

Abstract
E. coli is the main causative agent of mastitis in dairy cows, but the mechanism of molecular regulation underlying the occurrence and development of mastitis has not yet been fully elucidated. In this study, an E. coli-induced mastitis model was created and RNASeq technology was used to measure the miRNA expression profiles at different times post-infection (0, 1, 3, 5, 7 dpi), as well as to screen for differentially expressed miRNA. The results show detection of 2416 miRNAs, including 628 known miRNAs and 1788 newly discovered miRNAs. A total of 200 differentially expressed miRNAs were found at different time points. Bioinformatics analysis showed that these differentially expressed miRNAs may regulate the occurrence and development of mastitis in dairy cows through seven signal transduction pathways, namely cytokine-cytokine receptor interaction, MAPK signaling pathway, chemokine signaling pathway, leukocyte transendothelial migration, T cell receptor signaling pathway, Toll-like receptor signaling pathway, and cell adhesion molecules. In addition, bta-miR-200a, bta-miR-205, bta-miR-122, bta-miR-182 and the newly discovered conservative_15_7229 might be involved in immune process in late stage of E. coli-induced mastitis. The results of this study lay the foundation for molecular network analysis of mastitis and molecular breeding of dairy cows.
Introduction
Mastitis is an inflammatory disease caused by pathogenic microorganisms in mammary tissues. The high incidence of mastitis in dairy cows leads to decreased milk production and lowered dairy product quality, which can seriously affect the profitability of dairy farms1. E. coli is one of the most commonly observed pathogens in dairy cow mastitis. The occurrence and development of mastitis are the results of co-regulation by gene networks composed of multiple genes2.
MicroRNAs (miRNA) are recently discovered endogenous non-coding RNAs. They bind to their target mRNAs and degrade or downregulate the mRNAs, thereby regulating a number of biological processes such as growth, development and immunity in humans and animals3–5. Studies have shown that, after cells receive exogenous or endogenous signals, changes in expression of miRNA form part of the early response. For example, the expression levels of certain miRNAs change rapidly after disease occurs in an organism to control the scope and intensity of disease development6–8. Therefore, miRNA expression levels during disease onset or progression can be used as an early diagnostic biomarker of disease7,9. However, miRNA research in dairy mastitis is still in its infancy. In recent years, scholars have mainly used mammary tissues or mammary epithelial cells as research materials for miRNA isolation, identification and functional studies10–13. There have only been a few reports on blood miRNAs, for example, in the use of high-throughput techniques to detect miRNA expression profiles in clinical mastitis in dairy cows14,15 and in the detection of miRNA expression levels in mononuclear cells from dairy cows with mastitis caused by infection with S. uberis16. The changes in expression of blood miRNA in dairy cows caused by E. coli infection have not been previously reported. Therefore, in this study, RNAseq was used to determine the expression levels of blood miRNA in E. coli-induced mastitis at different times after infection, in order to provide a basis for the biotherapy, and molecular mechanism behind mastitis in dairy cows.
Results
Overview of sequencing data
To investigate the changes in the expression of blood miRNAs during bovine mastitis, we performed deep sequencing of small RNA libraries using the Illumina Hiseq2500 platform. Based on the raw reads, unreliable sequences were removed as described in the method to obtain high quality clean reads (Table 1). In addition, using Bowtie software17, we compared the clean reads with the bovine reference genome to obtain mapped small RNA reads. Quality control ensured that Q30 (%) was greater than 94% for each sample and clean data was greater than 11.89 M, indicating reliable sequencing data for subsequent sRNA.
Table 1.
Overview of miRNA sequencing data.
| dpi | Raw reads | Q30 (%) | Clean reads | Mapped Small RNA Reads |
|---|---|---|---|---|
| 0 | 30358145 | 94.33 | 26833388 | 16812993 |
| 1 | 18604382 | 97.23 | 16600883 | 10672762 |
| 3 | 16922373 | 94.16 | 14615093 | 9504913 |
| 5 | 14437756 | 97.15 | 12570752 | 8096806 |
| 7 | 12858078 | 97.45 | 11887296 | 7747705 |
sRNA classification
Bowtie software is a short sequence alignment software and is especially suited for alignment of reads obtained by high-throughput sequencing. Therefore, we used Bowtie software17 to align clean reads with the Silva, GtRNAdb, Rfam, and Repbase databases to remove ncRNA and repetitive sequences such as rRNA, tRNA, snRNA, and snoRNA, and obtained the unannotated reads for miRNA sequences (Table 2).
Table 2.
Distribution of the small RNA among different categories at different times of mastitis induction.
| dpi | Clean reads | rRNA | snRNA | snoRNA | tRNA | Repbase | Unannotated | miRNA | Unannotated small RNA |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 26,833,388 | 639,348 | 12 | 181,237 | 116,546 | 494,265 | 25,401,980 | 21,961,291 | 3,440,689 |
| 100.00% | 2.38% | 0.00% | 0.68% | 0.43% | 1.84% | 94.67% | 81.84% | 12.83% | |
| 1 | 16,600,883 | 518,804 | 10 | 123,445 | 89,050 | 317,135 | 15,552,439 | 13,350,150 | 2,202,289 |
| 100.00% | 3.13% | 0.00% | 0.74% | 0.54% | 1.91% | 93.68% | 80.42% | 13.26% | |
| 3 | 14,615,093 | 501,037 | 12 | 109,114 | 127,350 | 270,833 | 13,606,747 | 11,593,688 | 2,013,059 |
| 100.00% | 3.43% | 0.00% | 0.75% | 0.87% | 1.85% | 93.10% | 79.33% | 13.77% | |
| 5 | 12,570,752 | 362,417 | 12 | 99,001 | 45,138 | 262,799 | 11,801,385 | 10,157,070 | 1,644,315 |
| 100.00% | 2.88% | 0.00% | 0.79% | 0.36% | 2.09% | 93.88% | 80.80% | 13.08% | |
| 7 | 11,887,296 | 382,122 | 3 | 66,238 | 44,194 | 134,766 | 11,259,973 | 9,842,917 | 1,417,056 |
| 100.00% | 3.21% | 0.00% | 0.56% | 0.37% | 1.13% | 94.72% | 82.80% | 11.92% |
miRNA identification
To identify the miRNA sequences in the unannotated reads described above, miRDeep2 software package was used. Our results show that, among the five groups of sequencing results, the miRNA reads accounted for 81.84%, 80.42%, 79.33%, 80.80%, and 82.80% of clean reads (Table 2). In addition, unknown small RNAs make up 12.83%, 13.26%, 13.77%, 13.08% and 11.92% of clean reads (Table 2), and their roles need to be further explored.
After miRNA sequencing of the five sets of sequencing data, a total of 2416 miRNAs were obtained, including 628 known miRNAs (included in the miRBase database) and 1788 novel miRNAs (Supplementary Table S2). The results of miRNA fragment distribution analysis showed that no matter if the identified miRNA was previously known or newly predicted, the length of the fragment was primarily 22 nt, followed by 21, 23, 20 and 24 nt (Fig. 1). It is noteworthy that, through comparative analysis of the number of differentially expressed miRNA in the five groups, 14, 17, 11, 10, and 16 known miRNAs were specifically expressed in peripheral blood on the 0th, 1st, 3rd, 5th, and 7th day following infection with E. coli, respectively. Meanwhile, there was specific expression of 54, 43, 105, 96 and 92 novel miRNAs (Fig. 2), respectively, suggesting that miRNAs may play important roles in inducing different activities at different times.
Figure 1.

Length distribution of the mapped miRNAs in peripheral blood of dairy cows. (a) Known miRNA, (b) Predicted miRNA.
Figure 2.

Comparison of miRNA expression in peripheral blood of dairy cows at different times post induction (venn diagram). BE0, BE1, BE3, BE5 and BE7 in the figure represent the groups 0d, 1d, 3d, 5d and 7d after E. coli induction.
miRNA expression overall distribution
Based on our deep sequencing data, we calculated the TPM (transcripts per million) values of the miRNAs from the five sample groups. The overall expression pattern of miRNAs is shown in Fig. 3. As shown in Fig. 3, there are differences in the expression patterns of miRNAs between the five samples, suggesting that these differentially expressed miRNAs may play different roles in the development of mastitis. In addition, the abundance of expressed miRNAs (TPM >1000) (Supplementary Table S3) shows that the top 10 highly expressed miRNAs in all samples are: bta-miR-486, bta-miR-451, bta-miR-92a, bta-let-7f, bta-miR-25, bta-let-7i, bta-let-7g, bta-miR-26a, bta-miR-21-5p and bta-miR-191. In addition, out of all the new miRNAs, unconservative_3_22065 had the highest level of expression in all five samples with a TPM of over 1200.
Figure 3.
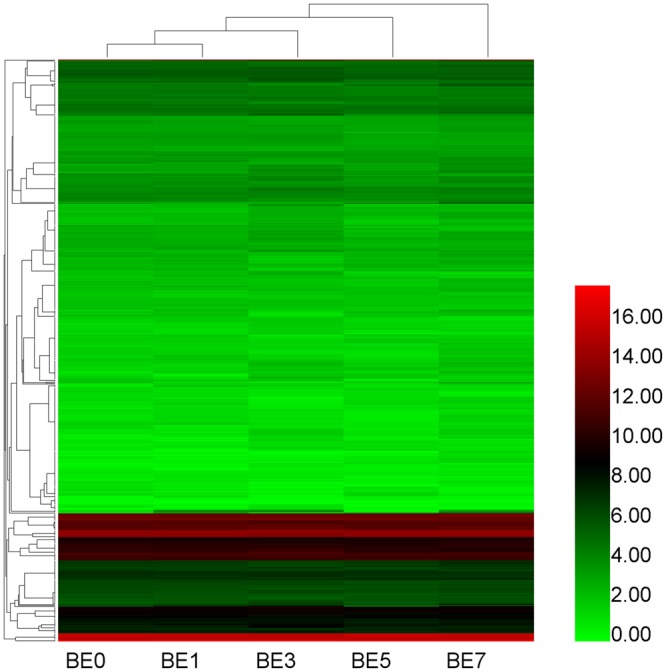
miRNA expression profiles. Heat map is based on the expression of miRNAs TPM-based data, drawn with HemI1.0 software.
miRNA differential expression analysis
Based on 5 sets of TPM data for sequencing expression, we performed paired comparative analysis, using |log2(FC)| ≥ 1, FDR ≤ 0.01, TPM > 1 as the selection criteria, and obtained 200 differentially expressed miRNAs (DIE-miRNAs) (Fig. 4 and Supplementary Table S4), of which 76 are known miRNAs and 124 are novel miRNAs (see Supplementary Table S5 for sequence information). Pairwise comparisons revealed that, compared to the control group (BE0), there were fewer miRNAs differentially expressed in BE1 and BE5, while more miRNAs were significantly differentially expressed in BE3 and BE7 (Fig. 4). It is noteworthy that the expression of bta-miR-200a and bta-miR-205 in BE1, BE5 and BE7 groups were significantly increased (bta-miR-200a log2FC between 4.36 and 7.13, bta-miR- 205 log2FC between 1.93 and 4.70). At the same time, the expression of bta-miR-122 was significantly downregulated in BE1 and BE3 (log2FC was -1.82), while it was significantly upregulated in BE5 and BE7 (log2FC 0.91 and 1.82, respectively). This indicates that bta-miR-122 plays complex roles in mastitis. The expression level of bta-miR-200a and bta-miR-205 in blood significantly increased on the first day after infection, and decreased to pre-infection level on the 3rd day after infection, while on the 5th and 7th day it was significantly increased (Fig. 5). In addition, our study found that compared with the BE0 group, the expression of the novel miRNA conservative_15_7229 in BE5 and BE7 groups was significantly decreased. The above results indicate that these 4 miRNAs (bta-miR-122, bta-miR-200a, bta-miR-205 and conservative_15_7229) may be involved in the immune process in the late stage of dairy cow mastitis induced by E. coli, which still needs further study.
Figure 4.

Analysis of differentially expressed miRNA in dairy cow blood with 0 dpi group as control. Based on the sequenced TPM data, heat map was drawn using HemI1.0 software with |log2FC| ≥ 1 and FDR ≤ 0.01 as the screening standard. BE0, BE1, BE3, BE5 and BE7 represent the 0, 1, 3, 5 and 7 d groups after mammary gland infection with E. coli; the blue block in the heat map indicates the expression level is 0; the red font indicates that the expression of miRNA is significant increased; blue font indicates a significant down-regulation of expression; from top to bottom, the log2FC gradually decreased.
Figure 5.
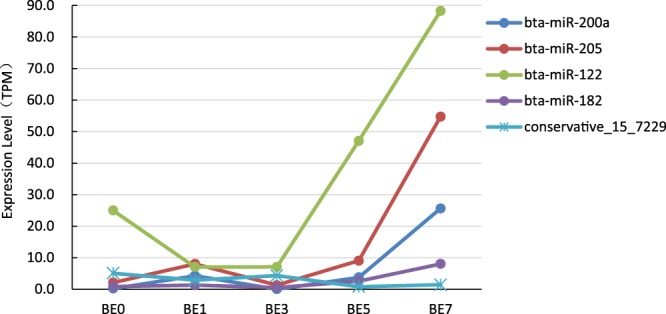
Five miRNAs expression changes.
Our results showed that, on days 1, 3, 5, and 7 following induction, there were many differentially expressed miRNAs in dairy cow blood following E. coli-induced mastitis. The differences in expression were very pronounced (Supplementary Table S4). Notably, bta-miR-182 expression significantly increased in BE1_vs_BE7, BE3_vs_BE5, BE3_vs_BE7 and BE5_vs_BE7 comparisons (log2FC of 2.65, 2.30, 3.95 and 1.64, respectively) (Supplementary Table S4). This indicates that bta-miR-182 is significantly upregulated at 5 or 7 dpi and may also participate in the immune process in the late stage of mastitis.
Differentially expressed miRNA qPCR validation
Ten differentially expressed miRNAs (five of which were upregulated and five of which were downregulated) were randomly selected to perform qPCR experiments and the qPCR results were compared with the high-throughput sequencing results as shown in Fig. 6. The results show that qPCR results are in complete agreement with the results of high-throughput sequencing, which indicates that our findings of differential expression in miRNAs through high-throughput sequencing are accurate and reliable.
Figure 6.
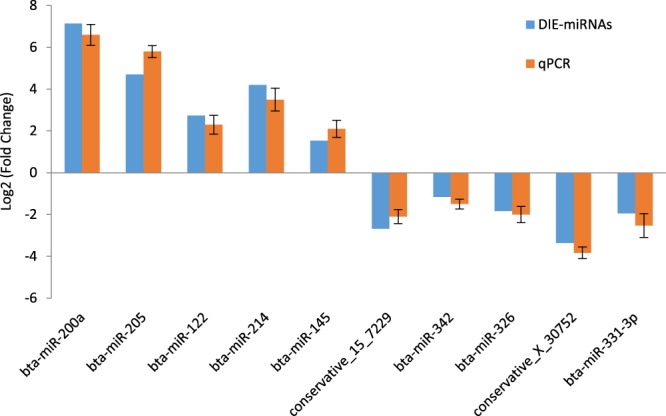
qPCR validation of differentially expressed miRNAs. qPCR expression was normalized with cel-miR-39. There were 10 differentially expressed miRNAs for qPCR validation, with bta-miR-200a, bta-miR-205 and bta-miR-342 from the BE0 vs BE7 comparison; conservative_15_7229 from BE0 vs BE5; bta-miR-326 and conservative_X_30752 from BE3 vs BE5; bta-miR-145 and bta-miR-331-3p from BE3 vs BE7; bta-miR-122 from BE1 vs BE5; bta- miR-214 from BE1 vs BE7 comparison.
DIE-miRNA target gene prediction and functional annotation and KEGG signaling pathway analysis
To explore the function of DIE-miRNA in the development of mastitis, we predicted the target genes of DIE-miRNAs, and our results show that the 200 DIE-miRNAs may have 18,369 target genes. The results of KEGG pathway enrichment analysis showed that these target genes may be involved in the following 6 aspects: disease, environmental information processing, cellular processes, metabolism, organismal systems, and genetic information processing, and encompass 50 significant signal pathways, of which 7 signal pathways (cytokine-cytokine receptor interaction, chemokine signaling pathway, leukocyte transendothelial migration, T cell receptor signaling pathway, Toll-like receptor signaling pathway, and cell adhesion molecules) are associated with immunity (Fig. 7). Therefore, we conclude that these DIE-miRNAs may participate in the disease development process by regulating their target genes during E. coli-induced mastitis.
Figure 7.

KEGG enrichment pathway analysis of DIE-miRNAs target genes.
Discussion
Dairy cow mastitis is an inflammatory disease caused by pathogenic microorganisms. Studies have shown that the expression level of immunity-related genes rapidly changes after pathogens infect tissues or cells, and the process of disease occurrence and development can be finely regulated18–20. Therefore, screening differentially expressed genes may play a very important role in the research of disease molecular mechanism or rapid diagnostic technology. In recent years, with the development of sequencing technology, high-throughput sequencing techniques are often used to mine new miRNAs related to immunity and disease and to screen for differentially expressed miRNAs. In the case of dairy cow mastitis, screening of differentially expressed miRNAs based on high-throughput sequencing has focused on both mammary epithelial cells and mammary tissue. For example, Jin et al.21 found 113 novel miRNAs in E. coli and S. aureus-infected bMECs and found 10 DIE-miRNAs. Lawless et al.13 screened 21 DIE-miRNAs from S. uberis infected bMECs. In addition, other researchers have also used RNASeq technology to measure the miRNA expression profiles of E. coli and S. aureus-infected mammary tissues and the results show that there are multiple differentially expressed miRNAs in infected mammary tissues10,22,23.
Although changes in the expression of blood miRNAs have been shown to be putative biomarkers for early diagnosis of disease7,9, the differential expression of miRNAs can also be used as biomarkers of animal performance24. However, little has been done so far on miRNAs in the blood of dairy cows affected by mastitis. Lawless et al.16 detected three DIE-miRNAs (bta-miR-146b, -451, and -411a) in blood monocytes in dairy cows infected with S. uberis in vivo. Li et al.14 and Chen et al.15 used qRT-PCR and Illumina’s Genome Analyzer IIe to detect DIE-miRNAs in the peripheral blood of mastitis-affected dairy cows in the clinic. Their results showed that, compared with healthy cows, there were 123 mastitis-related miRNAs that experience significant upregulation (such as bta-miR-16a, bta-miR-125b, bta-miR-15a, bta-miR-29b, bta-miR-301a, bta-miR-21-3p, bta-miR-181a and bta-miR-181b, etc.) and 50 miRNAs that undergo significant downregulation (such as bta-miR-223, bta-let-7f, bta-miR-375, bta-miR-148a, etc.). However, it is surprising that the results of the above two scholars vary greatly, and some findings are even in opposition to one another. For example, their results show that bta-miR-146a and bta-miR-146b are significantly downregulated in mastitis cow blood in one reference15, but are significantly upregulated in another reference14. Thus it is clear that the specific dairy cow population, feeding environment and the type of infection and time of infection (or the degree of mastitis) have a significant effect on blood miRNA expression profiles. Therefore, we felt that an in-depth study of E. coli-induced mastitis miRNA expression profile was essential to clarify the molecular mechanism of E. coli-induced mastitis. Therefore, we used a small dose of E. coli for a long-term infection of mammary tissue, and used RNASeq technology to detect miRNA expression in dairy cow blood at different times after infection. Our results show that 2416 miRNAs were detected, including 628 known miRNAs and 1788 newly predicted miRNA (Fig. 2 and Supplementary Table S2). The result of comparison of blood miRNA expression at different times revealed that 200 DIE-miRNAs (including 76 known miRNAs and 124 novel miRNAs) were differentially expressed, of which bta-miR-200a, bta-miR-205, bta-miR-122, bta-miR-182 expression were significantly upregulated in late mastitis, while the newly found conservative_15_7229 was significantly downregulated.
As for the function of the differentially expressed conservative_15_7229, thus far, there have been no reports on this miRNA. A closely related gene found in recent years is miR-200a. The expression of miR-200a is significantly increased in many types of disease, consistent with the results of this study. Zhao et al.25 showed that the expression of miR-200a-3p is increased in hepatocytes of alcoholic liver disease, and may target ZEB2 to induce apoptosis. Wang et al.26 have shown that the expression of miR-200a in CD4+ T cells correlates with the expression of Th17/Treg cells and related cytokines in psoriasis vulgaris. It is noteworthy that, Zarate-Neira et al.27 found that mouse miR-200a-3p expression was significantly increased in a systemic lupus erythematosus (SLE) model and can inhibit the TLR4/MyD88 signalling pathway and promote interferon production, thereby regulating the immune response in mice and proving its usefulness as a biomarker of SLE. In addition, the expression of miR-200a is significantly increased in fish lymphocytes28 and micro-vascular endothelial cells29 induced by LPS (cell surface component of E. coli), targeting the expression TLR128 and ZEB130 genes to regulate the development and progression of disease30,31. However, the current relationship between miR-200a and mastitis in dairy cows is unknown. Therefore, in order to explore the function of bovine miR-200a, we predicted its target gene, and the results indicated that ZEB1 gene may be the target gene of bovine miR-200a. In addition, we found that the 3′-UTR of the ZEB1 gene of other species, such as bovine and human, are highly homologous and that the sequences of bovine and human miR-200a are substantially identical (Supplementary Fig. S1). Therefore, we conclude that bovine miR-200a may also target the bovine ZEB1 gene to regulate the development of E. coli-induced dairy mastitis disease. The specifics remain to be further studied.
As for the expression of miR-122, the results of this study show that the expression level of bta-miR-122 in dairy cow blood is significantly increased in the late stage of E. coli infection (5 dpi and 7 dpi). Consistent with the trend of our results, miR-122 also significantly increases in the bloodstream late in the murine liver injury model and has been shown to be a biomarker of liver injury32. In addition, it has been shown that miR-122 is involved in the regulation of Toll-like receptor signaling pathway after Vibrio anguillarum infection of fish (miiuy croaker) by targeting TLR1433. Therefore, we hypothesize that bta-miR-122 may also be involved in inflammatory response in the late stage of E. coli-induced mastitis.
There are only a few studies on the roles of miR-205 and miR-182 in immune regulation. Studies have shown that the expression of miR-205 is significantly upregulated in patients with allergic rhinitis34, and that miR-205 can also regulate the expression of erbB2/erbB3 in breast cancer cells to promote apoptosis35. IL-2 cytokine stimulation of Th cells can result in a significant increase in the expression of miR-182, reaching its maximum three days after stimulation. Furthermore, in vitro and in vivo studies using the mouse arthritis model showed that IL-2 induced mouse miR-182 expression shows a dose-dependent increase, while inhibiting miR-182 may be beneficial in the treatment of arthritis36. Therefore, we hypothesized that the expression of IL-2 and other inflammatory cytokines in E. coli-induced dairy cow mastitis was low in early stages, but that during later stages IL-2 and other inflammatory cytokines continued to accumulate and induced a significant increase of miR-182 expression in the late stage of E. coli-induced mastitis.
In addition, in order to explore the function of DIE-miRNAs, we predicted the target genes of 200 DIE-miRNAs in E. coli-induced mastitis and analyzed their KEGG signaling pathway. The results indicated that these DIE-miRNAs may be involved in seven animal immunity-related signalling pathways, of which the Toll-like receptor signaling pathway37 and chemokine signaling pathway38 are closely related to innate immunity and inflammatory response, while the T cell receptor signaling pathway39 is related to adaptive immunity. Therefore, we concluded that these DIE-miRNAs may be involved in the innate or adaptive immunity in dairy cow mastitis and may thus regulate the development of mastitis.
In summary, this study used RNASeq technology to detect the miRNA expression profile in the peripheral blood of dairy cows infected with E. coli-induced mastitis at five different post-induction times. The results show that a total of 2416 miRNAs were detected, of which 628 miRNAs were known, and 1788 miRNAs were unknown. A total of 200 DIE-miRNAs were found to be involved in cytokine-cytokine receptor interaction, MAPK signaling pathway, chemokine signaling pathway, leukocyte transendothelial migration, T cell receptor signaling pathway, Toll-like receptor signaling pathway, and cell adhesion molecules in a total of seven signal transduction pathways that regulate the development of mastitis. In addition, bta-miR-200a, bta-miR-205, bta-miR-122, bta-miR-182 and the newly discovered conservative_15_7229 might be involved in immunity in late stage of dairy cow mastitis caused by E. coli. The results of this study have laid the foundation for molecular network analysis and molecular breeding of mastitis, and also provide new molecular materials for the biotherapy of mastitis.
Materials and Methods
Ethics Statement
All animal procedures were conducted with attention to animal welfare and humanitarian procedures according to guidelines laid down by the Institutional Animal Care and Use Committee of Northwest A&F University, and the protocols were approved by the Experimental Animal Manage Committee of Northwest A&F University.
Experimental animals and mastitis model induction
Experimental animals and methods used for the induction and verification of mastitis models have been previously described22. In brief, healthy 2-year-old half-sibling dairy cows were divided into control and experimental groups. Animals in the experimental group were injected with 5 mL of 105 CFU/mL E. coli (ATCC 25922) via the nipple. The control group was injected with an equivalent amount of PBS. We adopted a painless surgical method to collect mammary tissue and used hematoxylin-eosin staining and pathology analysis to ensure the reliability of the model.
Sample collection and RNA extraction
On days 0, 1, 3, 5, and 7 following infection with E. coli, 5 mL of blood was collected to extract total RNA using TRIzoL according to the instruction manual. The purity, concentration, and integrity of RNA were tested to ensure the use of qualified samples for sequencing (RIN value ≥8.0, 28S/18S ≥1.5; no baseline uplift; 5S peak normal).
Small RNA library construction and high-throughput sequencing
Based on protocols from previous publications22,40,41, 1.5 μg of qualified RNA was taken and a small RNA library was constructed using the method described in the NEBNext® Ultra™ Small RNA Sample Library Prep Kit for Illumina® (NEB, USA). After the library was constructed, the library concentration was tested using Qubit 2.0 and was adjusted to 1 ng/μl. Insert Size was measured with an Agilent 2100 bioanalyzer, and qPCR was used to accurately determine the effective concentration of the library to ensure library quality. After qualification of the library, high-throughput sequencing was performed with the Illumina HiSeq2500 of BioMerker Biotech Co., Ltd. (Beijing).
High-throughput sequencing quality control
Sequencing reads were single-end (SE) 50 nt. In order to ensure accurate and credible analysis results, the raw data from each sample after sequencing was screened as follows: (1) low quality sequences were removed; (2) sequences with unknown base N (unidentifiable base) content greater than or equal to 10% of reads were removed; (3) reads without 3′ linker sequences and insertions were removed; (4) reads contaminated with 5′ linkers were removed; (5) cleavage of 3′ adapter sequences and T/C/G reads were removed; (7) sequences shorter than 18 nt or longer than 30 nt were removed. This process yielded high-quality, clean, reliable data.
sRNA classification annotation
Clean reads were aligned using Silva, GtRNAdb, Rfam, and Repbase databases using Bowtie software17 to filter out non-coding RNAs such as rRNA, tRNA, snRNA and snoRNA and repeated sequences to obtain the unannotated reads.
UMD3.1 (http://www.ensembl.org/Bos_taurus/Info/Index) was used as the reference genome for sequence alignment and subsequent analysis. Using miRDeep2 software42, unannotated reads were aligned with the reference genome for positional information on the reference genome, these are subsequently referred to as mapped reads.
miRNA identification
To determine miRNA sequences in the unannotated reads, known miRNA identification and novel miRNA prediction were performed using the miRDeep2 software package42, i.e. reads and genomic sequences were aligned to obtain possible miRNA precursor sequences using RNAfold and randfold to calculate the energy of the precursor structure and to predict the secondary structure. Combined with information on subsets of sequences (e.g. miRNA production characteristics, mature, star and loop sequences), RNAfold and randfold were scored using the Bayesian model to finally identify miRNAs. MiRNAs with scores greater than 80% were considered to be reliable.
miRNA expression and differential expression analysis
Expression of miRNA in each sample was statistically analyzed and was normalized using the TPM algorithm43. TPM normalization formula is as follows:
The miRNA differential expression analysis between two samples was performed using IDEG6 software44. To remove statistical false positives, Benjamini Hochberg’s correction method was used to correct the p-value to the q-value during analysis45. Differentially expressed miRNAs were screened using |log2(Fold Change)| ≥ 1, FDR ≤ 0.01, and q-value < 0.005 as standard.
miRNA expression qPCR verification
Ten differentially expressed miRNAs (five upregulated and five downregulated miRNAs) were randomly selected from the sequencing data, and qPCR was used to verify the expression results from RNASeq. The specific method used was as follows: using miRcute miRNA isolation kit (Tiangen Biotech Co., Ltd. Beijing, China), miRNA extraction was performed according to the manufacturer’s instructions, and the purity and concentration of the miRNA was measured by a trace UV spectrophotometer and diluted to 50 ng/μl. In addition, at the time of miRNA extraction, exogenous cel-miR-39 (5′-UCACCGGGUGUAAAUCAGCUUG), which has no bovine homologue, was added to each tube for subsequent normalization of qPCR quantification results46,47. The reaction system consisted of 4 μl of miRNA template, 10 μl of 2× miRNA RT Reaction Buffer, 2 μl of miRNA RT Enzyme Mix, and 4 μl RNase-free H2O. The reaction conditions were 42 °C for 60 min and 95 °C for 3 min to produce the first strand cDNA, which was then diluted to a concentration of 80 ng/μl.
The qPCR reaction was performed using the miRcute Plus miRNA qPCR Detection Kit (SYBR Green) (Tiangen Biotech Co., Ltd. Beijing, China). The reaction system consisted of: 1 μL cDNA template, 10 μL 2× miRcute miRNA Premix (with SYBR & ROX), 0.4 μL Reverse Primer (10 μM), 0.4 μL Forward Primer, and 8.2 μL RNase-Free ddH2O. The reaction conditions were as follows: 95 °C for 15 min, 5 cycles of 94 °C for 20 s, 64 °C for 30 s, 72 °C for 34 s, followed by 45 cycles of 94 °C for 20 s, then 60 °C for 34 s. Experiments using qPCR followed the MIQE guidelines48: NTC and NRC groups were set up for each qPCR experiment to ensure that the entire experiment was free of DNA or RNA contamination. The qPCR primer sequences are shown in Supplementary Table S1. The qPCR amplification efficiency of each primer ranged from 99.8% to 103.9% (R2 > 0.99). To ensure the specificity of primer amplification, primer melting curve detection was performed at the end of qPCR. All quantitative experiments were performed in three technical duplicates.
The miRNA expression level was calculated using the 2−ΔΔCt method49, and the exogenous cel-miR-39 was used to normalize the quantitative results. The expression level was expressed as mean ± SD, and Student’s t-test was used to test the significance of the difference. A P value of less than 0.05 indicated a significant difference.
miRNA target gene prediction and functional annotation
Using known miRNAs and newly predicted miRNAs and their corresponding species, miRanda50 and RNAhybrid51 were used to perform target gene analysis. KEGG pathway enrichment analysis was performed using KOBAS software52.
Electronic supplementary material
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31501935), National Science-technology Support Plan Projects (2012BAD12B07), and the Construct Program of the Key Discipline in Hunan province.
Author Contributions
Zhuo-Ma Luoreng and Lin-Sen Zan designed the experiments; Zhuo-Ma Luoreng completed the experiments and wrote the manuscript; Xing-Ping Wang improved the manuscript. Zhuo-Ma Luoreng, Xing-Ping Wang, Chu-Gang Mei, and Lin-Sen Zan analyzed the data. All authors read and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-30518-2.
References
- 1.Cha E, et al. The cost and management of different types of clinical mastitis in dairy cows estimated by dynamic programming. J Dairy Sci. 2011;94:4476–4487. doi: 10.3168/jds.2010-4123. [DOI] [PubMed] [Google Scholar]
- 2.Wellnitz O, Bruckmaier RM. The innate immune response of the bovine mammary gland to bacterial infection. Vet J. 2012;192:148–152. doi: 10.1016/j.tvjl.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 3.Contreras J, Rao DS. MicroRNAs in inflammation and immune responses. Leukemia. 2011;26:404–413. doi: 10.1038/leu.2011.356. [DOI] [PubMed] [Google Scholar]
- 4.Guan L, et al. Bta-miR-23a involves in adipogenesis of progenitor cells derived from fetal bovine skeletal muscle. Sci Rep. 2017;7:43716. doi: 10.1038/srep43716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawless N, Vegh P, Oâ Farrelly C, Lynn DJ. The role of microRNAs in bovine infection and immunity. Front Immunol. 2014;5:611. doi: 10.3389/fimmu.2014.00611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hecker M, et al. MicroRNA sxpression changes during interferon-beta treatment in the peripheral blood of multiple sclerosis patients. Int J Mol Sci. 2013;14:16087–16110. doi: 10.3390/ijms140816087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertoli G, et al. MicroRNAs as biomarkers for diagnosis, prognosis and theranostics in prostate cancer. Int J Mol Sci. 2016;17:421. doi: 10.3390/ijms17030421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leidinger P, et al. What makes a blood cell based miRNA expression pattern disease specific? - A miRNome analysis of blood cell subsets in lung cancer patients and healthy controls. Oncotarget. 2014;5:9484–9497. doi: 10.18632/oncotarget.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farrell D, et al. The identification of circulating miRNA in bovine serum and their potential as novel biomarkers of early mycobacterium avium subsp paratuberculosis infection. Plos One. 2015;10:e134310. doi: 10.1371/journal.pone.0134310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li R, et al. Transcriptome MicroRNA profiling of bovine mammary glands infected with Staphylococcus aureus. Int J Mol Sci. 2015;16:4997–5013. doi: 10.3390/ijms16034997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song N, et al. MicroRNA-214 regulates immunity-related genes in bovine mammary epithelial cells by targeting NFATc3 and TRAF3. Mol Cell Probe. 2017;35:27–33. doi: 10.1016/j.mcp.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, et al. Bovine miR-146a regulates inflammatory cytokines of bovine mammary epithelial cells via targeting the TRAF6 gene. J Dairy Sci. 2017;100:7648–7658. doi: 10.3168/jds.2017-12630. [DOI] [PubMed] [Google Scholar]
- 13.Lawless N, et al. Next generation sequencing reveals the expression of a unique miRNA profile in response to a gram-positive bacterial infection. Plos One. 2013;8:e57543. doi: 10.1371/journal.pone.0057543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, et al. Identification and characterization of novel and differentially expressed microRNAs in peripheral blood from healthy and mastitis Holstein cattle by deep sequencing. Anim Genet. 2014;45:20–27. doi: 10.1111/age.12096. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, et al. Expression differences of miRNAs and genes on NF-κB pathway between the healthy and the mastitis Chinese Holstein cows. Gene. 2014;545:117–125. doi: 10.1016/j.gene.2014.04.071. [DOI] [PubMed] [Google Scholar]
- 16.Lawless N, et al. MicroRNA regulation of bovine monocyte inflammatory and metabolic networks in an in vivo infection model. G3 (Bethesda). 2014;4:957–971. doi: 10.1534/g3.113.009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langmead B, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strandberg Y, et al. Lipopolysaccharide and lipoteichoic acid induce different innate immune responses in bovine mammary epithelial cells. Cytokine. 2005;31:72–86. doi: 10.1016/j.cyto.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Gilbert FB, et al. Differential response of bovine mammary epithelial cells to Staphylococcus aureus or Escherichia coli agonists of the innate immune system. Vet Res. 2013;44:40. doi: 10.1186/1297-9716-44-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu CJ, Lu LF. MicroRNA in immune regulation. Curr Top Microbiol Immunol. 2017;410:249–267. doi: 10.1007/82_2017_65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin W, et al. Transcriptome microRNA profiling of bovine mammary epithelial cells challenged with Escherichia coli or Staphylococcus aureus bacteria reveals pathogen directed microRNA expression profiles. BMC Genomics. 2014;15:181. doi: 10.1186/1471-2164-15-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LuoReng ZM, et al. Comparison of microRNA profiles between bovine mammary glands infected with Staphylococcus aureus and Escherichia coli. Int J Biol Sci. 2018;14:87–99. doi: 10.7150/ijbs.22498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang XP, et al. Expression patterns of miR-146a and miR-146b in mastitis infected dairy cattle. Mol Cell Probe. 2016;30:342–344. doi: 10.1016/j.mcp.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Mach N, et al. Integrated mRNA and miRNA expression profiling in blood reveals candidate biomarkers associated with endurance exercise in the horse. Sci Rep. 2016;6:22932. doi: 10.1038/srep22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao, Y. X. et al. MicroRNA-200a induces apoptosis by targeting ZEB2 in alcoholic liver disease. Cell Cycle., 1–13 (2018). [DOI] [PMC free article] [PubMed]
- 26.Wang XY, et al. MiR-200a expression in CD4+ T cells correlates with the expression of Th17/Treg cells and relevant cytokines in psoriasis vulgaris: A case control study. Biomed Pharmacother. 2017;93:1158–1164. doi: 10.1016/j.biopha.2017.06.055. [DOI] [PubMed] [Google Scholar]
- 27.Zarate-Neira LA, et al. Dysregulation of miR-155-5p and miR-200-3p and the anti-non-nilayer phospholipid arrangement antibodies favor the development of lupus in three novel murine lupus models. J Immunol Res. 2017;2017:8751642. doi: 10.1155/2017/8751642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Xu G, Han J, Xu T. miR-200a-3p regulates TLR1 expression in bacterial challenged miiuy croaker. Dev Comp Immunol. 2016;63:181–186. doi: 10.1016/j.dci.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Chakraborty N, et al. An integrated omics analysis: impact of microgravity on host response to lipopolysaccharide in vitro. BMC Genomics. 2014;15:659. doi: 10.1186/1471-2164-15-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang WC, et al. The lncRNA H19 promotes epithelial to mesenchymal transition by functioning as miRNA sponges in colorectal cancer. Oncotarget. 2015;6:22513–22525. doi: 10.18632/oncotarget.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li SP, et al. LncRNA HULC enhances epithelial-mesenchymal transition to promote tumorigenesis and metastasis of hepatocellular carcinoma via the miR-200a-3p/ZEB1 signaling pathway. Oncotarget. 2016;7:42431–42446. doi: 10.18632/oncotarget.9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leelahavanichkul A, et al. Serum miRNA-122 in acute liver injury induced by kidney injury and sepsis in CD-1 mouse models. Hepatol Res. 2015;45:1341–1352. doi: 10.1111/hepr.12501. [DOI] [PubMed] [Google Scholar]
- 33.Cui J, et al. miR-122 involved in the regulation of toll-like receptor signaling pathway after Vibrio anguillarum infection by targeting TLR14 in miiuy croaker. Fish Shellfish Immunol. 2016;58:67–72. doi: 10.1016/j.fsi.2016.09.027. [DOI] [PubMed] [Google Scholar]
- 34.Suojalehto H, et al. MicroRNA profiles in nasal mucosa of patients with allergic and nonallergic rhinitis and asthma. Int Forum Allergy Rhinol. 2013;3:612–620. doi: 10.1002/alr.21179. [DOI] [PubMed] [Google Scholar]
- 35.Wang S, et al. Functional cooperation of miR-125a, miR-125b, and miR-205 in entinostat-induced downregulation of erbB2/erbB3 and apoptosis in breast cancer cells. Cell Death Dis. 2013;4:e556. doi: 10.1038/cddis.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stittrich AB, et al. The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol. 2010;11:1057–1062. doi: 10.1038/ni.1945. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, et al. MicroRNA-146a-5p negatively regulates pro-inflammatory cytokine secretion and cell activation in lipopolysaccharide stimulated human hepatic stellate cells through inhibition of Toll-like receptor 4 signaling pathways. Int J Mol Sci. 2016;17:e1076. doi: 10.3390/ijms17071076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stone MJ, et al. Mechanisms of regulation of the chemokine-receptor network. Int J Mol Sci. 2017;18:e342. doi: 10.3390/ijms18020342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li MO, Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol. 2016;16:220–233. doi: 10.1038/nri.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao L, et al. Profilings of microRNAs in the liver of Common Carp (Cyprinus carpio) infected with flavobacterium columnare. Int J Mol Sci. 2016;17:566. doi: 10.3390/ijms17040566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, et al. Parsing the regulatory network between small RNAs and target genes in Ethylene pathway in tomato. Front Plant Sci. 2017;8:527. doi: 10.3389/fpls.2017.00527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friedländer MR, et al. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40:37–52. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagner GP, et al. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131:281–285. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 44.Romualdi C, et al. IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiol Genomics. 2003;12:159–162. doi: 10.1152/physiolgenomics.00096.2002. [DOI] [PubMed] [Google Scholar]
- 45.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitchell PS, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ioannidis J, Donadeu FX. Circulating miRNA signatures of early pregnancy in cattle. BMC Genomics. 2016;17:184. doi: 10.1186/s12864-016-2529-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bustin SA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 49.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 50.Betel D, et al. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rehmsmeier M, et al. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mao X, et al. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.