

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
PGN infusion mimics the systemic inflammation and coagulopathy observed in late-stage B anthracis challenge.
PGN directly activates the extrinsic coagulation and promotes contact pathway amplification in nonhuman primates.
Abstract
Anthrax infections exhibit progressive coagulopathies that may contribute to the sepsis pathophysiology observed in fulminant disease. The hemostatic imbalance is recapitulated in primate models of late-stage disease but is uncommon in toxemic models, suggesting contribution of other bacterial pathogen-associated molecular patterns (PAMPs). Peptidoglycan (PGN) is a bacterial PAMP that engages cellular components at the cross talk between innate immunity and hemostasis. We hypothesized that PGN is critical for anthrax-induced coagulopathies and investigated the activation of blood coagulation in response to a sterile PGN infusion in primates. The PGN challenge, like the vegetative bacteria, induced a sepsis-like pathophysiology characterized by systemic inflammation, disseminated intravascular coagulation (DIC), organ dysfunction, and impaired survival. Importantly, the hemostatic impairment occurred early and in parallel with the inflammatory response, suggesting direct engagement of coagulation pathways. PGN infusion in baboons promoted early activation of contact factors evidenced by elevated protease-serpin complexes. Despite binding to contact factors, PGN did not directly activate either factor XII (FXII) or prekallikrein. PGN supported contact coagulation by enhancing enzymatic function of active FXII (FXIIa) and depressing its inhibition by antithrombin. In parallel, PGN induced de novo monocyte tissue factor expression in vitro and in vivo, promoting extrinsic clotting reactions at later stages. Activation of platelets further amplified the procoagulant state during PGN challenge, leading to DIC and subsequent ischemic damage of peripheral tissues. These data indicate that PGN may be a major cause for the pathophysiologic progression of Bacillus anthracis sepsis and is the primary PAMP behind the pathogen-induced coagulopathy in late-stage anthrax.
Visual Abstract
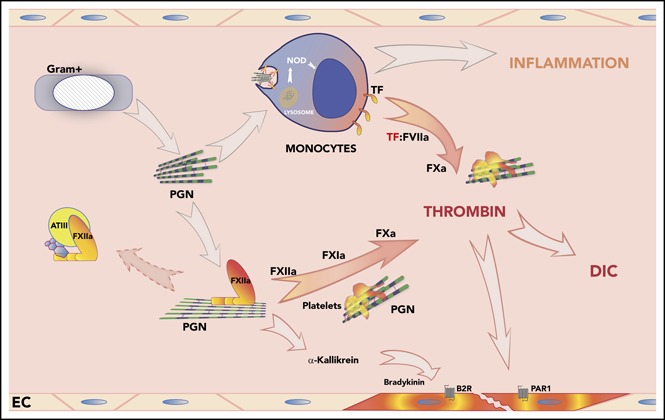
Introduction
Fulminant anthrax infections are characterized by high morbidity and mortality,1,2 stressing the need for better understanding of late-stage anthrax pathophysiology. Anthrax toxins are critical for the initial seeding and progression of the infection3-5 but toxemic models do not recapitulate the late-stage features, especially the progressive coagulopathy and associated organ damage observed in patients.6 We have previously shown that intravascular infusion of vegetative bacteria in primates induces a sepsis-like manifestation characterized by systemic inflammation and progressive coagulopathy, leading to disseminated intravascular coagulation (DIC), multiple organ failure (MOF), and death.7 Multiple bacterial pathogen-associated molecular patterns (PAMPs) may contribute to these manifestations. Although anthrax toxins also promote MOF,8 the mechanisms behind anthrax-induced coagulopathy and the associated tissue damage are poorly understood.
Overt DIC has been associated with high mortality rates in clinical settings including invasive infections, trauma, and cancer.9 Monocyte expression of tissue factor (TF) and activation of the extrinsic coagulation cascade are critical for DIC,10-12 whereas the contact coagulation pathway may contribute to the early hemostatic imbalance.13,14 Lipopolysaccharide (LPS) is the main driver of DIC in gram-negative sepsis,15-18 but gram-positive PAMPs that induce coagulopathies are less understood. Unlike anthrax toxins, peptidoglycan (PGN) has been shown to associate with coagulopathic markers in rats,19 but the underlying mechanisms have not been investigated. Systemic inflammation, shock, and MOF were observed in rodents challenged with Staphylococcus aureus cell wall preparations containing teichoic acids and PGN,20,21 but either component alone failed to fully replicate the pathophysiology.20-23
PGN, the main component of the cell wall of gram-positive bacteria,24 is a polymer of linear glycan chains with repeating monomeric disaccharide units of N-acetyl glucosamine and N-acetyl muramic acid. The glycan chains are crosslinked by a variable polypeptide stem bridge. In vitro, we have shown that, unlike mouse cells,25 human and nonhuman primate (NHP) monocytes respond vigorously to purified Bacillus anthracis PGN by producing proinflammatory cytokines,25-28 highlighting model-specific limitations.29 Anthrax PGN is not a Toll-like receptor (TLR) ligand30 but rather signals through intracellular pattern recognition nucleotide-binding oligomerization receptors 1 and 2 (NOD1 and NOD2).31,32 PGN signaling requires recognition of opsonized PGN particles,27,33 internalization, and lysosomal processing.25 Digested PGN fragments but not the polymeric structures activate NOD sensors.28 Alternatively, immunoglobulin G (IgG) or serum amyloid P opsonized PGN can activate cellular FcγR receptors27,33 and this mechanism is partially responsible for the platelet activation we reported.34 PGN-activated platelets exhibit procoagulant responses including aggregation and exposure of anionic phospholipids.34 Thus, PGN interacts with critical cellular components of hemostasis.
We hypothesized that PGN is the bacterial PAMP responsible for the coagulopathy we saw in the NHP model of late-stage anthrax.6,7 Here, we tested this hypothesis and investigated the mechanisms underlying clotting responses to anthrax PGN. We found that, like the vegetative bacteria, vascular infusion of anthrax PGN induced both systemic inflammation and a concomitant progressive coagulopathy leading to overt DIC. Early hemostatic imbalance involved activation of the contact pathway, whereas TF-dependent extrinsic coagulation dominated later stages. PGN did not activate the contact pathway directly but promoted proteolytic amplification of this cascade. Anthrax PGN by itself induced de novo monocyte TF expression and procoagulant activity. Platelet activation further amplified the procoagulant state leading to impaired hemostasis, intravascular and extravascular fibrin deposition, MOF, and ultimately death. Our data show that PGN is a major contributor to the septic pathophysiology in late-stage anthrax.
Methods
NHP model
Animal study protocols were approved by the Institutional Animal Care and Use Committees at both Oklahoma Medical Research Foundation (OMRF) and the University of Oklahoma Health Sciences Center (OUHSC). We used healthy Papio anubis baboons, males and females, 3 to 4 years old, 6- to 10-kg body weight, with normal leukocyte counts. Animals were challenged by IV infusion of highly pure B anthracis PGN, with no TLR-signaling capacity. PGN purification methodology and analytical assays were detailed recently30 and are summarized in supplemental Methodology (available on the Blood Web site). Two experimental groups were studied: (1) a high-dose PGN challenge (37.5 mg/kg, n = 6) and (2) a low-dose PGN challenge (8.5 mg/kg, n = 3). Blood samples and physiologic parameters were collected before (T0) and at defined time points after the PGN challenge. Animals were continuously monitored and humanely euthanized when conditions deteriorated or when they reached the primary end point (7 days survival).7,35 Blood and tissue samples were collected at end point for biochemical and histopathologic analysis.7,12,36 One high-dose animal deteriorated rapidly after the PGN challenge and was euthanized at 2 hours; it was excluded from the serial analysis of coagulopathy (first 24 hours) due to lack of paired samples, although it is included in the end-point histopathologic analysis.
Coagulation tests
Activated partial thromboplastin times (aPTTs), prothrombin times (PTs), fibrinogen, fibrin degradation products (FDPs), thrombin-antithrombin (TAT) complexes, and D-dimers were measured as reported.14,36 Other coagulation protease-antithrombin (AT) complexes were measured by enzyme-linked immunosorbent assay (ELISA) after coating with affinity-purified antibodies against the protease (clone 9G3 monoclonal anti-human factor XII [FXII], clone 1A6 monoclonal anti-human FXI,37 affinity-purified goat anti-human FVII [R&D Systems, Minneapolis, MN]) and detection with biotinylated sheep anti-human AT (Affinity Biologicals, Ancaster, ON, Canada). Kallikrein-C1 inhibitor (C1inh) complexes were detected after coating with the KOK12 monoclonal anti-human C1inh38,39 and detection with a biotinylated anti-human kallikrein monoclonal (clone 13G11; Pierce). The methodology is detailed in supplemental Methodology. Complexes were quantified using a standard curve obtained by in vitro activation of lepirudin-anticoagulated pooled normal baboon plasma. Protease-serpin complexes generated in vitro using aPTT (for FXIIa-AT, FXIa-AT, and kallikrein-C1inh) or PT reagents (FVIIa-AT) for 1 hour at 37°C were considered 1 U/mL.
PGN effect on contact coagulation
Purified human contact factors and AT were obtained from Enzyme Research Laboratories (South Bend, IN) or Haematologic Technologies (Essex Junction, VT). Biotinylated FXII and prekallikrein were incubated with PGN-coated plates and binding constants were estimated from the best-fit curves using Prism (GraphPad Software, La Jolla, CA). PGN effect on contact factors autoactivation, FXIIa, and kallikrein proteolytic activities, was assessed using the p-nitroaniline (pNA) containing S2302 chromogenic substrate (DiaPharma, West Chester, OH). Specific absorbance changes were converted into productivity (micromolar pNA) using pNA extinction coefficient 9920 M-1cm-1.40,41 Initial velocities (V0 expressed as micromolar pNA per minute) were assessed from the initial linear fit of productivity curves. The PGN effect on specific enzyme activities is reported as micromolar pNA per minute per picomole enzyme. Antithrombin (50-2000 nM) inhibition of FXIIa activity (100 nM) was assessed in the presence or absence of a constant concentration of heparin (200 µg/mL ≈ 40 inhibitory U/mL) by quantifying the residual FXIIa activity. The AT concentration that inhibited half of the FXIIa activity (IC50) was estimated from the nonlinear best-fit curves using Prism. The PGN effect on AT-heparin–mediated inhibition of FXIIa was assessed at 2 different PGN concentrations (50 and 200 μg/mL).
Biochemical tests
Serum metabolites indicative of organ function were measured using diagnostic profile rotors and the VetScan VS2 analyzer (Abaxis Veterinary Diagnostics, Union City, CA). Circulating cytokines were measured using the Milliplex Map NHP cytokine magnetic bead panel (EMD Millipore, Billerica, MA).14
Vascular permeability
Vascular permeability was investigated by IV injection of Alexa Fluor 594–conjugated bovine serum albumin (Invitrogen, Carlsbad, CA) 15 minutes before the PGN challenge. Residual circulating fluorescent albumin was quantified using a FLUOstar Omega microplate reader (BMG Labtech, Cary, NC).
Microscopic analysis
Histopathologic analysis and scoring were performed on formalin-fixed, paraffin-embedded sections stained with phosphotungstic acid–hematoxylin (PTAH), Prussian Blue, or hematoxylin and eosin (H&E) by a veterinary pathologist blinded to the experimental condition.7,14,36 Immunofluorescence microscopy was performed on paraformaldehyde-fixed, optimum cutting temperature media–embedded cryosections or methanol-fixed blood smears as described.14,36 Whole-mount en face staining of aortic endothelium was previously described.12,42 For comparison, we used time-paired (2 hours) historical control samples from a saline-infused baboon.12 For immunofluorescence microscopy, we used HTF-1 monoclonal anti-human TF (Miltenyi Biotec, Auburn, CA), PAC-1 monoclonal against active GPIIbIIIa (BD Biosciences, San Jose, CA), BV9 monoclonal anti-human vascular endothelial cadherin (VE-cadherin) (Abcam, Cambridge, MA). Primary antibodies were omitted in staining controls.
PGN stimulation of primary human monocytes
Studies on primary human cells were performed according to protocols approved by the OMRF Institutional Review Board. Peripheral blood mononuclear cells (PBMCs) were isolated from healthy volunteers by density gradient centrifugation (Histopaque-1077; Sigma). PBMCs were cultured in RPMI 1640 containing 1% autologous human serum and stimulated for up to 6 hours with either 10 µg/mL PGN or 1 µg/mL LPS (positive control). Messenger RNA (mRNA) was purified from Trizol-lysed PBMCs (Invitrogen), complementary DNA (cDNA) synthesized with Superscript III RT (Invitrogen), and quantified by quantitative reverse transcription polymerase chain reaction (qRT-PCR) using TF-validated primers (Origene Technologies, Rockville, MD). Surface TF antigen was quantified by flow cytometry using clone HTF-1 and was only detected on CD14+ monocytes (clone MAb11; eBioscience, San Diego, CA). Surface TF procoagulant activity on stimulated monocytes was assessed by a 2-stage Xa generation assay as described.43
Data analysis and representation
Data are depicted as mean plus or minus standard error of the mean (SEM). Statistical analysis was performed using GraphPad Prism. Differences between groups were analyzed using multiple Student t tests or repeated measures (RM) analysis of variance (ANOVA) with the Holm-Sidak multiple comparisons test. Significance threshold was set at P < .05. Differences in survival curves were evaluated with the log-rank (Mantel-Cox) test. Graphs were generated with Prism and figures collated with Adobe Illustrator (Adobe Systems, San Jose, CA).
Results
PGN induces septic manifestations in NHP
We previously reported the pathophysiology of anthrax sepsis in primates challenged with 5 × 107/kg to 5 × 109/kg vegetative B anthracis,7 which recapitulate clinical features observed with fulminant anthrax patients. In this study, we investigated PGN contributions at levels likely produced by these doses of bacteria,44 and we infused baboons with either a high-dose PGN (37.5 mg/kg), equivalent of the PGN burden in a lethal-dose B anthracis challenge, or a sublethal low PGN dose (8.5 mg/kg). The high-dose PGN challenge is similar with studies in other laboratory animal models.19 The PGN challenge triggered clinical signs of sepsis (neutropenia, elevated heart and respiration rates, reduced oxygen saturation, increased hematocrit) in a dose-dependent manner. PGN induced a systemic inflammatory response characterized by induction of both pro- and anti-inflammatory cytokines/chemokines. Among those, proinflammatory and procoagulant mediators (tumor necrosis factor α [TNFα], interleukin 6 [IL6], IL8)45-48 peaked early whereas regulatory anti-inflammatory cytokines were delayed (supplemental Figure 1).
PGN induces a progressive coagulopathy
Concomitant with induction of cytokines, high-dose challenged baboons exhibited a progressive hemostatic imbalance characterized by prolonged clotting times (aPTT, PT), prothrombin activation (TAT), consumption of fibrinogen, and elevated levels of FDP and D-dimers (Figure 1). APTT elevations occurred early, peaked at 2 to 4 hours and preceded PT elevations in paired samples, which peaked at 4 to 6 hours (Figure 1A-B). Elevated plasma levels of TAT show an extended thrombin generation throughout the first 8 hours of the study, which peaked at 4 hours when both contact and extrinsic clotting cascades converged (Figure 1C). Consistent with these features of consumptive coagulopathy, we observed a severe depletion of circulating fibrinogen (Figure 1D) and increases in circulating FDP and D-dimers (Figure 1E-F). These data indicate that a sterile infusion of PGN activates blood coagulation in vivo in a dose-dependent manner.
Figure 1.

PGN infusion induces DIC in baboons. Time-course dynamics of clotting times, aPTT (A) and PT (B), and hemostatic biomarkers, TAT complexes (C), fibrinogen consumption (D), FDP (E) and D-dimers (F) generation were evaluated in baboons challenged with either high-dose (n = 5) or low-dose PGN (n = 3). Data are represented as mean ± SEM. Multiple comparisons were performed by 2-way repeated measures ANOVA followed by Holm-Sidak post hoc test and statistically significant changes from baseline (T0) are depicted: *P < .05; **P < .01; ***P < .001.
PGN effect on the contact coagulation cascade
The contact pathway is initiated through transactivation of FXII and kallikrein, followed by clotting propagation through FXIIa-mediated activation of FXI. We found that in vivo PGN induced an acute elevation of FXIIa-AT, kallikrein-C1inh, and FXIa-AT complexes (Figure 2A-C), in a dose-dependent manner. Together with the prolonged aPTT, these data indicate activation of and propagation through the intrinsic clotting cascade in PGN-challenged baboons. In vitro, we found that both contact initiators, FXII and prekallikrein, bound to PGN (Figure 2D). The estimated binding affinities for FXII (KD ≈ 104 nM) and prekallikrein (KD ≈ 715 nM) were within physiologic range for both factors (∼500 nM). Nevertheless, PGN did not promote direct autoactivation of either FXII (Figure 2E) or prekallikrein (supplemental Figure 2) and had minimal effect on transactivation of purified zymogens (supplemental Figure 2). In contrast, PGN enhanced the enzymatic activity of active contact initiators, FXIIa and/or kallikrein, using small peptide chromogenic substrates (Figure 2F) or the transactivating physiologic substrate (supplemental Figure 2). Additionally, we found that PGN protected FXIIa against AT inhibition in vitro (Figure 2G). In the presence of heparin, AT inhibited FXIIa activity with an estimated IC50 of 130 to 180 nM depending on the AT lot potency. PGN reduced the potency of AT-heparin inhibition of FXIIa in a dose-dependent manner. At equivalent PGN and heparin concentrations (200 µg/mL), PGN doubled the AT-heparin IC50 (333 nM vs 134 nM, P = .0004, 1-way ANOVA). Overall, these data indicate that PGN does not activate the contact pathway directly but rather supports amplification of the clotting signal by enhancing the enzymatic function of initiating proteases and protecting against AT inhibition.
Figure 2.

PGN activates the intrinsic coagulation pathway in challenged baboons. (A-C) Activation of contact coagulation cascade in baboons challenged with either high-dose (n = 5) or low-dose PGN (n = 3). Time-course changes in FXIIa-AT (A), kallikrein-C1inh (B), and FXIa-AT (C) are shown. Statistical analysis and data representation are consistent with Figure 1. (D) Binding of purified contact pathway initiators to immobilized PGN was evaluated in vitro using biotinylated purified human FXII or prekallikrein. Data shown are mean ± SEM from 1 experiment run in quadruplicate. Dissociation constants (KD) were estimated from the best-fit binding curves using GraphPad Prism and average KD from 2 to 3 independent binding experiments are shown in the inset. (E) The effect of PGN (0.1-100 µg/mL) on autoactivation of purified FXII was evaluated in a continuous assay using the S2302 chromogenic substrate. No significant changes in the rate of chromophore (pNA) generation were observed. (F) The effect of PGN (1-200 µg/mL) on the enzymatic function of active proteases α-FXIIa (50 nM) and α-kallikrein (α-Kal, 25 nM). Specific activities are represented after normalization of initial velocities (V0) to the amount of enzyme used (micromolar pNA per minute per picomole enzyme). For both proteases, we found a strong linear correlation between the enzymatic activity and PGN concentration (Pearson r = 0.9878 (P < .0001) for α-FXIIa and r = 0.9344 (P = .0007) for α-Kal). The best fit linear regression (solid line) and 95% confidence interval (CI; dotted lines) are graphically depicted for each. (G) The effect of PGN on AT-heparin inhibition of α-FXIIa (100 nM) proteolysis of the S2302 chromogenic substrate. Observed residual α-FXIIa activity (micromolar pNA per minute) at different AT concentration (0-2 µM, represented on a log scale) in the presence of constant saturating levels of heparin (200 µg/mL) are shown (blue circles). PGN effect on AT-heparin inhibition of α-FXIIa was assessed at 50 µg/mL (green triangles) and 200 µg/mL (red squares). PGN increases AT-heparin IC50 in a dose-dependent manner (inset). (F-G) Data are represented as mean ± SEM (n = 3).
PGN challenge affects vascular permeability
Activated kallikrein can promote vascular permeability through proteolytic release of bradykinin from high-molecular-weight kininogen. Consistent with altered vascular permeability and concomitant with the activation of contact coagulation cascade, PGN challenge induced an acute drop in arterial pressure and a concurrent increase in hematocrit (Figure 3A-B). To directly investigate PGN-induced permeability changes, we injected fluorescently labeled albumin in a subset of animals (2 low dose, 4 high dose). Retention of fluorescent albumin in circulation was significantly reduced in high-dose vs low-dose challenged baboons (Figure 3C; P = .0035, RM 2-way ANOVA). We investigated endothelial integrity in tissues collected from the high-dose animal killed at 2 hours. En face microscopy of arterial endothelium showed cellular contraction and discontinuous VE-cadherin staining (Figure 3D PGN), indicating shape changes and impaired endothelial junctions. Similar junctional abnormalities and extravasation of the fluorescent albumin tracer were also observed in mesenteric vessels (supplemental Figure 3). These changes were not seen in time-matched saline-infused historic controls (Figure 3D control), and resolved by end point in other animals in the current study.
Figure 3.

PGN challenge affects vascular permeability in baboons. Time-course dynamics of mean systemic arterial pressure (MSAP; A) and normalized hematocrit (B) in baboons challenged with different doses of PGN as described in Figure 1. Statistical analysis and data representation are consistent with Figure 1. Not represented, MSAP changes in high-dose PGN challenge significantly deviated from T0 throughout the first 2 hours (P ≤ .05) whereas only the T+0.25 h in low-dose challenged primates reached significance threshold. (C) Vascular clearance of a fluorescent-albumin tracer was evaluated in a subset of animals (high-dose challenge, n = 4; low-dose challenge, n = 2). Data are represented as mean ± SEM. Multiple comparisons were performed by 2-way repeated measures ANOVA followed by the Holm-Sidak post hoc test. PGN dose significantly affected the clearance rates and accounted for 20.45% of the total variance observed (P = .0035). Paired time-point differences above significance threshold between the 2 PGN groups are represented (*P < .05, **P < .01). (D) En face immunofluorescence microscopy of arterial endothelial junctions revealed by VE-cadherin staining (green) in the presence of 4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstain. Micrographs from the time-paired (2 hours) early-euthanized high-dose challenged baboon (right) and a saline-infused historical control (left) are shown. Discontinued VE-cadherin staining (arrows) indicative of disorganized adherent junctions and endothelial cell contraction were observed in the PGN-challenged baboon only. Confocal images were captured on a Nikon Eclipse TE2000-U inverted microscope equipped with a Nikon C1 scanning head. Images were acquired and processed using EZ-C1 software (v 3.80; Nikon, Melville NY). Scale bar represents 20 µm.
PGN activates the extrinsic coagulation cascade
TF-dependent extrinsic coagulation is critical for the development of DIC. PGN induced monocyte TF expression in vivo, evidenced by immunofluorescence analysis of serial blood smears (Figure 4A). TF levels were elevated in high-dose as compared with low-dose PGN challenge, and peaked later (4 hours; Figure 4B) than markers of contact activation, coinciding with prolonged PT in these animals (Figure 1B). In parallel, we observed priming of FVII, the enzymatic component of the extrinsic tenase complex. FVII activation, indicated by FVIIa-AT complexes (Figure 4C), preceded the peak TF expression. PGN-dependent induction of TF was confirmed in primary human monocytes in vitro. Similar to LPS controls, PGN stimulation of monocytes induced de novo expression of TF mRNA, elevated expression on cell surface, and increased TF procoagulant activity (Figure 4D-F). Taken together, our data show that PGN induces the expression of monocyte TF both in vivo and in vitro and promotes FVII activation in vivo, which supports extrinsic coagulation events at later stages.
Figure 4.

PGN induces monocyte TF expression and extrinsic pathway activation in vivo and in vitro. (A-C) PGN challenge induces monocyte TF expression in vivo and priming of the extrinsic tenase protease. (A) Serial blood smears were methanol fixed, stained overnight at 4°C with biotinylated HTF-1 monoclonal anti-human TF, followed by detection with Cy3-labeled streptavidin (Jackson ImmunoResearch, West Grove, PA). Monocytes were identified morphologically. Confocal images were acquired on a Nikon Eclipse TE2000-U inverted microscope equipped with a Nikon C1 scanning head using EZ-C1 software. Representative micrographs of TF immunostaining in time-paired PGN-challenged baboons are shown (scale bar represents 20 µm). TF staining is stronger in high-dose challenged baboons (right panel) as opposed to low-dose challenge (middle panel). For comparison, no monocyte TF staining is observed on blood smears processed before the high-dose PGN challenge (left panel). (B) Time-course quantitation of TF immunostaining on serial blood smears depicted in panel A. Monocyte TF staining was quantified in at least 5 individual fields from each time point. (C) Time-course evaluation of FVIIa-AT complexes, as a marker of in vivo FVII activation, in PGN-challenged primates. Statistical analysis and representation are consistent with Figure 1. (D-F) PGN stimulation induces de novo TF expression in primary human monocytes in vitro. Time-course analysis of TF mRNA (D), surface antigen levels (E), and TF procoagulant activity (F) on monocytes stimulated with either 10 µg/mL PGN or 1 µg/mL LPS. Data are represented as mean ± SEM from 3 experiments using independent donors. Multiple comparisons were performed by 2-way repeated measures ANOVA followed by Holm-Sidak post hoc test and statistically significant changes from unstimulated (T0) controls are depicted: *P < .05; ** P < .01; ***P < .001.
PGN induces platelet activation in vivo
Overt DIC requires activation of the proteolytic coagulation cascades as well as activation of platelets, which provide procoagulant surfaces for clotting reactions. We previously reported that PGN activates human platelets in vitro, leading to aggregation and expression of phosphatidylserine-rich procoagulant surfaces.34 In vivo, PGN induced thrombocytopenia (Figure 5A), which was more pronounced in the high-dose compared with the low-dose group. In parallel, platelet aggregates, normally absent before challenge, were observed on blood smears as early as 2 hours after the PGN infusion (Figure 5B). Furthermore, conformational changes of integrin GPIIbIIIa characteristic of platelet activation49 were identified by immunofluorescence using peripheral tissues at end point (Figure 5C). Taken together, our data show that PGN challenge induces platelet activation in vivo, which promotes the hemostatic imbalance.
Figure 5.

PGN induces platelet activation in challenged baboons. (A) PGN challenge induces progressive thrombocytopenia in baboons in a dose-dependent manner. Data representation and statistical analysis are consistent with Figure 1. (B) Representative micrographs of Giemsa-stained serial blood smears from high-dose PGN group highlighting circulating platelet aggregates (arrows) in challenged baboons. Images were acquired on an upright Nikon Eclipse E800M microscope equipped with an Omax high-speed digital camera using the Omax ToupView software. (C) Immunofluorescence analysis of platelet activation in peripheral tissues of high-dose PGN-challenged primates at end point. Conformational changes in active GPIIbIIIa integrin were detected using the PAC-1 monoclonal followed by fluorescein isothiocyanate (FITC)-labeled donkey anti-mouse secondary (Jackson ImmunoResearch). DAPI was used for nuclear counterstaining. Confocal images were acquired on a Nikon Eclipse TE2000-U inverted microscope equipped with a Nikon C1 scanning head using EZ-C1 software. Representative micrographs from kidneys (left) and lung (right) sections from high-dose PGN-challenged baboons are shown. Scale bars represent 20 µm.
PGN induces thrombosis
Clinical features of consumptive coagulopathy, including skin and mucosal petechiae, were observed in high-dose challenged animals. In addition, histopathologic analysis of tissues collected at end point showed thrombosis and fibrin deposition in multiple organs. H&E and fibrin staining by PTAH on lung and kidney sections from median surviving high-dose animals are illustrated in Figure 6. Histologic analysis of lung sections revealed increased leukocyte infiltration, intravascular and extravascular fibrin deposition, and capillary leakage (Figure 6A-C). PGN-induced renal pathology included epithelial cell pyknosis and karyolysis within proximal tubes, glomerular necrosis, leukocyte accumulation in peritubular capillaries, and glomerular and tubular microthrombosis and fibrin deposition (Figure 6D-F). Overall, high-dose challenged baboons exhibited more pronounced pathologic modifications as compared with the low-dose group (supplemental Figure 4).
Figure 6.

PGN challenge induces peripheral organ congestion and fibrin deposition. Pathologic changes in peripheral tissues at end point were evaluated after histologic staining with H&E (top) or PTAH (middle and bottom) for detection of fibrin deposits. Images were acquired on an upright Nikon Eclipse E800M microscope equipped with an Omax high-speed digital camera using the Omax ToupView software. Representative lung and kidney micrographs from PGN-challenged baboons are shown. (A-C) Pulmonary pathologic changes are more prominent in the high-dose (left panel) than low-dose group (right panel). Liquid-filled (*) alveoli (av) and fibrin deposits (arrows) are highlighted throughout the figure. An example of leukocyte aggregates obstructing the pulmonary microvasculature in the high-dose PGN challenge is depicted in the left panel inset in panel A. Intravascular (left panel) and extravascular (right panel) fibrin deposits are easier identified at higher magnification in panel C. (D-F) Renal pathology is more prominent in the high-dose PGN challenge (left panels) than the low-dose group (right panels). Glomeruli (G) and tubules (T) are exemplified throughout the figure. Fibrin deposition (arrows) in either tubular (left) or glomerular (G) areas is highlighted at higher magnification in panel F. Scale bars represent 100 µm.
PGN induces multiple organ dysfunction
Similar to bacterial sepsis, high-dose PGN challenge induced a progressive multiorgan dysfunction as revealed by serum metabolite quantitation. Creatinine and blood urea nitrogen (BUN) levels (Figure 7A-B), indicative of kidney dysfunction, continually increased throughout the study, except in the surviving high-dose animal, which showed normalization of metabolites after 24 hours. Alanine aminotransferase levels (ALT) levels, indicative of liver dysfunction, increased during the first 24 hours of the study and, despite a normalizing trend afterward, it never returned to physiologic range (Figure 7C). In the low-dose challenge group, serum metabolites either did not change or quickly normalized. Overall, the high-dose challenge induced MOF that ultimately led to impaired survival (Figure 7D; log-rank test, P = .0394).
Figure 7.

PGN challenge induces progressive peripheral organ failure and ultimately death. Time-course evaluation of plasma biomarkers of organ dysfunction: creatinine (A), BUN (B) and ALT (C), in baboons challenged with different doses of PGN. The expected physiologic range for each biomarker is depicted by dotted lines. Data are represented as mean ± SEM. Paired time-point data are compared between the 2 groups using multiple Student t tests (*P < .05, **P < .01, ***P < .001). (D) Survival curves of baboons challenged with either high-dose or low-dose PGN. Mortality was significantly higher in high-dose compared with low-dose challenge (log-rank Mantel-Cox test, P = .0394).
Discussion
In this study, we investigated the role of cell wall PGN to the pathophysiologic progression of late-stage anthrax using an NHP model. In this model, PGN challenge by itself induces most of the features of live B anthracis infusion,7 highlighting PGN as an important PAMP for the septic development in late-stage disease. PGN, like the bacteria from which it derives, induces (1) cellular innate immune responses, (2) cytokine-mediated systemic inflammation, (3) progressive coagulopathies and DIC, (4) multiple organ dysfunction, and (5) impaired survival.
Due to its multifactorial etiology, a single animal model does not easily recapitulate all anthrax septic manifestations. Although systemic inflammation, MOF, and subsequent survival deficit have been recapitulated in rodent models,8,20 investigations of coagulopathic mechanisms are scarce and sometimes contradictory. Although Culley et al50 reported that anthrax lethal toxin induced DIC in mice, Qiu et al19 concluded that PGN challenge but not anthrax toxins associated with DIC markers. The differences may be due to model-specific limitations or temporal dissimilarities in study design. The end point DIC reported by Culley et al50 could be a secondary manifestation of the lethal toxin-induced organ damage and release of damage-associated molecular patterns (DAMPs) and not necessarily a PAMP-mediated event. To complicate matters, the intimate cross talk between inflammation and coagulation during infections can make it difficult to differentiate between pathogen-driven and host-driven coagulopathic responses during sepsis.
A growing body of evidence implicates PGN as an important PAMP in gram-positive infections. We focused our studies on PGN contribution to coagulopathy because PGN interacts with cellular components situated at the cross talk between innate immunity (neutrophils, monocytes) and hemostasis (platelets). PGN induced a robust proinflammatory response in monocyte and neutrophils25-28 and secretion of cytokines (TNFα, IL6, and IL8) that could contribute to inflammation-induced coagulopathy.45-48 These proinflammatory cytokines are similarly elevated in B anthracis–infused baboons.7
PGN-challenged baboons exhibited a progressive hemostatic imbalance, which started immediately after the PGN infusion. Prolonged aPTT and PT, indicative of consumption or inhibition of clotting factors, mimicked kinetics observed with live B anthracis infusions. Fibrin generation prompted fibrinolysis, leading to elevated levels of FDP and D-dimers. Importantly, clotting activation occurred as early as the induction of inflammatory mediators. These events suggest that activation of coagulation occurs concurrently with and not secondary to systemic inflammation. Consequently, they also confirm that PGN is the anthrax PAMP that engages the hemostatic system. In vivo, we observed the initial activation of contact coagulation confirmed by elevated levels of contact protease-serpin complexes. Coagulation proteases have a short lifetime in circulation where plasma serpins are abundantly present and complexes of serpins with coagulation enzymes have been reported in multiple sepsis studies.13,38,51-54 We were surprised by AT regulation of FXIIa because C1inh is considered the main regulatory serpin for both FXIIa and kallikrein.38,55,56 FXIIa-C1inh complexes were easily generated in vitro, but they were inconsistently observed during the in vivo PGN challenge (significantly elevated in only 2 of 5 high-dose challenged baboons). This could reflect contact activation by glycosaminoglycans that also augment AT, but not C1inh,57 regulation of FXIIa. Alternatively, C1inh consumption due to complement activation could promote similar outcomes. We did not detect significant α2-macroglobulin complexes13 in samples collected from PGN challenged baboons (not shown).
Although purified FXII and prekallikrein bind PGN, these interactions do not induce activation of either protein. This indicates that PGN does not directly support contact activation. However, PGN moderately enhanced the enzymatic activity of FXIIa and kallikrein. As these proteases are continuously generated in normal health,58 introducing PGN into the circulation may enhance their basal activities. FXIIa and kallikrein activities may have been amplified further by loss of AT regulation, either as a direct effect of PGN, or through AT depletion during the consumptive coagulopathy induced by PGN. It is also a strong possibility that contact activation, or a process similar to it, was induced indirectly by PGN infusion. For example, naturally occurring polyanions, such as polyphosphates from platelets59,60 and DNA/histones from neutrophils,61 support FXII and prekallikrein activation. Furthermore, the vascular leakage may allow thrombin generation by blood exposure to perivascular TF, which could bolster FXI activation.62 FXIa could in turn activate FXII, as suggested by work with mouse sepsis models.63 We conclude, therefore, that PGN most likely promotes activation and activity of FXIIa and kallikrein in vivo indirectly through (1) enhancement of protease activity, (2) reduction/loss of regulatory serpins, and (3) release of DAMPs that support contact activation. In vivo studies using specific inhibitors for the proposed pathways would be required to unequivocally establish the mechanisms by which FXII and prekallikrein operate in the PGN model.
Kallikrein-kininogen activation releases bradykinin with subsequent effects on vascular permeability. Plasmin-mediated bradykinin generation, downstream of coagulation-fibrinolytic events, has also been proposed,64 and circulating D-dimers indicative of intravascular plasmin activity were detected in PGN-challenged baboons. In vivo, we observed early acute changes in vascular permeability (Figure 3) in 4 of 5 high-dose challenged baboons. Analysis of a fluorescent albumin tracer confirmed the altered permeability in early stages of the PGN challenge, and immunofluorescence microscopy revealed contracted endothelial cells with impaired adherent junctions. Both bradykinin and thrombin-PAR1 signaling could potentially induce these changes65,66 and discriminating between their contributions to the observed vascular permeability changes during the PGN challenge will require further investigations.
The TF-dependent extrinsic coagulation cascade is critical for the development of DIC. In PGN-challenged baboons, elevated PT times suggest the activation of the extrinsic pathway as early as 2 hours. Monocyte TF expression in vivo was shown by immunostaining on serial blood smears and correlated with prolonged PT. In vitro studies confirmed that PGN induces TF expression in primary human monocytes directly. In addition, FVIIa-AT complexes in PGN-challenged baboons indicate priming of the extrinsic tenase protease, FVII, which binds TF and supports clotting. Interestingly, FVIIa-AT precedes maximal TF expression, suggesting that initial activation of downstream clotting enzymes, most likely FXa,67 activate FVII before monocyte TF expression.
Activated platelets provide a procoagulant surface critical for the amplification of clotting reactions.68 We previously reported that PGN activates platelets in vitro through engagement of FcγR and complement activation.34 In vivo, PGN induced progressive thrombocytopenia; platelet aggregates observed on blood smears and conformational changes in platelet GPIIbIIIa confirmed platelet activation in vivo. Sustained thrombin activation in response to the PGN challenge could contribute to platelet activation downstream of coagulation events and further amplify the hemostatic imbalance. Overall, our data show that in vivo the PGN challenge promotes clotting reactions and activates platelets, requirements for a clinical diagnosis of overt DIC. The observed hemostatic imbalance can impair organ function through hypoperfusion and ischemia/reperfusion insults. Ultimately, impaired function of multiple organs compromised survival and initiated euthanasia protocols in 5 of 6 high-dose challenged baboons, which mimicked the lethal dose of live B anthracis infusion.
In summary, we offer the first characterization of PGN-induced pathology in an in vivo primate model of late-stage anthrax. PGN from B anthracis, and possibly other gram-positive pathogens, induces systemic inflammation in primates and activates clotting cascades leading to DIC, ischemic injury of multiple organs, and, ultimately, death. These results support the hypothesis that PGN can largely account for the sepsis-like pathology accompanying gram-positive infections and position PGN as an important but underappreciated PAMP.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors are grateful for the help of S. Kosanke, University of Oklahoma, for aid in grading the pathology slides from the challenged baboons.
This work was supported by grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (U19AI062629 and 1R21 AI113020 [K.M.C. and F.L.]) and National Institute of General Medical Sciences (GM116184, GM121601, and P30GM114731 [F.L.]).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: N.I.P., R.S., and R.S.K. performed animal experiments and histological and biochemical analysis; N.I.P., A. Girton, and T.B. purified and characterized the PGN; D.G. and N.I.P. investigated the PGN effect on contact pathway in vitro; S.S.Z. and A. Gruber contributed critical reagents and assay designs; F.L. and K.M.C. conceived and supervised the study; N.I.P., F.L., and K.M.C. analyzed the results and wrote the manuscript; all authors read and approved the manuscript.
Conflict-of-interest disclosure: A. Gruber and Oregon Health & Science University may have a financial interest in the results of this study. The remaining authors declare no competing financial interests.
Correspondence: K. Mark Coggeshall, Oklahoma Medical Research Foundation, 825 NE 13th St, Oklahoma City, OK 73104; e-mail: mark-coggeshall@omrf.org.
REFERENCES
- 1.Sweeney DA, Hicks CW, Cui X, Li Y, Eichacker PQ. Anthrax infection. Am J Respir Crit Care Med. 2011;184(12):1333-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jernigan JA, Stephens DS, Ashford DA, et al. ; Anthrax Bioterrorism Investigation Team. Bioterrorism-related inhalational anthrax: the first 10 cases reported in the United States. Emerg Infect Dis. 2001;7(6):933-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moayeri M, Haines D, Young HA, Leppla SH. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J Clin Invest. 2003;112(5):670-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Firoved AM, Miller GF, Moayeri M, et al. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am J Pathol. 2005;167(5):1309-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu S, Zhang Y, Moayeri M, et al. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature. 2013;501(7465):63-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coggeshall KM, Lupu F, Ballard J, et al. The sepsis model: an emerging hypothesis for the lethality of inhalation anthrax. J Cell Mol Med. 2013;17(7):914-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stearns-Kurosawa DJ, Lupu F, Taylor FB Jr, Kinasewitz G, Kurosawa S. Sepsis and pathophysiology of anthrax in a nonhuman primate model. Am J Pathol. 2006;169(2):433-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu S, Moayeri M, Leppla SH. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol. 2014;22(6):317-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levi M, Ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999;341(8):586-592. [DOI] [PubMed] [Google Scholar]
- 10.Taylor FB Jr, Chang A, Ruf W, et al. Lethal E. coli septic shock is prevented by blocking tissue factor with monoclonal antibody. Circ Shock. 1991;33(3):127-134. [PubMed] [Google Scholar]
- 11.Levi M, ten Cate H, Bauer KA, et al. Inhibition of endotoxin-induced activation of coagulation and fibrinolysis by pentoxifylline or by a monoclonal anti-tissue factor antibody in chimpanzees. J Clin Invest. 1994;93(1):114-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lupu C, Westmuckett AD, Peer G, et al. Tissue factor-dependent coagulation is preferentially up-regulated within arterial branching areas in a baboon model of Escherichia coli sepsis. Am J Pathol. 2005;167(4):1161-1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pixley RA, De La Cadena R, Page JD, et al. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia. In vivo use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J Clin Invest. 1993;91(1):61-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keshari RS, Silasi R, Popescu NI, et al. Inhibition of complement C5 protects against organ failure and reduces mortality in a baboon model of Escherichia coli sepsis. Proc Natl Acad Sci USA. 2017;201706818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Deventer SJ, Büller HR, ten Cate JW, Aarden LA, Hack CE, Sturk A. Experimental endotoxemia in humans: analysis of cytokine release and coagulation, fibrinolytic, and complement pathways. Blood. 1990;76(12):2520-2526. [PubMed] [Google Scholar]
- 16.Mészáros K, Aberle S, Dedrick R, et al. Monocyte tissue factor induction by lipopolysaccharide (LPS): dependence on LPS-binding protein and CD14, and inhibition by a recombinant fragment of bactericidal/permeability-increasing protein. Blood. 1994;83(9):2516-2525. [PubMed] [Google Scholar]
- 17.Franco RF, de Jonge E, Dekkers PE, et al. The in vivo kinetics of tissue factor messenger RNA expression during human endotoxemia: relationship with activation of coagulation. Blood. 2000;96(2):554-559. [PubMed] [Google Scholar]
- 18.Taylor FB, Haddad PA, Hack E, et al. Two-stage response to endotoxin infusion into normal human subjects: correlation of blood phagocyte luminescence with clinical and laboratory markers of the inflammatory, hemostatic response. Crit Care Med. 2001;29(2):326-334. [DOI] [PubMed] [Google Scholar]
- 19.Qiu P, Li Y, Shiloach J, et al. Bacillus anthracis cell wall peptidoglycan but not lethal or edema toxins produces changes consistent with disseminated intravascular coagulation in a rat model. J Infect Dis. 2013;208(6):978-989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Kimpe SJ, Kengatharan M, Thiemermann C, Vane JR. The cell wall components peptidoglycan and lipoteichoic acid from Staphylococcus aureus act in synergy to cause shock and multiple organ failure. Proc Natl Acad Sci USA. 1995;92(22):10359-10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kengatharan KM, De Kimpe S, Robson C, Foster SJ, Thiemermann C. Mechanism of gram-positive shock: identification of peptidoglycan and lipoteichoic acid moieties essential in the induction of nitric oxide synthase, shock, and multiple organ failure. J Exp Med. 1998;188(2):305-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Himanen JP, Pyhälä L, Olander RM, et al. Biological activities of lipoteichoic acid and peptidoglycan-teichoic acid of Bacillus subtilis 168 (Marburg). J Gen Microbiol. 1993;139(11):2659-2665. [DOI] [PubMed] [Google Scholar]
- 23.Wray GM, Foster SJ, Hinds CJ, Thiemermann C. A cell wall component from pathogenic and non-pathogenic gram-positive bacteria (peptidoglycan) synergises with endotoxin to cause the release of tumour necrosis factor-alpha, nitric oxide production, shock, and multiple organ injury/dysfunction in the rat. Shock. 2001;15(2):135-142. [DOI] [PubMed] [Google Scholar]
- 24.Schleifer KH, Kandler O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev. 1972;36(4):407-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iyer JK, Khurana T, Langer M, et al. Inflammatory cytokine response to Bacillus anthracis peptidoglycan requires phagocytosis and lysosomal trafficking. Infect Immun. 2010;78(6):2418-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langer M, Malykhin A, Maeda K, et al. Bacillus anthracis peptidoglycan stimulates an inflammatory response in monocytes through the p38 mitogen-activated protein kinase pathway. PLoS One. 2008;3(11):e3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun D, Raisley B, Langer M, et al. Anti-peptidoglycan antibodies and Fcγ receptors are the key mediators of inflammation in Gram-positive sepsis. J Immunol. 2012;189(5):2423-2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iyer JK, Coggeshall KM. Cutting edge: primary innate immune cells respond efficiently to polymeric peptidoglycan, but not to peptidoglycan monomers. J Immunol. 2011;186(7):3841-3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seok J, Warren HS, Cuenca AG, et al. ; Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110(9):3507-3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langer M, Girton AW, Popescu NI, Burgett T, Metcalf JP, Coggeshall KM. Neither Lys- and DAP-type peptidoglycans stimulate mouse or human innate immune cells via Toll-like receptor 2. PLoS One. 2018;13(2):e0193207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chamaillard M, Hashimoto M, Horie Y, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4(7):702-707. [DOI] [PubMed] [Google Scholar]
- 32.Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278(11):8869-8872. [DOI] [PubMed] [Google Scholar]
- 33.Girton AW, Popescu NI, Keshari RS, Burgett T, Lupu F, Coggeshall KM. Serum amyloid P and IgG exhibit differential capabilities in the activation of the innate immune system in response to Bacillus anthracis peptidoglycan. Infect Immun. 2018;86(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun D, Popescu NI, Raisley B, et al. Bacillus anthracis peptidoglycan activates human platelets through FcγRII and complement. Blood. 2013;122(4):571-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taylor FB Jr, Kinasewitz GT, Lupu F. Pathophysiology, staging and therapy of severe sepsis in baboon models. J Cell Mol Med. 2012;16(4):672-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silasi-Mansat R, Zhu H, Popescu NI, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. 2010;116(6):1002-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tucker EI, Marzec UM, White TC, et al. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood. 2009;113(4):936-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nuijens JH, Huijbregts CC, Eerenberg-Belmer AJ, et al. Quantification of plasma factor XIIa-Cl(-)-inhibitor and kallikrein-Cl(-)-inhibitor complexes in sepsis. Blood. 1988;72(6):1841-1848. [PubMed] [Google Scholar]
- 39.Minnema MC, Pajkrt D, Wuillemin WA, et al. Activation of clotting factor XI without detectable contact activation in experimental human endotoxemia. Blood. 1998;92(9):3294-3301. [PubMed] [Google Scholar]
- 40.Lottenberg R, Jackson CM. Solution composition dependent variation in extinction coefficients for p-nitroaniline. Biochim Biophys Acta. 1983;742(3):558-564. [DOI] [PubMed] [Google Scholar]
- 41.Ivanov I, Matafonov A, Sun MF, et al. Proteolytic properties of single-chain factor XII: a mechanism for triggering contact activation. Blood. 2017;129(11):1527-1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jelev L, Surchev L. A novel simple technique for en face endothelial observations using water-soluble media -‘thinned-wall’ preparations. J Anat. 2008;212(2):192-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Popescu NI, Lupu C, Lupu F. Extracellular protein disulfide isomerase regulates coagulation on endothelial cells through modulation of phosphatidylserine exposure. Blood. 2010;116(6):993-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spika JS, Peterson PK, Wilkinson BJ, et al. Role of peptidoglycan from Staphylococcus aureus in leukopenia, thrombocytopenia, and complement activation associated with bacteremia. J Infect Dis. 1982;146(2):227-234. [DOI] [PubMed] [Google Scholar]
- 45.van der Poll T, Büller HR, ten Cate H, et al. Activation of coagulation after administration of tumor necrosis factor to normal subjects. N Engl J Med. 1990;322(23):1622-1627. [DOI] [PubMed] [Google Scholar]
- 46.Bauer KA, ten Cate H, Barzegar S, Spriggs DR, Sherman ML, Rosenberg RD. Tumor necrosis factor infusions have a procoagulant effect on the hemostatic mechanism of humans. Blood. 1989;74(1):165-172. [PubMed] [Google Scholar]
- 47.Neumann FJ, Ott I, Marx N, et al. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler Thromb Vasc Biol. 1997;17(12):3399-3405. [DOI] [PubMed] [Google Scholar]
- 48.Lauw FN, Dekkers PE, te Velde AA, et al. Interleukin-12 induces sustained activation of multiple host inflammatory mediator systems in chimpanzees. J Infect Dis. 1999;179(3):646-652. [DOI] [PubMed] [Google Scholar]
- 49.Shattil SJ, Hoxie JA, Cunningham M, Brass LF. Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. J Biol Chem. 1985;260(20):11107-11114. [PubMed] [Google Scholar]
- 50.Culley NC, Pinson DM, Chakrabarty A, Mayo MS, LeVine SM. Pathophysiological manifestations in mice exposed to anthrax lethal toxin. Infect Immun. 2005;73(10):7006-7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abbink JJ, Nuijens JH, Eerenberg AJ, et al. Quantification of functional and inactivated alpha 2-macroglobulin in sepsis. Thromb Haemost. 1991;65(1):32-39. [PubMed] [Google Scholar]
- 52.de Boer JP, Creasey AA, Chang A, et al. Alpha-2-macroglobulin functions as an inhibitor of fibrinolytic, clotting, and neutrophilic proteinases in sepsis: studies using a baboon model. Infect Immun. 1993;61(12):5035-5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koyama K, Madoiwa S, Nunomiya S, et al. Combination of thrombin-antithrombin complex, plasminogen activator inhibitor-1, and protein C activity for early identification of severe coagulopathy in initial phase of sepsis: a prospective observational study. Crit Care. 2014;18(1):R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wuillemin WA, Fijnvandraat K, Derkx BH, et al. Activation of the intrinsic pathway of coagulation in children with meningococcal septic shock. Thromb Haemost. 1995;74(6):1436-1441. [PubMed] [Google Scholar]
- 55.Pixley RA, Schapira M, Colman RW. The regulation of human factor XIIa by plasma proteinase inhibitors. J Biol Chem. 1985;260(3):1723-1729. [PubMed] [Google Scholar]
- 56.Schapira M, Scott CF, Colman RW. Contribution of plasma protease inhibitors to the inactivation of kallikrein in plasma. J Clin Invest. 1982;69(2):462-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wuillemin WA, Eldering E, Citarella F, de Ruig CP, ten Cate H, Hack CE. Modulation of contact system proteases by glycosaminoglycans. Selective enhancement of the inhibition of factor XIa. J Biol Chem. 1996;271(22):12913-12918. [DOI] [PubMed] [Google Scholar]
- 58.Revenko AS, Gao D, Crosby JR, et al. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood. 2011;118(19):5302-5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R, Morrissey JH. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci USA. 2006;103(4):903-908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gajsiewicz JM, Smith SA, Morrissey JH. Polyphosphate and RNA differentially modulate the contact pathway of blood clotting. J Biol Chem. 2017;292(5):1808-1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Noubouossie DF, Whelihan MF, Yu YB, et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood. 2017;129(8):1021-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matafonov A, Sarilla S, Sun MF, et al. Activation of factor XI by products of prothrombin activation. Blood. 2011;118(2):437-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bane CE Jr, Ivanov I, Matafonov A, et al. Factor XI deficiency alters the cytokine response and activation of contact proteases during polymicrobial sepsis in mice. PLoS One. 2016;11(4):e0152968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hofman Z, de Maat S, Hack CE, Maas C. Bradykinin: inflammatory product of the coagulation system. Clin Rev Allergy Immunol. 2016;51(2):152-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279-367. [DOI] [PubMed] [Google Scholar]
- 66.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258-264. [DOI] [PubMed] [Google Scholar]
- 67.Butenas S, Mann KG. Kinetics of human factor VII activation. Biochemistry. 1996;35(6):1904-1910. [DOI] [PubMed] [Google Scholar]
- 68.Mann KG, Bovill EG, Krishnaswamy S. Surface-dependent reactions in the propagation phase of blood coagulation. Ann N Y Acad Sci. 1991;614(1):63-75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.