

Abstract
Extracellular nucleotides can regulate the production/drainage of the aqueous humor via activation of P2 receptors, thus affecting the intraocular pressure (IOP). We evaluated 5-OMe-UDP(α-B), 1A, a potent P2Y6-receptor agonist, for reducing IOP and treating glaucoma. Cell viability in the presence of 1A was measured using [3-(4, 5-dimethyl-thiazol-2-yl) 2, 5-diphenyl-tetrazolium bromide] (MTT) assay in rabbit NPE ciliary non-pigmented and corneal epithelial cells, human retinoblastoma, and liver Huh7 cells. The effect of 1A on IOP was determined in acute glaucomatous rabbit hyaluronate model and phenol-induced chronic glaucomatous rabbit model. The origin of activity of 1A was investigated by generation of a homology model of hP2Y6-R and docking studies. 1A did not exert cytotoxic effects up to 100 mM vs. trusopt and timolol in MTT assay in ocular and liver cells. In normotensive rabbits, 100 μM 1A vs. xalatan, trusopt, and pilocarpine reduced IOP by 45 vs. 20–30%, respectively. In the phenol animal model, 1A (100 μM) showed reduction of IOP by 40 and 20%, following early and late administration, respectively. Docking results suggest that the high activity and selectivity of 1A is due to intramolecular interaction between Pα-BH3 and C5-OMe which positions 1A in a most favorable site inside the receptor. P2Y6-receptor agonist 1A effectively and safely reduces IOP in normotense, acute, and chronic glaucomatous rabbits, and hence may be suggested as a novel approach for the treatment of glaucoma.
Electronic supplementary material
The online version of this article (10.1007/s11302-018-9614-7) contains supplementary material, which is available to authorized users.
Keywords: Human P2Y6 receptor, P2Y6 receptor agonist, Glaucoma, Intraocular pressure, 5-OMe-uridine-5′-α-borano-diphosphate
Introduction
Extracellular nucleotides and dinucleotides have been shown to play a role in ocular physiology and pathophysiology. Nucleotides and dinucleotides activate P2Y- and P2X-receptors (P2YRs/P2XRs) expressed in all ocular tissues. Neuronal P2XRs in the inner and outer retina contribute to visual processing, as well as cell death in retinal ganglia [1]. Thus, in chronic glaucoma, P2X7R activated by ATP increases cell death in the retina, whereas the P2X1R mediates ATP release in trabecular meshwork and ciliary epithelial cells. Yet, the presence of P2XRs heterotrimers complicates the pharmacology of P2XRs and their application as drug targets.
P2Y-Rs have multiple actions in the eye: they control tear production, corneal wound healing, aqueous humor dynamics, and retinal physiology [1]. Therefore, nucleotides have been suggested as therapeutic agents for dry eye disease, retinal detachment, and glaucoma [2].
Ocular hypertension, the most common cause of glaucoma, is a target for agents that reduce intraocular pressure (IOP) [3]. Trabecular meshwork cells (TMC) form a tissue in the eye that is responsible for draining the aqueous humor, thus reducing IOP. This tissue expresses receptors for extracellular nucleotides, including P2Y1, P2Y2, P2Y4, and P2Y6 receptors [4]. Indeed, IOP is reduced by administration of ATP, adenosine tetraphosphate (Ap4) [5], and diadenosine tetraphosphate (Ap4A) [6]. Likewise, the synthetic P2Y1-R selective agonist, 2-MeS-ADP, increases aqueous humor outflow in bovines [7]. However, the therapeutic potential of endogenous nucleotides for the treatment of glaucoma is limited, since they are degraded by extracellular enzymes, hence reducing their potency, efficacy, and duration of action [8].
Recently, P2Y6-R was shown to be a key target for the treatment of glaucoma [9]. Activation of P2Y6-R changes aqueous humor dynamics, thereby reducing IOP levels. Moreover, the P2Y6-R was shown to be critical for lowering IOP and that ablation of the P2Y6-R gene in mice results in hypertensive glaucoma-like optic neuropathy. Topical application of UDP, the endogenous agonist of P2Y6-R, decreased IOP [9, 10].
Although P2Y6-R plays an important role in reduction of intraocular pressure, this receptor lacked stable, potent, and selective agonists to be used as potential drugs for the treatment of glaucoma. For this purpose, we previously developed 5-OMe-UDP(α-B), Rp isomer, 1A (Fig. 1), the structure of which is based on the P2Y6-R endogenous ligand, UDP, 2 [11]. Analogue 1A was shown to be the most potent and P2Y6-R selective agonist currently known (EC50 0.008 μM). It was 19-fold more potent than UDP, and showed no activity at other uridine nucleotide receptors, i.e., P2Y2- and P2Y4-R. Notably, analogue 1A exhibited chemical and metabolic stability. It was highly chemically stable even under conditions mimicking gastric juice acidity, resisted hydrolysis by nucleotide pyrophosphatases (NPP1,3) up to three-fold more than UDP, and was significantly metabolically stable in human blood serum (t1/2 17 vs. 2.5, 12, and 17 h of UDP; 5-OMe-UDP; and UDP(α-B), respectively).
Fig. 1.
Structures of 5-OMe-UDP(α-B), Rp isomer, 1A, and related UDP analogues used for docking studies
Although P2Y receptors are involved in the regulation of IOP, they have not been targeted so far for therapeutics of glaucoma. Currently used drugs for the treatment of glaucoma target other receptors and enzymes such as prostaglandin receptor, β2-adrenergic receptor, α-receptor, and carbonic anhydrase.
The limitations of current drugs for the treatment of glaucoma, on the one hand, and the involvement of P2Y6-R in aqueous humor drainage, on the other hand, encouraged us to explore the application of this new mechanism for the reduction of IOP.
Here, we describe the ability of 1A to reduce IOP and potentially treat glaucoma. Specifically, we evaluated the efficacy of 1A for IOP reduction in normotensive rabbits. Next, this drug candidate was evaluated at several acute and chronic animal models of glaucoma, including sodium hyaluronate- and phenol-induced models. In addition, we conducted cytotoxicity studies. The efficacy and cytotoxicity effects of 1A were compared to several current drugs for the treatment of glaucoma. To explore the origin of the activity of 1A, we constructed hP2Y6-R homology model, and analyzed the molecular recognition pattern of 1A vs. other hP2Y6-R agonists. In particular, we rationalized the origin of high activity of 1A and diastereoselectivity of hP2Y6 by docking studies [12, 13].
Methods
General
Rabbit NPE ciliary non-pigmented epithelial cells, rabbit corneal epithelial cells (SIRC), and human retinoblastoma cell line (Y-79; kindly provided by Prof. Perelman, Technion, Haifa, Israel) were depicted as three ocular cell lines for the in vitro studies. NPE cells were cultured in DMEM high glucose (4.5 g/L) medium supplemented with 50 mg/L Gentamicin (Sigma-Aldrich), 2 mM l-glutamine, 1 mM sodium pyruvate, 1500 mg/L sodium bicarbonate, and 10% (v/v) heat-inactivated FBS (reagents from Biological Industries Israel Beit-Haemek Ltd.). SIRC cells were cultured in Eagle’s minimum essential medium (EMEM) supplemented with 15 nonessential amino acids (NEAA), 2 mM l-glutamine, 1 mM sodium pyruvate, 1500 mg/L sodium bicarbonate, and 10% (v/v) heat-inactivated FBS (reagents from Biological Industries Israel Beit-Haemek Ltd.). Y-79 cells were cultured in DMEM high glucose (4.5 g/L) medium supplemented with 1% (v/v) penicillin-streptomycin, 2 mM l-glutamine and 10% (v/v) heat-inactivated FBS (reagents from Biological Industries Israel Beit-Haemek Ltd.).
Human hepatocellular carcinoma Huh7 cells were cultured in DMEM high glucose (4.5 g/L) medium supplemented with 1% (v/v) penicillin-streptomycin, 2 mM l-glutamine, and 10% (v/v) heat-inactivated FBS (reagents from Biological Industries Israel Beit-Haemek Ltd.).
Cytotoxicity and cell viability analyses
For cytotoxicity and cell viability studies, cells were incubated with its respective media, containing 2% FBS. All cells were maintained in 5% CO2-humidified incubator at 37 °C.
Three ocular cell lines, Y-79, NPE, and SIRC as well as liver Huh7 cells, were harvested and cultured in 96-well plates at a density of ~1 × 104 cells per well, and maintained in a humidified incubator at 37 °C for 24 h. Then, increased concentrations (0.1–100 mM) of the compounds: 1A, trusopt (carbonic anhydrase inhibitor), and timolol (beta blocker) were added for additional 24 h. Cell viability was measured by quantitative colorimetric assay with MTT [3-(4, 5-dimethyl-thiazol-2-yl) 2,5-diphenyl-tetrazolium bromide] (Sigma-Aldrich), as described previously (Bar-Am et al. 2005). Briefly, 10 μL of the MTT labeling reagent (5 mg/mL in PBS) was added to each well and the plate incubated at 37 °C with 5% CO2 and 95% air (v/v) for an additional 2 h. Next, the insoluble formazan was dissolved for 24 h with 100 μL of 10% (v/v) SDS in 0.01 M HCl; colorimetric determination of MTT reduction was measured with a Tecan Sunrise Eliza-Reader (Switzerland) at λ = 570/650 nm after automatic subtraction of background readings.
Morphology of NPE and SIRC cells treated with 1A and timolol (10 and 50 mM) were examined in inverted microscope connected to a digital camera (× 10 objective) 24 h after exposure to the drugs.
Animal studies
All procedures were carried out in accordance with the National Institutes of Health Guide for care and Use of Laboratory Animals, and were approved by the Animal Ethics Committee of the Technion, Haifa, Israel. New Zealand white (NZW) rabbits obtained from Harlan (Jerusalem, Israel), weighing 3.0–3.5 kg, were housed at an ambient temperature of 22 °C with a 12 h light/dark cycle and humidity-controlled environment. Food and water were available ad libitum.
IOP measurements
IOP levels were measured by using a TONOVET rebound tonometer supplied by Tiolat Oy (Helsinki, Finland). In all experiments, animals were trained for the IOP measurement procedure during acclimation period in order to reduce stress and obtaining more accurate measurements during the study. IOP measurements were performed 5–10 consecutive times and always obtained in the right eye first. In order to avoid the effect of the circadian rhythm, the IOP was always tested at the same time of day in the morning. The average pre-dosing IOP of the experimental and control eyes was 12.3 mmHg.
Normotensive rabbit studies
The effect of 1A on IOP was tested over a range of doses (10−9–10−3 M) in normotensive rabbits (4–6 animals in each group). 1A was administered bilaterally in drops to the cornea of rabbits, at a single dose of a fixed volume of 10 μL. IOP was measured up to 3 h after treatment; two IOP measurements were taken before compound was administered.
For time-dependent effect on IOP of a single dose of 100 μM, 1A was employed in normotensive rabbits. The drug was administered to the cornea at volume of 10 μL. Two IOP measurements were taken before the drug was instilled. IOP was followed up to 6 h to study the time course of the effect.
For comparative analysis of the effect on IOP, different hypotensive compounds were administered unilaterally to the cornea of rabbits at a fixed volume of 10 μL: 1A (100 μM), UDP (100 μM), xalatan (0.005%), trusopt (2%), and pilocarpine (2%). The contralateral eye received the same volume of saline solution (0.9% NaCl, vehicle). Two IOP measurements were taken before any compound was administered. Experiments were performed following a blinded design where no visible indication was given to the experimenter as to the nature of the applied solution. IOP was followed up to 2 h after treatment.
Acute glaucomatous models in rabbits
Rabbits were subjected to anesthesia by using a mixture of Ketamine (35 mg/kg) and Xylazine (5 mg/kg) intramuscularly administered. In addition, 1–2 drops of local anesthetic (Localin®) were instilled to the eyes, prior to the experiment initiation.
Sodium hyaluronate-induced acute ocular hypertensive model
Eight rabbits were subjected to unilateral injection of 2.3% sodium hyaluronate into the anterior chamber of the eye [14]. Eight rabbits were divided to two groups and administered either with saline (vehicle) or with 1A (100 μM) at a dose volume of 30 μL/eye, starting at 2 h post 2.3% sodium hyaluronate injection. IOP levels were measured up to 24 h after administration.
Phenol-induced chronic model of glaucoma in rabbits
Early-phenol administration
Thirty rabbits were subjected to one subconjunctival injection of 0.4 mL of 5% phenol in almond oil into the right eye [15]. Two rabbits were not treated and used as baseline measurements for IOP fluctuation. Animals were subjected to two times daily repeated ocular instillations of 1A 100 μM, timolol (0.5%) , or saline vehicle; control) at a dose volume of 10 μL/eye), [15] performed during the entire observation period for 11 days, starting on day 1 of the experiment. Baseline IOP levels were measured in both eyes prior to the beginning of the experiment, and thereafter, IOP levels were measured twice a day.
Late-phenol administration
Thirty rabbits were subjected to one subconjunctival injection of 0.4 mL of 5% phenol in almond oil into the right eye. Two rabbits were not treated and used as baseline measurements for IOP fluctuation. Animals were subjected to two times daily repeated ocular instillations of 1A (100 μM) timolol (0.5%) or saline (vehicle; control) at a dose volume of 10 μL/eye, starting at day 7 when the ∆IOP level was 40% above the basal levels (days 7–11). Baseline IOP levels were taken in both eyes on day 1before phenol injection. IOP levels were measured twice a day throughout the entire observation period for 11 days.
Statistical analysis
All data are presented as the mean ± SEM of 3–4 independent experiments in vitro and animal studies. Significant differences were determined by two-way ANOVA test with Bonferroni/Dunnett’s post-tests using GraphPad Prism 5 software. Probability values of P < 0.05 or less were considered statistically significant.
Homology modeling of hP2Y6R
Selection of templates
The hP2Y6-R model was computed based on several crystal structures of GPCRs used as templates. First, we used two crystal structures of the human P2Y1R (pdb codes: 4XNV and 4XNW) [16, 17] and their combination (P2Y1-all). Second, we used two sets of crystal structures of the human P2Y12R (pdb codes: 4XNV and 4XNW [18] and their combination (P2Y12-all). Finally, we combined the P2Y1R and P2Y12R (All (P2Y1 + P2Y12)) and used these structures as template.
The search and alignments were done with the Discovery Studio (DS) 2016 software (Biovia, Inc.) [19].
Initial preparation of the homology model
Before the creation of the homology model, the GPCR templates which included T4L insertions (β2, A2A, CXCR4) were treated by removal of the insertion and addition of the missing amino acid residues and then refinement of the loop region(s). The homology model was created with Modeler program [20] (in Discovery Studio 4.0) based on the several GPCR templates. The modeled structure can be further evaluated using the Verify Protein (Profile-3D) protocol, which assesses the compatibility of the 3D structure of a protein model with the sequence of residues it contains.
The scores for pairing a residue i with an environment j is given by the information value [21],
| 1 |
Here, P(i:j) is the probability of finding residue i in environment j and Pi is the overall probability of finding residue i in any environment. These probabilities were determined from a database of 16 known protein structures and sets of homologous sequences aligned to the sequence of known structure as described by Bowie et al. [21]. For each position in the aligned set of sequences, we determined the environment category of the position from the known structure and counted the number of each residue type found at the position within the set of aligned sequences. At the end, we plot the difference between the verify score and expected verify high score. The closer the verify score is to the expected verify score, the better the quality of the model receptor.
The definition of the binding site was done using the “define and edit binding site” tool in DS 4.0. Prior to docking the ligands into the receptor, we generated a library of various ligand conformations (maximum 100) using the FAST or BEST conformation generation method with a maximum energy threshold of 20 kcal/mol. All the conformations were subjected to docking using a high throughput protocol designed within Pipeline Pilot (Pipeline Pilot 2014) [22]. The grid-based molecular docking program CDOCKER [23, 24] with the CHARMm force field [25] was employed to dock all the generated conformations into the P2Y1-R. We also fine-tuned the force constant for OS-PO3-B251 angle, which was set to 200 kcal/mol/Å2 for present study. Detailed discussion can be found in the SI. The docked pose with the highest negative interaction energy (i.e., lowest energy) was considered for further analysis. The solvation energy of the ligands was calculated using the Delphi [26] program. The solvation energy of each ligand is computed by averaging the solvation energy of the 100 lowest energy conformations obtained from the conformational search mentioned above.
To facilitate comparison with experiment, we predicted EC50 values of analogues 1A and 2–8 (Fig. 1) using multi-regression analysis of the solvation energy, the CDOCKER interaction energy, and of both the solvation energy and the CDOCKER interaction energy. To relax the pose of 5-OMe-UDP, 3, in the initial model, we performed short molecular dynamics simulations. A low energy conformation was chosen from the dynamics production phase and was further refined by rotating Arg266, Arg103, and Tyr283; re-minimization; and short molecular dynamics simulations and re-docking of agonists 1A and 2–8.
Docking selected nucleotides in hP2Y6R model
Since the CHARMm force field does not include parameters for the borano-bearing compounds, the initial docking of these ligands were done using the “CDOCKER with QM charges” protocol which calculated the atomic charges for the docked structure using DMol3 (charge method ESP with quality set to medium). The partial charges assigned were used for all further simulations and energy calculations.
Results
Cytotoxic effects of 1A compared with anti-glaucomatous drugs in ocular and liver hepatoma cell cultures
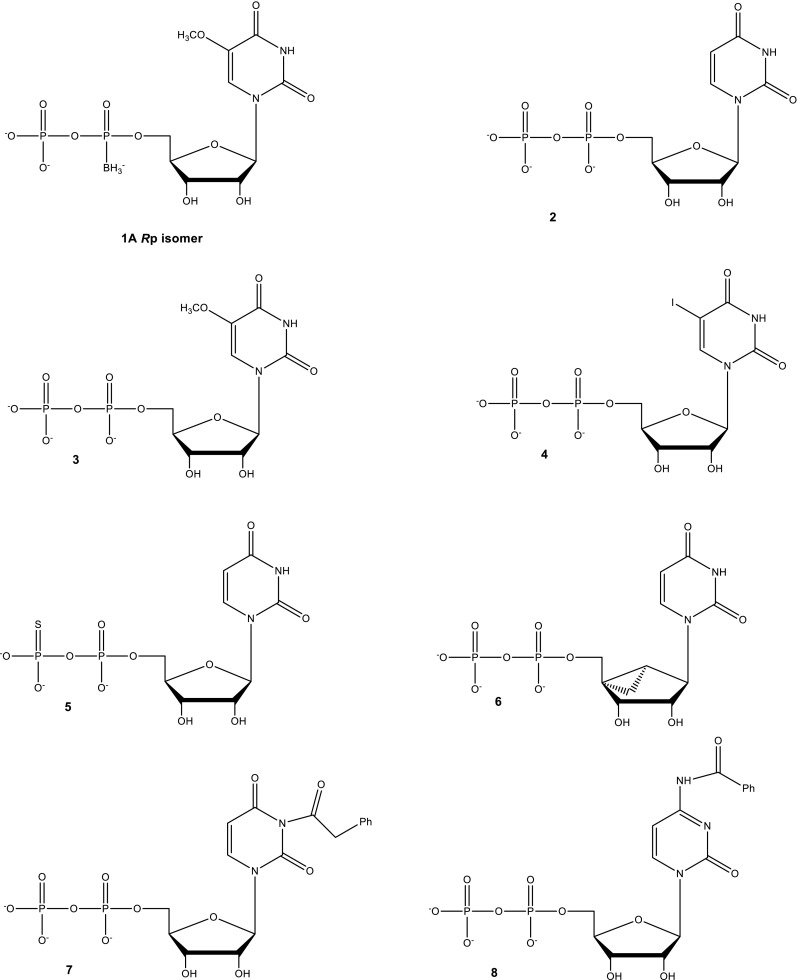
The cytotoxic effects of 1A (0.1–100 mM) were examined compared to two commercial anti-glaucomatous medications, trusopt (carbonic anhydrase inhibitor) and timolol (beta blocker) in three ocular cell lines: human Y-79 retinoblastoma cells, rabbit NPE non-pigmented ciliary, and SIRC corneal epithelial cells (Fig. 2, Table 1). Compound 1A significantly exerted less toxic effects than timolol and trusopt in all three ocular cell cultures (Fig. 2, Table 1). As shown in Fig. 2a, at 0.1–10 mM, both 1A and trusopt were not toxic in Y79 retinoblastoma cells, unlike timolol. At high concentrations (50 and 100 mM), 1A was significantly less toxic vs. trusopt in Y79 retinoblastoma cells (at 50 mM: 81.3 ± 1.6 vs. 65.1 ± 1.7% of control, respectively; and at 100 mM: 70.4 ± 0.8 vs. 41.4 ± 1.7% of control, respectively). In NPE cells, both 1A and trusopt were not toxic at 100 mM (Fig. 2a).
Fig. 2.
Cytotoxicity effects of 1A compared with anti-glaucomatous drugs in ocular cell cultures. Human Y-79 retinoblastoma cells and rabbit NPE non-pigmented ciliary and SIRC corneal epithelial cells were treated with 1A and anti-glaucomatous drugs, trusopt and timolol at 0.1–100 mM for 24 h. a Cell viability was assessed by MTT test. The results are mean ± S.E.M of a representative experiment of 3–4 identical assays and expressed as percent of respective control. (Asterisk) vs. vehicle (control); (number sign) vs. 1A; one sign P < 0.05, two signs P < 0.01, three signs P < 0.001. b Effect of 1A and timolol on morphology changes in rabbit (a) NPE and (b) SIRC cells. Cells were cultured in DMEM and EMEM, respectively, for 24 h. Vehicle (control), 10, and 50 mM of 1A or timolol, were added for additional 24 h. Images were acquired using an inverted microscope, connected to a digital camera (× 10 objective)
Table 1.
Effect of 1A on cell viability, compared with timolol and trusopt in drug-treated human Y-79 retinoblastoma cells, rabbit NPE, SIRC cells, and human Huh7 cell cultures
| Drug | Cell viability; IC50 (mM) | |||
|---|---|---|---|---|
| Human Y79 RB | Rabbit NPE | Rabbit SIRC | Human Huh7 | |
| 1A | 216.3 ± 5.4 | 264.7 ± 5.2 | 216.3 ± 5.4 | 320.8 ± 3.2 |
| Trusopt | 70.8 ± 1.8* | 193.7 ± 3.1* | nd | 123.8 ± 8.8* |
| Timolol | 19. 6 ± 0.8* | 22.2 ± 0.6* | 30.8 ± 0.3* | 42.1 ± 0.6* |
IC50 values expressed as mM ± S.E.M, following administration of increasing concentrations (0.001–500 mM) of 1A, trusopt, and timolol. *P < 0.001 vs. 1A; one-way ANOVA with Tukey post-tests. nd not determined
In Y-79 retinoblastoma and SIRC cells, a significant reduction in cell viability was observed with 50 mM timolol (4.3 ± 0.3 and 48.12 ± 0.9% of control, respectively) and 100 mM timolol (2.3 ± 0.4 and 1.3 ± 0.2% of control, respectively), while 1A at both concentrations (50 and 100 mM) was significantly less toxic (70–85% of control) (Fig. 2a, b). Similar nontoxic effects of 1A were observed in NPE cells (Fig. 2a, b).
In addition, in liver hepatoma Huh7 cells, 1A (50 and 100 mM) was three- and eightfold less toxic than trusopt and timolol, respectively (Fig. 3 and Table 1).
Fig. 3.

Cytotoxicity effects of 1A compared with anti-glaucomatous drugs in human Huh7 liver hepatoma cells. Cells treated with 1A and anti-glaucomatous drugs, trusopt, and timolol at 0.1–100 mM for 24 h. Cell viability was assessed by MTT test. The results are mean ± S.E.M of a representative experiment of three identical experimental assays and expressed as percentage of respective control. (Asterisk) vs. vehicle (control); (number sign) vs. 1A; one sign P < 0.05, two signs P < 0.01, three signs P < 0.001
Compound 1A potently and efficaciously reduced IOP in normotensive NZW rabbits
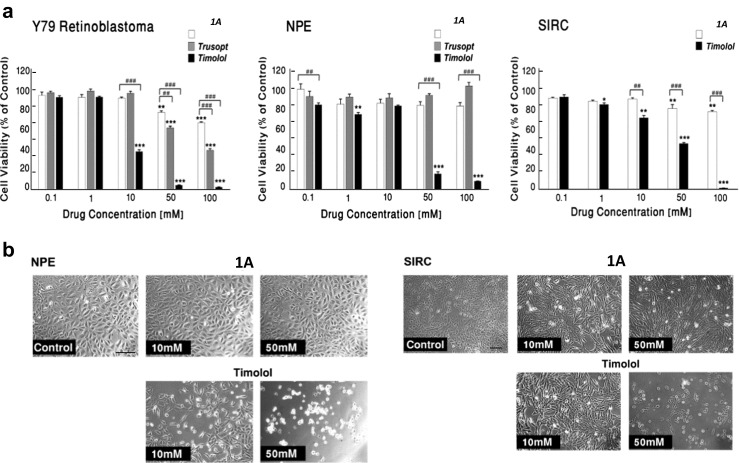
The effect of compound 1A on IOP in normotensive rabbits was evaluated at 10‑9to 10‑3 M concentration range. Compound 1A at 10–100 μM significantly reduced IOP (~45%) 2 h after administration (Fig. 4a, concentration-dependent effect). We have further examined the time-dependent effect of 1A to modify IOP in normotensive rabbits. A single dose of 100 μM of 1A was able to significantly reduce IOP (44 ± 4%) at 2 h after the instillation of the compound and the effect lasted 4 h (Fig. 4b, time-dependent effect).
Fig. 4.
Effect of 1A on IOP levels in normotensive rabbits. a Dose-dependent effect on IOP of a single administration of 1A in normotensive NWZ rabbits. IOP was measured following 2 h. b Time-dependent effect on IOP of a single dose of 1A (100 μM) in normotensive NWZ rabbits. c Comparative analysis the maximal hypotensive effect of UDP and 1A (100 μM) in normotensive NWZ rabbits. d Comparative analysis of the effect on IOP of 1A (100 μM) and commercial drugs, xalatan (0.005%), trusopt (2%), and pilocarpine (2%), in normotensive NWZ rabbits. All values are the mean ± S.E.M. (n = 4–6) of three independent experiments and expressed as percentage of respective control. *P < 0.05 vs. control; **P < 0.01 vs. control; and ***P < 0.001 vs. control
Comparing the maximal hypotensive effect of 1A (100 μM) with UDP (100 μM), the endogenous P2Y6 receptor agonist, we observed that 1A is able to produce a more robust effect on IOP than the naturally occurring compound, UDP, 45% IOP reduction vs. 15%, respectively (Fig. 4c).
In addition, the efficacy of 1A to reduce IOP was also compared to the effect of current drugs in normotensive rabbits. Figure 4d demonstrated that 1A markedly reduced IOP by 45% (of control), as compared with xalatan (prostaglandin analogue), trusopt (carbonic anhydrase inhibitor), or pilocarpine (cholinergic agonist), exhibiting IOP reduction by 20, 17, and 30%, respectively. This highly effective hypotensive capability of 1A makes it a potential alternative to currently available treatments.
Compound 1A reduced IOP in acute glaucomatous rabbit models
Effect of 1A treatment on IOP in acute ocular hypertensive rabbit model induced by sodium hyaluronate
We have further assessed the efficacy of 1A to reduce IOP in an established acute ocular experimental glaucoma model induced by an intraocular injection of 2.3% sodium hyaluronate in NZW rabbits [14]. IOP measurements revealed a marked level of hypertension, 2 h post 2.3% hyaluronate injection, compared to the baseline measurements and the respective contralateral untreated eyes (Fig. 5). At this time point of 2 h, 1A (100 μM) was locally administered to rabbit eyes treated by hyaluronate, and the IOP levels were examined up to 24 h. 1A reduced IOP levels by 40% in sodium hyaluronate-treated rabbits vs. vehicle-treated animals at 4 h from beginning of the study (Fig. 5; 4 h 293 ± 52 vs. 495 ± 42%, respectively; *P < 0.01).
Fig. 5.

Effect of 1A on IOP in acute glaucomatous rabbit model. Effect of 1A treatment on IOP in acute ocular hypertensive model induced by sodium hyaluronate in NZW rabbits. NZW rabbits were subjected to unilateral injection of 2.3% sodium hyaluronate into the anterior chamber. Animals were topically administered with 1A (100 μM), starting at 2 h following sodium hyaluronate injection. Values represent the mean ± S.E.M (n = 4) of three independent experiments and expressed as percentage of respective control. *P < 0.05 vs. hyaluronic acid + saline. #P < 0.05 1A vs. vehicle. Two-way ANOVA test with Bonferroni post tests
Effect of 1A early- and late-treatment in chronic ocular hypertensive model induced by 5% phenol in NWZ rabbits
In order to investigate the effect of 1A in chronic hypertensive condition, we have used rabbits treated with subconjunctival injections of 5% phenol. Under the condition of 5% phenol, rabbits treated with saline exerted significantly high levels of IOP from day 3 of the beginning of the experiment (day 10, 149 ± 18.6%; P < 0.01), compared to the basal levels in control eyes (Fig. 6a). Other phenol-treated rabbits were topically treated with hypotensive compounds, 1A (100 μM) and timolol (5%) 1 day after phenol administration. Both drugs have significantly attenuated IOP elevation and markedly reduced IOP by ~40% from day 2 until end of the experiment (day 9, 105 ± 7.6 and 102 ± 2.7%, respectively; P < 0.01), compared with vehicle-treated eyes-induced by phenol (147 ± 23.3%) (Fig. 6a).
Fig. 6.

Effect of 1A pre- and post-treatment on chronic ocular hypertensive glaucomatous model induced by 5% phenol in NZW rabbits. Rabbits were subjected to one subconjunctival injection of 5% phenol in the right eye. a Animals were topically administered with 1A (100 μM), 0.5% timolol or vehicle (control), performed throughout the entire observation period, starting on day 1 pre-phenol administration period. b Animals were topically administered with 1A (100 μM), 0.5% timolol, or vehicle (control), performed throughout the entire observation period, starting on day 7 post-phenol administration period. Values represent the mean ± S.E.M (n = 6–7) and expressed as percentage of respective control. *P < 0.05 vs. vehicle-treated right eyes; **P < 0.01 vs. vehicle-treated right eyes two-way ANOVA test with Bonferroni post-tests
In addition, we have investigated the effect of post-treatment of 1A (100 μM) and timolol (5%) in phenol-induced rabbits. At day 7 of the experiment when IOP levels increased by 40% above the basal levels of control eyes (143.9 ± 5.6%), rabbits were randomly assigned to the treatment of 1A and timolol (Fig. 6b). Both drugs have significantly attenuated IOP elevation and markedly reduced IOP by ~20% from day 8 until end of the experiment (day 11, 120.5 ± 5.0 and 126.3 ± 9.9%, respectively; P < 0.01), compared with vehicle-treated eyes-induced by phenol (139.6 ± 5.6%) (Fig. 6b).
Elucidation of the origin of the high potency of 5-OMe-UDP(α-B), 1A
The remarkably high potency and selectivity of 1A at hP2Y6-R (EC50 8 nM), [11] resulted in a highly significant reduction of IOP by 1A of both glaucomatous and normotensive animals. This high potency of 1A, related to P2Y6-R activation, prompted us to explore the origin of activity of 1A at the molecular level. Specifically, we attempted to elucidate the binding-mode of 1A at hP2Y6-R as compared to a series of related UDP analogues, 2–8 (Fig. 1).
Construction of hP2Y6-R homology model
Current approaches used for studying the structure and function of P2Y-Rs include mutational analysis of amino acid residues [27–30] and molecular modeling of these receptors and their ligand complexes [31–35]. Progress has been made in the field of GPCR crystallography, and to date, medium- to high-resolution crystal structures have been solved [16, 17]. In the current work, we constructed a 3-D model of the hP2Y6-R using homology modeling [36, 37], and the crystal structures of hP2Y1-R and hP2Y12-R as templates (see Supporting Information for details).
Analysis of the hP2Y6-R binding-site and the recognition mode of P2Y6-R agonists
We performed docking studies using the protocol described in the “Methods” section and in the Supporting Information. Our docking results suggest that the experimental EC50 values of hP2Y6-R agonists 1A and 2–8 correlate well with a combination of desolvation and docking energies of these agonists, but not with either energetic or desolvation term alone (Fig. 7). This is in agreement with our earlier docking studies of P2Y1-R [38].
Fig. 7.

Experimental and predicted EC50 values: a predicted EC50 values using solvation energy; b predicted EC50 values using ligand CDOCKER interaction energy with the P2Y6 receptor; c predicted EC50 values using both ligand CDOCKER interaction energy with the P2Y6 receptor and solvation energy
Based on the hP2Y6-R model and docking results, we propose that the phosphate chain of the endogenous hP2Y6-R agonist, UDP, 2, is tightly held by the three cationic residues (Arg103, Arg284, and Lys25) and a tyrosine residue (Tyr262) (Fig. 8). The 2′-hydroxyl group of the Southern ribose conformer of UDP is H-bonded to Tyr107 and 3′-hydroxyl group is H-bonded to Lys284, and the uracil base interacts with Arg103 and the backbone of Cys177 via two-oxygen atom. Based on docking the more active P2Y6-R agonist 5-OMe-UDP, 3, we suggest that there are additional interactions of the base 5-OMe-substituent with hydrophobic binding site residues (Met190, Met194, and Phe104). Specifically, Met194, which faces the C5-methoxy group, may be involved in hydrophobic interactions with the methyl group of the C5-methoxy substitution. Investigation of the molecular recognition of the Pα-borano group bearing 5-OMe-UDP-α-B analogues, 1A and 1B, by hP2Y6-R revealed that the active isomer, 1A (Rp isomer), [11] forms hydrophobic intra-molecular interaction between Pα-BH3 and the methyl group of the C5 substitution. This intramolecular interaction positions 1A in a most favorable mode inside the binding pocket. In this position, the nucleotide phosphate chain is involved in additional H-bonding interactions with the polar side chain Thr175, which do not occur with 5-OMe-UDP. We speculate that these favorable interactions are not possible for the inactive Sp isomer. Interestingly for the Sp isomer, our docking results suggest no H-bonding interactions with Thr175 and weakening of H-bonding with the Val176 backbone (see Supporting Information for details). The current model is not sufficiently accurate to categorically discriminate between the isomers, and future mutational studies of binding site residues or a crystal structure will be necessary to refine the current hP2Y6-R model.
Fig. 8.
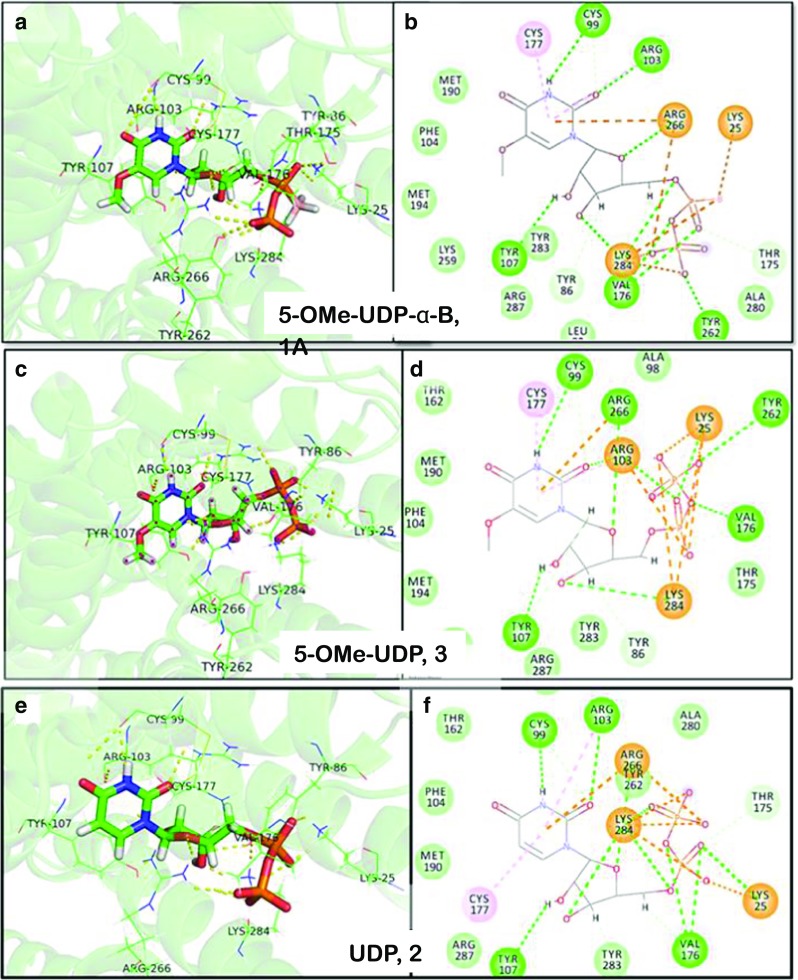
3D and 2D description of the binding site in the hP2Y6-R model with bound 5-OMe-UDP (α-B), 1A (a, b, respectively), 5-OMe-UDP, 3 (c, d, respectively), and UDP, 2 (e, f, respectively)
Discussion
It is well accepted that an increase in IOP is a major factor for the development of glaucoma, a pathology that leads to a progressive optic neuropathy which may result in the loss of vision [39]. Most drugs for the treatment of glaucoma either decrease the production aqueous humor or improve its outflow, thus lower IOP and reduce the damage of the optic nerve [40].
Currently used drugs for the treatment of glaucoma include the following agents, and their combinations, for reduction of IOP. Prostaglandin analogs (e.g., xalatan) are first line drugs. They lower IOPs in glaucoma patients, providing potent IOP control throughout the day, by increasing the outflow of fluid from the eye. The most common adverse effects associated with prostaglandin analogs are conjunctival hyperemia and eyelash growth [41].
Topical β-blockers, such as timolol, are widely used IOP-reducing agents [42]. These agents decrease the production of aqueous humor by blocking the β-adrenergic receptors in the ciliary body [43, 44]. Although applied locally, topical β-blockers are partially absorbed into the systemic circulation via the conjunctiva or the nasolacrimal system [45]. Contrary to their beneficial ocular effect, β-blockers may induce severe systemic adverse effects by blocking the β1-adrenoceptors of the heart, and by blocking the bronchial smooth muscle’s β2-adrenergic receptors [46]. For instance, timolol may cause pulmonary side effects, such as bronchospasm and asthma exacerbation [47, 48] by blocking the bronchial smooth muscle’s β2-adrenergic receptors [49]. Indeed, glaucomatous patients with obstructive pulmonary disease which were treated with topical β-blockers, mostly non-cardioselective (timolol), were more prone to be hospitalized or visit the emergency room while on the medication. [42]
Furthermore, timolol may cause bradycardia, arrhythmia, congestive heart failure, and syncope [50–53]. Hence, timolol should not be used also in patients with myasthenia gravis [54, 55] or those with diabetic mellitus with hypoglycemic attacks [56].
Carbonic anhydrase inhibitors (e.g., trusopt) are also used for reduction of IOP by decreasing the production of intraocular fluid, while α-receptor agonists (e.g., brimonidine) both decrease production of aqueous humor and increase its drainage. Combination therapy is suggested for patients who need more than one type of drug.
Currently used drugs for the treatment of glaucoma target prostaglandin receptors, β2-adrenergic receptor, α-receptor, and carbonic anhydrase. Although, synthetic agonists of various P2Y receptors have promise as agents for the treatment of ocular diseases, including glaucoma [1], currently no drug on the market targets P2Y receptors for the regulation of IOP.
The limitations of current drugs for the treatment of glaucoma, on the one hand, and the involvement of P2Y-Rs in aqueous humor drainage, on the other hand, encouraged us to explore the application of this new mechanism for the reduction of IOP. Specifically, our finding of a most potent, stable, and selective P2Y6R agonist, 5-OMe-UDP(α-B), Rp isomer, 1A [11], prompted us to explore the potential of activation of P2Y6R, present in the trabecular meshwork, for the enhancement of aqueous humor drainage, and consequently IOP reduction.
The trabecular meshwork, a tissue in the eye that is responsible for draining the aqueous humor, expresses P2Y-Rs which are activated by extracellular nucleotides. Specifically, P2Y1, P2Y2, P2Y4, and P2Y6 receptors play a role in IOP regulation [1, 4, 7]. P2Y6 receptor was shown to be expressed in the retina [57, 58]. Decrease in the expression levels of P2Y6-R in glaucoma was suggested using in-vitro ocular hypertension model (i.e., glaucoma model) [59]. Hence, we assumed that activation of P2Y6R by compound 1A may have a beneficial effect on reducing IOP in glaucoma. Indeed, previous reports indicated that UDP, P2Y6R endogenous agonist, reduced rabbit’s IOP by 17%. Likewise, P2YR agonists, including P2Y6R agonists, lower IOP [10, 60].
Specifically, we found here that compound 1A significantly reduced IOP in normotensive rabbits. A single dose of 100 μM of 1A was able to significantly reduce IOP (44%) at 2 h after the instillation of the compound and the effect lasted 4 h. Importantly, 1A markedly reduced IOP as compared to the endogenous P2Y6-R agonist, UDP, and currently used drugs—xalatan, trusopt, and pilocarpine (by 44% vs. 15, 20, 17, and 30%, respectively).
Encouraged by these promising results, we explored the effect of 1A on IOP reduction in animal glaucoma models as compared to timolol. In the sodium hyaluronate acute ocular hypertensive rabbit model and phenol-induced rabbits (pre-treatment), 1A reduced IOP by ~40%, proving to be equi-efficacious to timolol. Further work is necessary to determine the retinal ganglion cell (RGC) integrity at the completion of the study by histological studies.
Next, to get a preliminary indication of the safety of 1A, as compared to that of timolol, we evaluated their effects in several ocular cell lines. Notably, 1A was significantly less toxic in ocular Y-79 retinoblastoma and SIRC cells than timolol. Similar nontoxic effects of 1A were observed in NPE cells. Importantly, 1A was significantly less toxic than trusopt and timolol, also in liver hepatoma Huh7 cells. Although the P2Y6-receptor has a widespread distribution including heart, blood vessels, and brain [61], pre-clinical safety assessments demonstrated that instillation of 1A at 500 mM into rabbit eyes did not exert any eye irritation or noticeable clinical signs or toxic/mutagenic effects (GlaucoPharm company annual report, 2016; data not shown).
To decipher the origin of the beneficial activity of 1A at the molecular level, we constructed a homology model of hP2Y6-R using recently published high-resolution crystal structures of P2Y1/12-Rs as templates. Docking studies allowed analysis of the molecular recognition of 5-OMe-UDP-α-B isomers, 1A and 1B, by hP2Y6-R. We revealed that the active isomer, 1A (Rp isomer), forms hydrophobic intra-molecular interaction between Pα-BH3 and the methyl group of the C5-OMe substitution. This intramolecular interaction positions 1A in a most favorable site inside the binding pocket. In this position, the nucleotide phosphate chain is involved in additional H-bonding interactions with polar side chain Thr175, which do not occur with P2Y6-R agonist, 5-OMe-UDP.
We concluded that 1A is a safe drug candidate which markedly reduced IOP as compared to the endogenous P2Y6-R agonist, UDP. Compound 1A was equi-efficacious to timolol in animal glaucoma models (hyaloronate and phenol-induced glaucoma). Yet, the severe limitations of timolol, which prevent its use for treatment of glaucomatous patients with obstructive pulmonary disease, diabetes, and heart disease, imply that a selective P2Y6-R agonist may be a promising therapeutic alternative to timolol.
Electronic supplementary material
Supporting Information includes: Homology modeling description; binding-site description and ligand recognition mode; binding interactions of hP2Y6-R and agonists; uracil moiety binding mode; and binding modes of 5-OMe-UDP(α-B) diastereoisomers. 3 Tables and 11 Figures are submitted as part of the Supporting Information. (DOCX 31124 kb)
Acknowledgements
The authors gratefully acknowledge the support of the Office of the Israeli Chief Scientist, and Youdim Pharmaceuticals Ltd. Israel.
Abbreviations
- IOP
Intraocular pressure
- MTT
[3-(4, 5-dimethyl-thiazol-2-yl) 2,5-diphenyl-tetrazolium bromide]
- UDP
Uridine 5′-diphosphate
- hP2Y6-R
Human P2Y6 receptor
Conflict of interest
Authors Orly Weinreb and Tali Fishman-Jacob received research grants from the Office of the Israeli Chief Scientist, and Youdim Pharmaceuticals Ltd. Israel.
Ethical approval
This study does not contain any studies with human participants.
Footnotes
₤ Patent—US Application No. 61/344,972 of 01/12/2010 and US Application No. 61/467,108 of 24/03/2011 International Filing: e:01/12/2011
Contributor Information
Dan T. Major, Phone: 972-3-5317392, Email: majort@biu.ac.il
Orly Weinreb, Phone: +972-4-9090007, Email: orly.weinreb@youdim.com.
Bilha Fischer, Phone: 972-3-5318303, Email: bilha.fischer@biu.ac.il.
References
- 1.Jacobson KA, Civan MM. Ocular purine receptors as drug targets in the eye. J Ocul Pharmacol Ther. 2016;32(8):534–547. doi: 10.1089/jop.2016.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guzmán-Aranguez A, Crooke A, Peral A, Hoyle CH, Pintor J. Dinucleoside polyphosphates in the eye: from physiology to therapeutics. Prog Retin Eye Res. 2007;26(6):674–687. doi: 10.1016/j.preteyeres.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Pintor J. Adenine nucleotides and dinucleotides as new substances for the treatment of ocular hypertension and glaucoma. Curr Opin Investig Drugs. 2005;6(1):76–80. [PubMed] [Google Scholar]
- 4.Crooke A, Guzman-Aranguez A, Carracedo G, de Lara MJP, Pintor J. Understanding the presence and roles of Ap4A (Diadenosine tetraphosphate) in the eye. J Ocul Pharmacol Ther. 2017;33(6):426–434. doi: 10.1089/jop.2016.0146. [DOI] [PubMed] [Google Scholar]
- 5.Pintor J, Peláez T, Peral A. Adenosine tetraphosphate, Ap4, a physiological regulator of intraocular pressure in normotensive rabbit eyes. J Pharmacol Exp Ther. 2004;308(2):468–473. doi: 10.1124/jpet.103.058669. [DOI] [PubMed] [Google Scholar]
- 6.Pintor J, Peral A, Peláez T, Martín S, Hoyle CH. Presence of diadenosine polyphosphates in the aqueous humor: their effect on intraocular pressure. J Pharmacol Exp Ther. 2003;304(1):342–348. doi: 10.1124/jpet.102.041368. [DOI] [PubMed] [Google Scholar]
- 7.Soto D, Pintor J, Peral A, Gual A, Gasull X. Effects of dinucleoside polyphosphates on trabecular meshwork cells and aqueous humor outflow facility. J Pharmacol Exp Ther. 2005;314(3):1042–1051. doi: 10.1124/jpet.105.085274. [DOI] [PubMed] [Google Scholar]
- 8.El-Tayeb A, Qi A, Müller CE. Synthesis and structure− activity relationships of uracil nucleotide derivatives and analogues as agonists at human P2Y2, P2Y4, and P2Y6 receptors. J Med Chem. 2006;49(24):7076–7087. doi: 10.1021/jm060848j. [DOI] [PubMed] [Google Scholar]
- 9.Shinozaki Y, Kashiwagi K, Namekata K, Takeda A, Ohno N, Robaye B, Harada T, Iwata T, Koizumi S (2017) Purinergic dysregulation causes hypertensive glaucoma–like optic neuropathy. JCI insight 2(19) [DOI] [PMC free article] [PubMed]
- 10.Markovskaya A, Crooke A, Guzmán-Aranguez AI, Peral A, Ziganshin AU, Pintor J. Hypotensive effect of UDP on intraocular pressure in rabbits. Eur J Pharmacol. 2008;579(1):93–97. doi: 10.1016/j.ejphar.2007.10.040. [DOI] [PubMed] [Google Scholar]
- 11.Ginsburg-Shmuel T, Haas M, Grbic D, Arguin G, Nadel Y, Gendron F-P, Reiser G, Fischer B. UDP made a highly promising stable, potent, and selective P2Y6-receptor agonist upon introduction of a boranophosphate moiety. Bioorg Med Chem. 2012;20(18):5483–5495. doi: 10.1016/j.bmc.2012.07.042. [DOI] [PubMed] [Google Scholar]
- 12.Major DT, Fischer B. Molecular recognition in purinergic receptors. 1. A comprehensive computational study of the h-P2Y1-receptor. J Med Chem. 2004;47(18):4391–4404. doi: 10.1021/jm049772m. [DOI] [PubMed] [Google Scholar]
- 13.Ecke D, Tulapurkar M, Nahum V, Fischer B, Reiser G. Opposite diastereoselective activation of P2Y1 and P2Y11 nucleotide receptors by adenosine 5′-O-(α-boranotriphosphate) analogues. Br J Pharmacol. 2006;149(4):416–423. doi: 10.1038/sj.bjp.0706887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kivalo M, Hollmen T, Sukura A, Mononen T. The effect of intraoperative injection of hyaluronan under a one-piece glaucoma filtration implant in the rabbit eye. Acta Vet Scand. 1997;38(3):235–242. doi: 10.1186/BF03548486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monem AS, Ali FM, Ismail MW. Prolonged effect of liposomes encapsulating pilocarpine HCl in normal and glaucomatous rabbits. Int J Pharm. 2000;198(1):29–38. doi: 10.1016/S0378-5173(99)00348-8. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Zhang K, Gao Z-G, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Muller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q. Agonist-bound structure of the human P2Y12 receptor. Nature. 2014;509(7498):119–122. doi: 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang D, Gao ZG, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, Han GW, Liu H, Cherezov V, Katritch V, Jiang H, Stevens RC, Jacobson KA, Zhao Q, Wu B. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature. 2015;520(7547):317–321. doi: 10.1038/nature14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Liu J, Fang X, Nie G. Effects of surface piezoelectricity and nonlocal scale on wave propagation in piezoelectric nanoplates. European J Mechanics-A/Solids. 2014;46:22–29. doi: 10.1016/j.euromechsol.2014.01.005. [DOI] [Google Scholar]
- 19.Wang Q, He J, Wu D, Wang J, Yan J, Li H. Interaction of α-cyperone with human serum albumin: determination of the binding site by using discovery studio and via spectroscopic methods. J Lumin. 2015;164:81–85. doi: 10.1016/j.jlumin.2015.03.025. [DOI] [Google Scholar]
- 20.Šali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 21.Bowie JU, Lüthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 22.Papadatos G, Overington JP. The ChEMBL database: a taster for medicinal chemists. Future. 2014;6(4):361–364. doi: 10.4155/fmc.14.8. [DOI] [PubMed] [Google Scholar]
- 23.Wu G, Robertson DH, Brooks CL, Vieth M. Detailed analysis of grid-based molecular docking: a case study of CDOCKER—A CHARMm-based MD docking algorithm. J Comput Chem. 2003;24(13):1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
- 24.Tjong Kim Sang EF, De Meulder F Introduction to the CoNLL-2003 shared task: language-independent named entity recognition. In: Proceedings of the seventh conference on Natural language learning at HLT-NAACL 2003-Volume 4, 2003. Association for Computational Linguistics, pp 142–147
- 25.Brooks BR, Brooks CL, MacKerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sitkoff D, Sharp KA, Honig B. Accurate calculation of hydration free energies using macroscopic solvent models. J Phys Chem. 1994;98(7):1978–1988. doi: 10.1021/j100058a043. [DOI] [Google Scholar]
- 27.Ding Z, Tuluc F, Bandivadekar KR, Zhang L, Jin J, Kunapuli SP. Arg333 and Arg334 in the COOH terminus of the human P2Y1 receptor are crucial for Gq coupling. Am J Physiol Cell Physiol. 2005;288:C559–C567. doi: 10.1152/ajpcell.00401.2004. [DOI] [PubMed] [Google Scholar]
- 28.Hoffmann C, Moro S, Nicholas RA, Harden TK, Jacobson KA. The role of amino acids in extracellular loops of the human P2Y1 receptor in surface expression and activation processes. J Biol Chem. 1999;274:14639–14647. doi: 10.1074/jbc.274.21.14639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Q, Guo D, Lee BX, Van Rhee AM, Kim YC, Nicholas RA, Schachter JB, Harden TK, Jacobson KA. A mutational analysis of residues essential for ligand recognition at the human P2Y1 receptor. Mol Pharmacol. 1997;52:499–507. doi: 10.1124/mol.52.3.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moro S, Guo D, Camaioni E, Boyer JL, Harden TK, Jacobson KA. Human P2Y1 receptor: molecular modeling and site-directed mutagenesis as tools to identify agonist and antagonist recognition sites. J Med Chem. 1998;41:1456–1466. doi: 10.1021/jm970684u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa MJ, Marquez VE, Harden TK, Jacobson KA. Human P2Y6 receptor: molecular modeling leads to the rational design of a novel agonist based on a unique conformational preference. J Med Chem. 2005;48(26):8108–8111. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moro S, Jacobson KA. Molecular modeling as a tool to investigate molecular recognition in P2Y receptors. Curr Pharm Des. 2002;8:2401–2413. doi: 10.2174/1381612023392892. [DOI] [PubMed] [Google Scholar]
- 33.Costanzi S, Mamedova L, Gao ZG, Jacobson KA. Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J Med Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hillmann P, Ko G-Y, Spinrath A, Raulf A, von Kügelgen I, Wolff SC, Nicholas RA, Kostenis E, Höltje H-D, Müller CE. Key determinants of nucleotide-activated G protein-coupled P2Y2 receptor function revealed by chemical and pharmacological experiments, mutagenesis and homology modeling. J Med Chem. 2009;52(9):2762–2775. doi: 10.1021/jm801442p. [DOI] [PubMed] [Google Scholar]
- 35.Major DT, Fischer B. Molecular recognition in purinergic receptors. 1. A comprehensive computational study of the h-P2Y1-receptor. J Med Chem. 2004;47:4391–4404. doi: 10.1021/jm049772m. [DOI] [PubMed] [Google Scholar]
- 36.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 37.Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2014;1137:1–15. doi: 10.1007/978-1-4939-0366-5_1. [DOI] [PubMed] [Google Scholar]
- 38.Azran S, Danino O, Förster D, Kenigsberg S, Reiser G, Dixit M, Singh V, Major DT, Fischer B. Identification of highly promising antioxidants/neuroprotectants based on nucleoside 5′-phosphorothioate scaffold. Synthesis, activity, and mechanisms of action. J Med Chem. 2015;58(21):8427–8443. doi: 10.1021/acs.jmedchem.5b00575. [DOI] [PubMed] [Google Scholar]
- 39.Davson H (1993) The aqueous humour and the intraocular pressure, physiology of the eye. 5th Edn. Pergamon Press, New York
- 40.Gupta S, Galpalli Niranjan D, Agrawal S, Srivastava S, Saxena R. Recent advances in pharmacotherapy of glaucoma. Indian J Pharmacology. 2008;40(5):197–208. doi: 10.4103/0253-7613.44151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Higginbotham EJ, Schuman JS, Goldberg I, Gross RL, VanDenburgh AM, Chen K, Whitcup SM. One-year, randomized study comparing bimatoprost and timolol in glaucoma and ocular hypertension. Arch Ophthalmol. 2002;120(10):1286–1293. doi: 10.1001/archopht.120.10.1286. [DOI] [PubMed] [Google Scholar]
- 42.Kaiserman I, Fendyur A, Vinker S. Topical beta blockers in asthmatic patients–is it safe? Curr Eye Res. 2009;34(7):517–522. doi: 10.1080/02713680902989337. [DOI] [PubMed] [Google Scholar]
- 43.Frishman WH, Fuksbrumer MS, Tannenbaum M. Topical ophthalmic β-adrenergic blockade for the treatment of glaucoma and ocular hypertension. J Clin Pharmacol. 1994;34(8):795–803. doi: 10.1002/j.1552-4604.1994.tb02042.x. [DOI] [PubMed] [Google Scholar]
- 44.Brubaker R. Flow of aqueous humor in humans [the Friedenwald lecture] Invest Ophthalmol Vis Sci. 1991;32(13):3145–3166. [PubMed] [Google Scholar]
- 45.Tan AY, LeVatte TL, Archibald ML, Tremblay F, Kelly ME, Chauhan BC. Timolol concentrations in rat ocular tissues and plasma after topical and intraperitoneal dosing. J Glaucoma. 2002;11(2):134–142. doi: 10.1097/00061198-200204000-00009. [DOI] [PubMed] [Google Scholar]
- 46.Hoyng PF, van Beek LM. Pharmacological therapy for glaucoma. Drugs. 2000;59(3):411–434. doi: 10.2165/00003495-200059030-00003. [DOI] [PubMed] [Google Scholar]
- 47.Leuppi JD, Schnyder P, Hartmann K, Reinhart WH, Kuhn M. Drug-induced bronchospasm: analysis of 187 spontaneously reported cases. Respiration. 2001;68(4):345–351. doi: 10.1159/000050525. [DOI] [PubMed] [Google Scholar]
- 48.Kirwan JF, Nightingale JA, Bunce C, Wormald R. β blockers for glaucoma and excess risk of airways obstruction: population based cohort study. BMJ. 2002;325(7377):1396–1397. doi: 10.1136/bmj.325.7377.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carstairs J, Nimmo A, Barnes PJ (1985) Autoradiographic visualization of beta-adrenoceptor subtypes in human lung 1–3. Am Rev Respir Dis 132 (3):541–547 [DOI] [PubMed]
- 50.McMahon CD, Shaffer RN, Hoskins HD, Hetherington J. Adverse effects experienced by patients taking timolol. Am J Ophthalmol. 1979;88(4):736–738. doi: 10.1016/0002-9394(79)90674-3. [DOI] [PubMed] [Google Scholar]
- 51.van Buskirk EM. Adverse reactions from timolol administration. Ophthalmology. 1980;87(5):447–450. doi: 10.1016/S0161-6420(80)35215-9. [DOI] [PubMed] [Google Scholar]
- 52.Fraunfelder FT. Interim report: national registry of possible drug-induced ocular side effects. Ophthalmology. 1979;86(1):126–130. doi: 10.1016/S0161-6420(79)35540-3. [DOI] [PubMed] [Google Scholar]
- 53.Dickstein K, AARSLAND T. Comparison of the effects of aqueous and gellan ophthalmic timolol on peak exercise performance in middle-aged men. Am J Ophthalmol. 1996;121(4):367–371. doi: 10.1016/S0002-9394(14)70432-5. [DOI] [PubMed] [Google Scholar]
- 54.Shaivitz SA. Timolol and myasthenia gravis. JAMA. 1979;242(15):1611–1612. doi: 10.1001/jama.1979.03300150017011. [DOI] [PubMed] [Google Scholar]
- 55.Coppeto JR. Timolol-associated myasthenia gravis. Am J Ophthalmol. 1984;98(2):244–245. doi: 10.1016/0002-9394(87)90366-7. [DOI] [PubMed] [Google Scholar]
- 56.Velde TM, Kaiser FE. Ophthalmic timolol treatment causing altered hypoglycemic response in a diabetic patient. Arch Intern Med. 1983;143(8):1627–1627. doi: 10.1001/archinte.1983.00350080149037. [DOI] [PubMed] [Google Scholar]
- 57.Zhang P, Yang X, Zhong Y. Cellular localization of P2Y6 receptor in rat retina. Neuroscience. 2012;220:62–69. doi: 10.1016/j.neuroscience.2012.06.032. [DOI] [PubMed] [Google Scholar]
- 58.Fries JE, Wheeler-Schilling TH, Kohler K, Guenther E. Distribution of metabotropic P2Y receptors in the rat retina: a single-cell RT-PCR study. Mol Brain Res. 2004;130(1–2):1–6. doi: 10.1016/j.molbrainres.2004.06.041. [DOI] [PubMed] [Google Scholar]
- 59.Taguchi M, Shinozaki Y, Kashiwagi K, Shigetomi E, Robaye B, Koizumi S. Müller cell-mediated neurite outgrowth of the retinal ganglion cells via P2Y6 receptor signals. J Neurochem. 2016;136(4):741–751. doi: 10.1111/jnc.13427. [DOI] [PubMed] [Google Scholar]
- 60.Haas M, Ginsburg-Shmuel T, Fischer B, Reiser G. 5-OMe-uridine-5′-O-(α-boranodiphosphate), a novel nucleotide derivative highly active at the human P2Y 6 receptor protects against death-receptor mediated glial apoptosis. Neurosci Lett. 2014;578:80–84. doi: 10.1016/j.neulet.2014.06.030. [DOI] [PubMed] [Google Scholar]
- 61.von Kügelgen I. Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol Ther. 2006;110(3):415–432. doi: 10.1016/j.pharmthera.2005.08.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information includes: Homology modeling description; binding-site description and ligand recognition mode; binding interactions of hP2Y6-R and agonists; uracil moiety binding mode; and binding modes of 5-OMe-UDP(α-B) diastereoisomers. 3 Tables and 11 Figures are submitted as part of the Supporting Information. (DOCX 31124 kb)