

Abstract
Despite the availability of potent chemotherapy regimens, such as 5-fluorouracil, folinic acid, irinotecan, and oxaliplatin (FOLFIRINOX) and nab-paclitaxel plus gemcitabine, treatment outcomes in metastatic pancreatic cancer (PC) remain unsatisfactory. The presence of an abundant fibrous stroma in PC is considered a crucial factor for its unfavorable condition. Apparently, stroma acts as a physical barrier to restrict intratumoral cytotoxic drug penetration and creates a hypoxic environment that reduces the efficacy of radiotherapy. In addition, stroma plays a vital supportive role in the development and progression of PC, which has prompted researchers to assess the potential benefits of agents targeting several cellular (e.g., stellate cells) and acellular (e.g., hyaluronan) elements of the stroma. This study aims to briefly review the primary structural properties of PC stroma and its interaction with cancer cells and summarize the current status of anti-stromal therapies in the management of metastatic PC.
Keywords: Pancreatic cancer, Stroma, Stellate cells, Hyaluronan, Secreted protein acidic and rich in cysteine
Core tip: The primary characteristic of pancreatic adenocarcinoma is the presence of an extensive desmoplastic stroma around neoplastic cells. In this study, we aim to briefly review the primary structural properties of pancreatic cancer (PC) stroma and its interaction with cancer cells and summarize the current status of anti-stromal therapies in the management of metastatic PC.
INTRODUCTION
The primary characteristic of pancreatic adenocarcinoma is the presence of an extensive desmoplastic stroma around neoplastic cells in both primary and metastatic lesions[1]. The structural organization of stroma is not entirely different from those in other solid tumors; in fact, it is a mixture of cellular and acellular [extracellular matrix (ECM) proteins] elements[2]. However, in contrast to several other solid tumors, stromal elements can occupy ≥ 80% of the total tumor volume in most pancreatic cancer (PC) cases[3].
Abundant accumulation of fibrous proteins, primarily collagen (types I and III), fibronectin, and secreted protein acidic and rich in cysteine (SPARC) in the ECM offers exceptional mechanical properties of pancreatic adenocarcinoma stroma, including considerably enhanced stiffness and reduced elasticity[4]. In addition, increased deposition of another crucial ECM element hyaluronan (HA) in the tumor stroma creates substantial swelling stress, which progressively increases the interstitial fluid pressure[5]. The occurrence of this condition besides increased tissue stiffness compresses intratumoral blood vessels, resulting in tumor hypoperfusion and hypoxia. Reportedly, hypoperfusion drastically reduces intratumoral delivery of chemotherapy drugs and, consequently, their efficacy[6,7]. Hypoxia confers a survival advantage for neoplastic cells and potentiates their invasion, stemness, and metastatic capacity primarily through the hypoxia-inducible factor-1α–mediated hepatocyte growth factor/c-Met pathway activation[8,9]. Moreover, hypoxia compromises the efficacy of radiotherapy.
In PC, ECM proteins are primarily produced by a distinct type of stromal cells called activated pancreatic stellate cells (PSCs). PSCs phenotypically resemble myofibroblasts and exhibit the α-smooth muscle actin expression. However, in contrast to myofibroblasts, PSCs are positively stained for selective markers such as desmin and glial fibrillary acidic protein. They also demonstrate increased proliferation and migration ability relative to myofibroblasts, and can produce large amounts of collagen and other ECM proteins[10,11]. PSCs possess the adequate capacity to interact with cancer cells and other stromal cells (i.e., immune cells, inflammatory cells, and endothelial cells) to extend stroma and promote cancer progression. Thus, both cellular (especially PSCs) and acellular (especially HA) components of PC stroma have been held accountable for unsatisfactory treatment outcomes in patients with PC. This condition has encouraged PC researchers to elucidate the potential beneficial effects of stroma disrupting agents alone or in combination with standard chemotherapy in the treatment of PC (Figure 1).
Figure 1.
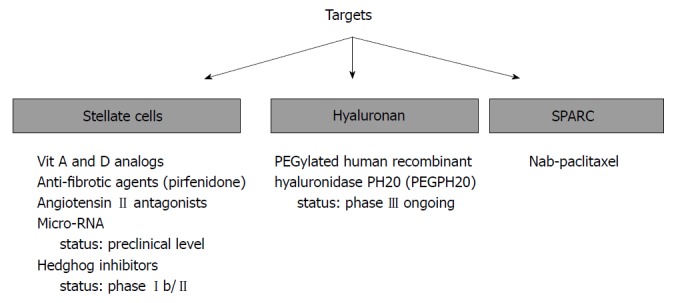
Stroma-targeting treatment strategies in pancreatic cancer. SPARC: Secreted protein acidic and rich in cysteine.
ROLES OF PSCS IN PC PROGRESSION
Despite being debatable, activated PSCs are deliberated to originate from their inactive (quiescent) forms that are primarily found in the periacinar space of the exocrine pancreas[12]. Reportedly, inflammatory (i.e., interleukin-1 and interleukin-6, and tumor necrosis factor-α) and mitogenic (i.e., transforming growth factor and platelet-derived growth factor) cytokines secreted by cancer cells are accountable for the PSC activation[13-18]. Perhaps, some intracellular pathways, including p38 mitogen-activated protein kinase, RhoA/Rho kinase, and cyclooxygenase-2, could play a vital role in this process[18-22].
In pancreatic carcinogenesis, activated PSCs seemingly serve two primary functions, to produce ECM molecules and regulate the formation of desmoplastic reaction and enable cancer cell proliferation and invasion[13]. The direct cell-to-cell contact between PSCs and PC cells has been demonstrated to result in the activation of the Notch signaling pathway in both cell types[23]. The Notch signaling plays a vital role in the proliferation, migration, differentiation, and apoptosis of cancer cells[24,25]. Apparently, PSCs can activate the mitogen-activated protein kinase and Akt pathways in tumor cells, causing enhanced tumor growth and metastasis[26]. PSCs secrete matrix metalloproteinase-2 into the tumor microenvironment in response to extracellular matrix metalloproteinase inducer (EMMPRIN) secreted by cancer cells to facilitate the tissue invasion and metastasis[27]. In addition, PSCs can accompany cancer cells to distant sites, where they stimulate angiogenesis, cancer cell seeding, survival, and proliferation and, thus, facilitate the metastasis formation[28]. Furthermore, PSCs can indirectly protect cancer cells from the immune system attack. A study demonstrated that PSCs secreted CXCL12 chemokine and sequestered CD8+ T cells to reduce their accumulation in the juxtatumoral compartments[29]. Mace et al[30] suggested that PSC-derived cytokines, such as interleukin-6, cause myeloid-derived suppressor cell expansion in the stroma, thereby indirectly inducing immune cell dysfunction.
Preclinical data indicated that PCSs might enhance stem-cell like phenotypes in PC cells[31]. Indirect co-culture of PSCs with PC cells increased the spheroid-forming capacity of tumor cells, and induced the expression of stem cell-related genes including Nestin, ABCG2 and LIN28[31]. Lonardo et al[32] showed that the secretion of transforming growth factor-β superfamily members Nodal and Activin from PCSs significantly promotes the self-renewal capacity and invasiveness of PC stem cells.
Recent studies have shown that extracellular vesicles (also known as exosomes) derived from PSCs may play a role in the progression of PC[33,34]. Takikawa et al[34] reported that immortalized human PSCs produce exosomes containing numerous microRNAs (miRNAs) that can induce chemokine gene expression in PC cell lines resulted in increased proliferation and migration. Leca et al[35] found that annexin 6A/receptor-related protein 1/thrombospondin-1 complex-containing exosomes released by PSCs could increase PC cell aggressiveness under physiopathologic conditions. In addition, exosomes have been suggested to contribute to chemoresistance of PC cells by promoting reactive oxygen species detoxification and by decreasing gemcitabine-metabolizing enzyme activity[36].
POTENTIAL THERAPEUTIC STRATEGIES TARGETING PSCS
Vitamin D and A analogs
Preclinical studies have reported that the PSC activation can be restricted or reversed by pharmacological interventions leading to substantial modulation of the tumor stroma[37,38]. Apparently, PSCs express higher levels of vitamin D receptors[37]. Sherman et al[37] reported that a potent vitamin D analog calcipotriol treatment decreased the expression of activation and cancer signature genes in cultured PSCs, stimulated lipid droplet formation, and reduced the α-smooth muscle actin expression, signifying their inactivation; this correlated with a decline in stromal inflammation and fibrosis. In addition, the authors compared the efficacy of gemcitabine plus calcipotriol treatment with gemcitabine alone in the KPC model of PC and reported that the combination therapy increased the intratumoral uptake of gemcitabine, reduced tumor volume, and exhibited 57% improvement in animal survival compared with gemcitabine monotherapy. These findings suggested that the tumor stromal modulation (reprogramming) by inactivating PSCs could be a reasonable treatment strategy for PC. Paricalcitol, a synthetic vitamin D analog is currently being tested in combination with conventional chemotherapy or immunotherapy in the treatment of metastatic PC (Table 1).
Table 1.
Summary of existing studies evaluating the efficacy of anti-stromal agents in the treatment of metastatic pancreatic cancer
| Agent | Target | Treatment arm (s) | Type of study | National clinical trial number | Status | Results |
| Paricalcitol | Vitamin D metabolic pathway | Gemcitabine and nab-paclitaxel plus paricalcitol or placebo | Phase I/II | NCT03520790 | Recruiting | |
| Nab-paclitaxel, cisplatin and gemcitabine plus paricalcitol | Phase II | NCT03415854 | Recruiting | |||
| Nivolimumab1, nab-paclitaxel, cisplatin, and gemcitabine plus paricalcitol | Phase II | NCT02754726 | Recruiting | |||
| Pembrolizumab1 plus paricalcitol or placebo | Phase II | NCT03331562 | Recruiting | |||
| All trans retinoic acid | Pancreatic stellate cells | Gemcitabine and nab-paclitaxel plus all trans retinoic acid | Phase I | NCT03307148 | Recruiting | |
| Vismodegib | Hedgehog signaling | Gemcitabine plus vismodegib or placebo | Phase I/II | NCT0106422 | Completed | Vismodegib did not improve ORR, PFS and OS |
| IPI-926 | Hedgehog signaling | FOLFIRINOX plus IPI-926 | Phase I | NCT01383538 | Completed | The combination treatment was safe but IP-926 was not beneficial |
| Gemcitabine plus IPI-926 or placebo | Phase I/II | NCT01130142 | Completed | The combination treatment was well tolerated, and showed promising activity | ||
| PEGPH20 | Hyaluronic acid | Gemcitabine and nab-paclitaxel plus PEGPH20 vs chemotherapy alone | Phase II | NCT01839487 | Completed | PEGPH20 significantly improved PFS, especially in patients having tumors with high-level hyaluronic acid |
| Gemcitabine and nab-paclitaxel plus PEGPH20 or placebo2 | Phase III | NCT02715804 | Recruiting | |||
| Modified FOLFIRINOX plus PEGPH20 vs chemotherapy alone | Phase I/II | NCT01959139 | Closed | PEGPH20 was found to have a detrimental effect on OS |
Nivolimumab and Pembrolizumab: PD-1-targeted T-cell checkpoint inhibitors;
This study included only patients whose tumors had high levels of hyaluronic acid. ORR: Overall response rate; PFS: Progression-free survival; OS: Overall survival; FOLFIRINOX: 5-fluorouracil, irinotecan and, oxaliplatin.
Quiescent PSCs store vitamin A-containing lipid droplets in their cytoplasm, which are lost during the activation process. Research has revealed that restoring vitamin A in PCSs by using vitamin A metabolites could reprogram these cells to a quiescent phase[39]. Jaster et al[40] reported that all-trans-retinoic acid (ATRA) could impede the proliferation and collagen synthesis of PSCs isolated from rat pancreas by hindering the AP-1 activation. Of note, AP-1 is a transcription factor that regulates cell growth, differentiation, and survival. McCarroll et al[41] described that ATRA and 9-cis retinoic acid could avert the activation of cultured activated PSCs by inhibiting the mitogen-activated protein kinase signaling pathway, and decreased collagen I, fibronectin, and laminin expression in these cells. In addition, a study reported that the reduction of Wnt-B-catenin signaling by ATRA in PC cells resulted in slower tumor progression[42]. Furthermore, Chronopoulos et al[43] determined that ATRA could reduce the actomyosin-dependent contractility, mechanosensing, and migration of PSCs in a retinoic acid receptor (RAR)-β–dependent manner. Likewise, Sarper et al[39] also reported similar findings. Overall, reprogramming of PSCs using vitamin A metabolites, such as ATRA or selective RAR-β agonists, in a clinical setting could open new avenues in the treatment of PC (Table 1).
Antifibrotic agents
Kozono et al[44] reported that the antifibrotic anti-inflammatory agent pirfenidone could impede the proliferation, invasiveness, migration, and ECM protein synthesis ability of PSCs in vitro. In mice bearing orthotopically implanted PC and PSCs, pirfenidone was shown to suppress the tumor growth and metastasis formation and displayed a synergistic antitumor effect with gemcitabine. In addition, Suklabaidya et al[45] reported that the effects of pirfenidone could be potentiated when co-administered with antioxidant N-acetyl cysteine. Thus, the potential effects of pirfenidone alone or in combination with N-acetyl cysteine in PC necessitate further assessment in human subjects.
Angiotensin II inhibitors
Previously, preclinical studies have demonstrated that angiotensin II plays a promoting role in the PSC proliferation, which seems to be controlled by induction of the Smad7 expression through a protein kinase C–dependent pathway, resulting in the inhibition of TGF-β1 signaling[46]. On the basis of these findings, several angiotensin II receptor antagonists have been investigated as a potential strategy to reduce PSC-mediated stromal fibrosis. Yamada et al[47] reported that candesartan considerably reduces the PSC proliferation and decreases the histological score of experimental pancreatic inflammation and fibrosis formation by avoiding the activation of TGF-β1 signaling. In addition, Masamune et al[48] investigated the effects of another angiotensin II antagonist, olmesartan, on PC-associated fibrosis in a subcutaneous xenograft model. Apparently, olmesartan could inhibit the PSC proliferation and collagen Iproduction, resulting in the tumor growth suppression. Nevertheless, further preclinical data are warranted before advancing these agents to clinical trials.
Upregulation of microRNAs in PSCs
miRNAs are small noncoding RNA molecules involved in RNA silencing and post-transcriptional gene expression regulation. A study reported that miR-21, a profibrotic miRNA, is upregulated in cancer–associated myofibroblasts and PSCs isolated from resected PC tissues[49]. In addition, PC cells have been assumed to induce miR-21 upregulation in these cells, expediting their invasion and metastasis[49]. Donahue et al[50] reported that a high stromal miR-21 level correlated with worse overall survival in patients with PC who received adjuvant 5-fluorouracil but not gemcitabine. A meta-analysis showed that miR-21 upregulation in tumor tissue and blood samples of patients with PC was significantly associated with poorer overall survival, disease-free survival, and progression-free survival. A significant correlation was detected between miR-21 expression and lymph node status and tumor grade[51]. Frampton et al[52] reported that, in addition to miR-21, other miRNAs, such as miR-10b, miR-34, miR-155, and miR-203 also appear to have prognostic significance in pancreatic ductal adenocarcinoma. The dysregulation of miR-320a, miR-365, miR-200, and miR-210 has been found to be involved in tumor invasion, epithelial to mesenchymal transition development, and chemotherapeutic drug resistance in PC[53]. Thus, silencing of specific miRNAs by chemically modified antisense oligonucleotides could be a novel therapeutic intervention for PC.
Inhibition of hedgehog signaling in PSCs
Bailey et al[54] were the first to report that sonic hedgehog (Hh) ligands secreted by PC can activate the canonical Hh signaling pathway in PSCs, resulting in their activation, differentiation, and proliferation. In addition, sonic Hh has been shown to promote desmoplasia in orthotopic mouse models of PC, and inhibiting sonic Hh with monoclonal antibody 5E1 markedly decreased the degree of desmoplasia[54].
In their groundbreaking preclinical study, Olive et al[55] assessed the effects of orally administered smoothened antagonist IPI-926 (or saridegib, a derivative of Hh inhibitor cyclopamine) on the tumor stroma and intratumoral uptake of gemcitabine in pancreatic tumor-bearing KPC mice. The result revealed that IPI-926 treatment considerably reduced the proliferation of stromal myofibroblasts, considerably depleted stromal components, and resulted in a transient increase in the intratumoral vascular density and intratumoral concentration of gemcitabine, facilitating transient disease stabilization. On the basis, in part, of these findings, a phase I/III clinical study was commenced to assess the safety and efficacy of IPI-926 and gemcitabine combination treatment in metastatic PC[56] (Table 1). The initial outcomes revealed that this combination was well tolerated and resulted in a partial response in 5 of 16 patients in the phase 1b portion of the study.
In another phase I study, IPI-926 was used in combination with 5-fluorouracil, folinic acid, irinotecan, and oxaliplatin (FOLFIRINOX), a potent and intensive chemotherapy regimen, in the first-line treatment of advanced PC[57]. The preliminary outcomes revealed that the unsubstantiated overall response rate was 66.7%, and that treatment-related toxicities were acceptable and tolerable. However, the initial findings of a phase Ib/II study conducted by Catenacci et al[58] questioned the efficacy of Hh inhibition in advanced PC. The authors evaluated the synergistic activity of vismodegib, a small-molecule inhibitor of smoothened, and gemcitabine in patients with metastatic PC. They observed no safety concerns in the phase 1b portion of the study. In the phase II portion of the study, they randomized 106 patients into gemcitabine plus vismodegib or gemcitabine plus placebo groups, but observed no significant differences in the progression-free (P = 0.30) and overall survival (P = 0.84) between the two treatment groups. Moreover, the response rates were not significantly different (Table 1).
OTHER TARGETABLE ELEMENTS OF STROMA
Hyaluronan
Reportedly, the PC stroma might comprise a considerable amount of HA, which is a high-molecule glycosaminoglycan comprising repeating units of D-glucuronic acid and N-acetyl-glucosamine[59,60]. Reportedly, HA levels in PC tissue might reach 12-fold higher than that found in healthy pancreatic tissue[61]. In addition, PC cells typically express high levels of the primary HA receptor, CD44[62,63]. When HA binds to CD44, four major signaling pathways activated in PC cells are as follows: RAS, Rac, MAPK, and phosphatidylinositol-3-kinase. In fact, signaling through these pathways accelerates the proliferation, epithelial-to-mesenchymal transition, stemness, and metastatic capacity of PC cells and increases their resistance against chemotherapeutic drugs[64-70]. Besides its significant tumor-promoting effects, HA is a crucial contributor to the impaired blood perfusion of tumor cells, increased tumor hypoxia, and, more crucially, insufficient drug delivery to the tumor, as mentioned previously[1,60,69,70].
Some preclinical studies have reported that the enzymatic degradation of HA using PEGylated human recombinant hyaluronidase PH20 (PEGPH20) in genetically engineered mouse models of PC could prompt the re-expansion of collapsed tumor vessels and promote doxorubicin and gemcitabine delivery. Furthermore, the combined use of gemcitabine and PEGPH20 exhibited a synergistic effect and substantially inhibited the tumor growth, resulting in the upgraded survival of animals. Conversely, gemcitabine monotherapy only modestly affected the tumor growth compared with PEGPH20 alone[60]. Provenzano et al[71] reported similar findings and observed that PEGPH20 effectively ablated HA from metastatic deposits as with primary tumors and reinstated the vascular pattern.
Consequently, a phase 1b study by Hingorani et al[72] evaluated the safety and efficacy of escalating doses of intravenous PEGPH20 combined with gemcitabine in patients with metastatic PC. The treatment was well tolerated by patients (n = 28) and exhibited a promising clinical activity. However, patients with tumors comprising higher HA levels seemingly benefited more from this treatment than those whose tumors had lower HA levels. In addition, the median progression-free and overall survival durations were 7.2 and 13 mo for patients with high HA levels and 3.5 and 5.7 mo for patients with low HA levels, respectively. Notably, these results encouraged further clinical research.
The final outcomes of phase 2 HALO-109-202 study, in which PEGPH20 was administered together with nab-paclitaxel plus gemcitabine regimen, were presented at the 2017 American Society of Clinical Oncology Annual Meeting[73]. The study randomized 279 patients with untreated metastatic PC to receive either PEGPH20 plus chemotherapy (100 patients treated) or chemotherapy alone (160 patients treated). The combination therapy substantially improved the median progression-free survival (primary endpoint: 6.0 mo vs 5.3 mo; P = 0.045) in unselected patients. In HA-high patients (34% of enrolled patients), a significant increase was again noted in the progression-free survival with PEGPH20 plus chemotherapy compared with chemotherapy alone (median: 9.2 mo vs 5.2 mo; P = 0.48). However, no significant difference was observed between the two treatment arms regarding the overall survival (median: 11.5 mo vs 8.5 mo; HR, 0.96). Apparently, thromboembolic events pose a primary complication of PEGPH20 treatment. In the first stage of this phase 2 study, none of the patients randomized to PEGFP20 arm was provided thromboprophylaxis, and 43% of these developed thrombosis, causing a temporary cessation in the treatment. However, in the second stage, the rate of this complication was decreased to 28% with the administration of enoxaparin prophylaxis. PEGPH20 treatment was also associated with increased incidence and severity of other manageable side effects, such as painful muscle spasms, arthralgia, peripheral edema, and neutropenia. Overall, PEGPH20 is the first stroma-targeting agent that has demonstrated its efficacy in a clinical setting. Currently, a phase III study (HALO Pancreatic 301; NCT02715804) is recruiting patients with stage IV PC whose tumors have a high level of HA to validate phase II results.
In contrast, a recently presented randomized phase I/II study evaluating the efficacy of PEGPH20 and modified FOLFIRINOX in patients with metastatic PC who have a good performance status suggested that PEGPH20 can have a detrimental effect on OS (HR = 0.48). Therefore, further studies are needed to clarify whether the benefit from the use of PEGPH20 is restricted to patients treated with gemcitabine and nab-paclitaxel[74] .
Secreted protein acidic and rich in cysteine
SPARC (also known as osteonectin or basement membrane protein 40) is a member of the matricellular proteins group and plays regulatory roles in cellular proliferation and adhesion. Guweidhi et al[75] described that primary and metastatic lesions of PC expressed SPARC 31-fold more compared with normal pancreatic tissue. In addition, PC cells fail to produce SPARC because of aberrant hypermethylation in their SPARC gene. Thus, almost all SPARC in PC tissue is produced by PSCs[75-78]. Reportedly, SPARC can increase the migration ability and invasive properties of PC cells[78-80]. In addition, SPARC can stimulate the MMP production in neoplastic cells, thereby enhancing their metastatic potential[75,77,81,82]. Accordingly, patients with PC whose tumors contain elevated amounts of SPARC have been reported to have worse survival compared with those whose tumors contain lower SPARC levels following radical surgery or chemoradiotherapy[80,83-85].
Owing to its high affinity for albumin, stromal SPARC, perhaps, increases the intratumoral delivery and efficacy of the chemotherapeutic drug albumin-bound paclitaxel (nab-paclitaxel) in patients with PC[86]. In their phase I/II study, Von Hoff et al[86] examined the efficacy of escalating doses of nab-paclitaxel in combination with fixed doses gemcitabine in 67 patients with previously untreated metastatic PC. The treatment resulted in an overall response rate of 48%, and the median overall survival duration of 12.2 mo. In the study, the SPARC status was assessed in 36 patients, and patients whose tumors had high SPARC expression (n = 19) exhibited better overall survival than patients whose tumors displayed low SPARC expression (median: 17.8 mo vs 8.1 mo; P = 0.0431). In addition, the study established a significant correlation between the stromal SPARC level and the patients’ survival (P = 0.013). However, SPARC in tumor cells did not exert any effect on survival (P = 0.15). Besides, the authors assessed the treatment-related stromal changes and intratumoral penetration of the drugs in a patient-derived xenograft mouse model of PC and demonstrated that tumors resected from mice treated with gemcitabine alone demonstrated an extensive desmoplastic stroma. However, tumors in mice treated with nab-paclitaxel alone or in combination with gemcitabine exhibited the reduced stromal content, which was accompanied by dilated tumor blood vessels. Thus, the intratumoral concentration of gemcitabine was determined to be 2.8-fold higher in nab-paclitaxel plus gemcitabine-treated mice compared with mice receiving gemcitabine alone.
On the basis of these results, Von Hoff et al[87] conducted a phase III study in which 861 patients with metastatic PC were randomly allotted to receive either nab-paclitaxel plus gemcitabine or gemcitabine alone. Their findings established the superiority of the combination regimen over gemcitabine monotherapy. In addition, patients receiving nab-paclitaxel plus gemcitabine exhibited longer median overall survival compared with those receiving gemcitabine alone (8.5 mo vs 6.7 mo; P < 0.001). Furthermore, they demonstrated a better response rate (23% vs 7%; P < 0.001). Hence, it could be speculated that the tumor SPARC level could be used as a predictive marker to determine patients with advanced PC most likely to benefit from nab-paclitaxel–based chemotherapy.
CONCLUSION
Despite the determination of active chemotherapeutic regimens, such as nab-paclitaxel plus gemcitabine and FOLFIRINOX, in metastatic PC, the overall treatment outcomes remain inadequate. Perhaps, stroma-depletion strategies could provide novel treatment opportunities for patients with this formidable disease. Among them, the enzymatic degradation of stromal HA by PEGPH20 is currently the only effective method in the clinical setting. After the announcement of the final outcomes of the phase III HALO Pancreatic 301 study, PEGPH20 could be incorporated into standard-of-care treatment regimens in metastatic PC. Of note, promising preclinical effects of Hh inhibitors await clinical confirmation; however, these could exhibit a stronger activity and synergy when they are combined with potent chemotherapy combinations rather than gemcitabine monotherapy. Moreover, agents that have demonstrated promising anti-stromal activity in preclinical models, especially vitamin A and D analogs, warrant clinical testing and could extend the therapeutic armamentarium in the future.
Footnotes
Conflict-of-interest statement: No potential conflicts of interest relevant to this article were reported.
Manuscript source: Invited manuscript
Peer-review started: March 30, 2018
First decision: April 23, 2018
Article in press: June 27, 2018
Specialty type: Oncology
Country of origin: Turkey
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): D
Grade E (Poor): 0
P- Reviewer: Matsuda Y, Negoi I, Ramasamy TS S- Editor: Ji FF L- Editor: A E- Editor: Tan WW
Contributor Information
Ozkan Kanat, Department of Medical Oncology, Faculty of Medicine, Uludag University, Bursa 16059, Turkey. ozkanat@uludag.edu.tr.
Hulya Ertas, Department of Medical Oncology, Faculty of Medicine, Uludag University, Bursa 16059, Turkey.
References
- 1.Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, Hostetter G, Shepard HM, Von Hoff DD, Han H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin Cancer Res. 2015;21:3561–3568. doi: 10.1158/1078-0432.CCR-14-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bahrami A, Khazaei M, Bagherieh F, Ghayour-Mobarhan M, Maftouh M, Hassanian SM, Avan A. Targeting stroma in pancreatic cancer: Promises and failures of targeted therapies. J Cell Physiol. 2017;232:2931–2937. doi: 10.1002/jcp.25798. [DOI] [PubMed] [Google Scholar]
- 3.Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–868. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 4.Kota J, Hancock J, Kwon J, Korc M. Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Lett. 2017;391:38–49. doi: 10.1016/j.canlet.2016.12.035. [DOI] [PubMed] [Google Scholar]
- 5.Voutouri C, Polydorou C, Papageorgis P, Gkretsi V, Stylianopoulos T. Hyaluronan-Derived Swelling of Solid Tumors, the Contribution of Collagen and Cancer Cells, and Implications for Cancer Therapy. Neoplasia. 2016;18:732–741. doi: 10.1016/j.neo.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, Smith BL, Ferrone CR, Hornicek FJ, Boucher Y, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci USA. 2012;109:15101–15108. doi: 10.1073/pnas.1213353109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205–2218. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ide T, Kitajima Y, Miyoshi A, Ohtsuka T, Mitsuno M, Ohtaka K, Miyazaki K. The hypoxic environment in tumor-stromal cells accelerates pancreatic cancer progression via the activation of paracrine hepatocyte growth factor/c-Met signaling. Ann Surg Oncol. 2007;14:2600–2607. doi: 10.1245/s10434-007-9435-3. [DOI] [PubMed] [Google Scholar]
- 9.Kitajima Y, Ide T, Ohtsuka T, Miyazaki K. Induction of hepatocyte growth factor activator gene expression under hypoxia activates the hepatocyte growth factor/c-Met system via hypoxia inducible factor-1 in pancreatic cancer. Cancer Sci. 2008;99:1341–1347. doi: 10.1111/j.1349-7006.2008.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masamune A, Shimosegawa T. Pancreatic stellate cells--multi-functional cells in the pancreas. Pancreatology. 2013;13:102–105. doi: 10.1016/j.pan.2012.12.058. [DOI] [PubMed] [Google Scholar]
- 11.Habisch H, Zhou S, Siech M, Bachem MG. Interaction of stellate cells with pancreatic carcinoma cells. Cancers (Basel) 2010;2:1661–1682. doi: 10.3390/cancers2031661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bynigeri RR, Jakkampudi A, Jangala R, Subramanyam C, Sasikala M, Rao GV, Reddy DN, Talukdar R. Pancreatic stellate cell: Pandora’s box for pancreatic disease biology. World J Gastroenterol. 2017;23:382–405. doi: 10.3748/wjg.v23.i3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vonlaufen A, Joshi S, Qu C, Phillips PA, Xu Z, Parker NR, Toi CS, Pirola RC, Wilson JS, Goldstein D, et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 2008;68:2085–2093. doi: 10.1158/0008-5472.CAN-07-2477. [DOI] [PubMed] [Google Scholar]
- 14.Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, Ramm GA, Buchler M, Friess H, McCarroll JA, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas. 2004;29:179–187. doi: 10.1097/00006676-200410000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Bachem MG, Schünemann M, Ramadani M, Siech M, Beger H, Buck A, Zhou S, Schmid-Kotsas A, Adler G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005;128:907–921. doi: 10.1053/j.gastro.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 16.Dunér S, Lopatko Lindman J, Ansari D, Gundewar C, Andersson R. Pancreatic cancer: the role of pancreatic stellate cells in tumor progression. Pancreatology. 2010;10:673–681. doi: 10.1159/000320711. [DOI] [PubMed] [Google Scholar]
- 17.Bachem MG, Zhou S, Buck K, Schneiderhan W, Siech M. Pancreatic stellate cells--role in pancreas cancer. Langenbecks Arch Surg. 2008;393:891–900. doi: 10.1007/s00423-008-0279-5. [DOI] [PubMed] [Google Scholar]
- 18.Jaster R. Molecular regulation of pancreatic stellate cell function. Mol Cancer. 2004;3:26. doi: 10.1186/1476-4598-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999;44:534–541. doi: 10.1136/gut.44.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masamune A, Shimosegawa T. Signal transduction in pancreatic stellate cells. J Gastroenterol. 2009;44:249–260. doi: 10.1007/s00535-009-0013-2. [DOI] [PubMed] [Google Scholar]
- 21.Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, Friess H. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol. 2012;9:454–467. doi: 10.1038/nrgastro.2012.115. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida S, Ujiki M, Ding XZ, Pelham C, Talamonti MS, Bell RH Jr, Denham W, Adrian TE. Pancreatic stellate cells (PSCs) express cyclooxygenase-2 (COX-2) and pancreatic cancer stimulates COX-2 in PSCs. Mol Cancer. 2005;4:27. doi: 10.1186/1476-4598-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujita H, Ohuchida K, Mizumoto K, Egami T, Miyoshi K, Moriyama T, Cui L, Yu J, Zhao M, Manabe T, et al. Tumor-stromal interactions with direct cell contacts enhance proliferation of human pancreatic carcinoma cells. Cancer Sci. 2009;100:2309–2317. doi: 10.1111/j.1349-7006.2009.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang TP. Notch, apoptosis and cancer. Adv Exp Med Biol. 2012;727:199–209. doi: 10.1007/978-1-4614-0899-4_15. [DOI] [PubMed] [Google Scholar]
- 25.Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S, Wu GS, Wu K. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369:20–27. doi: 10.1016/j.canlet.2015.07.048. [DOI] [PubMed] [Google Scholar]
- 26.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang W, Erkan M, Abiatari I, Giese NA, Felix K, Kayed H, Büchler MW, Friess H, Kleeff J. Expression of extracellular matrix metalloproteinase inducer (EMMPRIN/CD147) in pancreatic neoplasm and pancreatic stellate cells. Cancer Biol Ther. 2007;6:218–227. doi: 10.4161/cbt.6.2.3623. [DOI] [PubMed] [Google Scholar]
- 28.Xu Z, Vonlaufen A, Phillips PA, Fiala-Beer E, Zhang X, Yang L, Biankin AV, Goldstein D, Pirola RC, Wilson JS, et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am J Pathol. 2010;177:2585–2596. doi: 10.2353/ajpath.2010.090899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, Marshall JF, Chin-Aleong J, Chelala C, Gribben JG, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145:1121–1132. doi: 10.1053/j.gastro.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, Fuchs JR, Eubank TD, Frankel WL, Bekaii-Saab T, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73:3007–3018. doi: 10.1158/0008-5472.CAN-12-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamada S, Masamune A, Takikawa T, Suzuki N, Kikuta K, Hirota M, Hamada H, Kobune M, Satoh K, Shimosegawa T. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem Biophys Res Commun. 2012;421:349–354. doi: 10.1016/j.bbrc.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 32.Lonardo E, Frias-Aldeguer J, Hermann PC, Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle. 2012;11:1282–1290. doi: 10.4161/cc.19679. [DOI] [PubMed] [Google Scholar]
- 33.Qiu J, Yang G, Feng M, Zheng S, Cao Z, You L, Zheng L, Zhang T, Zhao Y. Extracellular vesicles as mediators of the progression and chemoresistance of pancreatic cancer and their potential clinical applications. Mol Cancer. 2018;17:2. doi: 10.1186/s12943-017-0755-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takikawa T, Masamune A, Yoshida N, Hamada S, Kogure T, Shimosegawa T. Exosomes Derived From Pancreatic Stellate Cells: MicroRNA Signature and Effects on Pancreatic Cancer Cells. Pancreas. 2017;46:19–27. doi: 10.1097/MPA.0000000000000722. [DOI] [PubMed] [Google Scholar]
- 35.Leca J, Martinez S, Lac S, Nigri J, Secq V, Rubis M, Bressy C, Sergé A, Lavaut MN, Dusetti N, et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J Clin Invest. 2016;126:4140–4156. doi: 10.1172/JCI87734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel GK, Khan MA, Bhardwaj A, Srivastava SK, Zubair H, Patton MC, Singh S, Khushman M, Singh AP. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br J Cancer. 2017;116:609–619. doi: 10.1038/bjc.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hah N, Sherman MH, Yu RT, Downes M, Evans RM. Targeting Transcriptional and Epigenetic Reprogramming in Stromal Cells in Fibrosis and Cancer. Cold Spring Harb Symp Quant Biol. 2015;80:249–255. doi: 10.1101/sqb.2015.80.027185. [DOI] [PubMed] [Google Scholar]
- 39.Sarper M, Cortes E, Lieberthal TJ, Del Río Hernández A. ATRA modulates mechanical activation of TGF-β by pancreatic stellate cells. Sci Rep. 2016;6:27639. doi: 10.1038/srep27639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaster R, Hilgendorf I, Fitzner B, Brock P, Sparmann G, Emmrich J, Liebe S. Regulation of pancreatic stellate cell function in vitro: biological and molecular effects of all-trans retinoic acid. Biochem Pharmacol. 2003;66:633–641. doi: 10.1016/s0006-2952(03)00390-3. [DOI] [PubMed] [Google Scholar]
- 41.McCarroll JA, Phillips PA, Santucci N, Pirola RC, Wilson JS, Apte MV. Vitamin A inhibits pancreatic stellate cell activation: implications for treatment of pancreatic fibrosis. Gut. 2006;55:79–89. doi: 10.1136/gut.2005.064543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Froeling FE, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, Clevers H, Hart IR, Kocher HM. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-β-catenin signaling to slow tumor progression. Gastroenterology. 2011;141:1486–1497, 1497.e1-1497.14. doi: 10.1053/j.gastro.2011.06.047. [DOI] [PubMed] [Google Scholar]
- 43.Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D, Attwood S, García R, Ghassemi S, Fabry B, et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun. 2016;7:12630. doi: 10.1038/ncomms12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kozono S, Ohuchida K, Eguchi D, Ikenaga N, Fujiwara K, Cui L, Mizumoto K, Tanaka M. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 2013;73:2345–2356. doi: 10.1158/0008-5472.CAN-12-3180. [DOI] [PubMed] [Google Scholar]
- 45.Suklabaidya S, Das B, Ali SA, Jain S, Swaminathan S, Mohanty AK, Panda SK, Dash P, Chakraborty S, Batra SK, et al. Characterization and use of HapT1-derived homologous tumors as a preclinical model to evaluate therapeutic efficacy of drugs against pancreatic tumor desmoplasia. Oncotarget. 2016;7:41825–41842. doi: 10.18632/oncotarget.9729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hama K, Ohnishi H, Aoki H, Kita H, Yamamoto H, Osawa H, Sato K, Tamada K, Mashima H, Yasuda H, et al. Angiotensin II promotes the proliferation of activated pancreatic stellate cells by Smad7 induction through a protein kinase C pathway. Biochem Biophys Res Commun. 2006;340:742–750. doi: 10.1016/j.bbrc.2005.12.069. [DOI] [PubMed] [Google Scholar]
- 47.Yamada T, Kuno A, Masuda K, Ogawa K, Sogawa M, Nakamura S, Ando T, Sano H, Nakazawa T, Ohara H, et al. Candesartan, an angiotensin II receptor antagonist, suppresses pancreatic inflammation and fibrosis in rats. J Pharmacol Exp Ther. 2003;307:17–23. doi: 10.1124/jpet.103.053322. [DOI] [PubMed] [Google Scholar]
- 48.Masamune A, Hamada S, Kikuta K, Takikawa T, Miura S, Nakano E, Shimosegawa T. The angiotensin II type I receptor blocker olmesartan inhibits the growth of pancreatic cancer by targeting stellate cell activities in mice. Scand J Gastroenterol. 2013;48:602–609. doi: 10.3109/00365521.2013.777776. [DOI] [PubMed] [Google Scholar]
- 49.Kadera BE, Li L, Toste PA, Wu N, Adams C, Dawson DW, Donahue TR. MicroRNA-21 in pancreatic ductal adenocarcinoma tumor-associated fibroblasts promotes metastasis. PLoS One. 2013;8:e71978. doi: 10.1371/journal.pone.0071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donahue TR, Nguyen AH, Moughan J, Li L, Tatishchev S, Toste P, Farrell JJ. Stromal microRNA-21 levels predict response to 5-fluorouracil in patients with pancreatic cancer. J Surg Oncol. 2014;110:952–959. doi: 10.1002/jso.23750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Negoi I, Hostiuc S, Sartelli M, Negoi RI, Beuran M. MicroRNA-21 as a prognostic biomarker in patients with pancreatic cancer - A systematic review and meta-analysis. Am J Surg. 2017;214:515–524. doi: 10.1016/j.amjsurg.2017.03.049. [DOI] [PubMed] [Google Scholar]
- 52.Frampton AE, Krell J, Jamieson NB, Gall TM, Giovannetti E, Funel N, Mato Prado M, Krell D, Habib NA, Castellano L, et al. microRNAs with prognostic significance in pancreatic ductal adenocarcinoma: A meta-analysis. Eur J Cancer. 2015;51:1389–1404. doi: 10.1016/j.ejca.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 53.Xiong G, Feng M, Yang G, Zheng S, Song X, Cao Z, You L, Zheng L, Hu Y, Zhang T, et al. The underlying mechanisms of non-coding RNAs in the chemoresistance of pancreatic cancer. Cancer Lett. 2017;397:94–102. doi: 10.1016/j.canlet.2017.02.020. [DOI] [PubMed] [Google Scholar]
- 54.Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, Ouellette MM, Hollingsworth MA. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14:5995–6004. doi: 10.1158/1078-0432.CCR-08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stephenson J, Richards DA, Wolpin BM, Becerra C, Hamm JT, Messersmith WA, Devens S, Cushing J, Goddard J, Schmalbach T, et al. The safety of IPI-926, a novel hedgehog pathway inhibitor, in combination with gemcitabine in patients (pts) with metastatic pancreatic cancer. J Clin Oncol. 2011;29:Abstract 4114. [Google Scholar]
- 57.Ko AH, LoConte N, Tempero MA, Walker EJ, Kate Kelley R, Lewis S, Chang WC, Kantoff E, Vannier MW, Catenacci DV, et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas. 2016;45:370–375. doi: 10.1097/MPA.0000000000000458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, Marsh R, Wallace J, Kozloff M, Rajdev L, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol. 2015;33:4284–4292. doi: 10.1200/JCO.2015.62.8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mahlbacher V, Sewing A, Elsässer HP, Kern HF. Hyaluronan is a secretory product of human pancreatic adenocarcinoma cells. Eur J Cell Biol. 1992;58:28–34. [PubMed] [Google Scholar]
- 60.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–120. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Theocharis AD, Tsara ME, Papageorgacopoulou N, Karavias DD, Theocharis DA. Pancreatic carcinoma is characterized by elevated content of hyaluronan and chondroitin sulfate with altered disaccharide composition. Biochim Biophys Acta. 2000;1502:201–206. doi: 10.1016/s0925-4439(00)00051-x. [DOI] [PubMed] [Google Scholar]
- 62.Takada M, Yamamoto M, Saitoh Y. The significance of CD44 in human pancreatic cancer: I. High expression of CD44 in human pancreatic adenocarcinoma. Pancreas. 1994;9:748–752. doi: 10.1097/00006676-199411000-00013. [DOI] [PubMed] [Google Scholar]
- 63.Immervoll H, Hoem D, Steffensen OJ, Miletic H, Molven A. Visualization of CD44 and CD133 in normal pancreas and pancreatic ductal adenocarcinomas: non-overlapping membrane expression in cell populations positive for both markers. J Histochem Cytochem. 2011;59:441–455. doi: 10.1369/0022155411398275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sato N, Kohi S, Hirata K, Goggins M. Role of hyaluronan in pancreatic cancer biology and therapy: Once again in the spotlight. Cancer Sci. 2016;107:569–575. doi: 10.1111/cas.12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Teranishi F, Takahashi N, Gao N, Akamo Y, Takeyama H, Manabe T, Okamoto T. Phosphoinositide 3-kinase inhibitor (wortmannin) inhibits pancreatic cancer cell motility and migration induced by hyaluronan in vitro and peritoneal metastasis in vivo. Cancer Sci. 2009;100:770–777. doi: 10.1111/j.1349-7006.2009.01084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kultti A, Zhao C, Singha NC, Zimmerman S, Osgood RJ, Symons R, Jiang P, Li X, Thompson CB, Infante JR, et al. Accumulation of extracellular hyaluronan by hyaluronan synthase 3 promotes tumor growth and modulates the pancreatic cancer microenvironment. Biomed Res Int. 2014;2014:817613. doi: 10.1155/2014/817613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng XB, Kohi S, Koga A, Hirata K, Sato N. Hyaluronan stimulates pancreatic cancer cell motility. Oncotarget. 2016;7:4829–4840. doi: 10.18632/oncotarget.6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shepard HM. Breaching the Castle Walls: Hyaluronan Depletion as a Therapeutic Approach to Cancer Therapy. Front Oncol. 2015;5:192. doi: 10.3389/fonc.2015.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Provenzano PP, Hingorani SR. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br J Cancer. 2013;108:1–8. doi: 10.1038/bjc.2012.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wong KM, Horton KJ, Coveler AL, Hingorani SR, Harris WP. Targeting the Tumor Stroma: the Biology and Clinical Development of Pegylated Recombinant Human Hyaluronidase (PEGPH20) Curr Oncol Rep. 2017;19:47. doi: 10.1007/s11912-017-0608-3. [DOI] [PubMed] [Google Scholar]
- 71.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM, Tjulandin SA, Gladkov OA, Holcombe RF, Korn R, et al. Phase Ib Study of PEGylated Recombinant Human Hyaluronidase and Gemcitabine in Patients with Advanced Pancreatic Cancer. Clin Cancer Res. 2016;22:2848–2854. doi: 10.1158/1078-0432.CCR-15-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hingorani SR, Bullock AJ, Seery TE, Zheng L, Sigal D, Ritch PS. Randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine (PAG) vs AG in patients (Pts) with untreated, metastatic pancreatic ductal adenocarcinoma (mPDA) J Clin Oncol. 2017;15:Abstract 4008. doi: 10.1200/JCO.2017.74.9564. [DOI] [PubMed] [Google Scholar]
- 74.Ramanathan RK, McDonough S, Philip PA, et al. A phase IB/II randomized study of mFOLFIRINOX (mFFOX) + pegylated recombinant human hyaluronidase (PEGPH20) versus mFFOX alone in patients with good performance status metastatic pancreatic adenocarcinoma (mPC): SWOG S1313 (NCT #01959139) Oral presentation at: 2018. pp. Gastrointestinal Cancers Symposium; January 18–20, 2018; San Francisco, CA. [Google Scholar]
- 75.Guweidhi A, Kleeff J, Adwan H, Giese NA, Wente MN, Giese T, Büchler MW, Berger MR, Friess H. Osteonectin influences growth and invasion of pancreatic cancer cells. Ann Surg. 2005;242:224–234. doi: 10.1097/01.sla.0000171866.45848.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Neuzillet C, Tijeras-Raballand A, Cros J, Faivre S, Hammel P, Raymond E. Stromal expression of SPARC in pancreatic adenocarcinoma. Cancer Metastasis Rev. 2013;32:585–602. doi: 10.1007/s10555-013-9439-3. [DOI] [PubMed] [Google Scholar]
- 77.Vaz J, Ansari D, Sasor A, Andersson R. SPARC: A Potential Prognostic and Therapeutic Target in Pancreatic Cancer. Pancreas. 2015;44:1024–1035. doi: 10.1097/MPA.0000000000000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rossi MK, Gnanamony M, Gondi CS. The ‘SPARC’ of life: Analysis of the role of osteonectin/SPARC in pancreatic cancer (Review) Int J Oncol. 2016;48:1765–1771. doi: 10.3892/ijo.2016.3417. [DOI] [PubMed] [Google Scholar]
- 79.Ting DT, Wittner BS, Ligorio M, Vincent Jordan N, Shah AM, Miyamoto DT, Aceto N, Bersani F, Brannigan BW, Xega K, et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014;8:1905–1918. doi: 10.1016/j.celrep.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mantoni TS, Schendel RR, Rödel F, Niedobitek G, Al-Assar O, Masamune A, Brunner TB. Stromal SPARC expression and patient survival after chemoradiation for non-resectable pancreatic adenocarcinoma. Cancer Biol Ther. 2008;7:1806–1815. doi: 10.4161/cbt.7.11.6846. [DOI] [PubMed] [Google Scholar]
- 81.Koshiba T, Hosotani R, Wada M, Miyamoto Y, Fujimoto K, Lee JU, Doi R, Arii S, Imamura M. Involvement of matrix metalloproteinase-2 activity in invasion and metastasis of pancreatic carcinoma. Cancer. 1998;82:642–650. doi: 10.1002/(sici)1097-0142(19980215)82:4<642::aid-cncr5>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 82.Bloomston M, Zervos EE, Rosemurgy AS 2nd. Matrix metalloproteinases and their role in pancreatic cancer: a review of preclinical studies and clinical trials. Ann Surg Oncol. 2002;9:668–674. doi: 10.1007/BF02574483. [DOI] [PubMed] [Google Scholar]
- 83.Gundewar C, Sasor A, Hilmersson KS, Andersson R, Ansari D. The role of SPARC expression in pancreatic cancer progression and patient survival. Scand J Gastroenterol. 2015;50:1170–1174. doi: 10.3109/00365521.2015.1024281. [DOI] [PubMed] [Google Scholar]
- 84.Infante JR, Matsubayashi H, Sato N, Tonascia J, Klein AP, Riall TA, Yeo C, Iacobuzio-Donahue C, Goggins M. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 2007;25:319–325. doi: 10.1200/JCO.2006.07.8824. [DOI] [PubMed] [Google Scholar]
- 85.Han W, Cao F, Chen MB, Lu RZ, Wang HB, Yu M, Shi CT, Ding HZ. Prognostic Value of SPARC in Patients with Pancreatic Cancer: A Systematic Review and Meta-Analysis. PLoS One. 2016;11:e0145803. doi: 10.1371/journal.pone.0145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias JL, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29:4548–4554. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]