

Abstract
DNA photolyase is a pyrimidine-dimer repair enzyme that uses visible light. Photolyase generally contains two chromophore cofactors. One is a catalytic cofactor directly contributing to the repair of a pyrimidine-dimer. The other is a light-harvesting cofactor, which absorbs visible light and transfers energy to the catalytic cofactor. Photolyases are classified according to their second cofactor into either a folate- or deazaflavin-type. The native structures of both types of photolyases have already been determined, but the mechanism of substrate recognition remains largely unclear because of the lack of structural information regarding the photolyase-substrate complex. Photolyase from Thermus thermophilus, the first thermostable class I photolyase found, is favorable for function analysis, but even the type of the second cofactor has not been identified. Here, we report the crystal structures of T. thermophilus photolyase in both forms of the native enzyme and the complex along with a part of its substrate, thymine. A structural comparison with other photolyases suggests that T. thermophilus photolyase has structural features allowing for thermostability and that its light-harvesting cofactor binding site bears a close resemblance to a deazaflavin-type photolyase. One thymine base is found at the hole, a putative substrate-binding site near the catalytic cofactor in the complex form. This structural data for the photolyase-thymine complex allow us to propose a detailed model for the pyrimidine-dimer recognition mechanism.
DNA is damaged by UV sunlight. Photoproducts such as a cyclobutane pyrimidine dimer (CPD) affect DNA replication and transcription and create serious problems. All living organisms, therefore, have various DNA repair mechanisms. Photoreactivation is an efficient and direct repair mechanism against CPD and (6–4) photoproducts. CPD photolyase, one of the DNA repair enzymes, independently binds to a damaged DNA, including CPD, and repairs it by using visible light without the aid of any other proteins (1, 2). CPD photolyases are classified into two classes, I and II, based on amino acid sequence similarity. Class I photolyases are found in many microorganisms, whereas most of class II photolyases are found in higher eukaryotes (3). Photolyase generally has two kinds of chromophores. One is a catalytic cofactor, which directly interacts with substrate (CPD) in the photo-repair reaction. The other is a light-harvesting cofactor, which acts as an antenna to harvest light energy transferred to the catalytic cofactor. All known photolyases contain a reduced flavin-adenine dinucleotide (FADH2) as a catalytic cofactor. It has been shown in experiments with other CPD photolyases that the pyrimidine-dimer is repaired by electron transfer from FADH2 (4, 5). Class I photolyases are classified according to their second chromophore into either a deazaflavin- or folate-type. A deazaflavin-type photolyase has an 8-hydroxy-5-deazaflavin (8-HDF) as a light-harvesting cofactor; in contrast, a folate-type photolyase has a 5,10-methenyltetrahydrofolic acid (MTHF). Until now, only two crystal structures of the photolyases from Escherichia coli (deazaflavin-type; ref. 6) and Anidulans nidulans (folate-type; ref. 7) have been solved at atomic resolution. They are the first three-dimensional structures for each type of photolyase, in which the geometry of the two cofactors became clear and an analysis of the energy-transfer process was made. Both types of photolyases have a similar backbone structure of the apoprotein, but show completely different binding modes for the light-harvesting cofactors, which account for the more efficient energy transfer in A. nidulans photolyase than in E. coli photolyase (4, 5, 7). For the substrate recognition, the base-flipping mechanism was proposed on the basis of the structure of E. coli photolyase (6). However, we know little about the mechanism of substrate recognition because both molecular structures have been determined in the absence of the substrate.
DNA photolyase from Thermus thermophilus was characterized as the first thermostable class I photolyase (8, 9). This enzyme is stable up to 60°C and in guanidine·HCl up to 2.5 M at neutral pH. The thermostable and physically stable characters are favorable for functional analysis, but even the type of light-harvesting cofactor is not known. Crystal structures of E. coli and A. nidulans photolyases have revealed that the common catalytic cofactor FAD is bound at the C-terminal regions, in which ≈30% of the residues are identical in these photolyases, and that the light-harvesting cofactor is almost at the N-terminal half, which is less conserved in general (Fig. 1). The primary structure of T. thermophilus photolyase does not show any similarity to either the deazaflavin- or folate-type with respect to the amino acid residues playing a key role in the light-harvesting cofactor binding. It is difficult to elucidate the type of light-harvesting cofactor only from the amino acid sequences. Spectroscopic properties indicate that the purified recombinant T. thermophilus photolyase from E. coli contains only FAD as a chromophore (8). This finding may indicate that T. thermophilus photolyase has no second chromophores or that is has 8-HDF as a light-harvesting cofactor, because E. coli cannot synthesize deazaflavin (8-HDF). However, it does not still rule out the possibility that MTHF or another chromophore might bind to the protein in vivo, as a second chromophore is known to be easily removed (10).
Figure 1.

Sequence alignment of photolyases from A. nidulans, E. coli, and T. thermophilus. The conserved residues are highlighted in blue. The residues in contact with FAD, 8-HDF, and MTHF are shown in yellow, green, and blue, respectively (6, 7). The residues in the active site and the positive residues around the hole are shown in red and highlighted in pink, respectively. The secondary structures, helices, and strands of T. thermophilus photolyase are shown in white and gray boxes under the sequences. The sequence of photolyase from T. thermophilus HB8 has been deposited in DNA Data Base in Japan (accession no. AB064548).
To understand the structural features of a thermostable DNA photolyase, we have performed an x-ray crystal structure analysis. Here we describe the three-dimensional structure of DNA photolyase from extremely thermophilic bacterium, T. thermophilus HB8, determined at 2.1 Å resolution. This structure reveals the thermostable features and identifies the second chromophore type. Furthermore, the structure of photolyase complexed with a part of its substrate, the thymine base, suggests a detailed mechanism of substrate recognition and provides a structural basis for the specific substrate recognition. This is the first direct evidence, to our knowledge, that CPD photolyases can recognize flipped-out DNA bases.
Materials and Methods
Crystallization and Structure Determination.
DNA photolyase from T. thermophilus HB8 was purified from overproducing E. coli, as described (8). The hexagonal crystals were obtained by the vapor-diffusion method with 100 mM sodium acetate buffer (pH 4.6), 1.0 M NH4H2PO4, and 100 mM Li2SO4 as a precipitation buffer (11). The crystals of the derivative (HG) and the complex with thymine (THYMINE) were prepared by soaking in the precipitation buffer containing 1 mM methylmercury chloride (CH3HgCl) and 10 mM thymine, respectively. The diffraction data were collected by using the synchrotron-radiation source, SPring-8 (Harima, Japan), at the BL40B2 beamline with an Area Detector System Corporation (Poway, CA) charge-coupled device (CCD) detector for the derivative crystals (HG) and at the BL44B2 beamline with a MarCCD detector for the other crystals (NATI1, NATI2, and THYMINE). All data were processed with the mosflm and scala programs (12). Data collection and refinement statistics are summarized in Table 1. The single isomorphous replacement with anomalous scattering (SIRAS) phases were calculated with NATI1 and HG data sets by the mlphare program (12). The initial model was built with the SIRAS map with the molecular graphic program o (13). Most of the residues could be identified and fitted in this electron density map. A simulated annealing refinement was carried out with the cns program (14) by using the initial native data set (NATI1). Afterward, the second native data set, NATI2, was used for refinement and model-building. After the rigid-body refinement, further refinement and model-building were carried out alternately to fit the complete model by using 2Fo-Fc and Fo-Fc maps. The progress and validity of the refinement protocol were checked by monitoring the Rfree value for 5% (1,444 reflections) of the total reflections (28,567 reflections; ref. 15). The final R and Rfree factors are 0.214 and 0.251, respectively, for reflections in the resolution range of 25–2.1 Å. The final native model consists of 415 residues, 73 water molecules, 1 phosphate ion, and 1 FAD. Model geometry was analyzed with the procheck program (16), and 90.2% of residues were found to be in the most favored region of the Ramachandran plot. In the same manner, the model of the complex with thymine was refined with the THYMINE data set. The rms deviation was calculated by using the lsqman program (17).
Table 1.
Crystallographic statistics
| Data set | NATI1 | HG | NATI2 | THYMINE |
|---|---|---|---|---|
| Resolution, Å | 25–2.4 (2.54–2.40) | 25–2.1 (2.21–2.10) | 25–2.1 (2.21–2.10) | 25–2.2 (2.28–2.20) |
| Completeness, I > 1σ, % | 86.9 (69.7) | 79.0 (74.0) | 89.5 (74.0) | 88.9 (69.8) |
| Mosaicity, ° | 0.07 | 0.08 | 0.110 | 0.07 |
| Redundancy | 3.6 | 8.9 | 3.8 | 11.5 |
| I/σ(I) | 13.3 (2.8) | 10.2 (3.0) | 8.5 (2.8) | 36.8 (3.5) |
| *Rmerge, % | 4.9 (26.3) | 4.8 (22.7) | 6.1 (27.7) | 5.8 (33.6) |
| Unit cell parameters (P6122) a=b=, c=, Å | 113.60, 141.99 | 113.51, 141.68 | 112.89, 142.64 | 113.10, 142.55 |
| Phasing (NATI1-HG) | ||||
| Resolution, Å | 15–2.4 (2.72–2.40) | |||
| Mean figure of merit | 0.578 (0.393) | |||
| Cullis R, Cullis Rano | 0.64 (0.83), 0.63 (0.88) | |||
| Refinement | ||||
| Resolution limits, Å | 25–2.1 | 25–2.2 | ||
| Number of reflections | 28,567 | 25,481 | ||
| †R (%), ‡Rfree (%) | 21.4, 25.1 | 21.1, 24.3 | ||
| Rmsd for bond distances (Å), angles (°) | 0.006, 1.20 | 0.006, 1.30 | ||
| Average B factor, Å2 | 44.1 | 47.4 | ||
| Model | 3,497 atoms | 3,501 atoms | ||
| Protein | 415 residues | 415 residues | ||
| Cofactor | FAD | FAD | ||
| Solvent molecules | 73 waters, 1 PO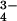
|
68 waters, 1 PO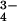 , thymine , thymine |
Rmerge = ∑|Ii − 〈Ii〉|/∑〈Ii〉, where Ii is the observed intensity and 〈Ii〉 is the average intensity over symmetry equivalent measurements.
R = ∑∥Fo| − |Fc∥/∑|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
Rfree was calculated by using 5% of randomly selected reflections that were excluded from the refinement (15). Rmsd, rms deviation.
Results and Discussion
Overall Structure.
The crystal structure of DNA photolyase from T. thermophilus was determined by the single isomorphous replacement with anomalous scattering method at 2.1 Å resolution (Table 1). The R factor was finally converged to 0.214 for the refined model comprising 1 phosphate ion, 73 waters, and residues 2–416 of all 420 amino acids. The overall structure is shown in Fig. 2a. It consists of two domains, the α/β domain in the N-terminal region and the helical domain in the C-terminal region. The α/β domain is folded into 5 β-strands (β1–β5) and 5 helices (α1–α5) with an open α/β-sheet structure as a typical dinucleotide-binding fold (18). The helical domain contains 14 helices (α6–α19) and three 310 helices. A very long loop, including the α6 helix, connects the α/β domain and the helical domain. The topology is almost the same as those of photolyases from E. coli and A. nidulans, with the exception of a few additions and deletions of secondary structures. The position and number of α and 310 helices are different from each other. The backbone structure of T. thermophilus photolyase shows a high similarity in overall folding, including both α/β and helical domains, compared with those of E. coli and A. nidulans photolyases as shown in Fig. 2b. The rms deviations were found to be 1.54, 1.60, and 1.12 Å, for Cα atoms common to T. thermophilus–E. coli photolyases (388 Cα atoms), T. thermophilus–A. nidulans photolyases (355 Cα atoms), and E. coli–A. nidulans photolyases (413 Cα atoms), respectively.
Figure 2.

Crystal structure of T. thermophilus photolyase. (a) Overall structure of T. thermophilus photolyase. FAD is shown in yellow. The N and C termini are labeled N and C, respectively. (b) A comparison of the folding of photolyases from T. thermophilus, A. nidulans, and E. coli. The Cα traces and FAD molecules for T. thermophilus, A. nidulans, and E. coli photolyases are shown in red, green, and blue, respectively. Circles indicate more compact regions in T. thermophilus than the others. The figures in a and b were prepared by using the programs MOLSCRIPT (24) and RASTER3D (25). (c) The proline-rich region at the very long interdomain loop between α6 and α7 with an experimental single isomorphous replacement with anomalous scattering electron-density map. The figure in c was drawn by the O program (13).
A remarkable feature of T. thermophilus photolyase is that loop regions, especially those between α6-α7 and α18-α19 helices, are extremely shortened, as shown in Fig. 2b. Furthermore, one helix observed in both E. coli and A. nidulans photolyases is absent between α18 and α19 helices. These shortenings and deletions make the structure more compact compared with the other photolyases. In addition, the proline-rich sequence (a series of five prolines with an insertion of one leucine residue) is found at the very long loop region connecting the N-terminal α/β domain and the C-terminal helical domain (Fig. 2c). These structural features indicate that the three-dimensional structure of this enzyme is less flexible than the others and might be efficient for the thermostable and physically stable characteristics also found in the structure of hyperthermostable triosephosphate isomerase (19).
FAD Binding Site.
The catalytic cofactor FAD is a common cofactor of photolyases. It accepts excitation energy from the light-harvesting cofactor and repairs the pyrimidine-dimer directly by donation of an electron. FAD lies deeply buried in the center of a helical domain, which is in the same position as the photolyases from E. coli and A. nidulans (Fig. 2b). The conformation of FAD is also the same among these photolyases. The sequence alignment (Fig. 1) shows the amino acid residues interacting with FAD, which are conserved at exactly the same positions in these three photolyases with the exception of Phe-307 and Val-346, which are replaced by tryptophan and alanine, respectively, in photolyases from E. coli and A. nidulans.
Light-Harvesting Cofactor.
The light-harvesting cofactor serves as an antenna for the light energy transferred to the catalytic site. Besides the species of the cofactor, the binding position and conformation are important to the efficiency of the photoreaction (4, 5, 7). The light-harvesting cofactor of T. thermophilus photolyase is unknown, and the spectroscopic analysis indicates there is no chromophore other than FAD in T. thermophilus photolyase overproduced in E. coli (8). In the crystal structures of photolyases from E. coli and A. nidulans, light-harvesting cofactors are nestled into a shallow cleft between the α/β and helical domains (Fig. 2b). In the case of E. coli photolyase, MTHF as a light-harvesting cofactor is bound to the exterior of the cleft between the two domains, partially sticking out of the enzyme (6). In contrast, A. nidulans photolyase has sufficient space to contain the 8-HDF as a light-harvesting cofactor at the interior cleft, and the whole of 8-HDF is buried inside of the cleft (7).
The crystal structure of T. thermophilus photolyase reveals a large cavity inside of the cleft between the two domains, which is large enough to accommodate an 8-HDF but has no space for an MTHF at the corresponding position (Fig. 3a). This cavity is bigger than that of A. nidulans. However, there is no significant bulky electron density for the cofactor but only for a solvent molecule, including a PO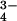 , in the crystal structure. Fig. 3b shows the detailed geometry of the amino acid residues inside of the cleft. In comparison with the 8-HDF binding site in A. nidulans photolyase, the amino acid residues interacting with 8-HDF are found at almost the same position in T. thermophilus photolyase. The 8-HDF model could be fitted to the same position as that of T. thermophilus photolyase, maintaining suitable interactions similar to A. nidulans photolyase, although only two residues, Arg-47 and Arg-114, are identical (Fig. 1). This result strongly suggests that T. thermophilus photolyase can be a deazaflavin-type photolyase. The high similarity of the binding mode of 8-HDF predicts that the interchromophoric energy transfer in T. thermophilus photolyase might have high efficiency similar to that of A. nidulans photolyase.
, in the crystal structure. Fig. 3b shows the detailed geometry of the amino acid residues inside of the cleft. In comparison with the 8-HDF binding site in A. nidulans photolyase, the amino acid residues interacting with 8-HDF are found at almost the same position in T. thermophilus photolyase. The 8-HDF model could be fitted to the same position as that of T. thermophilus photolyase, maintaining suitable interactions similar to A. nidulans photolyase, although only two residues, Arg-47 and Arg-114, are identical (Fig. 1). This result strongly suggests that T. thermophilus photolyase can be a deazaflavin-type photolyase. The high similarity of the binding mode of 8-HDF predicts that the interchromophoric energy transfer in T. thermophilus photolyase might have high efficiency similar to that of A. nidulans photolyase.
Figure 3.

Light-harvesting cofactor binding site. (a) Cavities at the light-harvesting cofactor binding sites in the crystal structures of T. thermophilus, A. nidulans, and E. coli photolyases. Solvent-accessible cavities are shown in yellow wire mesh. 8-HDF and MTHF are shown in red and blue, respectively. 8-HDF and MTHF in T. thermophilus, MTHF in A. nidulans, and 8-HDF in E. coli are hypothetical models, which are drawn transparently. These figures were prepared with the programs MOLSCRIPT (24), RASTER3D (25), and CONSCRIPT (26). (b) Schematic diagram of 8-HDF binding sites in A. nidulans and T. thermophilus photolyases. 8-HDF in T. thermophilus is a hypothetical model.
It is difficult to predict whether the CPD photolyases are deazaflavin- or folate-type only from their amino acids sequences. Three-dimensional structures indicate that differences of several amino acids can cause different local conformations, which are suitable for binding different cofactors, although the overall folding is quite similar among all of the CPD photolyases.
Pyrimidine-Dimer Recognition Mechanism.
Base-flipping out of double-helical DNA has been observed in many DNA repair systems, in which the enzymes need to approach to target DNA bases to perform reaction on it (20). It is also postulated to occur in the case of photolyases, because the modeling of normal double-helical B-DNA into its native structure fails to bring the target pyrimidine bases close to the active site around FAD. Examination of the solvent-accessible surface of the photolyases revealed that the FAD cofactor was accessible to the pyrimidine-dimer only through a hole leading from the isoalloxazine ring of flavin to the surface (Fig. 4a). This hole in T. thermophilus photolyase is at the same location and is almost the same size as in E. coli and A. nidulans photolyases. The positively charged concave surface around the hole, which is also found in other photolyases, is in agreement with the DNA binding site. To clarify whether the bases flipped out of the double-helical DNA can bind into this hole and to understand the detailed substrate-binding mechanism, we also determined the crystal structure of the complex with a part of its substrate, the thymine base. Photolyases bind not only damaged DNA but also nondamaged DNA. We tried to make a complex with various substrates (thymidine-dimer, thymidylyl-thymidine, thymidine-monophosphate, thymidine, and thymine), and finally succeeded in making a stable complex only with thymine. Thymine is not a genuine substrate, but it might reflect the intermediate state after the photoreaction. It is natural to consider that thymine base can bind to the active site similarly to its binding to the genuine substrate, thymidine-dimer.
Figure 4.

Substrate-binding site. (a) A view from the left side of Fig. 2a showing the putative substrate-binding site of T. thermophilus photolyase. The positive and negative charges are shown in blue and red, respectively. FAD buried in the hole is shown in yellow. The charge distribution of the molecular surface was calculated and represented by using the GRASP program (27). (b) Stereo view of the thymine binding site with the difference Fourier map of the native and thymine-complex. The Fo(THYMINE) − Fo(NATI2) electron density contoured at 4σ is shown in black wire mesh. Thymine, FAD, and amino acid residues in the active site are shown in red, yellow, and green, respectively. (c) Schematic diagram of thymine interactions with T. thermophilus photolyase. The C5-CH3 group of thymine also takes part in van der Waals contacts, indicating that thymine-containing dimers have marginally higher affinities than uracil-containing dimers. The gray circle indicates the hypothetical position for the other thymine of the thymidine-dimer. (d) Stereo view of the active site compared with other photolyases. The residues in the crystal structures of T. thermophilus, A. nidulans, and E. coli photolyases are shown in red, green, and blue, respectively. The figures in b and d were prepared with the programs MOLSCRIPT (24), RASTER3D (25), and CONSCRIPT (26).
In the crystal structure of the complex with thymine, one thymine base is found in the hole, the putative substrate-binding site (Fig. 4b). Although the electron density of thymine is partially disordered owing to the partial occupancy and high B-factor, the most suitable binding form can be assigned. Thymine is accommodated deeply within the hole and is very close to the active site, making van der Waals contact with the isoalloxazine ring of FAD. It forms a stacking interaction with Trp-353 and van der Waals interactions with other residues, Trp-247, Met-314, and Gln-349 (Fig. 4c). In particular, Trp-247 and Trp-353 seem to sandwich the thymine. Thymine also forms hydrogen bonds with Asn-310 and adenine of FAD. All these residues are conserved within the CPD photolyases, except for Gln-349, which is replaced by tyrosine in E. coli and A. nidulans photolyases (Fig. 1). The positively charged residues, Arg-201, Arg-311, Arg-363, and Arg-376, which are also conserved within the other photolyases, are found immediately outside of the thymine-binding site (Fig. 4c). Based on their structures, these residues are likely to interact with the DNA sugar-phosphate backbone flanking the pyrimidine-dimer. These interactions are almost consistent with those observed in a previous mutagenesis study (21). It has been reported that the residues in photolyase from yeast corresponding with Trp-353, Trp-247, Met-314, Glu-244, and adenine of FAD contribute to the binding affinity to substrate discrimination and to maintaining the dimer in the flipped state (21). In the complex structure, Glu-244 does not interact with the thymine. However, we expect that the other 5′-thymine base of the pyrimidine-dimer would fall into the space between Trp-247 and the thymine, and that the substrate could form a hydrogen bond with Glu-244, as predicted (Fig. 4c). In the crystal structure of the complex, the thymine base, which is much smaller than pyrimidine-dimer, nestles into the hole deeply and much closer to FAD than expected by the molecular dynamics simulations (22). However, because the inner part of the hole is too small for two thymine bases, the thymine bases might be located at the outer side in the case of the genuine substrate, pyrimidine-dimer. A comparison between native and complex structures reveals significant conformational changes around the active site. Trp-353 and Met-314 residues are found at slightly different positions, as shown in the difference Fourier map (Fig. 4b). Furthermore, compared with A. nidulans and E. coli photolyases, the Trp-353 residue is located at a considerably different position in these structures (Fig. 4d). The flexibility of Trp-353 and other residues, such as Met-314 and Trp-247, would expand the hole fitting for the pyrimidine-dimer. This model will be approximately in agreement with a previous mutagenesis study (21). This thymine-binding mode in the complex structure indicates that photolyases recognize the substrate completely flipped out of the double helix. We therefore suggest that the Trp-353 residue plays an important role in the binding of the substrate (23). Sequence conservation of the residues interacting with the thymine strongly suggests that these enzymes bind to DNA and repair the substrate in the similar manner.
Acknowledgments
We thank Drs. S. Adachi and K. Miura of SPring-8 for their kind help in the x-ray diffraction experiment. This work was partly supported by the Research for the Future program from the Japan Society for the Promotion of Science (JSPS-RFTF) Grant 97L00501 (to K.M.) and by a Grant-in-Aid (No. 4741) for the Japan Society for the Promotion of Science fellows (to H.K.).
Abbreviations
- CPD
cyclobutane pyrimidine dimer
- FAD
flavin-adenine dinucleotide
- 8-HDF
8-hydroxy-5-deazaflavin
- MTHF
5,10-methenyltetrahydrofolic acid
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org [PDB ID codes 1IQR (native) and 1IQU (complex)].
References
- 1.Sancar A, Sancar G B. Annu Rev Biochem. 1988;57:29–67. doi: 10.1146/annurev.bi.57.070188.000333. [DOI] [PubMed] [Google Scholar]
- 2.Sancar A. Biochemistry. 1994;33:2–9. doi: 10.1021/bi00167a001. [DOI] [PubMed] [Google Scholar]
- 3.Yasui A, Eker A P, Yasuhira S, Yajima H, Kobayashi T, Takao M, Oikawa A. EMBO J. 1994;13:6143–6151. doi: 10.1002/j.1460-2075.1994.tb06961.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim S T, Heelis P F, Okamura T, Hirata Y, Mataga N, Sancar A. Biochemistry. 1991;30:11262–11270. doi: 10.1021/bi00111a011. [DOI] [PubMed] [Google Scholar]
- 5.Kim S T, Heelis P F, Sancar A. Biochemistry. 1992;31:11244–11248. doi: 10.1021/bi00160a040. [DOI] [PubMed] [Google Scholar]
- 6.Park H W, Kim S T, Sancar A, Deisenhofer J. Science. 1995;268:1866–1872. doi: 10.1126/science.7604260. [DOI] [PubMed] [Google Scholar]
- 7.Tamada T, Kitadokoro K, Higuchi Y, Inaka K, Yasui A, de Ruiter P E, Eker A P, Miki K. Nat Struct Biol. 1997;4:887–891. doi: 10.1038/nsb1197-887. [DOI] [PubMed] [Google Scholar]
- 8.Kato R, Hasegawa K, Hidaka Y, Kuramitsu S, Hoshino T. J Bacteriol. 1997;179:6499–6503. doi: 10.1128/jb.179.20.6499-6503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oshima T. Seikagaku. 1974;46:887–907. [PubMed] [Google Scholar]
- 10.Payne G, Wills M, Walsh C, Sancar A. Biochemistry. 1990;29:5706–5711. doi: 10.1021/bi00476a010. [DOI] [PubMed] [Google Scholar]
- 11.Komori, H., Tsujiuchi, H., Masui, R., Kuramitsu, S., Yokoyama, S., Shibata, T., Inoue, Y. & Miki, K. (2001) Protein Peptide Lett., in press. [DOI] [PMC free article] [PubMed]
- 12.Collaborative Computational Project, Number 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 13.Jones, T. A. & Kjeldgaard, M. (1994) O, The Manual (Uppsala University, Uppsala).
- 14.Brunger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 15.Brünger A T. Nature (London) 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 16.Laskowski R J, Macarthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–290. [Google Scholar]
- 17.Kleywegt G J, Jones T A. Eur Sci Found/CCP4 Newsl. 1994;31:9–14. [Google Scholar]
- 18.Rossmann M G, Moras D, Olsen K W. Nature (London) 1974;250:194–199. doi: 10.1038/250194a0. [DOI] [PubMed] [Google Scholar]
- 19.Walden H, Bell G S, Russell R J, Siebers B, Hensel R, Taylor G L. J Mol Biol. 2001;306:745–757. doi: 10.1006/jmbi.2000.4433. [DOI] [PubMed] [Google Scholar]
- 20.Roberts R J, Cheng X. Annu Rev Biochem. 1998;67:181–198. doi: 10.1146/annurev.biochem.67.1.181. [DOI] [PubMed] [Google Scholar]
- 21.Vande Berg B J, Sancar G B. J Biol Chem. 1998;273:20276–20284. doi: 10.1074/jbc.273.32.20276. [DOI] [PubMed] [Google Scholar]
- 22.Sanders D B, Wiest O. J Am Chem Soc. 1999;121:5127–5134. [Google Scholar]
- 23.Li Y F, Heelis P F, Sancar A. Biochemistry. 1991;30:6322–6329. doi: 10.1021/bi00239a034. [DOI] [PubMed] [Google Scholar]
- 24.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 25.Merrit E A, Murphy M E. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 26.Lawrence M C, Bourke P. J Appl Crystallogr. 2000;33:990–991. [Google Scholar]
- 27.Nicholls A, Sharp K A, Honig B. Proteins Struct Funct Genet. 1991;11:282. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]