single strand breaks are seen in the renal cortex, correlating with the increase in copper concentration. Of note, DNA damage in the kidneys occurs prior to the onset of liver disease (Hayashi et al. 2005).
Treatment studies
Penicillamine given at a dose of 100 mg/kg/day over 12 weeks prevents the onset of hepatitis in LEC rats with reduction of hepatic and serum copper and increased urinary copper excretion (Togashi et al. 1992). Trientine hydrochloride given from 6 weeks of age at 1500 parts per million (ppm) in drinking water also prevents the development of hepatitis. When given at the same dose from 8 to 35 weeks and at 750 ppm thereafter, trientine prevents the development of hepatocellular carcinoma up to 87 weeks of age (Sone et al. 1996). Hepatic copper is reduced by approximately 50% with trientine, but remains at levels significantly higher than controls. Trientine also reduces hepatic and renal DNA damage and renal copper (Hayashi et al. 2004; Sone et al. 1996; Hayashi et al. 2005).
Tetrathiomolybdate detoxifies copper via the formation of molybdenum-copper complexes. These complexes are initially deposited in the liver but subsequently result in the excretion of copper in bile (George et al. 2003; Lai and Sugawara 1997; Ogra et al. 2000). Tetrathiomolybdate given as a single intraperitoneal injection of 10mg/kg after the onset of hepatitis results in clinical improvement of hepatitis and reduced hepatic copper within 4 days (Klein et al. 2004). Given subcutaneously for 65 days from 8 weeks of age (before the onset of hepatitis), at a weekly dose of 5mg/kg, tetrathiomolybdate lowers hepatic copper but does not improve liver function parameters or weight. Furthermore the treatment appeared to increase hepatic iron and redistribute hepatic copper to the kidney, spleen and testes (Sugawara et al. 1999a).
Zinc reduces hepatic, renal, and intestinal copper and iron (Medici et al. 2002; Medici et al. 2005; Santon et al. 2003). It acts via upregulation of metallothionein in the liver and intestines, resulting in reduced copper absorption in the gut due to increased binding of copper to metallothionein in the intestinal cells lining the gut walls (Santon et al. 2002; Santon et al. 2003). 8 weeks of oral zinc acetate (50 mg/ml/day) initiated before the onset of hepatitis at 5 weeks of age prevents the onset of acute hepatitis (Medici et al. 2005).
Experimental treatments beneficial in the prevention of hepatitis in LEC rats through protection against free-radicals include D-mannitol, N-acetylcysteine (NAC), D-galactosaminehydrochloride (GalN), proline solution, ascorbic acid, alpha-lipoic acid and thioredoxin (Hawkins et al. 1995; Otsuka et al. 2006; Fu et al. 2014; Yamamoto et al. 2001). Combined treatment with proline and ascorbic acid have an additive protective effect in the prevention of hepatitis (Hawkins et al. 1995).
Chelation treatment with N-benzyl-D-glucamine dithiocarbamate (BGD) is also protective in the LEC rat. It stimulates biliary copper excretion and prevented rises in serum ALT and bilirubin and protected against histological changes in the liver. With this treatment, no copper build up is seen in the brain and hepatic iron accumulation was prevented (Shimada et al. 2005). Similarly, excess dietary histidine stimulates urinary excretion of copper and decreases hepatic copper in the LEC rat (Xu et al. 2003).
LEC rats have been repeatedly used in the development of protocols for hepatocyte transplantation as a treatment of WD and other metabolic liver diseases. Different methods of preconditioning and transplantation have been attempted in this rodent model of WD with varying degrees of success (Yoshida et al. 1996; Irani et al. 2001; Malhi et al. 2002; Malhi et al. 2008; Joseph et al. 2009; Sauer et al. 2012). Similarly, Katsuda et al developed a hyaluronic acid sponge as a scaffold for healthy hepatocytes which they embedded into the mesentery of LEC rats with reduction in liver pathology, lowered plasma copper and prevention of jaundice (Katsuda et al. 2010), while Chen et al transplanted ATP7B-transduced mesenchymal stem cells with improvement in hepatic copper levels and liver function (Chen et al. 2014). In contrast to studies aiming at the prevention of hepatitis, Ueyama et al developed a strategy of treating established hepatitis in LEC rats with a single blood exchange. All animals treated with blood exchange survived the hepatitis, compared to a 50% mortality in untreated controls (Ueyama et al. 2010).
Novel investigations
New imaging modalities assessed in LEC rats include: (99) Tc-mebrofenin scintigraphy, in which the handling of (99) Tc-mebrofenin correlates with liver morphology, function and copper accumulation (Malhi et al. 2002); whole-body positron-emission tomography (PET) using radio-isotope 64Cu bound to L-histidine, demonstrating hepatic uptake of radiolabelled copper with failure of biliary excretion in LEC rats (Bahde et al. 2012); and single-photon emission computed tomography (SPECT) imaging of radio-labelled prion protein injected into the tail vein demonstrating increased radioactivity in the LEC rat livers (Yezdimer et al. 2013).
Relative exchangeable copper (REC) has been proposed as a serum diagnostic marker for WD. Exchangeable copper is the fraction of copper in the serum that is labile and predominantly bound to albumin. Its derivative, REC, is the ratio of exchangeable copper to total copper in the serum, expressed as a percentage. In the rat, a REC of 19% discriminated between LEC rats and controls at a sensitivity of 97.3% and a specificity of 100% (Schmitt et al. 2013). Serum metallothionein levels rise in the serum and urine during the hepatitis stage (Nakazato et al. 2012). This finding was more recently echoed in human WD and has been proposed as a further potential serum marker (Nakazato et al. 2014).
Limitations
LEC rats have additional mutations in the gene for serotonin N-acetyltransferase (NAT), which is involved in melatonin production and a further unspecified mutation with Mendelian inheritance responsible for their cinnamon coat colour (Ahmed et al. 2005). Lymphoid abnormalities are also present in the LEC rat and include splenic and thymic hypoplasia. These are apparently unrelated to Atp7b deficiency and due to deletions in Ptprk (encoding protein tyrosine phosphatase kappa) and Themis. There is maturational arrest of CD4+ cells in the thymus, resulting in T-helper immunodeficiency (thid) with unknown implications on the hepatic phenotype (Jung et al. 2001; Chai et al. 1996; Iwata et al. 2010; Asano et al. 2007). Other features of the LEC rat limiting its use in modelling WD include the frequent occurrence of hepatocellular and cholangiocarcinoma. This is not concordant with human disease, in which progression of liver damage to carcinoma occurs in only 1.2% of patients (Pfeiffenberger et al. 2015). Also unlike human disease, Kayser-Fleischer rings are absent in the LEC rat (Li et al. 1991).The absence of a convincing neurological phenotype is another limitation that prevents the use of the LEC rat in the modelling of neurological WD.
LPP rats
To eliminate contributions of non-Atp7b mutations to the phenotype, Ahmed et al crossed LEC rats with PVG rats, which are wild-type for NAT, Atp7b and coat colour. This procedure yielded a new strain of rat with wild-type NAT and coat colour but with mutated Atp7b, termed the LPP rat (Ahmed et al. 2005). LPP rats progressively accumulate hepatic copper and show biochemical signs of liver disease developing from around 90 days of age. Once hepatic disease becomes evident, untreated LPP rats die within 30 days (Zischka et al. 2011). The microscopic appearance of LPP livers mimic those of human disease, with progressive fibrosis and mitochondrial abnormalities such as widened intramembranous spaces and cristae dilatation (Lichtmannegger et al. 2016).
Treatment studies
LPP rats were used in treatment studies using the bacterial peptide methanobactin. Methanobactin removed copper from LPP livers, especially the mitochondrial compartment. Furthermore methanobactin was effective as a long-term chelator and in the treatment of acute hepatic failure, whereby twice daily injections for 1 week restored hepatic copper levels, liver function and body weight and prevented death in the otherwise moribund animals (Lichtmannegger et al. 2016).
Larger mammal models
A high incidence of hereditary copper toxicosis is found in several purebred canine populations, including the Dalmatian, the West Highland White terrier, the Dobermann, the Bedlington terrier and Labrador retrievers. Hereditary canine copper toxicosis shares several phenotypic similarities with WD including copper accumulation in the liver, onset in adolescence to middle-age and response to treatment with D-penicillamine (Fieten et al. 2012b). Until recently, none of these were established as models of WD. Bedlington terrier copper toxicosis was originally hypothesised to be a canine model of WD, however identification of the causal mutation instead resulted in the discovery of a new protein involved in copper homeostasis, COMMD-1 (van De Sluis et al. 2002).
Labrador retrievers
In 2016 a Genome wide association study (GWAS) study of copper toxicosis in the Labrador retriever demonstrated that an arginine to glutamine mutation in the C-terminus ATP7B was associated with increased hepatic copper levels, thus establishing it as a further animal model of WD (Fieten et al. 2016).
Expression of the mutant protein in a cell line demonstrated impaired trafficking from the endoplasmic reticulum in high copper conditions. The mutation also results in failure of copper excretion in polarised hepatocytes (Fieten et al. 2016). Labrador retriever copper toxicosis is more common in female dogs (Fieten et al. 2012b). In Labrador retrievers, it appears that a concurrent mutation in ATP7A modifies the effects of the causative ATP7B mutation by reducing hepatic copper, particularly in male dogs. It is therefore possible that similar mutations in human ATP7A in WD patients could explain some of the phenotypic variance seen in human WD (Fieten et al. 2016).
Hepatic disease
The mean age of presentation of hepatitis in Labrador retrievers is 7 years (Hoffmann et al. 2006). Affected dogs present with signs of liver disease including weight loss, jaundice and ascites, developing over days to weeks. Without treatment, affected dogs rapidly deteriorate, however the condition responds well to penicillamine (Hoffmann et al. 2006). Hepatic copper is elevated, accumulating in the centrolobular region (Hoffmann et al. 2006). Electron microscopy of affected livers demonstrates copper-laden lysosomes and fibrosis, comparable to human WD (Fieten et al. 2016). However, there are histological differences, including the absence of Mallory bodies and fatty degeneration (Fieten et al. 2016). Hepatic copper is further elevated by increased dietary copper and low dietary zinc (Fieten et al. 2012a).
Similar to other models, metallothionein gene expression is altered throughout disease. MT1A is upregulated in high copper conditions but downregulated in chronic hepatitis. Gene expression analysis also demonstrates upregulation of amyloid precursor protein genes in high copper chronic hepatitis and upregulation of COMMD1 in early disease (Dirksen et al. 2017).
Neurological disease
In common with other models, neurological disease has not been identified in the Labrador retriever model and Keiser Fleischer rings are absent (Fieten et al. 2016).
Conclusion
The existing models of WD all show good similarity to human hepatic WD and have been helpful in developing an improved understanding of the human disease. These animals offer a spectrum of liver disease ranging from mild liver disease in tx mice to more pronounced pathology in the Atp7b−/− mice to fulminant liver failure and death in the LEC rat (Czachor et al. 2002; Howell and Mercer 1994; Huster et al. 2006; Yoshida et al. 1987). As mammals, the mouse, rat and canine models also benefit from high homology to the human genome. However, the time necessary for the assessment of any compound on the phenotype, the resulting cost and ethical considerations prevent their use in high-throughput experiments (Williams and Hong 2011). Furthermore, important differences exist between these mammal models and human disease, particularly the absence of a convincing neurological phenotype. As neurological worsening can be observed in all currently-available treatments for WD (Merle et al. 2007), the need for more research in this area is evident. These factors suggest the need for additional models of WD, especially a model which would accurately reflect the neurological pathology seen in WD patients and could be utilised in high-throughput screens for the identification of neuroprotective compounds.
Acknowledgments
This is a summary of independent research carried out at the National Institute for Health Research (NIHR) Sheffield Biomedical Research Centre (Translational Neuroscience). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. SL acknowledges support from NIDDK (grant R01DK071865). No other funding sources were available.
Abbreviations
- Ala
Alanine
- Arg
Arginine
- Akr1b
aldo-keto reductase family 1, member B7
- ATP7A
P1B-type copper-transporting adenosine ATPase 1
- ATP7B
P1B-type copper-transporting adenosine ATPase 2
- BGD
N-benzyl-D-glucamine dithiocarbamate
- C. elegans
Caenorhabiditis elegans
- COMMD-1
copper metabolism MURR1 domain-containing protein 1
- Cu
copper
- Cu/Zn-SOD1
Copper/Zinc superoxide dismutase
- CUA-1
C. elegans homolog of ATP7A/B
- DBH
Dopamine beta-hydroxylase
- DmATP7
Drosophila melanogaster orthologue of ATP7
- dpf
Days post fertilization
- GaIN
D-galactosaminehydrochloride
- Gly
glycine
- GWAS
Genome wide association study
- Hsp70
Heat shock protein 70
- LEC rat
Long-Evan’s Cinnamon rat
- Menkes disease
MD
- mRNA
messenger ribonucleic acid
- MRI
Magnetic resonance imaging
- MT1A
Metallothionein 1A
- NAC
N-acetylcysteine
- NAT
N-acetyltransferase
- PET/CT
positron emission tomography/computer tomography
- PNPLA3
Patatin-like phospholipase domain-containing protein 3
- Ptprk
protein tyrosine phosphatase kappa
- S. cerevisiae
saccharomyces cerevisiae
- Ser
Serine
- Sod-2
Mn-superoxide dismutase
- SPECT
single-photon emission computed tomography (SPECT)
- REC
relative exchangeable copper
- TNF-α
Tumour necrosis factor alpha
- Tx
toxic milk mice
- Val
Valine
- WD
Wilson disease
- Thid
T-helper immunodeficiency
Footnotes
DR. OLIVER BANDMANN (Orcid ID: 0000-0003-3149-0252)
Conflict of interest:
N/A.
Involves human subjects:
If yes: Informed consent & ethics approval achieved:
=> if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
ARRIVE guidelines have been followed:
Yes
=> if No or if it is a Review or Editorial, skip complete sentence => if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.” unless it is a Review or Editorial
Conflicts of interest: None
=> if ‘none’, insert “The authors have no conflict of interest to declare.”
=> otherwise insert info unless it is already included
References
- Abe S, Yamazaki K, Takikawa S, Suzuki K. Impaired biliary excretion of copper and lysosomal enzymes in Long-Evans cinnamon. Tohoku J Exp Med. 1994;172:355–367. doi: 10.1620/tjem.172.355. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Deng J, Borjigin J. A new strain of rat for functional analysis of PINA. Brain Res Mol Brain Res. 2005;137:63–69. doi: 10.1016/j.molbrainres.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Ahn TB, Cho SS, Kim DW, Jeon BS. Absence of nigrostriatal degeneration in LEC rats up to 20 weeks of age. Neurol Res. 2005;27:409–411. doi: 10.1179/016164105X48851. [DOI] [PubMed] [Google Scholar]
- Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- Allen KJ, Buck NE, Cheah DMY, Gazeas S, Bhathal P, Mercer JFB. Chronological changes in tissue copper, zinc and iron in the toxic milk mouse and effects of copper loading. Biometals. 2006;19:555–564. doi: 10.1007/s10534-005-5918-5. [DOI] [PubMed] [Google Scholar]
- Asano A, Tsubomatsu K, Jung C-G, Sasaki N, Agui T. A deletion mutation of the protein tyrosine phosphatase kappa (Ptprk) gene is responsible for T-helper immunodeficiency (thid) in the LEC rat. Mamm Genome. 2007;18:779–786. doi: 10.1007/s00335-007-9062-0. [DOI] [PubMed] [Google Scholar]
- Bahadorani S, Bahadorani P, Marcon E, Walker DW, Hilliker AJ. A Drosophila model of Menkes disease reveals a role for DmATP7 in copper absorption and neurodevelopment. Dis Model Mech. 2010;3:84–91. doi: 10.1242/dmm.002642. [DOI] [PubMed] [Google Scholar]
- Bahde R, Kapoor S, Bhargava KK, Schilsky ML, Palestro CJ, Gupta S. PET with 64Cu-histidine for noninvasive diagnosis of biliary copper excretion in Long-Evans cinnamon rat model of Wilson disease. J Nucl Med. 2012;53:961–968. doi: 10.2967/jnumed.111.092361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balamurugan K, Egli D, Hua H, Rajaram R, Seisenbacher G, Georgiev O, Schaffner W. Copper homeostasis in Drosophila by complex interplay of import, storage and behavioral avoidance. EMBO J. 2007;26:1035–1044. doi: 10.1038/sj.emboj.7601543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015;14:103–113. doi: 10.1016/S1474-4422(14)70190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes N, Tsivkovskii R, Tsivkovskaia N, Lutsenko S. The copper-transporting ATPases, menkes and wilson disease proteins, have distinct roles in adult and developing cerebellum. J Biol Chem. 2005;280:9640–9645. doi: 10.1074/jbc.M413840200. [DOI] [PubMed] [Google Scholar]
- Boaru SG, Merle U, Uerlings R, Zimmermann A, Flechtenmacher C, Willheim C, Eder E, Ferenci P, Stremmel W, Weiskirchen R. Laser ablation inductively coupled plasma mass spectrometry imaging of metals in experimental and clinical Wilson’s disease. J Cell Mol Med. 2015;19:806–814. doi: 10.1111/jcmm.12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boaru SG, Merle U, Uerlings R, Zimmermann A, Weiskirchen S, Matusch A, Stremmel W, Weiskirchen R. Simultaneous monitoring of cerebral metal accumulation in an experimental model of Wilson’s disease by laser ablation inductively coupled plasma mass spectrometry. BMC Neurosci. 2014;15:98. doi: 10.1186/1471-2202-15-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braiterman L, Nyasae L, Guo Y, Bustos R, Lutsenko S, Hubbard A. Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B. Am J Physiol Gastrointest Liver Physiol. 2009;296:G433–44. doi: 10.1152/ajpgi.90489.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buiakova OI, Xu J, Lutsenko S, Zeitlin S, Das K, Das S, Ross BM, Mekios C, Scheinberg IH, Gilliam TC. Null mutation of the murine ATP7B (Wilson disease) gene results in intracellular copper accumulation and late-onset hepatic nodular transformation. Hum Mol Genet. 1999;8:1665–1671. doi: 10.1093/hmg/8.9.1665. [DOI] [PubMed] [Google Scholar]
- Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- Burke R, Commons E, Camakaris J. Expression and localisation of the essential copper transporter DmATP7 in Drosophila neuronal and intestinal tissues. Int J Biochem Cell Biol. 2008;40:1850–1860. doi: 10.1016/j.biocel.2008.01.021. [DOI] [PubMed] [Google Scholar]
- Burkhead JL, Ralle M, Wilmarth P, David L, Lutsenko S. Elevated copper remodels hepatic RNA processing machinery in the mouse model of Wilson’s disease. J Mol Biol. 2011;406:44–58. doi: 10.1016/j.jmb.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai JG, Sakai T, Hisaeda H, Nagasawa H, Yasutomo K, Furukawa A, Ishikawa H, et al. Development of functional rat-derived T cells in SCID mice engrafted with the fetal thymus of LEC rats which are defective in CD4+ T cells. Microbiol Immunol. 1996;40:659–664. doi: 10.1111/j.1348-0421.1996.tb01124.x. [DOI] [PubMed] [Google Scholar]
- Chan HW, Liu T, Verdile G, Bishop G, Haasl RJ, Smith MA, Perry G, Martins RN, Atwood CS. Copper Induces Apoptosis of Neuroblastoma Cells Via Post-translational Regulation of the Expression of Bcl-2-family Proteins and the tx Mouse is a Better Model of Hepatic than Brain Cu Toxicity. Int J Clin Exp Med. 2008;1:76–88. [PMC free article] [PubMed] [Google Scholar]
- Cheah DMY, Deal YJ, Wright PFA, Buck NE, Chow CW, Mercer JFB, Allen KJ. Heterozygous tx mice have an increased sensitivity to copper loading: implications for Wilson’s disease carriers. Biometals. 2007;20:751–757. doi: 10.1007/s10534-006-9038-7. [DOI] [PubMed] [Google Scholar]
- Chen D-B, Feng L, Lin X-P, Zhang W, Li F-R, Liang X-L, Li XH. Penicillamine increases free copper and enhances oxidative stress in the brain of toxic milk mice. PLoS ONE. 2012;7:e37709. doi: 10.1371/journal.pone.0037709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Shao C, Dong T, Chai H, Xiong X, Sun D, Zhang L, Yu Y, Wang P, Cheng F. Transplantation of ATP7B-transduced bone marrow mesenchymal stem cells decreases copper overload in rats. PLoS ONE. 2014;9:e111425. doi: 10.1371/journal.pone.0111425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun H, Sharma AK, Lee J, Chan J, Jia S, Kim B-E. The Intestinal Copper Exporter CUA-1 Is Required for Systemic Copper Homeostasis in Caenorhabditis elegans. J Biol Chem. 2017;292:1–14. doi: 10.1074/jbc.M116.760876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronado V, Nanji M, Cox DW. The Jackson toxic milk mouse as a model for copper loading. Mamm Genome. 2001;12:793–795. doi: 10.1007/s00335-001-3021-y. [DOI] [PubMed] [Google Scholar]
- Czachor JD, Cherian MG, Koropatnick J. Reduction of copper and metallothionein in toxic milk mice by tetrathiomolybdate, but not deferiprone. J Inorg Biochem. 2002;88:213–222. doi: 10.1016/s0162-0134(01)00383-x. [DOI] [PubMed] [Google Scholar]
- Davies LP, Macintyre G, Cox DW. New mutations in the Wilson disease gene, ATP7B: implications for molecular testing. Genet Test. 2008;12:139–145. doi: 10.1089/gte.2007.0072. [DOI] [PubMed] [Google Scholar]
- Dening TR, Berrios GE. Wilson’s disease: a longitudinal study of psychiatric symptoms. Biol Psychiatry. 1990;28:255–265. doi: 10.1016/0006-3223(90)90581-l. [DOI] [PubMed] [Google Scholar]
- Dirksen K, Spee B, Penning LC, van den Ingh TSGAM, Burgener IA, Watson AL, Groot Koerkamp M, Rothuizen J, van Steenbeek FG, Fieten H. Gene expression patterns in the progression of canine copper-associated chronic hepatitis. PLoS ONE. 2017;12:e0176826. doi: 10.1371/journal.pone.0176826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Shi S-S, Chen S, Ni W, Zhu M, Wu Z-Y. The discrepancy between the absence of copper deposition and the presence of neuronal damage in the brain of Atp7b(−/−) mice. Metallomics. 2015;7:283–288. doi: 10.1039/c4mt00242c. [DOI] [PubMed] [Google Scholar]
- Fieten H, Gill Y, Martin AJ, Concilli M, Dirksen K, van Steenbeek FG, Spee B, et al. The Menkes and Wilson disease genes counteract in copper toxicosis in Labrador retrievers: a new canine model for copper-metabolism disorders. Dis Model Mech. 2016;9:25–38. doi: 10.1242/dmm.020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieten H, Hooijer-Nouwens BD, Biourge VC, Leegwater PAJ, Watson AL, van den Ingh TSGAM, Rothuizen J. Association of dietary copper and zinc levels with hepatic copper and zinc concentration in Labrador Retrievers. J Vet Intern Med. 2012a;26:1274–1280. doi: 10.1111/j.1939-1676.2012.01001.x. [DOI] [PubMed] [Google Scholar]
- Fieten H, Leegwater PAJ, Watson AL, Rothuizen J. Canine models of copper toxicosis for understanding mammalian copper metabolism. Mamm Genome. 2012b;23:62–75. doi: 10.1007/s00335-011-9378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong RN, Gonzalez BPE, Fuentealba IC, Cherian MG. Role of tumor necrosis factor-alpha in the development of spontaneous hepatic toxicity in Long-Evans Cinnamon rats. Toxicol Appl Pharmacol. 2004;200:121–130. doi: 10.1016/j.taap.2004.03.023. [DOI] [PubMed] [Google Scholar]
- Fu Y, Nath RG, Dyba M, Cruz IM, Pondicherry SR, Fernandez A, Schultz CL, et al. In vivo detection of a novel endogenous etheno-DNA adduct derived from arachidonic acid and the effects of antioxidants on its formation. Free Radic Biol Med. 2014;73:12–20. doi: 10.1016/j.freeradbiomed.2014.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George GN, Pickering IJ, Harris HH, Gailer J, Klein D, Lichtmannegger J, Summer KH. Tetrathiomolybdate causes formation of hepatic copper-molybdenum clusters in an animal model of Wilson’s disease. J Am Chem Soc. 2003;125:1704–1705. doi: 10.1021/ja029054u. [DOI] [PubMed] [Google Scholar]
- Gerbasi V, Lutsenko S, Lewis EJ. A mutation in the ATP7B copper transporter causes reduced dopamine beta-hydroxylase and norepinephrine in mouse adrenal. Neurochem Res. 2003;28:867–873. doi: 10.1023/a:1023219308890. [DOI] [PubMed] [Google Scholar]
- Gray LW, Peng F, Molloy SA, Pendyala VS, Muchenditsi A, Muzik O, Lee J, Kaplan JH, Lutsenko S. Urinary copper elevation in a mouse model of Wilson’s disease is a regulated process to specifically decrease the hepatic copper load. PLoS ONE. 2012;7:e38327. doi: 10.1371/journal.pone.0038327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Nyasae L, Braiterman LT, Hubbard AL. NH2-terminal signals in ATP7B Cu-ATPase mediate its Cu-dependent anterograde traffic in polarized hepatic cells. Am J Physiol Gastrointest Liver Physiol. 2005;289:G904–16. doi: 10.1152/ajpgi.00262.2005. [DOI] [PubMed] [Google Scholar]
- Hachmoller O, Aichler M, Schwamborn K, Lutz L, Werner M, Sperling M, Walch A, Karst U. Element bioimaging of liver needle biopsy specimens from patients with Wilson’s disease by laser ablation-inductively coupled plasma-mass spectrometry. J Trace Elem Med Biol. 2016;35:97–102. doi: 10.1016/j.jtemb.2016.02.001. [DOI] [PubMed] [Google Scholar]
- Hamilton JP, Koganti L, Muchenditsi A, Pendyala VS, Huso D, Hankin J, Murphy RC, et al. Activation of LXR/RXR pathway ameliorates liver disease in atp7b(−/−) (wilson disease) mice. Hepatology. 2015:n/a–n/a. doi: 10.1002/hep.28406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JP, Koganti L, Muchenditsi A, Pendyala VS, Huso D, Hankin J, Murphy RC, et al. Activation of liver X receptor/retinoid X receptor pathway ameliorates liver disease in Atp7B(−/−) (Wilson disease) mice. Hepatology. 2016;63:1828–1841. doi: 10.1002/hep.28406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada M. Pathogenesis and management of Wilson disease. Hepatol Res. 2014;44:395–402. doi: 10.1111/hepr.12301. [DOI] [PubMed] [Google Scholar]
- Harada M, Kawaguchi T, Kumemura H, Terada K, Ninomiya H, Taniguchi E, Hanada S, et al. The Wilson disease protein ATP7B resides in the late endosomes with Rab7 and the Niemann-Pick C1 protein. Am J Pathol. 2005;166:499–510. doi: 10.1016/S0002-9440(10)62272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman B, Manuelpillai U, Wallace EM, van de Waasenburg S, Cater M, Mercer JFB, Ackland ML. Expression and localization of menkes and Wilson copper transporting ATPases in human placenta. Placenta. 2004;25:512–517. doi: 10.1016/j.placenta.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Hawkins RL, Mori M, Inoue M, Torii K. Proline, ascorbic acid, or thioredoxin affect jaundice and mortality in Long Evans cinnamon rats. Pharmacol Biochem Behav. 1995;52:509–515. doi: 10.1016/0091-3057(95)00118-g. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Fuse S, Endoh D, Horiguchi N, Nakayama K, Kon Y, Okui T. Accumulation of copper induces DNA strand breaks in brain cells of Long-Evans Cinnamon (LEC) rats, an animal model for human Wilson Disease. Exp Anim. 2006;55:419–426. doi: 10.1538/expanim.55.419. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Kuge T, Endoh D, Nakayama K, Arikawa J, Takazawa A, Okui T. Hepatic copper accumulation induces DNA strand breaks in the liver cells of Long-Evans Cinnamon strain rats. Biochem Biophys Res Commun. 2000;276:174–178. doi: 10.1006/bbrc.2000.3454. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Miyane K, Hirooka T, Endoh D, Higuchi H, Nagahata H, Nakayama K, Kon Y, Okui T. Inhibitory effects of trientine, a copper-chelating agent, on induction of DNA strand breaks in hepatic cells of Long-Evans Cinnamon rats. Biochim Biophys Acta. 2004;1674:312–318. doi: 10.1016/j.bbagen.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Miyane K, Senou M, Endoh D, Higuchi H, Nagahata H, Nakayama K, Kon Y, Okui T. Inhibitory effects of trientine, a copper-chelating agent, on induction of DNA strand breaks in kidney cells of Long-Evans Cinnamon (LEC) rats. Exp Anim. 2005;54:403–412. doi: 10.1538/expanim.54.403. [DOI] [PubMed] [Google Scholar]
- Hoffmann G, van den Ingh TSGAM, Bode P, Rothuizen J. Copper-associated chronic hepatitis in Labrador Retrievers. J Vet Intern Med. 2006;20:856–861. doi: 10.1892/0891-6640(2006)20[856:cchilr]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Howell JM, Mercer JF. The pathology and trace element status of the toxic milk mutant mouse. J Comp Pathol. 1994;110:37–47. doi: 10.1016/s0021-9975(08)80268-x. [DOI] [PubMed] [Google Scholar]
- Huster D. Wilson disease. Best Pract Res Clin Gastroenterol. 2010;24:531–539. doi: 10.1016/j.bpg.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Huster D, Finegold MJ, Morgan CT, Burkhead JL, Nixon R, Vanderwerf SM, Gilliam CT, Lutsenko S. Consequences of copper accumulation in the livers of the Atp7b−/− (Wilson disease gene) knockout mice. Am J Pathol. 2006;168:423–434. doi: 10.2353/ajpath.2006.050312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huster D, Purnat TD, Burkhead JL, Ralle M, Fiehn O, Stuckert F, Olson NE, Teupser D, Lutsenko S. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J Biol Chem. 2007;282:8343–8355. doi: 10.1074/jbc.M607496200. [DOI] [PubMed] [Google Scholar]
- Irani AN, Malhi H, Slehria S, Gorla GR, Volenberg I, Schilsky ML, Gupta S. Correction of liver disease following transplantation of normal rat hepatocytes into Long-Evans Cinnamon rats modeling Wilson’s disease. Mol Ther. 2001;3:302–309. doi: 10.1006/mthe.2001.0271. [DOI] [PubMed] [Google Scholar]
- Iwata R, Sasaki N, Agui T. Contiguous gene deletion of Ptprk and Themis causes T-helper immunodeficiency (thid) in the LEC rat. Biomed Res. 2010;31:83–87. doi: 10.2220/biomedres.31.83. [DOI] [PubMed] [Google Scholar]
- Jackson Laboratory T. Mouse strain data sheet - 001576 [Google Scholar]
- Jia G, Takahashi R, Zhang Z, Tsuji Y, Sone H. Aldo-keto reductase 1 family B7 is the gene induced in response to oxidative stress in the livers of Long-Evans Cinnamon rats. Int J Oncol. 2006;29:829–838. [PMC free article] [PubMed] [Google Scholar]
- Joseph B, Kapoor S, Schilsky ML, Gupta S. Bile salt-induced pro-oxidant liver damage promotes transplanted cell proliferation for correcting Wilson disease in the Long-Evans Cinnamon rat model. Hepatology. 2009;49:1616–1624. doi: 10.1002/hep.22792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CG, Miyamoto T, Tsumagari T, Agui T. Genetic association between low expression phenotype of CD62L (L-selectin) in peripheral CD4+ T cells and the thid (T-helper immunodeficiency) phenotype in the LEC rat. Exp Anim. 2001;50:337–340. doi: 10.1538/expanim.50.337. [DOI] [PubMed] [Google Scholar]
- Kato J, Kobune M, Kohgo Y, Sugawara N, Hisai H, Nakamura T, Sakamaki S, Sawada N, Niitsu Y. Hepatic iron deprivation prevents spontaneous development of fulminant hepatitis and liver cancer in Long-Evans Cinnamon rats. J Clin Invest. 1996;98:923–929. doi: 10.1172/JCI118875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuda T, Teratani T, Ochiya T, Sakai Y. Transplantation of a fetal liver cell-loaded hyaluronic acid sponge onto the mesentery recovers a Wilson’s disease model rat. J Biochem. 2010;148:281–288. doi: 10.1093/jb/mvq063. [DOI] [PubMed] [Google Scholar]
- Kawano H, Takeuchi Y, Yoshimoto K, Matsumoto K, Sugimoto T. Histological changes in monoaminergic neurons of Long-Evans Cinnamon rats. Brain Res. 2001;915:25–31. doi: 10.1016/s0006-8993(01)02818-9. [DOI] [PubMed] [Google Scholar]
- Kim DW, Ahn T-B, Kim JM, Jeon GS, Seo JH, Jeon BS, Cho SS. Enhanced Mn-SOD immunoreactivity in the dopaminergic neurons of long-evans cinnamon rats. Neurochem Res. 2005a;30:475–478. doi: 10.1007/s11064-005-2683-3. [DOI] [PubMed] [Google Scholar]
- Kim JM, Ko SB, Kwon SJ, Kim HJ, Han MK, Kim DW, Cho SS, Jeon BS. Ferrous and ferric iron accumulates in the brain of aged Long-Evans Cinnamon rats, an animal model of Wilson’s disease. Neurosci Lett. 2005b;382:143–147. doi: 10.1016/j.neulet.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Kinnier Wilson SA. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. The Lancet. 1912;179:1115–1119. [Google Scholar]
- Klein D, Arora U, Lichtmannegger J, Finckh M, Heinzmann U, Summer KH. Tetrathiomolybdate in the treatment of acute hepatitis in an animal model for Wilson disease. J Hepatol. 2004;40:409–416. doi: 10.1016/j.jhep.2003.11.034. [DOI] [PubMed] [Google Scholar]
- Klein D, Lichtmannegger J, Finckh M, Summer KH. Gene expression in the liver of Long-Evans cinnamon rats during the development of hepatitis. Arch Toxicol. 2003;77:568–575. doi: 10.1007/s00204-003-0493-4. [DOI] [PubMed] [Google Scholar]
- Klein D, Lichtmannegger J, Heinzmann U, Müller-Höcker J, Michaelsen S, Summer KH. Association of copper to metallothionein in hepatic lysosomes of Long-Evans cinnamon (LEC) rats during the development of hepatitis [se e comments] Eur J Clin Invest. 1998;28:302–310. doi: 10.1046/j.1365-2362.1998.00292.x. [DOI] [PubMed] [Google Scholar]
- Klein D, Michaelsen S, Sato S, Luz A, Stampfl A, Summer KH. Binding of Cu to metallothionein in tissues of the LEC rat with inherited abnormal copper accumulation. Arch Toxicol. 1997;71:340–343. doi: 10.1007/s002040050396. [DOI] [PubMed] [Google Scholar]
- Komatsu Y, Ogra Y, Suzuki KT. Copper balance and ceruloplasmin in chronic hepatitis in a Wilson disease animal model, LEC rats. Arch Toxicol. 2002;76:502–508. doi: 10.1007/s00204-002-0370-6. [DOI] [PubMed] [Google Scholar]
- Koropatnick J, Cherian MG. A mutant mouse (tx) with increased hepatic metallothionein stability and accumulation. Biochem J. 1993;296(Pt 2):443–449. doi: 10.1042/bj2960443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Fontaine S, Theophilos MB, Firth SD, Gould R, Parton RG, Mercer JF. Effect of the toxic milk mutation (tx) on the function and intracellular localization of Wnd, the murine homologue of the Wilson copper ATPase. Hum Mol Genet. 2001;10:361–370. doi: 10.1093/hmg/10.4.361. [DOI] [PubMed] [Google Scholar]
- Lai YR, Sugawara N. Outputs of hepatic copper and cadmium stimulated by tetrathiomolybdate (TTM) injection in Long-Evans Cinnamon (LEC) rats pretreated with cadmium, and in Fischer rats pretreated with copper and cadmium. Toxicology. 1997;120:47–54. doi: 10.1016/s0300-483x(97)03636-6. [DOI] [PubMed] [Google Scholar]
- Lalioti V, Peiro R, Perez-Berlanga M, Tsuchiya Y, Munoz A, Villalba T, Sanchez C, Sandoval IV. Basolateral sorting and transcytosis define the Cu+-regulated translocation of ATP7B to the bile canaliculus. J Cell Sci. 2016;129:2190–2201. doi: 10.1242/jcs.184663. [DOI] [PubMed] [Google Scholar]
- Lee BH, Kim JH, Kim J-M, Heo SH, Kang M, Kim G-H, Choi J-H, Yoo HW. The early molecular processes underlying the neurological manifestations of an animal model of Wilson’s disease. Metallomics. 2013;5:532–540. doi: 10.1039/c3mt20243g. [DOI] [PubMed] [Google Scholar]
- Lee BH, Kim J-M, Heo SH, Mun JH, Kim J, Kim JH, Jin HY, Kim G-H, Choi J-H, Yoo HW. Proteomic analysis of the hepatic tissue of Long-Evans Cinnamon (LEC) rats according to the natural course of Wilson disease. Proteomics. 2011;11:3698–3705. doi: 10.1002/pmic.201100122. [DOI] [PubMed] [Google Scholar]
- Lenartowicz M, Krzeptowski W, Lipinski P, Grzmil P, Starzynski R, Pierzchala O, Møller LB. Mottled Mice and Non-Mammalian Models of Menkes Disease. Front Mol Neurosci. 2015;8:72. doi: 10.3389/fnmol.2015.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy E, Brunet S, Alvarez F, Seidman E, Bouchard G, Escobar E, Martin S. Abnormal hepatobiliary and circulating lipid metabolism in the Long-Evans Cinnamon rat model of Wilson’s disease. Life Sci. 2007;80:1472–1483. doi: 10.1016/j.lfs.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Li Y, Togashi Y, Sato S, Emoto T, Kang JH, Takeichi N, Kobayashi H, Kojima Y, Une Y, Uchino J. Spontaneous hepatic copper accumulation in Long-Evans Cinnamon rats with hereditary hepatitis. A model of Wilson’s disease. J Clin Invest. 1991;87:1858–1861. doi: 10.1172/JCI115208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtmannegger J, Leitzinger C, Wimmer R, Schmitt S, Schulz S, Kabiri Y, Eberhagen C, et al. Methanobactin reverses acute liver failure in a rat model of Wilson disease. J Clin Invest. 2016;126:2721–2735. doi: 10.1172/JCI85226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggi M, Murgia D, Civolani A, Demelia E, Sorbello O, Demelia L. The relationship between copper and steatosis in Wilson’s disease. Clin Res Hepatol Gastroenterol. 2013;37:36–40. doi: 10.1016/j.clinre.2012.03.038. [DOI] [PubMed] [Google Scholar]
- Linz R, Barnes NL, Zimnicka AM, Kaplan JH, Eipper B, Lutsenko S. Intracellular targeting of copper-transporting ATPase ATP7A in a normal and Atp7b−/− kidney. Am J Physiol Renal Physiol. 2008;294:F53–61. doi: 10.1152/ajprenal.00314.2007. [DOI] [PubMed] [Google Scholar]
- Lutsenko S. Atp7b−/− mice as a model for studies of Wilson’s disease. Biochem Soc Trans. 2008;36:1233–1238. doi: 10.1042/BST0361233. [DOI] [PubMed] [Google Scholar]
- Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- Machado A, Chien HF, Deguti MM, Cançado E, Azevedo RS, Scaff M, Barbosa ER. Neurological manifestations in Wilson’s disease: Report of 119 cases. Mov Disord. 2006;21:2192–2196. doi: 10.1002/mds.21170. [DOI] [PubMed] [Google Scholar]
- Malhi H, Bhargava KK, Afriyie MO, Volenberg I, Schilsky ML, Palestro CJ, Gupta S. 99mTc-mebrofenin scintigraphy for evaluating liver disease in a rat model of Wilson’s disease. J Nucl Med. 2002;43:246–252. [PubMed] [Google Scholar]
- Malhi H, Joseph B, Schilsky ML, Gupta S. Development of cell therapy strategies to overcome copper toxicity in the LEC rat model of Wilson disease. Regen Med. 2008;3:165–173. doi: 10.2217/17460751.3.2.165. [DOI] [PubMed] [Google Scholar]
- Masuda R, Yoshida MC, Sasaki M, Dempo K, Mori M. Hereditary hepatitis of LEC rats is controlled by a single autosomal recessive gene. Lab Anim. 1988;22:166–169. doi: 10.1258/002367788780864402. [DOI] [PubMed] [Google Scholar]
- Medici V, Kieffer DA, Shibata NM, Chima H, Kim K, Canovas A, Medrano JF, et al. Wilson Disease: Epigenetic effects of choline supplementation on phenotype and clinical course in a mouse model. Epigenetics. 2016;11:804–818. doi: 10.1080/15592294.2016.1231289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici V, Santon A, Sturniolo GC, D’Incà R, Giannetto S, Albergoni V, Irato P. Metallothionein and antioxidant enzymes in Long-Evans Cinnamon rats treated with zinc. Arch Toxicol. 2002;76:509–516. doi: 10.1007/s00204-002-0377-z. [DOI] [PubMed] [Google Scholar]
- Medici V, Sturniolo GC, Santon A, D’Incà R, Bortolami M, Cardin R, Basso D, Albergoni V, Irato P. Efficacy of zinc supplementation in preventing acute hepatitis in Long-Evans Cinnamon rats. Liver Int. 2005;25:888–895. doi: 10.1111/j.1478-3231.2005.01108.x. [DOI] [PubMed] [Google Scholar]
- Mendelsohn BA, Yin C, Johnson SL, Wilm TP, Solnica-Krezel L, Gitlin JD. Atp7a determines a hierarchy of copper metabolism essential for notochord development. Cell Metab. 2006;4:155–162. doi: 10.1016/j.cmet.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study. Gut. 2007;56:115–120. doi: 10.1136/gut.2005.087262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merle U, Tuma S, Herrmann T, Muntean V, Volkmann M, Gehrke SG, Stremmel W. Evidence for a critical role of ceruloplasmin oxidase activity in iron metabolism of Wilson disease gene knockout mice. J Gastroenterol Hepatol. 2010;25:1144–1150. doi: 10.1111/j.1440-1746.2009.06173.x. [DOI] [PubMed] [Google Scholar]
- Michalczyk AA, Rieger J, Allen KJ, Mercer JF, Ackland ML. Defective localization of the Wilson disease protein (ATP7B) in the mammary gland of the toxic milk mouse and the effects of copper supplementation. Biochem J. 2000;352(Pt 2):565–571. [PMC free article] [PubMed] [Google Scholar]
- Michalczyk A, Bastow E, Greenough M, Camakaris J, Freestone D, Taylor P, Linder M, Mercer J, Ackland ML. ATP7B expression in human breast epithelial cells is mediated by lactational hormones. J Histochem Cytochem. 2008;56:389–399. doi: 10.1369/jhc.7A7300.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchenditsi A, Yang H, Hamilton JP, Koganti L, Bhattacharjee A, Housseau F, Aronov L, et al. Targeted inactivation of copper-transporter ATP7B in hepatocytes causes liver steatosis and obesity in mice. Am J Physiol Cell Physiol. 2017 doi: 10.1152/ajpgi.00312.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu Y, Yamada T, Moralejo DH, Suzuki Y, Matsumoto K. Fetal copper uptake and a homolog (Atp7b) of the Wilson’s disease gene in rats. Res Commun Mol Pathol Pharmacol. 1998;101:225–231. [PubMed] [Google Scholar]
- Murata Y, Yamakawa E, Iizuka T, Kodama H, Abe T, Seki Y, Kodama M. Failure of copper incorporation into ceruloplasmin in the Golgi apparatus of LEC rat hepatocytes. Biochem Biophys Res Commun. 1995;209:349–355. doi: 10.1006/bbrc.1995.1510. [DOI] [PubMed] [Google Scholar]
- Murillo O, Luqui DM, Gazquez C, Martinez-Espartosa D, Navarro-Blasco I, Monreal JI, Guembe L, et al. Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy. J Hepatol. 2016;64:419–426. doi: 10.1016/j.jhep.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Nair J, Strand S, Frank N, Knauft J, Wesch H, Galle PR, Bartsch H. Apoptosis and age-dependant induction of nuclear and mitochondrial etheno-DNA adducts in Long-Evans Cinnamon (LEC) rats: enhanced DNA damage by dietary curcumin upon copper accumulation. Carcinogenesis. 2005;26:1307–1315. doi: 10.1093/carcin/bgi073. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Nakayama K, Shishido N, Yumino K, Ohyama T. Metal-induced hydroxyl radical generation by Cu(+)-metallothioneins from LEC rat liver. Biochem Biophys Res Commun. 1997;231:549–552. doi: 10.1006/bbrc.1996.6044. [DOI] [PubMed] [Google Scholar]
- Nakazato K, Nakajima K, Nakano T, Kodaira T, Nakayama K, Satoh M, Nagamine T. Metallothionein (MT) 1/2 expression in MT 1/2 and MT 3 knock-out mice and Long-Evans Cinnamon (LEC) rats. J Toxicol Sci. 2012;37:169–175. doi: 10.2131/jts.37.169. [DOI] [PubMed] [Google Scholar]
- Nakazato K, Tomioka S, Nakajima K, Saito H, Kato M, Kodaira T, Yatsuzuka SI, et al. Determination of the serum metallothionein (MT)1/2 concentration in patients with Wilson’s disease and Menkes disease. J Trace Elem Med Biol. 2014;28:441–447. doi: 10.1016/j.jtemb.2014.07.013. [DOI] [PubMed] [Google Scholar]
- Norgate M, Lee E, Southon A, Farlow A, Batterham P, Camakaris J, Burke R. Essential roles in development and pigmentation for the Drosophila copper transporter DmATP7. Mol Biol Cell. 2006;17:475–484. doi: 10.1091/mbc.E05-06-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogra Y, Chikusa H, Suzuki KT. Metabolic fate of the insoluble copper/tetrathiomolybdate complex formed in the liver of LEC rats with excess tetrathiomolybdate. J Inorg Biochem. 2000;78:123–128. doi: 10.1016/s0162-0134(99)00218-4. [DOI] [PubMed] [Google Scholar]
- Okayasu T, Tochimaru H, Hyuga T, Takahashi T, Takekoshi Y, Li Y, Togashi Y, Takeichi N, Kasai N, Arashima S. Inherited copper toxicity in Long-Evans cinnamon rats exhibiting spontaneous hepatitis: a model of Wilson’s disease. Pediatr Res. 1992;31:253–257. doi: 10.1203/00006450-199203000-00011. [DOI] [PubMed] [Google Scholar]
- Ono S, Koropatnick DJ, Cherian MG. Regional brain distribution of metallothionein, zinc and copper in toxic milk mutant and transgenic mice. Toxicology. 1997;124:1–10. doi: 10.1016/s0300-483x(97)00133-9. [DOI] [PubMed] [Google Scholar]
- Ostapowicz G, Fontana RJ, Schiødt FV, Larson A, Davern TJ, Han SHB, McCashland TM, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–954. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- Otsuka T, Izumi K, Tokunaga I, Gotohda T, Ipposhi K, Takiguchi Y, Kaneda S, et al. Prevention of lethal hepatic injury in Long-Evans Cinnamon (LEC) rats by D-galactosamine hydrochloride. J Med Invest. 2006;53:81–86. doi: 10.2152/jmi.53.81. [DOI] [PubMed] [Google Scholar]
- Peng F, Lutsenko S, Sun X, Muzik O. Positron emission tomography of copper metabolism in the Atp7b−/− knock-out mouse model of Wilson’s disease. Mol Imaging Biol. 2012a;14:70–78. doi: 10.1007/s11307-011-0476-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng F, Lutsenko S, Sun X, Muzik O. Imaging copper metabolism imbalance in Atp7b (−/−) knockout mouse model of Wilson’s disease with PET-CT and orally administered 64CuCl2. Mol Imaging Biol. 2012b;14:600–607. doi: 10.1007/s11307-011-0532-0. [DOI] [PubMed] [Google Scholar]
- Petrukhin K, Lutsenko S, Chernov I, Ross BM, Kaplan JH, Gilliam TC. Characterization of the Wilson disease gene encoding a P-type copper transporting ATPase: genomic organization, alternative splicing, and structure/function predictions. Hum Mol Genet. 1994;3:1647–1656. doi: 10.1093/hmg/3.9.1647. [DOI] [PubMed] [Google Scholar]
- Pfeiffenberger J, Mogler C, Gotthardt DN, Schulze-Bergkamen H, Litwin T, Reuner U, Hefter H, et al. Hepatobiliary malignancies in Wilson disease. Liver Int. 2015;35:1615–1622. doi: 10.1111/liv.12727. [DOI] [PubMed] [Google Scholar]
- Pierson H, Muchenditsi A, Kim B-E, Ralle M, Zachos N, Huster D, Lutsenko S. The Function of ATPase Copper Transporter ATP7B in Intestine. Gastroenterology. 2018;154:168–180.e5. doi: 10.1053/j.gastro.2017.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polishchuk EV, Concilli M, Iacobacci S, Chesi G, Pastore N, Piccolo P, Paladino S, et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev Cell. 2014;29:686–700. doi: 10.1016/j.devcel.2014.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyłkowski A, Gromadzka G, Wawer A, Bulska E, Jabłonka-Salach K, Grygorowicz T, Schnejder-Pachołek A, Członkowski A. Neurochemical and behavioral characteristics of toxic milk mice: an animal model of Wilson’s disease. Neurochem Res. 2013a;38:2037–2045. doi: 10.1007/s11064-013-1111-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyłkowski A, Gromadzka G, Wawer A, Grygorowicz T, Cybulska A, Członkowska A. Intestinal expression of metal transporters in Wilson’s disease. Biometals. 2013b;26:925–934. doi: 10.1007/s10534-013-9668-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralle M, Huster D, Vogt S, Schirrmeister W, Burkhead JL, Capps TR, Gray L, Lai B, Maryon E, Lutsenko S. Wilson disease at a single cell level: intracellular copper trafficking activates compartment-specific responses in hepatocytes. J Biol Chem. 2010;285:30875–30883. doi: 10.1074/jbc.M110.114447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch H. Toxic milk, a new mutation affecting copper metabolism in the mouse. J Hered. 1983;74:141–144. doi: 10.1093/oxfordjournals.jhered.a109751. [DOI] [PubMed] [Google Scholar]
- Rauch H, Wells AJ. The toxic milk mutation, tx, which results in a condition resembling Wilson disease in humans, is linked to mouse chromosome 8. Genomics. 1995;29:551–552. doi: 10.1006/geno.1995.9968. [DOI] [PubMed] [Google Scholar]
- Reed V, Williamson P, Bull PC, Cox DW, Boyd Y. Mapping of the mouse homologue of the Wilson disease gene to mouse chromosome 8. Genomics. 1995;28:573–575. doi: 10.1006/geno.1995.1191. [DOI] [PubMed] [Google Scholar]
- Roberts EA, Robinson BH, Yang S. Mitochondrial structure and function in the untreated Jackson toxic milk (tx-j) mouse, a model for Wilson disease. Mol Genet Metab. 2008;93:54–65. doi: 10.1016/j.ymgme.2007.08.127. [DOI] [PubMed] [Google Scholar]
- Roelofsen H, Wolters H, Van Luyn MJ, Miura N, Kuipers F, Vonk RJ. Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology. 2000;119:782–793. doi: 10.1053/gast.2000.17834. [DOI] [PubMed] [Google Scholar]
- Rosencrantz R, Schilsky M. Wilson disease: pathogenesis and clinical considerations in diagnosis and treatment. Semin Liver Dis. 2011;31:245–259. doi: 10.1055/s-0031-1286056. [DOI] [PubMed] [Google Scholar]
- Roybal JL, Endo M, Radu A, Gray L, Todorow CA, Zoltick PW, Lutsenko S, Flake AW. Early gestational gene transfer with targeted ATP7B expression in the liver improves phenotype in a murine model of Wilson’s disease. Gene Ther. 2012;19:1085–1094. doi: 10.1038/gt.2011.186. [DOI] [PubMed] [Google Scholar]
- Saito T, Itoh T, Fujimura M, Saito K. Age-dependent and region-specific differences in the distribution of trace elements in 7 brain regions of Long-Evans Cinnamon (LEC) rats with hereditary abnormal copper metabolism. Brain Res. 1995;695:240–244. doi: 10.1016/0006-8993(95)00819-c. [DOI] [PubMed] [Google Scholar]
- Saito T, Nagao T, Okabe M, Saito K. Neurochemical and histochemical evidence for an abnormal catecholamine metabolism in the cerebral cortex of the Long-Evans Cinnamon rat before excessive copper accumulation in the brain. Neurosci Lett. 1996;216:195–198. doi: 10.1016/0304-3940(96)13041-x. [DOI] [PubMed] [Google Scholar]
- Sambongi Y, Wakabayashi T, Yoshimizu T, Omote H, Oka T, Futai M. Caenorhabditis elegans cDNA for a Menkes/Wilson disease gene homologue and its function in a yeast CCC2 gene deletion mutant. J Biochem. 1997;121:1169–1175. doi: 10.1093/oxfordjournals.jbchem.a021711. [DOI] [PubMed] [Google Scholar]
- Samuele A, Mangiagalli A, Armentero M-T, Fancellu R, Bazzini E, Vairetti M, Ferrigno A, Richelmi P, Nappi G, Blandini F. Oxidative stress and pro-apoptotic conditions in a rodent model of Wilson’s disease. Biochim Biophys Acta. 2005;1741:325–330. doi: 10.1016/j.bbadis.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Santon A, Giannetto S, Sturniolo GC, Medici V, D’Incà R, Irato P, Albergoni V. Interactions between Zn and Cu in LEC rats, an animal model of Wilson’s disease. Histochem Cell Biol. 2002;117:275–281. doi: 10.1007/s00418-002-0380-8. [DOI] [PubMed] [Google Scholar]
- Santon A, Sturniolo GC, Albergoni V, Irato P. Metallothionein-1 and metallothionein-2 gene expression and localisation of apoptotic cells in Zn-treated LEC rat liver. Histochem Cell Biol. 2003;119:301–308. doi: 10.1007/s00418-003-0515-6. [DOI] [PubMed] [Google Scholar]
- Sauer SW, Merle U, Opp S, Haas D, Hoffmann GF, Stremmel W, Okun JG. Severe dysfunction of respiratory chain and cholesterol metabolism in Atp7b(−/−) mice as a model for Wilson disease. Biochim Biophys Acta. 2011;1812:1607–1615. doi: 10.1016/j.bbadis.2011.08.011. [DOI] [PubMed] [Google Scholar]
- Sauer V, Siaj R, Stöppeler S, Bahde R, Spiegel HU, Köhler G, Zibert A, Schmidt HH-J. Repeated transplantation of hepatocytes prevents fulminant hepatitis in a rat model of Wilson’s disease. Liver Transpl. 2012;18:248–259. doi: 10.1002/lt.22466. [DOI] [PubMed] [Google Scholar]
- Sawaki M, Enomoto K, Takahashi H, Nakajima Y, Mori M. Phenotype of preneoplastic and neoplastic liver lesions during spontaneous liver carcinogenesis of LEC rats. Carcinogenesis. 1990;11:1857–1861. doi: 10.1093/carcin/11.10.1857. [DOI] [PubMed] [Google Scholar]
- Schaefer M, Hopkins RG, Failla ML, Gitlin JD. Hepatocyte-specific localization and copper-dependent trafficking of the Wilson’s disease protein in the liver. Am J Physiol. 1999;276:G639–46. doi: 10.1152/ajpgi.1999.276.3.G639. [DOI] [PubMed] [Google Scholar]
- Schmitt F, Podevin G, Poupon J, Roux J, Legras P, Trocello J-M, Woimant F, Laprévote O, Nguyen TH, Balkhi El S. Evolution of exchangeable copper and relative exchangeable copper through the course of Wilson’s disease in the Long Evans Cinnamon rat. PLoS ONE. 2013;8:e82323. doi: 10.1371/journal.pone.0082323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada H, Takahashi M, Shimada A, Okawara T, Yasutake A, Imamura Y, Kiyozumi M. Protection from spontaneous hepatocellular damage by N-benzyl-d-glucamine dithiocarbamate in Long-Evans Cinnamon rats, an animal model of Wilson’s disease. Toxicol Appl Pharmacol. 2005;202:59–67. doi: 10.1016/j.taap.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Shishido N, Nakayama K, Takazawa A, Ohyama T, Nakamura M. Cu-metallothioneins (Cu(I)8-MTs) in LEC rat livers 13 weeks after birth still act as antioxidants. Arch Biochem Biophys. 2001;387:216–222. doi: 10.1006/abbi.2000.2233. [DOI] [PubMed] [Google Scholar]
- Siaj R, Sauer V, Stöppeler S, Spiegel HU, Köhler G, Zibert A, Schmidt HH-J. Dietary copper triggers onset of fulminant hepatitis in the Long-Evans cinnamon rat model. World J Gastroenterol. 2012;18:5542–5550. doi: 10.3748/wjg.v18.i39.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sintusek P, Dhawan A. Lipid and copper metabolism in humans with wilson disease: Enigmatic relationship. Hepatology. 2017;65:753–755. doi: 10.1002/hep.28936. [DOI] [PubMed] [Google Scholar]
- Sone K, Maeda M, Wakabayashi K, Takeichi N, Mori M, Sugimura T, Nagao M. Inhibition of hereditary hepatitis and liver tumor development in Long-Evans cinnamon rats by the copper-chelating agent trientine dihydrochloride. Hepatology. 1996;23:764–770. doi: 10.1053/jhep.1996.v23.pm0008666330. [DOI] [PubMed] [Google Scholar]
- Southon A, Burke R, Norgate M, Batterham P, Camakaris J. Copper homoeostasis in Drosophila melanogaster S2 cells. Biochem J. 2004;383:303–309. doi: 10.1042/BJ20040745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stattermayer AF, Traussnigg S, Dienes HP, Aigner E, Stauber R, Lackner K, Hofer H, et al. Hepatic steatosis in Wilson disease–Role of copper and PNPLA3 mutations. J Hepatol. 2015;63:156–163. doi: 10.1016/j.jhep.2015.01.034. [DOI] [PubMed] [Google Scholar]
- Sternlieb I. Mitochondrial and fatty changes in hepatocytes of patients with Wilson’s disease. Gastroenterology. 1968;55:354–367. [PubMed] [Google Scholar]
- Sugawara N, Ikeda T, Lai YR, Sugawara C. The effect of subcutaneous tetrathiomolybdate administration on copper and iron metabolism, including their regional redistribution in the brain, in the Long-Evans Cinnamon rat, a bona fide animal model for Wilson’s disease. Pharmacol Toxicol. 1999a;84:211–217. doi: 10.1111/j.1600-0773.1999.tb01485.x. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Ikeda T, Sugawara C, Kohgo Y, Kato J, Takeichi N. Regional distribution of copper, zinc and iron in the brain in Long-Evans Cinnamon (LEC) rats with a new mutation causing hereditary hepatitis. Brain Res. 1992;588:287–290. doi: 10.1016/0006-8993(92)91587-5. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Ohta T, Lai YR, Sugawara C, Yuasa M, Nakamura M, Tamura M. Iron depletion prevents adenine nucleotide decomposition and an increase of xanthine oxidase activity in the liver of the Long Evans Cinnamon (LEC) rat, an animal model of Wilson’s disease. Life Sci. 1999b;65:1423–1431. doi: 10.1016/s0024-3205(99)00378-1. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Sugawara C, Katakura M, Takahashi H, Mori M. Copper metabolism in the LEC rat: involvement of induction of metallothionein and disposition of zinc and iron. Experientia. 1991;47:1060–1063. doi: 10.1007/BF01923342. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Miyazawa N, Nakata T, Seo HG, Sugiyama T, Taniguchi N. High copper and iron levels and expression of Mn-superoxide dismutase in mutant rats displaying hereditary hepatitis and hepatoma (LEC rats) Carcinogenesis. 1993;14:1881–1884. doi: 10.1093/carcin/14.9.1881. [DOI] [PubMed] [Google Scholar]
- Svetel M, Potrebić A, Pekmezović T, Tomić A, Kresojević N, Jesić R, Dragasević N, Kostić VS. Neuropsychiatric aspects of treated Wilson’s disease. Parkinsonism Relat Disord. 2009;15:772–775. doi: 10.1016/j.parkreldis.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, Romano DM, Parano E, Pavone L, Brzustowicz LM. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- Tarohda T, Yamamoto M, Amamo R. Regional distribution of manganese, iron, copper, and zinc in the rat brain during development. Anal Bioanal Chem. 2004;380:240–246. doi: 10.1007/s00216-004-2697-8. [DOI] [PubMed] [Google Scholar]
- Terada K, Aiba N, Yang XL, Iida M, Nakai M, Miura N, Sugiyama T. Biliary excretion of copper in LEC rat after introduction of copper transporting P-type ATPase, ATP7B. FEBS Lett. 1999;448:53–56. doi: 10.1016/s0014-5793(99)00319-1. [DOI] [PubMed] [Google Scholar]
- Terada K, Nakako T, Yang XL, Iida M, Aiba N, Minamiya Y, Nakai M, Sakaki T, Miura N, Sugiyama T. Restoration of holoceruloplasmin synthesis in LEC rat after infusion of recombinant adenovirus bearing WND cDNA. J Biol Chem. 1998;273:1815–1820. doi: 10.1074/jbc.273.3.1815. [DOI] [PubMed] [Google Scholar]
- Terwel D, Loschmann Y-N, Schmidt HH-J, Scholer HR, Cantz T, Heneka MT. Neuroinflammatory and behavioural changes in the Atp7B mutant mouse model of Wilson’s disease. J Neurochem. 2011a;118:105–112. doi: 10.1111/j.1471-4159.2011.07278.x. [DOI] [PubMed] [Google Scholar]
- Terwel D, Loschmann Y-N, Schmidt HH-J, Scholer HR, Cantz T, Heneka MT. Neuroinflammatory and behavioural changes in the Atp7B mutant mouse model of Wilson’s disease. J Neurochem. 2011b;118:105–112. doi: 10.1111/j.1471-4159.2011.07278.x. [DOI] [PubMed] [Google Scholar]
- Theophilos MB, Cox DW, Mercer JF. The toxic milk mouse is a murine model of Wilson disease. Hum Mol Genet. 1996;5:1619–1624. doi: 10.1093/hmg/5.10.1619. [DOI] [PubMed] [Google Scholar]
- Togashi Y, Li Y, Kang JH, Takeichi N, Fujioka Y, Nagashima K, Kobayashi H. D-penicillamine prevents the development of hepatitis in Long-Evans Cinnamon rats with abnormal copper metabolism. Hepatology. 1992;15:82–87. doi: 10.1002/hep.1840150116. [DOI] [PubMed] [Google Scholar]
- Tumer Z, Moller LB. Menkes disease. Eur J Hum Genet. 2010;18:511–518. doi: 10.1038/ejhg.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueyama J, Wakusawa S, Tatsumi Y, Hattori A, Yano M, Hayashi H. Preliminary study of spontaneous hepatitis in Long-Evans Cinnamon rats: a blood exchange may improve fetal hepatitis. Nagoya J Med Sci. 2010;72:173–177. [PMC free article] [PubMed] [Google Scholar]
- van De Sluis B, Rothuizen J, Pearson PL, van Oost BA, Wijmenga C. Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum Mol Genet. 2002;11:165–173. doi: 10.1093/hmg/11.2.165. [DOI] [PubMed] [Google Scholar]
- Voskoboinik I, Greenough M, La Fontaine S, Mercer JF, Camakaris J. Functional studies on the Wilson copper P-type ATPase and toxic milk mouse mutant. Biochem Biophys Res Commun. 2001;281:966–970. doi: 10.1006/bbrc.2001.4445. [DOI] [PubMed] [Google Scholar]
- Wang J, Yuan B, Guerrero C, Bahde R, Gupta S, Wang Y. Quantification of oxidative DNA lesions in tissues of Long-Evans Cinnamon rats by capillary high-performance liquid chromatography-tandem mass spectrometry coupled with stable isotope-dilution method. Anal Chem. 2011;83:2201–2209. doi: 10.1021/ac103099s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss KH, Stremmel W. Evolving perspectives in Wilson disease: diagnosis, treatment and monitoring. Curr Gastroenterol Rep. 2012;14:1–7. doi: 10.1007/s11894-011-0227-3. [DOI] [PubMed] [Google Scholar]
- Weiss KH, Thurik F, Gotthardt DN, Schafer M, Teufel U, Wiegand F, Merle U, et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013;11:1028–35. e1–2. doi: 10.1016/j.cgh.2013.03.012. [DOI] [PubMed] [Google Scholar]
- Williams CH, Hong CC. Multi-step usage of in vivo models during rational drug design and discovery. Int J Mol Sci. 2011;12:2262–2274. doi: 10.3390/ijms12042262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmarth PA, Short KK, Fiehn O, Lutsenko S, David LL, Burkhead JL. A systems approach implicates nuclear receptor targeting in the Atp7b(−/−) mouse model of Wilson’s disease. Metallomics. 2012;4:660–668. doi: 10.1039/c2mt20017a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooton-Kee CR, Jain AK, Wagner M, Grusak MA, Finegold MJ, Lutsenko S, Moore DD. Elevated copper impairs hepatic nuclear receptor function in Wilson’s disease. J Clin Invest. 2015;125:3449–3460. doi: 10.1172/JCI78991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Forbes JR, Chen HS, Cox DW. The LEC rat has a deletion in the copper transporting ATPase gene homologous to the Wilson disease gene. Nat Genet. 1994;7:541–545. doi: 10.1038/ng0894-541. [DOI] [PubMed] [Google Scholar]
- Xu H, Sakakibara S, Morifuji M, Salamatulla Q, Aoyama Y. Excess dietary histidine decreases the liver copper level and serum alanine aminotransferase activity in Long-Evans Cinnamon rats. Br J Nutr. 2003;90:573–579. doi: 10.1079/bjn2003939. [DOI] [PubMed] [Google Scholar]
- Yamada T, Agui T, Suzuki Y, Sato M, Matsumoto K. Inhibition of the copper incorporation into ceruloplasmin leads to the deficiency in serum ceruloplasmin activity in Long-Evans cinnamon mutant rat. J Biol Chem. 1993;268:8965–8971. [PubMed] [Google Scholar]
- Yamada T, Izumi K, Suzuki Y, Agui T, Matsumoto K. Correlation between a hepatic copper accumulation and an altered expression of glutathione S-transferase Ya/Yc subunits in LEC mutant rat. Res Commun Chem Pathol Pharmacol. 1992;76:113–116. [PubMed] [Google Scholar]
- Yamamoto H, Watanabe T, Mizuno H, Endo K, Hosokawa T, Kazusaka A, Gooneratne R, Fujita S. In vivo evidence for accelerated generation of hydroxyl radicals in liver of Long-Evans Cinnamon (LEC) rats with acute hepatitis. Free Radic Biol Med. 2001;30:547–554. doi: 10.1016/s0891-5849(00)00496-2. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Ohshima H, Asanuma T, Inukai N, Miyoshi I, Kasai N, Kon Y, Watanabe T, Sato F, Kuwabara M. The effects of alpha-phenyl-tert-butyl nitrone (PBN) on copper-induced rat fulminant hepatitis with jaundice. Free Radic Biol Med. 1996;21:755–761. doi: 10.1016/0891-5849(96)00222-5. [DOI] [PubMed] [Google Scholar]
- Yasuda J, Eguchi H, Fujiwara N, Ookawara T, Kojima S, Yamaguchi Y, Nishimura M, Fujimoto J, Suzuki K. Reactive oxygen species modify oligosaccharides of glycoproteins in vivo: a study of a spontaneous acute hepatitis model rat (LEC rat) Biochem Biophys Res Commun. 2006;342:127–134. doi: 10.1016/j.bbrc.2006.01.118. [DOI] [PubMed] [Google Scholar]
- Yezdimer EM, Umemoto T, Yamada H, Makino S, Tooyama I. Visualizing hepatic copper release in Long-Evans cinnamon rats using single-photon emission computed tomography. Appl Biochem Biotechnol. 2013;170:1138–1150. doi: 10.1007/s12010-013-0252-9. [DOI] [PubMed] [Google Scholar]
- Yoshida MC, Masuda R, Sasaki M, Takeichi N, Kobayashi H, Dempo K, Mori M. New mutation causing hereditary hepatitis in the laboratory rat. J Hered. 1987;78:361–365. doi: 10.1093/oxfordjournals.jhered.a110416. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Tokusashi Y, Lee GH, Ogawa K. Intrahepatic transplantation of normal hepatocytes prevents Wilson’s disease in Long-Evans cinnamon rats. Gastroenterology. 1996;111:1654–1660. doi: 10.1016/s0016-5085(96)70029-x. [DOI] [PubMed] [Google Scholar]
- Yurkova IL, Arnhold J, Fitzl G, Huster D. Fragmentation of mitochondrial cardiolipin by copper ions in the Atp7b−/− mouse model of Wilson’s disease. Chem Phys Lipids. 2011;164:393–400. doi: 10.1016/j.chemphyslip.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Zimbrean PC, Schilsky ML. Psychiatric aspects of Wilson disease: a review. Gen Hosp Psychiatry. 2014;36:53–62. doi: 10.1016/j.genhosppsych.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Zischka H, Lichtmannegger J, Schmitt S, Jägemann N, Schulz S, Wartini D, Jennen L, et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J Clin Invest. 2011;121:1508–1518. doi: 10.1172/JCI45401. [DOI] [PMC free article] [PubMed] [Google Scholar]