

Key Points
CRISPR-Cas9 library screening identifies CBS7/9 boundary that defines an aberrant HOXA chromatin domain and HOX gene transcription in AML.
Attenuation of CBS7/9 boundary impairs the leukemic transcription program and attenuates leukemic progressions in AML mouse models.
Abstract
HOX gene dysregulation is a common feature of acute myeloid leukemia (AML). The molecular mechanisms underlying aberrant HOX gene expression and associated AML pathogenesis remain unclear. The nuclear protein CCCTC-binding factor (CTCF), when bound to insulator sequences, constrains temporal HOX gene-expression patterns within confined chromatin domains for normal development. Here, we used targeted pooled CRISPR-Cas9–knockout library screening to interrogate the function of CTCF boundaries in the HOX gene loci. We discovered that the CTCF binding site located between HOXA7 and HOXA9 genes (CBS7/9) is critical for establishing and maintaining aberrant HOXA9-HOXA13 gene expression in AML. Disruption of the CBS7/9 boundary resulted in spreading of repressive H3K27me3 into the posterior active HOXA chromatin domain that subsequently impaired enhancer/promoter chromatin accessibility and disrupted ectopic long-range interactions among the posterior HOXA genes. Consistent with the role of the CBS7/9 boundary in HOXA locus chromatin organization, attenuation of the CBS7/9 boundary function reduced posterior HOXA gene expression and altered myeloid-specific transcriptome profiles important for pathogenesis of myeloid malignancies. Furthermore, heterozygous deletion of the CBS7/9 chromatin boundary in the HOXA locus reduced human leukemic blast burden and enhanced survival of transplanted AML cell xenograft and patient-derived xenograft mouse models. Thus, the CTCF boundary constrains the normal gene-expression program, as well as plays a role in maintaining the oncogenic transcription program for leukemic transformation. The CTCF boundaries may serve as novel therapeutic targets for the treatment of myeloid malignancies.
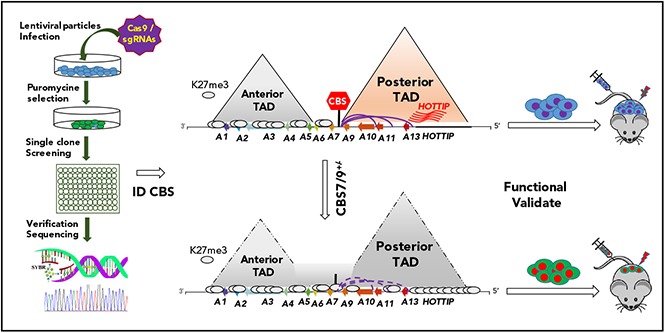
Visual Abstract
Introduction
Organization of the genome into separate topologically associated domains (TADs) modulates interactions between genes and regulatory elements. CCCTC-binding factor (CTCF) binds to TAD boundaries and constrains interactions of DNA elements that are located in neighboring TADs.1,2 Disruption of CTCF boundaries in the central region of the HOXA locus alters functional chromatin domain and gene expression in mouse embryonic stem (ES) cell differentiation. This suggests that CTCF-mediated TADs are structural components, as well as regulatory units that are required for proper enhancer action.3,4 Although CTCF-mediated TADs represent functional chromatin domains, genome-wide CTCF binding data have revealed that CTCF mostly interacts with the same DNA sites in different cell types.2,5,6 However, CTCF often functions as a chromatin barrier in one cell type but not in another.7 Whether and how these boundary elements (CTCF binding sites) are directly linked to their biological function remain largely unknown.
Abnormal HOX gene activation is a common feature of acute myeloid leukemia (AML).8,9 In healthy cells, the HOX genes, especially HOXA and HOXB genes, regulate ordinary hematopoietic stem and progenitor cell (HS/PC) function by controlling the balance between proliferation and differentiation.10-12 HOXA5–HOXA10 genes and anterior HOXB2–HOXB6 genes are highly expressed in HS/PCs and are downregulated during terminal differentiation.10,13-15 Dysregulation of HOXA9 or HOXA10 genes is a dominant mechanism of leukemic transformation by changing the self-renewal and differentiation properties of HS/PCs, thus leading to leukemic transformation.16,17 Additionally, overexpression of HOXA9 is a poor prognostic marker in leukemia patients,18,19 whereas low expression of HOXA9 and HOXB4 is a favorable predictor for AML patient outcome.9,20 Although HOXA9 and HOXA10 genes are aberrantly activated in many AML patients, the mechanism that establishes oncogenic expression patterns of HOXA genes and associated regulatory networks remains poorly understood.
In this study, we used a pooled CRISPR-Cas9–knockout (KO) screen to interrogate the CTCF binding motifs in all HOX loci. We identified a critical CTCF chromatin boundary (CBS7/9) located at the edge of a TAD encompassing the posterior HOXA genes. The CBS7/9 boundary maintains oncogenic expression of posterior HOXA genes. Reduced CBS7/9 boundary function leads to expansion of repressive chromatin structure into the posterior HOXA domain and blocks enhancer/promoter chromatin interaction networks, leading to decreases in posterior HOXA-associated oncogenic transcription and prolonged survival of transplanted AML mouse models. Thus, the CTCF boundary not only constrains the normal gene expression, but is hijacked to maintain an oncogenic transcription program for leukemic transformation. Because of this, we propose that CTCF boundaries may serve as novel therapeutic targets for the treatment of myeloid malignancies.
Methods
Patient samples and AML cell lines
Primary AML patient cells were obtained via approval of the Institutional Review Board of the University of Florida in accordance with the Declaration of Helsinki. MOLM13 and MV-4-11 cells were purchased from American Type Culture Collection repositories. All cell lines were verified by short tandem repeat analysis and tested for mycoplasma contamination.
CTCF sgRNA library design, cloning, and lentivirus production
The detailed protocol for CTCF single guide RNA (sgRNA) library generation is described in supplemental Materials and methods, available on the Blood Web site. The library consists of 1070 sgRNAs containing 303 random-gene targets, 500 nonhuman controls, 60 HOX loci-associated long intergenic noncoding RNA (lincRNA) targets, and 207 CTCF element targets in HOX loci. After generating high-titer viruses, MOLM13 cells were infected with sgRNA-pooled lentivirus at a multiplicity of infection of 0.3-0.4 and selected with puromycin for 2 days, and single cells were seeded into 96-well plates.
RNA, quantitative reverse transcription–polymerase chain reaction, RNA sequencing, and data analysis
Total RNA from AML cells and primary patient samples was extracted with TRIzol Reagent (Invitrogen). A total of 2 μg of RNA was subjected to reverse transcription with SuperScript II Reverse Transcriptase (Invitrogen) and analyzed by a Real-Time PCR Detection System (Bio-Rad). Paired-end RNA sequencing (RNA-seq) was performed by the University of Florida Interdisciplinary Center for Biotechnology Research core facility, according to standard protocols. All sequencing reads were processed and aligned to the human genome assembly (hg19) using TopHat (version 2.0) and Bowtie2.21-23 A detailed data analysis protocol is provided in supplemental Materials and methods. Sequence reads have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) under accession number GSE113191.
ChIP, ChIP-seq, ATAC-seq, and 4C-seq
Chromatin immunoprecipitation (ChIP) and ChIP sequencing (ChIP-seq) were performed as described previously.12,24 Assay for transposase-accessible chromatin using sequencing (ATAC-seq) was performed using a Nextera DNA Library Preparation Kit (Illumina), as described previously.25 4C sequencing (4C-seq) was performed as described previously.26 Additional methods and data analysis are provided in supplemental Materials and methods. All sequence reads have been deposited in the NCBI GEO under accession number GSE113191.
Xenotransplantation of human leukemic cells
Nonobese diabetic/LtSz severe combined immunodeficiency IL2Rγcnull (NSG) mice (6-8 weeks old; The Jackson Laboratory) were sublethally irradiated with 280 cGy total body irradiation and injected with wild-type (WT) control or CBS7/9+/− MOLM13 cells at a dose of 1 × 106 cells per mouse by tail vein injection. Primary AML cells (control or CBS7/9+/−) were injected at 1.8 × 105 cells per mouse. Peripheral blood (PB) was collected by retro-orbital bleeding. Bone marrow (BM) cells were isolated by flushing the bones. Spleens were mashed through a 70-μm mesh filter to prepare single-cell suspensions. PB was prepared for flow cytometry (fluorescence-activated cell sorting [FACS]) by ammonium chloride treatment to remove red cells. Human CD45 chimerism was analyzed by FACS (LSR II; BD Biosciences, San Jose, CA). Animals were used in accordance with a protocol approved by the Institutional Animal Care and Use Committees of the University of Miami and the University of Florida.
Results
Pooled CRISPR-Cas9–KO library genetic screening of CTCF boundaries
To gain new insights into the regulatory mechanisms associated with aberrant HOXA expression in AML, we examined histone-modification patterns across the HOXA locus in different subtypes of AML cells and primary patient samples. Active histone modifications, H3K4me3 (Figure 1A), H3K79me2 (Figure 1B), and H3K9ac (Figure 1C), are very low in the anterior HOXA domain and increased dramatically in the posterior HOXA domain, encompassing HOXA9 to HOXA13 genes in 3 subtypes of AML patients, including MLL rearrangement (Patient LPP4), NPM1C+/Flt3-ITD+ (Patient 974), and gain-of-copy MLL mutation (Patient 1306). Repressive H3K27me3 (Figure 1D) shows opposite patterns across the HOXA locus (Figure 1A-D). The transition from repressive to active chromatin occurs between the HOXA7 and HOXA9 genes (Figure 1; supplemental Figure 1A), where ChIP-seq revealed a strong CTCF binding peak (Figure 1E; supplemental Figure 1A-B).1 The unique chromatin patterns were further confirmed in MV4-11 cells with MLL-AF4 rearrangement (supplemental Figure 1D-E), which show consistent aberrant expression of posterior HOXA genes (Figure 1F; supplemental Figures 1C and 3A). These data suggest that the CTCF binding site located between HOXA7 and HOXA9 (CBS7/9) plays an important role in aberrant activation of posterior HOXA genes, which is required for AML pathogenesis.
Figure 1.

The CBS7/9 boundary demarcates active and repressive chromatin domain and maintains ectopic expression of posterior HOXA genes. ChIP analysis of H3K4me3 (A), H3K79me2 (B), H3K9ac (C), and H3K27me3 (D) across the HOXA locus in 3 subtypes of primary AML. Patient 974 possesses NPM1C+ and FLT3-ITD mutations, patient LPP4 contains MLL rearrangement, and patient 1306 has gain of MLL copy number. (E) ChIP-seq analysis of CTCF binding at the HOXA locus obtained from the NCBI GEO public database (accession number GSM1335528). (F) Quantitative reverse transcription–polymerase chain reaction (qRT-PCR) analysis of HOXA gene expression in 3 subtypes of primary AML (974, LPP4, and 1306).
To systematically examine the role of CTCF boundaries in HOX locus chromatin organization and gene transcription, we generated a pooled CRISPR-Cas9 lentivirus screening library targeting all potential CTCF sites in 4 HOX gene loci. We transduced MOLM13 cells carrying MLL-AF9 fusion at a low multiplicity of infection (0.3-0.4 MOI) to ensure that most selected cells contained a single sgRNA. After expansion, the resistant clones were screened for impairment of HOXA9 expression by reverse-transcriptase (RT)–droplet digital polymerase chain reaction (PCR) (Figure 2A). Of 528 survival clones screened, 10 clones exhibited a >50% reduction in HOXA9 levels (Figure 2B). sgRNAs integrated in the clones with reduced, unchanged, or increased HOXA9 levels were identified by PCR amplification of the sgRNA sequences using flanking vector primers, followed by Sanger sequencing (supplemental Table 1). Of 30 clones sequenced, 21 clones contained a single sgRNA (supplemental Table 1). They were grouped and analyzed for HOXA9 expression levels by the categories of sgRNA identified. A significant decrease in HOXA9 expression was found in the clones carrying sgRNA targeting the CBS7/9 site or HOTTIP lincRNA, but not in the nonhuman, random human genes, and other CTCF site controls (Figure 2C). Genotyping and SURVEYOR assays of the sgRNA-targeting CBS7/9 site revealed that the CBS7/9 mutation was present in 4 of 6 selected clones with reduced HOXA9 expression. Clones 5, 6, 28, and 121 were targeted by CBS7/9-specific sgRNA (Figure 2D; supplemental Figure 2A). However, clone 15 contained sgRNA specific to HOTTIP, whereas clone 31 contained several sgRNAs targeting HOAIRM1 lincRNA, HOTAIR lincRNA, and the HOXD9/10 CTCF site (Figure 2D; supplemental Figure 2A; supplemental Table 1). Among 10 clones with decreased levels of HOXA9 expression, 6 contained sgRNA targeting CBS7/9, and 3 contained sgRNA targeting HOTTIP (supplemental Table 1). It is known that HOTTIP lincRNA activates posterior HOXA gene expression by recruiting the mixed-lineage leukemia (MLL) complex.27
Figure 2.

Pooled CRISPR-Cas9–KO library screening identified a CBS7/9 boundary critical for posterior HOXA expression in AML cells. (A) Schematic diagram representing the pooled CRISPR-Cas9–KO library screening of the entire 4 HOX gene loci for CTCF boundary function in MLL-AF9–rearranged MOLM13 AML cells. (B) One-step RT-droplet digital PCR screening of HOXA9 expression in single clones infected with lentivirus containing the sgRNA library. Shown is the screening of 528 sgRNA library–infected clones for HOXA9 expression levels. (C) RT-droplet digital PCR analysis of HOXA9 levels in WT MOLM13 cells and the 21 clones containing single targeted sgRNA. HOXA9 expression data were grouped into 5 groups in accordance with the categories of sgRNA sequences: HOXA7/9 CTCF site, nonhuman targets, other CTCF sites in the HOX loci, HOX-associated lincRNAs, and other human targets. (D) The SURVEYOR nuclease assays of mutations occurred in the CBS7/9 site from the representative clones that exhibited reduced (red line), unchanged (blue line), or increased (purple line) levels of HOXA9 expression. The HOXA9-decreased clones 5, 6, 28, and 121 exhibited mutations in the CBS7/9 boundary. (E) ChIP analysis of CTCF binding across the HOXA locus in MOLM13 cells compared with the WT control and the CBS7/9+/− clone. Data are mean ± SD from 3 or 4 independent experiments. (F) Western blot analysis of CTCF protein levels compared with the WT control and the CBS7/9+/− MOLM13 clone. *P < .05, **P < .01, Student t test. ns, not significant.
Furthermore, ChIP analysis of 3 clones containing sgRNA targeting the CBS7/9 site (clones 5, 6, and 28) revealed a significant reduction in CTCF binding at the CBS7/9 site and other CTCF sites in the posterior domain (supplemental Figure 2B). Thus, CRISPR-Cas9 library screening revealed that the CBS7/9 site may act as a chromatin barrier to establish the posterior HOXA chromatin neighborhood and aberrant expression of HOXA genes in AML.
CBS7/9 is critical for maintaining the posterior HOXA chromatin neighborhood in AML
CTCF acts as a chromatin barrier to prevent the spreading of heterochromatin into euchromatin domains.7 To explore the role of the CBS7/9 site in posterior HOXA activation, we deleted the CBS7/9 boundary in MOLM13 and MV4-11 cells with 2 sgRNAs flanking 47 bp of the core CTCF motif of the CBS7/9 site (supplemental Figure 2C). MOLM13 and MV4-11 are MLL-rearranged AML cells that exhibit elevated levels of posterior HOXA genes (supplemental Figures 1C and 3A). ChIP analysis of CTCF recruitment revealed that heterozygous CBS7/9 KO (CBS7/9+/−) reduced CTCF binding in the CBS7/9 site, as well as in other CTCF sites in the posterior domain (Figure 2E). As a control, CBS7/9+/− does not alter global CTCF protein levels (Figure 2F). Although CBS7/9+/− also affects CTCF binding to the CBS5/6 site, CTCF binding in the CBS5/6 site is very weak in MOLM13 cells compared with other CTCF sites in the locus (Figure 2E). There was little to no effect on CTCF binding at the other anterior CTCF sites (Figure 2E). As a consequence, CBS7/9+/− significantly reduced the expression levels of posterior HOXA9-HOXA13 genes (Figure 3A; supplemental Figure 3A), whereas homozygous deletion of CBS7/9 resulted in a lethal phenotype in these 2 AML cells. In contrast, CBS5/6+/− affected CTCF binding to the CBS5/6 site but not HOXA gene expression in MOLM13 cells (supplemental Figures 2D and 3F-G).
Figure 3.

CBS7/9 boundary plays a critical role in maintaining the posterior HOXA chromatin neighborhood. (A) qRT-PCR analysis of HOXA gene expression in MOLM13 cells compared with the WT control and the CBS7/9+/− clone (upper panel). RNA-seq reads of HOXA gene transcripts comparing WT and CBS7/9+/− MOLM13 cells (lower panel). (B) ChIP-seq analysis of H3K27me3, H3K4me3, and H3K79me2 modifications in the HOXA locus in MOLM13 cells compared with the WT control and the CBS7/9+/− clone. (C) ChIP-seq analysis of H3K27me3, H3K4me3, and H3K79me2 modifications in the HOXD locus in MOLM13 cells compared with the WT control and the CBS7/9+/− clone.
We determined whether CBS7/9+/− affects TAD boundary and histone-modification patterns in the HOXA locus by ChIP-seq. Consistent with the loss of boundary function, deletion of 1 CBS7/9 allele expanded and elevated H3K27me3 levels from the anterior domain across the CBS7/9 boundary into posterior HOXA9-HOXA13 genes (Figure 3B), whereas H3K4me3 levels were significantly decreased in the same region in MLL-AF9–rearranged MOLM13 cells (Figure 3B). In contrast, CBS7/9+/− did not affect the anterior HOXA domain or HOXD locus (Figure 3B-C). ChIP assays confirmed the ChIP-seq data in MLL-AF4–rearranged MV4-11 cells (supplemental Figure 3B-E), indicating an invasion of facultative heterochromatin from the anterior domain into the active posterior domain that leads to transcriptional silencing of HOXA9-HOXA13 genes.
It was recently reported that subtypes of AML with MLL-AF rearrangements required DOT1L as a cofactor to aberrantly activate posterior HOXA genes by methylations of H3K79.28-30 We investigated whether partial loss of the CBS7/9 boundary affects H3K79me2 levels at the HOXA locus. Consistent with ectopic posterior HOXA gene expression in the MLL-rearranged AMLs, H3K79me2 levels were highly enriched in the posterior HOXA domain (Figure 3B; supplemental Figure 3D). CBS7/9+/− led to a significant decrease in H3K79me2 levels in this specific subtype of AML cells (Figure 3B; supplemental Figure 3D). Thus, the CBS7/9 boundary defines an oncogenic posterior HOXA chromatin domain for ectopic expression of HOXA9-HOXA13 genes in MLL-rearranged AML.
CBS7/9+/− perturbs enhancer/promoter chromatin accessibility and interaction networks
To test how the CBS7/9 boundary maintains ectopic posterior HOXA gene expression, we performed ATAC-seq, comparing WT and CBS7/9+/− AML cells. In MOLM13 cells, chromatin is primarily inaccessible in the HOXB, HOXC, and HOXD loci (supplemental Figure 4A-C). Partial deletion of the CBS7/9 site has little to no effect on chromatin accessibility in these loci (supplemental Figure 4A-C). In contrast, the HOXA locus is highly accessible in MOLM13 cells (Figure 4A). An attenuated CBS7/9 boundary resulted in a significant decrease in chromatin accessibility in the posterior HOXA domain but not in the anterior gene cluster (Figure 4A), suggesting that an altered CBS7/9 boundary perturbs chromatin structure and enhancer/promoter accessibility, leading to inhibition of posterior HOXA9-HOXA13 genes.
Figure 4.

CBS7/9+/− alters enhancer/promoter accessibility and interactome in the HOXA locus. (A-C) ATAC-seq analysis of the alteration in chromatin accessibility upon the heterozygous deletion of the CBS7/9 boundary site in MOLM13 cells. Shown are altered chromatin accessibility in the HOXA (A), RUNX1 (B), and ZEB1 (C) loci compared with the WT control and the CBS7/9+/− clone. (D) Long-range chromatin interactions from HOXA9 gene, as determined by 4C-seq analysis, compared with WT and CBS7/9+/− MOLM13 cells. The red arrow indicates that HOTTIP interacts with HOXA9 in WT cells, and the interaction is reduced in CBS7/9+/− cells. (E) Changed interaction between the HOXA9 and ZEB1 genes, as determined by 4C-seq analysis, compared with WT and CBS7/9+/− MOLM13 cells.
Although CBS7/9+/− does not significantly alter promoter accessibility globally, several genes involved in myeloid differentiation and proliferation were affected (Figure 4B-C). Among them, RUNX1 is a transcription factor that is critical for hematopoiesis and is mutated in 5% to 13% of AML cases.31,32 Distal (P1) and proximal (P2) promoters control transcription of RUNX1 isoforms that play nonredundant roles in hematopoiesis.33-35 The chromatin accessibility of P1 and P2 promoters was markedly decreased in CBS7/9+/− AML clones (Figure 4B). Furthermore, the promoter region and the 3′ untranslated region of the ZEB1 gene, which encodes an epithelial-mesenchymal transition (EMT) transcription factor involved in regulating MLL-rearranged AML blast migration and invasion,36 also lost chromatin accessibility in CBS7/9+/− cells (Figure 4C).
Given that CTCF-mediated chromatin organization plays an important role in regulating enhancer/promoter interaction networks within the confined TADs, we performed 4C-seq using the HOXA9 gene as a bait, comparing altered long-range chromatin interactions of WT and CBS7/9+/− MOLM13 cells. The 4C-seq data revealed that HOXA9 interacts with genes within the posterior TAD and also associates with genes within the anterior TAD (Figure 4D). The 4C-seq data were confirmed and quantitated by chromosome conformation capture (3C) quantitative PCR (supplemental Figure 4D-E). CBS7/9+/− decreases HOXA9 interaction networks, especially in HOXA9 proximal neighborhoods and the posterior domain (Figure 4D; supplemental Figure 4D). Consistent with the role of HOTTIP lincRNA in posterior HOXA gene activation,27 HOXA9 interacted with HOTTIP in WT control cells, and the interaction was significantly reduced upon CBS7/9+/− (Figure 4D; supplemental Figure 4D), suggesting that HOTTIP may cooperate with the CBS7/9 boundary to regulate posterior HOXA genes. In contrast, HOXA9 did not interact with the HOXB locus (Figure 4D, lower panel). Interestingly, an altered CBS7/9 boundary affected the interaction between HOXA9 and the ZEB1 upstream region (Figure 4E). Although the significance of the HOXA9 and ZEB1 interchromatin interaction remains unclear, the data suggest that HOXA9 and ZEB1 may be coregulated, because HOXA9 and EMT-related genes, including ZEB1, have been linked to a poor prognosis for AML.19,36 Nevertheless, our results demonstrate that the CBS7/9 boundary confines posterior topological domains to control enhancer/promoter chromatin accessibility and long-range chromatin interaction networks of the HOXA locus in AML cells.
Partial impairment of the CBS7/9 boundary prolongs survival of AML mouse models
HOXA9 and other posterior HOXA genes act as leukemic oncogenes that enhance self-renewal of leukemic stem cells in AML.37 To investigate the biological function of the CBS7/9 boundary in AML, we assessed the effects of CBS7/9+/− on leukemic cell growth and viability in AML cells carrying MLL rearrangements. Compared with WT AML cells, CBS7/9+/− led to a significant inhibition of cell proliferation in the different clones of MOLM13 and MV4-11 cells (Figure 5A; supplemental Figure 5A,C). Furthermore, cell cycle analysis revealed that CBS7/9+/− in MLL-AF9– and MLL-AF4–rearranged AML cells blocked cells in the G1 phase and significantly reduced the G2/M phases (Figure 5B; supplemental Figure 5B,D), suggesting that CBS7/9+/− inhibits AML cell proliferation by regulating cell cycle progression.
Figure 5.

Dysregulation of CBS7/9 boundary inhibits leukemic cell proliferation and prolongs survival time of transplanted NSG mice. (A) Proliferation curves of WT control and CBS7/9+/− MOLM13 cells were assessed using a cell viability count. (B) FACS analysis of the cell cycle was carried out using propidium iodide staining of WT control or CBS7/9+/− MOLM13 cells. (C) Survival of sublethally irradiated NSG mice (n = 6 per group) transplanted with WT control or CBS7/9+/− MOLM13 cells by tail vein injection. (D) FACS analysis of human CD45+ cell chimerism in BM, spleen, and PB from representative mice receiving control or CBS7/9+/− MOLM13 cells. (E) Percentages of human CD45+ cells in BM, spleen, and PB of NSG mice 16 days after transplantation of WT (n = 5) or CBS7/9+/− (n = 4) MOLM13 cells. (F) Anti-hCD45 immunohistochemical staining (IHS; brown) of femur and spleen sections from representative mice transplanted with WT or CBS7/9+/− MOLM13 cells for 16 days. Original magnification ×200. Data are mean ± SD from 3 or 4 independent experiments. *P < .05, **P < .01, Student t test.
To test whether CBS7/9+/− affects AML leukemogenesis in vivo, we used an AML transplantation mouse model in which WT or CBS7/9+/− MOLM13 cells were transplanted into irradiated NSG mice. All mice transplanted with WT MOLM13 cells died 13-18 days after transplantation, whereas the mice receiving CBS7/9+/− cells had a significantly prolonged survival time (24-30 days), presumably related to attenuated progression of myeloid leukemia (Figure 5C). Indeed, FACS analysis of human leukemic cells in different hematological tissues from recipients at day 16 after transplantation revealed that human CD45+ leukemic cell chimerism was significantly reduced in mice receiving CBS7/9+/− MOLM13 cells compared with WT-MOLM13 cells, from an average of 83.3% to 60.9% in BM, 67.0% to 25.1% in spleen, and 57.9% to 25.2% in PB (Figure 5D-E). Consistently, immunohistochemical staining of femur and spleen sections with anti-hCD45 antibody (brown, 200×) demonstrated that mice transplanted with CBS7/9+/− MOLM13 cells had dramatically decreased infiltration of human CD45+ AML blasts in BM and spleen compared with mice receiving WT MOLM13 cells (Figure 5F).
Further, the CBS7/9 site was also targeted in primary BM cells from an AML patient with MLL rearrangement (Patient LPP4) by direct transfection of CRISPR RNAs (crRNAs) targeting the CBS7/9 site and Cas9 nuclease using a Neon Transfection System. CBS7/9+/− was confirmed by PCR-based genotyping and sequencing (supplemental Figure 6A-B). Again, CBS7/9+/− in a primary AML patient sample impaired HOXA9-HOXA13 gene expression but not anterior genes (supplemental Figure 6C). We then transplanted 1.8 × 105 WT or CBS7/9+/− primary AML patient cells into NSG mice. Interestingly, mice receiving control AML patient cells died within 34 days after transplantation, whereas mice transplanted with CBS7/9+/− patient cells remained healthy for up to 60 days. FACS revealed that CBS7/9+/− dramatically decreased the human CD45+ cell chimerism in BM and PB of recipients (supplemental Figure 6D). Furthermore, hematoxylin and eosin staining (upper panels) and immunohistochemical staining with anti-hCD45 (lower panels; brown) of BM sections showed that CBS7/9+/− decreased the infiltration of human CD45+ AML cells in patient-derived xenograft mouse BM (supplemental Figure 6E). Thus, our AML patient-derived xenograft data indicated that attenuation of the CBS7/9-mediated chromatin boundary decreases tumor burden and attenuates leukemic progression in vivo, perhaps by disrupting the oncogenic posterior topological HOXA domain and gene expression required for pathogenesis of AML.
Dysregulation of the CBS7/9 boundary perturbs oncogenic transcription programs
Next, we sought to delineate mechanisms by which CBS7/9+/− attenuates leukemogenesis. We compared genome-wide transcriptome changes between WT control and CBS7/9+/− MOLM13 cells by performing RNA-seq analysis. A total of 865 genes exhibited more than twofold decreases in mRNA levels, whereas 623 genes had increased expression upon CBS7/9+/− (Figure 6A; supplemental Figure 7A). Gene ontology analysis revealed that pathways involved in myeloid/leukocyte activation and differentiation, as well as cellular differentiation and survival, are specifically affected (Figure 6B). Among downregulated genes, HOTTIP and posterior HOXA genes were significantly reduced (Figure 6A; supplemental Figure 7A-B). A subset of genes involved in myeloid malignancies was further verified by qRT-PCR (Figure 6C). In addition, genes involved in regulating the pluripotency of stem cells, the cell cycle, interleukin-2 signaling, and JAK-STAT signaling, which play a critical role in myeloid leukemia, are specifically impaired in CBS7/9+/− MOLM13 cells (Figure 6D). Perturbation of the cell cycle pathway is consistent with the observation that attenuation of the CBS7/9 boundary blocks cell cycle progression (Figure 5B; supplemental Figure 5B,D). When we subjected RNA-seq data to Gene Set Enrichment Analysis (GSEA) using the molecular signature database, the top-ranking genes are those involved in the MAPK signaling pathway and myeloid/leukocyte differentiation and migration (Figure 6E-F; supplemental Figure 7C). The MAPK signaling pathway is required for leukemic cell survival and uncontrolled proliferation and is downregulated in CBS7/9+/− cells (Figure 6E). It is particularly interesting that the downregulated genes enriched in CBS7/9+/− MOLM13 cells are involved in the CTCF functional pathway, which plays an essential role in genome organization and establishment of the chromatin boundary (supplemental Figure 7C, left). Intriguingly, upregulated genes enriched in CBS7/9+/− cells are those involved in myeloid/leukocyte differentiation and migration (Figure 6F; supplemental Figure 7C, right), suggesting that attenuation of the CBS7/9 boundary inhibits myeloid cell proliferation and promotes differentiation, perhaps through downregulating posterior HOXA genes.
Figure 6.

Dysregulation of CBS7/9 boundary perturbs myeloid oncogenic transcription programs. (A) Heat map of RNA-seq analysis shows upregulated and downregulated genes upon heterozygous deletion of CBS7/9 in MOLM13 cells. (B) The CBS7/9 boundary–regulated genes were analyzed and annotated by Gene Ontology analysis. (C) qRT-PCR analysis and confirmation of representative downregulated genes identified in the RNA-seq analysis. (D) Heat map analysis of the affected pathways critical for HS/PC function upon heterozygous deletion of CBS7/9 in MOLM13 cells. (E) Enrichment of downregulated target genes involved in the MAPK signaling pathway in CBS7/9+/− cells compared with WT control, as shown by GSEA. (F) Enrichment of upregulated target genes involved in the pathway important for myeloid and leukocyte migration and differentiation in CBS7/9+/− cells compared with WT control, as shown by GSEA. (G) Overlap between downregulated (left) and upregulated (right) genes by RNA-seq analysis, and global decreases (left) and increases (right) in promoter chromatin accessibility by ATAC-seq analysis.
Upon attenuation of the CBS7/9 boundary, 42% of downregulated genes exhibited loss of promoter chromatin accessibility (Figure 6G, left; supplemental Figure 7D), whereas 48% of upregulated genes showed gain of promoter chromatin accessibility (Figure 6G, right). When we compared decreased genes upon CBS7/9+/− with the publicly available HOXA9–knock-down (KD) downregulated gene list, we noted that a subset of genes downregulated by CBS7/9+/− is involved in the HOXA9 regulatory pathway (Figure 7A). About 16% of HOXA9-activated genes were downregulated by CBS7/9 attenuation (Figure 7B). Thus, attenuation of the chromatin boundary disrupts the active chromatin domain and perturbs oncogenic gene expression in AML, in part by disrupting the HOXA9 oncogenic pathway.
Figure 7.

CTCF boundary regulates HOXA locus chromatin structure and gene expression. (A) Enrichment of downregulated genes involved in the HOXA9 regulatory pathway in CBS7/9+/− MOLM13 cells compared with WT control, as shown by GSEA. (B) Overlap between downregulated genes, as determined by comparing RNA-seq data obtained from CBS7/9+/− cells and the HOXA9-KD gene expression data set (accession number GSE13714). (C) A model depicts that the CBS7/9 chromatin boundary defines an aberrant posterior HOXA chromatin domain and coordinates oncogenic transcription programs for leukemic transformation and invasion.
Discussion
In this study, we designed a targeted CRISPR-Cas9 library to screen for the function of CTCF chromatin boundaries in HOX gene regulation, with the goal of identifying a critical chromatin boundary for establishing and maintaining aberrant posterior HOXA gene expression. Our work provides novel insight into relationships among CTCF boundary, chromatin domain organization, and oncogenic transcription regulation in myeloid leukemogenesis.
The genome-scale CRISPR-Cas9–KO screen provides an effective loss-of-function validation of annotated genes attributed to their biological phenotypes when targeting coding regions.38,39 One challenge for functional genomics is to efficiently and functionally validate all annotated genetic noncoding regulatory elements in normal and disease states. CTCF binds to boundaries of topological domains and restricts the influence of neighboring genome regions by regulating enhancer action within a defined genome neighborhood.1 However, genome-wide CTCF binding data revealed that, although CTCF can bind to the same locus in different cell types, it often functions as a boundary in 1 cell type but not in another.7 How boundary elements are directly linked to its biological function remains largely unknown. Our data demonstrated that the CBS7/9 boundary located at the edge of the TAD encompassing the posterior HOXA genes establishes and maintains aberrant chromatin signatures and expression of the posterior HOXA genes to facilitate myeloid leukemogenesis (Figure 7).
The role of CTCF in genome organization and gene regulation has been well-established.2,5,40 In mammalian genomes, CTCF was widely implicated in demarcating the individual TAD boundaries in a way that is consistent with its ability to block enhancer/promoter interactions across its binding sites.1 Alteration of the CTCF-associated boundaries impairs limb and embryonic development by dysregulating gene interactions in TADs.3,6 In differentiated mouse ES cells, deletions of CBS5/6 and CBS7/9 boundaries led to expansion of active H3K4me3 into the adjacent repressive HOXA10 gene.4 In contrast to mouse ES cell differentiation, in which medial CTCF boundaries insulate facultative heterochromatin from invasion of impinging euchromatin for distinct anterior and posterior HOXA expression pattern,4 the CBS7/9 boundary prevents invasion of H3K27me3 facultative heterochromatin into the posterior euchromatin domain and maintains aberrant posterior HOXA gene expression in MLL-rearranged AML. Disruption of this oncogenic boundary resulted in major changes in chromatin structure, leading to silencing of HOXA9-HOXA13 genes that eventually blocks progression of AML (Figure 7). Thus, our data suggested that CTCF-mediated chromatin reorganization may also play a role in aberrant oncogene activation and leukemogenesis. Alternatively, as a chromatin boundary for normal HOXA gene regulation during development, the CBS7/9 boundary is hijacked by the oncogenic pathway to establish aberrant HOXA gene expression that is similar to the role of CTCF in the c-Myc locus.41
HOXA and HOXB genes are critical for maintaining the balance between self-renewal and differentiation of hematopoietic stem cells.10-12,42 Although overexpression of the posterior HOXA genes in AML has been attributed to specific chromosomal rearrangements involved in MLL and linked to leukemic transformation,17,43 the CBS7/9 boundary also plays a critical role in maintaining an AML-specific posterior chromatin domain that drives ectopic expression of HOXA9-HOXA13 genes. Interestingly, the CBS7/9 boundary modulates HOTTIP lincRNA (Figure 6), which recruits the MLL1 complex to maintain active H3K4me3 chromatin and activate posterior HOXA genes.27 Thus, 1 hypothesis is that the CBS7/9 boundary and HOTTIP cooperate to organize the chromatin domain and to coordinate posterior HOXA gene activation in AML. 4C-seq revealed that CBS7/9 attenuation blocks long-range interactions between HOXA9 and HOTTIP (Figure 4D), supporting the idea that HOTTIP may be involved in posterior HOXA gene regulation in AML.
Within posterior HOXA genes, HOXA9 is overexpressed in >50% of AML cases, and its ectopic expression is linked to poor outcome and prognosis.19 In MLL-AF9– or HOXA9-driven leukemia, HOXA9 acts to control myeloid leukemia stem cell self-renewal through recruiting epigenetic regulators.44 Activation of the HOXA13 gene also promotes tumorigenesis and metastasis in several solid tumors.45,46 The EMT genes were recently shown to control AML blast migration and invasion, linking to aggressiveness and poor prognostic outcomes for MLL-AF9–mediated AML.36 Thus, it is conceivable that a reduction in EMT genes and HOXA genes by CBS7/9 disruption prevents human AML invasion and progression, which prolongs the survival time of transplanted AML mouse models. However, it remains to be determined how the CBS7/9 boundary regulates chromatin structure and gene transcription of these non-HOXA genes.
Our data demonstrate that a normal CTCF boundary is hijacked to control oncogenic chromatin domain and transcription profiles for leukemic transformation and progression. Together, they constrain normal gene expression program, as well as coordinate oncogenic programs for leukemic transformation and invasion. The CTCF boundaries in the oncogene loci, such as the CBS7/9 boundary, may serve as novel therapeutic targets for treatment of myeloid malignancies.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the University of Florida Interdisciplinary Center for Biotechnology Research DNA sequencing core for Illumina sequencing, the Satellite Histological Core of the Sylvester Comprehensive Cancer Center Core facility at the University of Miami Miller School of Medicine for histological processing, and the Qiu and Huang laboratories for helpful discussions. The authors also thank Rachael Mills (Penn State Hershey College of Medicine) for editing the manuscript.
This work was supported by grants from the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (R01DK110108) (S.H.), National Cancer Institute (R01CA204044 [S.H.] and R01CA172408 [M.X.]), and National Heart, Lung, and Blood Institute (R01HL112294) (M.X.), the American Heart Association (16GRANT31020032) (S.H.), as well as a University of Florida Health Cancer Center Bridge grant (S.H.) and a Florida Academic Cancer Center Alliance Award (S.H. and M.X.). B.C. is supported by a key Medical Project of Jiangsu Province (BL2014078).
Footnotes
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE113191).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: H. Luo, F.W., B.C, Y.Q., M.X., and S.H. conceived and designed experiments; H. Luo, F.W., B.Y., J.Z, and Q.D. performed experiments; H. Luo, H. Li, and I.C. performed bioinformatics and statistical analyses; Q.D., B.Y., F.Y., and M.X. established the transplantation AML mouse model and performed in vivo assays; H. Luo, A.S., and C.V. designed and established the CRISPR-Cas9 sgRNA library and screening; L.D., B.X., and C.C. provided critical reagents and analyzed human patient samples with guidance from S.D.N. and J.L; H. Luo, S.H., Y.Q., and M.X. wrote the original draft, with input from S.D.N.; and J.L. revised and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for H. Luo and S.H. is Department of Pediatrics, Pennsylvania State University Hershey College of Medicine, Hershey, PA.
Correspondence: Boan Chen, Department of Hematology and Oncology, Zhongda Hospital, Southeast University Medical School, DingJiaQiao 87, Nanjing, Jiangsu 210009, China; e-mail: bachen@seu.edu.cn; Mingjiang Xu, Department of Biochemistry and Molecular Biology, University of Miami Miller School of Medicine, 1011 NW 15th St, Room 411, Gautier Building, MC R629, Miami, FL 33136-1019; e-mail: mxx51@miami.edu; and Suming Huang, Department of Pediatrics, Division of Pediatric Hematology/Oncology, Pennsylvania State University College of Medicine, H085, Suite C7830J, 500 University Dr, Hershey, PA 17033; e-mail: shuang4@pennstatehealth.psu.edu.
REFERENCES
- 1.Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137(7):1194-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Narendra V, Bulajić M, Dekker J, Mazzoni EO, Reinberg D. CTCF-mediated topological boundaries during development foster appropriate gene regulation [published correction appears in Genes Dev. 2016;31(16):1714]. Genes Dev. 2016;30(24):2657-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narendra V, Rocha PP, An D, et al. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science. 2015;347(6225):1017-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang Z, Luo OJ, Li X, et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell. 2015;163(7):1611-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lupiáñez DG, Kraft K, Heinrich V, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161(5):1012-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19(1):24-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drabkin HA, Parsy C, Ferguson K, et al. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia. 2002;16(2):186-195. [DOI] [PubMed] [Google Scholar]
- 9.Andreeff M, Ruvolo V, Gadgil S, et al. HOX expression patterns identify a common signature for favorable AML. Leukemia. 2008;22(11):2041-2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dou DR, Calvanese V, Sierra MI, et al. Medial HOXA genes demarcate haematopoietic stem cell fate during human development. Nat Cell Biol. 2016;18(6):595-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawrence HJ, Christensen J, Fong S, et al. Loss of expression of the Hoxa-9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood. 2005;106(12):3988-3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng C, Li Y, Liang S, et al. USF1 and hSET1A mediated epigenetic modifications regulate lineage differentiation and HoxB4 transcription. PLoS Genet. 2013;9(6):e1003524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rawat VP, Humphries RK, Buske C. Beyond Hox: the role of ParaHox genes in normal and malignant hematopoiesis. Blood. 2012;120(3):519-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spencer DH, Young MA, Lamprecht TL, et al. Epigenomic analysis of the HOX gene loci reveals mechanisms that may control canonical expression patterns in AML and normal hematopoietic cells. Leukemia. 2015;29(6):1279-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taghon T, Thys K, De Smedt M, et al. Homeobox gene expression profile in human hematopoietic multipotent stem cells and T-cell progenitors: implications for human T-cell development. Leukemia. 2003;17(6):1157-1163. [DOI] [PubMed] [Google Scholar]
- 16.Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013;27(5):1000-1008. [DOI] [PubMed] [Google Scholar]
- 17.Rice KL, Licht JD. HOX deregulation in acute myeloid leukemia. J Clin Invest. 2007;117(4):865-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531-537. [DOI] [PubMed] [Google Scholar]
- 19.Collins CT, Hess JL. Role of HOXA9 in leukemia: dysregulation, cofactors and essential targets. Oncogene. 2016;35(9):1090-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zangenberg M, Grubach L, Aggerholm A, et al. The combined expression of HOXA4 and MEIS1 is an independent prognostic factor in patients with AML. Eur J Haematol. 2009;83(5):439-448. [DOI] [PubMed] [Google Scholar]
- 21.Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Schulz VP, Deng C, et al. Setd1a and NURF mediate chromatin dynamics and gene regulation during erythroid lineage commitment and differentiation. Nucleic Acids Res. 2016;44(15):7173-7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109:21.29.1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stadhouders R, Kolovos P, Brouwer R, et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat Protoc. 2013;8(3):509-524. [DOI] [PubMed] [Google Scholar]
- 27.Wang KC, Yang YW, Liu B, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472(7341):120-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deshpande AJ, Deshpande A, Sinha AU, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell. 2014;26(6):896-908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117(25):6912-6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuntimaddi A, Achille NJ, Thorpe J, et al. Degree of recruitment of DOT1L to MLL-AF9 defines level of H3K79 di- and tri-methylation on target genes and transformation potential. Cell Reports. 2015;11(5):808-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marcucci G, Haferlach T, Döhner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29(5):475-486. [DOI] [PubMed] [Google Scholar]
- 32.Goyama S, Schibler J, Cunningham L, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013;123(9):3876-3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bee T, Swiers G, Muroi S, et al. Nonredundant roles for Runx1 alternative promoters reflect their activity at discrete stages of developmental hematopoiesis. Blood. 2010;115(15):3042-3050. [DOI] [PubMed] [Google Scholar]
- 34.Pozner A, Lotem J, Xiao C, et al. Developmentally regulated promoter-switch transcriptionally controls Runx1 function during embryonic hematopoiesis. BMC Dev Biol. 2007;7(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sroczynska P, Lancrin C, Kouskoff V, Lacaud G. The differential activities of Runx1 promoters define milestones during embryonic hematopoiesis. Blood. 2009;114(26):5279-5289. [DOI] [PubMed] [Google Scholar]
- 36.Stavropoulou V, Kaspar S, Brault L, et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell. 2016;30(1):43-58. [DOI] [PubMed] [Google Scholar]
- 37.Thorsteinsdottir U, Mamo A, Kroon E, et al. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood. 2002;99(1):121-129. [DOI] [PubMed] [Google Scholar]
- 38.Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343(6166):80-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dowen JM, Fan ZP, Hnisz D, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159(2):374-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuijers J, Manteiga JC, Weintraub AS, et al. Transcriptional dysregulation of MYC reveals common enhancer-docking mechanism. Cell Reports. 2018;23(2):349-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng C, Li Y, Zhou L, et al. HoxBlinc RNA recruits Set1/MLL complexes to activate Hox gene expression patterns and mesoderm lineage development. Cell Reports. 2016;14(1):103-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer C, Kowarz E, Hofmann J, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23(8):1490-1499. [DOI] [PubMed] [Google Scholar]
- 44.Zhu N, Chen M, Eng R, et al. MLL-AF9- and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J Clin Invest. 2016;126(3):997-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin C, Wang Y, Wang Y, et al. Transcriptional and posttranscriptional regulation of HOXA13 by lncRNA HOTTIP facilitates tumorigenesis and metastasis in esophageal squamous carcinoma cells. Oncogene. 2017;36(38):5392-5406. [DOI] [PubMed] [Google Scholar]
- 46.Li Z, Zhao X, Zhou Y, et al. The long non-coding RNA HOTTIP promotes progression and gemcitabine resistance by regulating HOXA13 in pancreatic cancer. J Transl Med. 2015;13(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.