

Abstract
Sunitinib is a multitargeted tyrosine kinase inhibitor associated with idiosyncratic hepatotoxicity. The mechanisms of this toxicity are unknown. We hypothesized that sunitinib undergoes metabolic activation to form chemically reactive, potentially toxic metabolites which may contribute to development of sunitinib-induced hepatotoxicity. The purpose of this study was to define the role of cytochrome P450 (P450) enzymes in sunitinib bioactivation. Metabolic incubations were performed using individual recombinant P450s, human liver microsomal fractions, and P450-selective chemical inhibitors. Glutathione (GSH) and dansylated GSH were used as trapping agents to detect reactive metabolite formation. Sunitinib metabolites were analyzed by liquid chromatography–tandem mass spectrometry. A putative quinoneimine–GSH conjugate (M5) of sunitinib was detected from trapping studies with GSH and dansyl–GSH in human liver microsomal incubations, and M5 was formed in an NADPH-dependent manner. Recombinant P450 1A2 generated the highest levels of defluorinated sunitinib (M3) and M5, with less formation by P450 3A4 and 2D6. P450 3A4 was the major enzyme forming the primary active metabolite N-desethylsunitinib (M1). In human liver microsomal incubations, P450 3A inhibitor ketoconazole reduced formation of M1 by 88%, while P450 1A2 inhibitor furafylline decreased generation of M5 by 62% compared to control levels. P450 2D6 and P450 3A inhibition also decreased M5 by 54 and 52%, respectively, compared to control. In kinetic assays, recombinant P450 1A2 showed greater efficiency for generation of M3 and M5 compared to that of P450 3A4 and 2D6. Moreover, M5 formation was 2.7-fold more efficient in human liver microsomal preparations from an individual donor with high P450 1A2 activity compared to a donor with low P450 1A2 activity. Collectively, these data suggest that P450 1A2 and 3A4 contribute to oxidative defluorination of sunitinib to generate a reactive, potentially toxic quinoneimine. Factors that alter P450 1A2 and 3A activity may affect patient risk for sunitinib toxicity.
Graphical abstract
INTRODUCTION
Sunitinib malate (Sutent; Pfizer) is an orally administered small molecule tyrosine kinase inhibitor approved for the treatment of metastatic renal cell carcinoma, imatinib-resistant gastro-intestinal stromal tumor, and pancreatic neuroendocrine tumors.1–3 Sunitinib inhibits multiple receptor tyrosine kinases, including vascular endothelial growth factor receptors, platelet-derived growth factor receptors, Fms-like tyrosine kinase 3, and stem cell factor receptor KIT.4,5 Sunitinib exhibits potent antitumor and antiangiogenic activity.4
Mechanism-based adverse events associated with sunitinib include hypertension, fatigue, skin toxicities, thrombocytopenia, neutropenia, and hypothyroidism.6,7 These toxicities are thought to be related to the interaction of sunitinib or its active metabolite with pharmacological targets.6 Moreover, sunitinib carries a black box warning for rare but potentially life-threatening idiosyncratic hepatotoxicity.3 Clinically significant liver injury was reported to occur in 3–4% of patients taking sunitinib, and liver failure occurred in 0.3% of patients.3 The underlying mechanisms for sunitinib-induced liver injury remain unknown.
Sunitinib is primarily metabolized in vitro and in vivo via N-deethylation by cytochrome P450 3A4 to the active metabolite N-desethylsunitinib (M1).1,6,8,9 M1 has activity equal to that of the parent drug, and M1 is the major circulating metabolite of sunitinib in humans.1,3,6,9 Other P450 enzymes, including P450 3A5, 1A2, and 1A1, have also been suggested to play a role in sunitinib metabolism; however, their relative contributions have not been established.10–12 Additional metabolic pathways identified for sunitinib include aromatic and aliphatic hydroxylation, N-oxidation, and oxidative defluorination.9 Recent evidence suggests that sunitinib may undergo metabolic activation to form chemically reactive, potentially toxic metabolites, similar to other tyrosine kinase inhibitors.13–17 The bioactivation pathways of sunitinib have not been characterized.
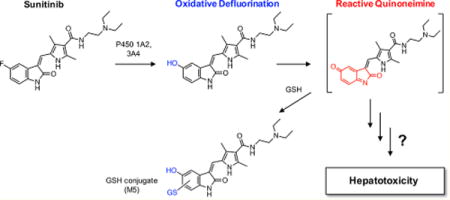
Understanding the metabolic pathways of sunitinib is important to gain insight into the potential mechanisms and risk factors of drug toxicity. The goals of this investigation were to (1) characterize the metabolism and bioactivation pathways of sunitinib in vitro and (2) identify which enzymes are involved. We present evidence that sunitinib undergoes P450 1A2 and 3A4-mediated bioactivation to form a reactive quinoneimine trapped as a GSH conjugate.
MATERIALS AND METHODS
General Reagents
Sunitinib free base (S-8877) and sunitinib malate (S-8803) were purchased from LC Laboratories (Woburn, MA). Deuterium-labeled sunitinib (N-[2-diethylamino(ethyl-d4)]-5-[(Z)-(5-fluoro-1,2-dihydro-2-oxo-3H-indo-3-yldiene)methyl]-2,4-di-methyl-1H-pyrrole-3-carboxamide, sunitinib-d4, S820003), N-desethyl-sunitinib (D289650), sunitinib N-oxide (S820005), furafylline, (+)-N-3-benzylnirvanol, 2-phenyl-2-(1-piperidinyl)propane (PPP), and CY-P3cide were purchased from Toronto Research Chemicals (Toronto, Canada). GSH, α-naphthoflavone (7,8-benzoflavone), ticlopidine·HCl, sulfaphenazole, quinidine, 4-methylpyrazole·HCl, and ketoconazole were purchased from Sigma Aldrich (St. Louis, MO). All other chemicals and reagents were of analytical grade or higher and were purchased from standard commercial sources. Stock solutions of sunitinib, sunitinib-d4 (internal standard), N-desethylsunitinib, and chemical inhibitors were prepared in dimethyl sulfoxide (DMSO) and diluted into acetonitrile to make up a working solution in 1/9 DMSO/acetonitrile (v/v), unless otherwise stated.
Recombinantly expressed P450 enzymes and human liver microsomal preparations used in this study were similar to those described by Towles et al.18 Briefly, Supersomes (baculovirus-infected insect cell microsomes) containing cDNA-expressed human P450 1A1, 1A2, 1B1, 2B6, 2C8, 2C9*1, 2C19, 2D6*1, 2E1, 2J2, 3A4, and 3A5 coexpressed with P450 reductase and cytochrome b5, except P450 1A2 and 2D6*1 (expressed without cytochrome b5), were purchased from Corning Discovery Labware (Woburn, MA). Pooled (150-Donor Ultra Pooled, mixed gender) and single-donor human liver microsomes (Lots HH741 and HH581) were purchased from Corning Discovery Labware. The NADPH-regenerating system (Solution A: 26 mM NADP+, 66 mM glucose 6-phosphate, 66 mM MgCl2 in water; Solution B: 40 U/mL glucose 6-phosphate dehydrogenase in 5 mM sodium citrate) and UDP-glucuronic acid, with alamethacin (UDP-glucuronosyltransferase, UGT solutions A and B), were purchased from Corning Discovery Labware.
Human Liver Microsomal Incubations for Metabolite Identification
Sunitinib (10 μM) was incubated with pooled human liver microsomes (0.5 mg/mL) in 100 mM potassium phosphate buffer, pH 7.4, in the presence of an NADPH-regenerating system for 30 min at 37 °C in a shaking temperature-controlled water bath. Control experiments were performed without the cofactor. The final reaction volume was 0.2 mL. After the incubation period, reactions were quenched with the addition of 2 volumes (0.4 mL) of ice-cold acetonitrile containing sunitinib-d4 (100 ng/mL) and 0.2% formic acid (v/v). Samples were mixed with a vortex device for 10 s, placed on ice for 5–10 min, and centrifuged for 20 min at 20000g at 4 °C. The supernatant was transferred to a clean microcentrifuge tube, and the solvent was dried under a gentle stream of nitrogen gas using a Biotage TurboVap system (Charlotte, NC) to concentrate the samples. Samples were then redissolved in 100 μL of 3/7 acetonitrile/water (v/v), mixed using a vortex device, and centrifuged for 5 min. The clear supernatant was transferred to vials for LC–MS and LC–MS/MS analysis.
GSH trapping experiments were conducted to determine the potential for sunitinib to form reactive electrophilic metabolites. For these studies, sunitinib (10–50 μM) was incubated with pooled human liver microsomes (0.5 mg/mL) in 100 mM potassium phosphate buffer, pH 7.4, in the presence of an NADPH-regenerating system and supplemented with GSH (5 mM) for 30–60 min. Samples were prepared for LC–MS/MS analysis as described above. Control experiments were performed without NADPH-regenerating system.
To examine sunitinib glucuronidation, sunitinib (10 μM) was incubated with pooled human liver microsomes (0.5 mg/mL) in 100 mM potassium phosphate buffer, pH 7.4, in the presence of UDP-glucuronic acid with alamethacin (UGT solutions A and B) for 30 min at 37 °C in a shaking temperature-controlled water bath. Control experiments were performed without cofactor. The final reaction volume was 0.2 mL. After the incubation period, reactions were quenched with the addition of 2 volumes (0.4 mL) of ice-cold acetonitrile containing sunitinib-d4 (100 ng/mL) and 0.2% formic acid (v/v). Samples were mixed using a vortex device for 10 s, placed on ice for 5–10 min, and centrifuged for 20 min at 20000g at 4 °C. The supernatant was subjected to LC–MS/MS analysis.
Reaction Phenotyping with Recombinant P450s
An initial screen of recombinant human P450 Supersomes was performed with sunitinib (25 μM). Subsequent screening was conducted with sunitinib (10 μM) using a panel of human P450 Supersomes: P450s 1A2, 2B6, 2C8, 2C9*1, 2C19, 2D6*1, 2E1, 3A4, and 3A5. For each screen, sunitinib was incubated with individual recombinant P450s (20 nM each) in 100 mM potassium phosphate buffer, pH 7.4, in the presence of an NADPH-regenerating system and supplemented with GSH (5 mM) for 10 min in a final reaction volume of 0.2 mL. Additional experiments were performed to evaluate sunitinib product formation by extrahepatic P450s 1A1, 1B1, and 2J2 along with P450s 3A4 and 3A5. For these experiments, sunitinib (25 μM) was incubated with recombinant P450s 1A1, 1B1, 2J2, 3A4, and 3A5 (20 nM) in the presence of an NADPH regenerating system and supplemented with GSH (5 mM) for 10 min in a final reaction volume of 0.2 mL. Control experiments were performed without NADPH-regenerating system. Samples were prepared for LC–MS/MS analysis as described above. Relative levels of sunitinib metabolites were measured by LC–MS/MS analysis.
Reaction Phenotyping with P450 Chemical Inhibitors
Because P450s 1A2 and 3A4 appeared to be involved in sunitinib product formation from phenotyping screens using recombinant enzymes, inhibition of sunitinib metabolism was initially evaluated by coincubation of sunitinib with α-naphthoflavone (P450 1A inhibitor), furafylline (P450 1A2 inhibitor), or ketoconazole (P450 3A inhibitor). Control experiments were performed with vehicle control (1/9 DMSO/acetonitrile) without inhibitor. Sunitinib (10 μM) was incubated with pooled human liver microsomes (0.1 mg protein/mL) in 100 mM potassium phosphate buffer, pH 7.4, supplemented with an NADPH-regenerating system and GSH (5 mM) in the presence or absence of P450 chemical inhibitors for 10 min in a final reaction volume of 0.2 mL.
Next, inhibition assays were performed with a panel of chemical inhibitors of P450 1A2, 2B6, 2C9, 2C19, 2D6, 2E1, and 3A to determine the effect of P450 inhibition on metabolite formation, similar to the methods described previously.18 Co-incubation of sunitinib (10 μM) in pooled human liver microsomes (0.1 mg protein/mL) was carried out with the following P450 chemical inhibitors: 1 μM α-naphthoflavone (P450 1A2), 25 μM furafylline (P450 1A2), 15 μM phenyl-piperidinyl propane (PPP) (P450 2B6), 5 μM ticlopidine (P450 2B6 and 2C19), 5 μM sulfaphenazole (P450 2C9), 5 μM (+)-N-3-benzylnirvanol (P450 2C19), 2 μM quinidine (P450 2D6), 100 μM 4-methylpyrazole (P450 2E1), 1 μM ketoconazole (P450 3A), and 2 μM CYP3cide (P450 3A4). Working solutions of each inhibitor were prepared in 1/9 DMSO/acetonitrile, except CYP3cide, which was prepared as a solution in methanol. Control incubations were carried out with vehicle (1/9 DMSO/acetonitrile, v/v, or methanol) in the absence of inhibitor.
Final organic solvent concentrations for each incubation were ≤1% (v/v). Reactions were initiated by addition of the NADPH-regenerating system (final reaction volume 0.2 mL) and continued for 10 min. After the incubation period, reactions were quenched by addition of 2 volumes (0.4 mL) of ice-cold acetonitrile containing 10 ng/mL of sunitinib-d4 (internal standard). Samples were mixed using a vortex device for 10 s, placed on ice for 5–10 min, and centrifuged for 20 min at 20000g at 4 °C. The clear supernatant was transferred to LC–MS vials, and aliquots (10 μL) were injected for LC–MS/MS analysis of metabolites.
Kinetic Determinations of Sunitinib Metabolite Formation
For kinetic assays with recombinant P450 enzymes, preliminary experiments were conducted to determine the linearity of product formation with respect to P450 concentration (5–20 nM) and incubation time (2.5–20 min). Product formation was found to be linear with P450 concentration 20 nM up to 10 min. Recombinant P450s 1A1, 1A2, 2D6, and 3A4 (20 nM) were incubated with a range of 10 sunitinib concentrations (final concentrations 0.05–50 μM) in the presence of GSH (5 mM) in 100 mM potassium phosphate buffer, pH 7.4. Reactions were initiated with addition of an NADPH-regenerating system. The total incubation volume was 0.2 mL. Incubations were carried out for 10 min. Reactions were quenched by the addition of 2 volumes (0.4 mL) of cold acetonitrile containing 10 ng/mL sunitinib-d4 (internal standard) with 0.2% formic acid (v/v). Formation of N-desethylsunitinib (M1) was quantified by LC–MS/MS MRM analysis using a standard curve. Relative levels of defluorosunitinib (M3) and quinoneimine–GSH conjugates (M5) were measured by LC–MS/MS analysis.
For kinetic assays with human liver microsomal preparations, preliminary experiments were conducted to determine the linearity of product formation with respect to microsomal protein concentration (0.05–0.25 mg/mL) and incubation time (2.5–20 min). Product formation was found to be linear with protein concentration 0.25 mg/mL up to 10 min. To determine the relative kinetic parameters (Km, Vmax) of metabolite formation in pooled human liver microsomes, a range of 10 sunitinib concentrations (final concentrations 0.05–50 μM) was incubated with pooled human liver microsomes (0.25 mg/mL) in 100 mM potassium phosphate buffer, pH 7.4, in the presence of GSH (5 mM). Reactions were initiated with addition of an NADPH-regenerating system. The total incubation volume was 0.2 mL. Following a 10 min incubation period, reactions were quenched by the addition of 2 volumes (0.4 mL) of cold acetonitrile containing 10 ng/mL sunitinib-d4 (internal standard) with 0.2% formic acid (v/v). As described above, formation of M1 was quantified by LC–MS/MS MRM analysis using a standard curve, and relative levels of M3 and M5 were measured by LC–MS/MS analysis.
In a second series of experiments, the kinetic parameters of sunitinib metabolite formation were determined with human liver microsomes from two individual donors characterized for high vs low P450 1A2 activity. P450 activities in single donor lots were characterized by Corning-Gentest using probe substrates: P450 1A2 (phenacetin O-deethylase) and P450 3A4 (testosterone 6β-hydroxylase). Lot HH581 (male, age 58) had a phenacetin O-deethylase activity of 2100 pmol/min/mg protein and testosterone 6β-hydroxylase activity of 5400 pmol/min/mg protein; lot HH741 (male, age 68) had a phenacetin O-deethylase activity of 240 pmol/min/mg protein and testosterone 6β-hydroxylase activity of 7400 pmol/min/mg protein (Corning Gentest inventory datasheet). Similar to the method described above, human liver microsomes from donor HH581 and HH741 (0.25 mg/mL) were incubated with a range of 10 sunitinib concentrations (final concentrations 0.05–50 μM) in the presence of GSH (5 mM). Reactions were initiated with addition of an NADPH-regenerating system. The total incubation volume was 0.2 mL. Incubations were carried out for 10 min. Reactions were quenched by the addition of 2 volumes (0.4 mL) of cold acetonitrile containing 10 ng/mL sunitinib-d4 (internal standard) with 0.2% formic acid (v/v). Levels of M1, M3, and M5 were measured by LC–MS/MS analysis.
Quantitation of N-Desethylsunitinib (M1)
A commercially available authentic standard of M1 (Toronto Research Chemicals) was used for the quantitation of M1 using a standard curve. A 10- to 12-point standard curve was prepared by adding known concentrations of M1 (final concentrations 1.2 ng/mL to 5000 ng/mL; final concentration of organic solvent, 0.1% DMSO, 0.9% acetonitrile, v/v) to 100 mM potassium phosphate buffer (pH 7.4) with pooled human liver microsomes (0.25 mg/mL) or recombinant P450 3A4 (20 nM). This standard mix was prepared in duplicate for each concentration with a final volume of 0.2 mL. Then, 2 volumes (0.4 mL) of cold acetonitrile containing 10 ng/mL sunitinib-d4 (internal standard) with 0.2% formic acid (v/v) were added to each sample. Calibration standards of M1 were prepared on three independent days to assess accuracy and intra- and interday variability. Authentic chemical standards of M3 and M5 were not available; therefore, relative levels of metabolite formation were measured by calculating the peak area ratio of metabolite to internal standard.
Synthesis and Characterization of Dansyl-Glutathione (dGSH)
The synthesis of dansyl–glutathione (dGSH) was similar to the methods described by Gan et al.19 Briefly, first, dansylated oxidized glutathione (dGSSG) was prepared by reaction of GSSG with dansyl chloride. The product was purified by preparative HPLC. Next, the disulfide bond of dGSSG was reduced to yield dGSH. The product was purified by preparative HPLC, and the identify was confirmed by LC–UV–MS analysis and fluorescence detection (excitation 340 nm, emission 525 nm).19 The dried preparation of dGSH was redissolved in methanol to prepare a 100 mM solution. LC–MS characterization of dGSH was performed using positive electrospray ionization. The precursor ion (m/z 541) of dGSH was confirmed by LC–MS full scan analysis (m/z 300–1100). MS/MS fragmentation of the precursor ion m/z 541 utilizing collision-induced dissociation yielded the major product ions m/z 466 (loss of glycine), 438, 363 (cleavage of the pyroglutamic acid-dansyl moiety), and 234 (cleavage of the dansyl moiety). This fragmentation pattern is consistent with the structure of dGSH.19,20
Identification of Dansyl–GSH Reactive Metabolite Conjugates
To evaluate the generation of reactive metabolite–dGSH conjugates, sunitinib (25 and 50 μM) was incubated with recombinant P450 1A2 Supersomes (20 nM) or pooled human liver microsomes (1 mg/mL) in the presence of 1 mM dGSH in 100 mM potassium phosphate buffer, pH 7.4. Reactions were initiated by addition of the NADPH-regenerating system and continued for 30 min. The final reaction volume was 0.2 mL, and the final concentration of organic solvent in each incubation was 0.05% DMSO, 0.45% acetonitrile (v/v). Following the 30 min incubation, reactions were terminated by addition of 2 volumes (0.4 mL) of ice-cold acetonitrile containing 10 ng/mL of sunitinib-d4 (internal standard) with 0.1% formic acid (v/v). Samples were mixed with a vortex device for 10 s, placed on ice 5–10 min, and centrifuged for 20 min at 20000g at 4 °C. The supernatant was transferred to separate vials, and 10–20 μL was analyzed by LC–MS/MS for detection of reactive metabolite–dGSH conjugates. Control incubations were without NADPH-regenerating system, dGSH, or substrate.
UPLC–UV–Fluorescence Analysis of Metabolic Incubations with Dansyl–GSH
Additional experiments were conducted to quantify the formation of dGSH conjugates of the putative sunitinib quinoneimine based on the methods described by Gan et al. with slight modifications.19,20 Briefly, sunitinib (25 μM) was incubated with pooled human liver microsomes (1 mg/mL) in the presence of dGSH (1 mM, containing 0.01 mM tris(2-carboxyethyl)phosphine hydrochloride, TCEP). The reaction mixture was prewarmed for 5 min at 37 °C. Reactions were initiated by the addition of an NADPH regenerating system, and incubations were carried out for 30 min at 37 °C in a shaking water-bath. The final reaction volume was 0.2 mL. Control incubations were without NADPH, without dGSH, or without drug substrate (blank). Each reaction condition was performed in triplicate. Reactions were terminated after 30 min by the addition of 2 volumes of ice-cold acetonitrile containing 10 ng/mL sunitinib-d4, and 5 mM dithiothreitol (DTT). Samples were mixed using a vortex mixer, placed on ice for 5–10 min, and centrifuged for 20 min at 14000 rpm. The supernatant was transferred to separate LC–MS vials for UPLC–UV–fluorescence analysis. Samples were analyzed using a Waters Acquity ultra performance liquid chromatography (UPLC)–UV–fluorescence system equipped with a Binary Solvent Manager, a Sample Manager autosampler, a photodiode array (PDA) detector, and a fluorescence (FLR) detector. The PDA was operated over the wavelength range 200–500 nm, and fluorescence detection was carried out at excitation 340 nm, emission 525 nm. Analyte separation was achieved using a Waters Acquity UPLC BEH C18 octadecylsilane column (1.7 μm, 2.1 × 100 mm) with a flow rate of 0.3 mL/min. Mobile phases were (A) 0.1% formic acid in LC/MS-grade water and (B) 0.1% formic acid in LC/MS-grade acetonitrile (all v/v). The following LC gradient program was used: 15% B (0–4.0 min), linear gradient from 15 to 40% B (4.0–14.0 min), 40 to 95% B (14.0–15.0 min), 95% B (15–16.0 min), and returned to 15% B (16.0–16.5 min) (all v/v). The total run time was 20 min. The sample injection volume was 20 μL. An external standard of dansyl–GSH (0.0098–10 μM) was prepared and analyzed by UPLC–UV–fluorescence. The limit of detection for dGSH above the background was 0.313 μM dGSH using this method.
Analysis of Sunitinib and Metabolites by LC–MS/MS
Analysis of sunitinib and sunitinib metabolites was carried out by LC–MS/MS using similar methods on two different systems. System A consisted of a Thermo Accela ultrahigh performance liquid chromatography (UHPLC) system equipped with a Thermo PAL autoinjector and a column oven coupled to a Thermo TSQ Quantum Triple Quadrupole mass spectrometer (Thermo Fisher Scientific, Waltham, MA), as described previously.18 Analyte separation was achieved using a Kinetex C18 or EVO C18 octadecylsilane column (2.6 μm, 50 mm × 2.1 mm, 100 Å) (Phenomenex, Torrance, CA) with a flow rate of 0.3 mL/min and column oven temperature 30 °C. Mobile phases were (A) 0.1% formic acid in LC/MS-grade water and (B) 0.1% formic acid in LC/MS-grade acetonitrile (all v/v). The following LC gradient program was used: 5% B (0–1.0 min), linear gradient from 5 to 95% B (1.0–3.5 min), 95% B (4.0–4.5 min), and returned to 5% B (4.5–5.0 min) (all v/v). The sample injection volume was 10–20 μL, and the eluent was introduced directly into the mass spectrometer via electrospray ionization in the positive ion mode (ESI+). The MS spectral data were analyzed and deconvoluted using Thermo Xcalibur software 2.0.
System B consisted of a Shimadzu Prominence XR UHPLC system, equipped with two Shimadzu LC-20ADXR pumps, a SIL-20ACXR autosampler, and a CTO-20A column oven, coupled to a Shimadzu LC–MS/MS 8030 triple quadrupole mass spectrometer (Shimadzu, Columbia, MD), similar to that described previously.21 As described above, analyte separation was achieved using a Kinetex C18 or EVO C18 octadecylsilane column (2.6 μm, 50 × 2.1 mm, 100 Å) (Phenomenex) with a flow rate of 0.3 mL/min and column oven temperature 32 °C. The following LC gradient program was used: 5% B (0–1.0 min), linear gradient from 5 to 95% B (1.0–2.0 min), 95% B (2.0–3.0 min), 95 to 5% B (3.0–3.5 min), and returned to 5% B (3.5–5.0 min) until the end of the run (all v/v). The injection volume was 10–20 μL, and the eluent was introduced directly into the mass spectrometer via ESI in positive ion mode. MS data were acquired and analyzed using Shimadzu LabSolutions software.
Structural Analysis and Quantitation
Sunitinib was initially subjected to LC–MS/MS analysis to observe its characteristic fragmentation. When analyzed by ESI in positive mode, sunitinib had the expected molecular ion m/z 399.22 The major product ions generated were m/z 326 and m/z 283, which correspond to loss of the diethylamine moiety and cleavage of the aliphatic amide bond, respectively.9 In the initial LC–MS method development for sunitinib, 2 chromatographic peaks were detected at retention times 3.02 and 3.86 min, with the same molecular ion at m/z 399 and similar fragmentation patterns (product ions m/z 326 and m/z 283). The observation of two chromatographic peaks suggested two isomers of sunitinib. Previous studies have described light-induced isomerization of sunitinib from the Z- to E-isomer.9,23 Thus, all subsequent experiments were performed under light protection.
Sunitinib metabolites were analyzed by LC–MS/MS in the product ion scan mode to permit structural elucidation. The predicted metabolites and corresponding precursor ions were as follows: M1 (m/z 371), M3 (m/z 397), monooxygenated metabolites (M2) (m/z 415), and glucuronide conjugate (M4) (m/z 575). Detection of M5 was performed by LC–MS/MS analysis using neutral loss scans for loss of 129 Da, corresponding to loss of the pyroglutamate portion of GSH.24 Structural characterization of M5 was conducted in product ion scan mode for the precursor ion m/z 702, corresponding to the predicted structure. MS/MS analysis of precursor ion m/z 935 was performed for characterization of quinoneimine–dGSH conjugates. In addition, MS/MS analysis of the precursor ions m/z 706 and m/z 573 was used for detection and structural elucidation of direct sunitinib–GSH conjugates and quinoneimine cysteinyl–glycine conjugates, respectively.
Relative levels of sunitinib metabolites were measured by LC–MS/MS analysis utilizing multiple reaction monitoring (MRM). The MRM transitions for the metabolites indicated were as follows: M1 (m/z 371 > 283), M2 (m/z 415 > 299), M3 (m/z 397 > 281), M5 (m/z 702 > 586), direct sunitinib–GSH conjugates (Sun–SG) (m/z 706 > 577), quinoneimine–dGSH conjugate(s) (m/z 935 > 862 and m/z 935 > 313), sunitinib–dGSH conjugate(s) (m/z 939 > 399), and M1–dGSH conjugate(s) (m/z 911 > 371).
High Resolution Mass Spectrometric Analysis of Sunitinib GSH Conjugates
GSH conjugates of sunitinib (Sun–SG) and the putative quinoneimine reactive metabolite (M5) were analyzed by high resolution mass spectrometry (HRMS) to confirm the predicted structures. HRMS analysis was performed using a Waters Acquity UPLC system coupled to an LTQ Orbitrap XL (Thermo). Analyte separation was achieved with a Kinetex EVO C18 octadecylsilane column (2.6 μm, 50 × 2.1 mm, 100 Å) (Phenomenex) with a flow rate of 0.3 mL/min and column oven temperature 32 °C. Mobile phases were (A) 0.1% formic acid in LC/MS-grade water and (B) 0.1% formic acid in LC/MS-grade acetonitrile (all v/v). Two UPLC gradient programs were used: one with a total run time of 5 min, and one with a total run time of 10 min. The 5 min UPLC gradient program was as follows: 5% B (0–1.0 min), linear gradient from 5 to 95% B (1.0–2.0 min), 95% B (2.0–3.0 min), 95 to 5% B (3.0–3.5 min), and returned to 5% B (3.5–5.0 min) (all v/v). The 10 min UPLC gradient program was as follows: 5% B (0–1.0 min), linear gradient from 5 to 95% B (1.0–6.0 min), 95% B (6.0–6.5 min), 95 to 5% B (6.5–7.0 min), and returned to 5% B (7.0–10.0 min) (all v/v). For both methods, the injection volume was 20 μL, and the eluent was introduced directly into the mass spectrometer via ESI in positive ion mode. Full scan HRMS spectra were obtained in positive ion ESI mode over the mass range m/z 150–800. HRMS spectra were collected with a resolution of 60000. MS/MS analysis was performed by ion trap mass spectrometry (ITMS) using higher energy collisional dissociation (collision energy 25 V). The following precursor ions were analyzed by MS/MS: m/z 702.29, 704.29, and 706.30.
Data Analysis
Kinetic parameters (Km, Vmax, kcat) were estimated by nonlinear regression analysis using GraphPad Prism Software 7.0. Data were fit to the Michaelis–Menten equation for estimation of Km, Vmax, kcat. Comparison of the effect of P450 inhibitors on metabolite formation vs control incubations without inhibitor was performed using a two-tailed unpaired t test. Each experiment was performed in triplicate, and at least three independent experiments were performed for each assay unless otherwise stated. Data are reported as mean ± standard deviation (SD) or standard error of the mean (SEM) where indicated. Statistical analysis was performed using GraphPad Prism Software 7.0. Statistical significance was defined as p < 0.05.
RESULTS
Sunitinib Metabolite Identification
Incubation of sunitinib (10 μM) with human liver microsomal preparations generated drug products similar to the metabolites described in vivo9 based on LC–MS/MS analysis. The N-desethyl metabolite of sunitinib (M1) has a molecular ion at m/z 371, which is 28 Da lower than that of the parent drug, indicating loss of one ethyl group (Figure S1A). The major product ions of M1 were m/z 326 and 283, which are similar to the fragment ions produced from sunitinib itself. Assignment of this metabolite as M1 was confirmed by LC–MS/MS analysis of an authentic chemical standard. Multiple mono-oxygenated metabolites of sunitinib (M2) were detected, each having a molecular ion at m/z 415 (Figure S1B), 16 Da higher than the parent drug and corresponding to addition of oxygen. The major product ions of m/z 415 were m/z 342 and 299, which indicate oxidation of the indolylidene/dimethylpyrrole moiety of sunitinib.9 LC–MS/MS analysis of additional metabolites with the precursor ion m/z 415 generated the product ions m/z 326 and 283 (data not shown). The difference in fragmentation pattern indicates addition of oxygen within the diethylamine portion of the molecule.9
Evidence for a defluorinated metabolite of sunitinib (M3) was also observed from incubations with human liver microsomes. The precursor ion for this metabolite was m/z 397, 2 Da lower than sunitinib (Figure S1C). The major product ions generated from m/z 397 were m/z 324 and 281. This fragmentation pattern is consistent with a defluorinated metabolite of sunitinib formed via oxidative defluorination and substitution with oxygen, as reported by Speed et al.9 The identity of M3 (C22H29N4O3+) was confirmed by HRMS analysis; the observed accurate mass was m/z 397.2239 (data not shown). A glucuronide conjugate of sunitinib (M4) was detected following incubation of human liver microsomes with sunitinib in the presence of the glucuronidation cofactor UDP-glucuronic acid. The precursor ion was m/z 575, which is 176 Da higher than sunitinib (Figure S1D). This corresponds to the addition of a glucuronic acid moiety. The major product ions of m/z 575 were m/z 502, 459, and 326. This metabolite and its MS/MS fragmentation pattern were reported in vivo by Speed et al.9
Other minor products observed from human liver microsomal incubations with sunitinib had molecular ions at m/z 343, 387, and 401 (data not shown). These likely represent bis-desethylsunitinib, monooxygenated N-desethylsunitinib, and dihydrosunitinib, respectively, as described previously.9
Detection of GSH Conjugates of Sunitinib
GSH trapping studies were performed to assess formation of electrophilic metabolites. Incubation of sunitinib (10–50 μM) with pooled human liver microsomes supplemented with GSH and the NADPH-regenerating system yielded a product consistent with a GSH conjugate of a putative quinoneimine metabolite (M5) (Figure 1A). When analyzed by LC–MS/MS in positive ion mode, this metabolite had a molecular ion at m/z 702, which is 305 Da higher than that of M3.13 The major product ions of m/z 702 were m/z 629 and 586, consistent with cleavage of the diethylamine moiety and the aliphatic amide bond of the drug portion of the molecule, respectively. Other fragment ions were m/z 573 (loss of 129 Da, characteristic of neutral loss of the pyroglutamic acid moiety of GSH)24 and m/z 500 (cleavage of the diethyl amine moiety on the drug portion of molecule and loss of the pyroglutamic acid moiety of GSH). M5 was formed in an NADPH-dependent manner, in that no GSH conjugates with the m/z 702 were detected in control incubations without the NADPH-generating system. A product consistent with a cysteinyl–glycine conjugate was also observed from incubation of sunitinib with human liver microsomes. The precursor ion was m/z 573, and the observed product ions were m/z 500 and m/z 457, corresponding to loss of the diethylamine moiety and cleavage of the amine bond of sunitinib, respectively (data not shown).
Figure 1.
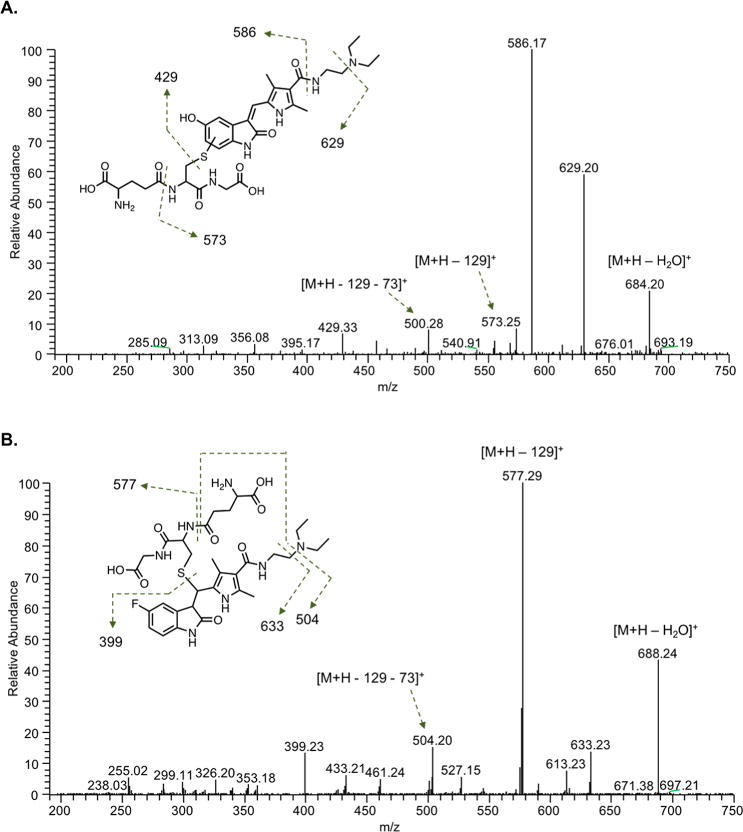
LC–MS/MS analysis of sunitinib GSH conjugates. Sunitinib (50 μM) was incubated with pooled human liver microsomes (0.5 mg/mL) in the presence of an NADPH-regenerating system and supplemented with GSH (5 mM) for 30 min. LC–MS/MS analysis using electrospray ionization was performed in positive ion mode. Shown are representative product ion spectra from UPLC–MS/MS analysis of (A) the putative quinoneimine–GSH conjugate of sunitinib (M5) with precursor ion m/z 702.29 and (B) the direct sunitinib–GSH conjugate (Sun–SG) with precursor ion m/z 706.30. Higher-energy collisional dissociation MS/MS analysis was performed using an LTQ Oribitrap XL (Thermo) in ESI positive ion mode (collision energy 25 V). The predicted structures of (A) M5 and (B) Sun–SG are shown.
In addition to detection of M5, evidence for the direct conjugation of GSH to the parent drug sunitinib (Sun–SG) was found from microsomal incubations in the absence of NADPH supplementation. The predicted precursor ion of the Sun–SG is m/z 706 (399 + 307). A major product ion of m/z 706 was m/z 577 (neutral loss of 129 Da, the pyroglutamic acid moiety, characteristic of GSH adducts)24 (Figure 1B).
HRMS Analysis of Sunitinib GSH Conjugates
HRMS analysis was used to confirm the identities of GSH conjugates derived from the putative quinoneimine metabolite and the parent drug sunitinib, M5 and Sun–SG, respectively. The calculated exact mass of the protonated quinoneimine–GSH conjugate (M5) is 702.2916 (C32H44N7O9S+). The observed mass was m/z 702.2911 (Δ0.7 ppm) (Figure S2A). The calculated exact mass of protonated product ions of M5 are 629.2024 (C28H33N6O9S+), 586.1602 (C26H28N5O9S+), and 573.2490 (C27H37N6O6S+). MS/MS analysis of m/z 702.29 yielded the product ions: m/z 684.20 (loss of water), 629.20 (cleavage of the diethylamine), 586.17 (cleavage of the aliphatic amide bond of the drug portion of the molecule), 573.25 (neutral loss of 129 Da, corresponding to the pyroglutamic acid moiety of GSH),24 and 500.28 (cleavage of the diethylamine and neutral loss of 129) (Figure 1A). The calculated exact mass of the protonated sunitinib–GSH conjugate (Sun–SG) is 706.3029 (C32H45FN7O8S+). The observed mass was m/z 706.3023 (Δ0.8 ppm) (Figure S2A). The calculated exact mass of major product ions of Sun–SG are 633.2137 (C28H34FN6O8S+), 577.2603 (C27H38FN6O5S+), 504.1711 (C23H27FN5O5S+), and 399.2191 (C22H28FN4O2+). MS/MS analysis of m/z 706.30 yielded the product ions: m/z 688.24 (loss of water), 633.23 (cleavage of the diethylamine), 577.29 (neutral loss of 129 Da, corresponding to the pyroglutamic acid moiety of GSH),24 504.20 (cleavage of the diethylamine and neutral loss of 129), 461.24 (cleavage of the aliphatic amide bond and neutral loss of 129), and 399.23 (cleavage of GSH) (Figure 1B).
During HRMS analysis of GSH conjugates, an additional molecular ion at m/z 704.2874 was observed (Figure S2A). This is 2 mass units lower than Sun–SG, and like Sun–SG, m/z 704 was present in incubations with and without NADPH (Figure S2B). MS/MS analysis of m/z 704.29 yielded the product ions: m/z 686.26, 631.20, 575.22, 502.17, and 459.12. This fragmentation pattern is similar to the product ions of Sun–SG but 2 Da lower (Figure S2C). This suggests that the compound at m/z 704 is a GSH conjugate structurally similar to Sun–SG. Common product ions from MS/MS analysis of m/z 704 and 706 were m/z 299, 255, and 238. The accurate mass m/z 704.2874 is consistent with a GSH conjugate of sunitinib in which the sunitinib portion of the molecule has undergone loss of −2H (C32H43FN7O8S+, Δ0.3 ppm); however, the exact identity of this product is unknown.
Multiple chromatographic peaks were observed from LC–MRM analysis of the precursor-to-product transition m/z 706 > 577, corresponding to Sun–SG (Figure S3). M5 was observed as a single chromatographic peak by LC–MRM analysis of the precursor-to-product transition m/z 702 > 586 (Figure S3).
Reaction Phenotyping with Recombinant P450 Enzymes
Reaction phenotyping experiments were performed with recombinant P450s (Supersomes) to determine which enzymes are responsible for sunitinib metabolite formation. Levels of M1, M3, and M5 were determined by LC–MS/MS analysis utilizing MRM. Representative LC–MRM chromatograms from metabolite detection are shown in Figure 2. In screens with recombinant P450 enzymes, P450 3A4 exhibited the highest activity for formation of M1 (Figure 3A). Lower levels of M1 were formed by P450 1A2 and 3A5. Incubations with recombinant P450 1A2 generated the highest levels of M3 and M5 (Figures 3B and C). Lower levels of M5 were also formed by P450 3A4 and 2D6.
Figure 2.
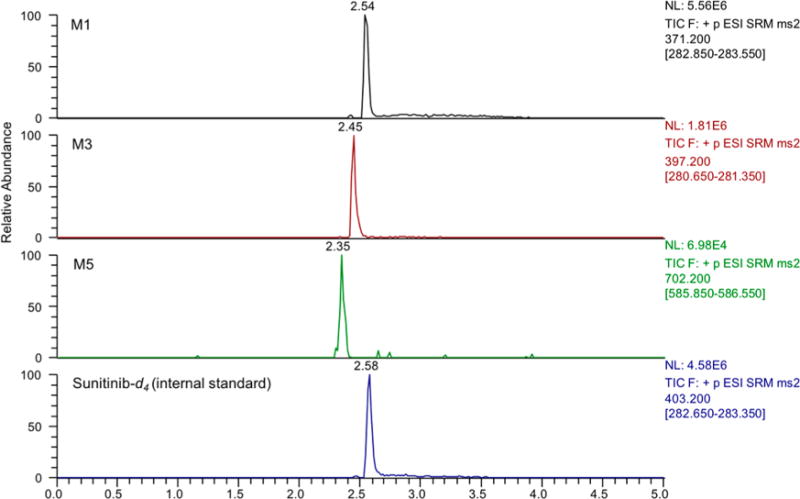
LC–MS/MS chromatograms of selected sunitinib metabolites. Sunitinib metabolites were analyzed by LC–MS/MS utilizing MRM. Shown are representative LC–MRM chromatograms of M1 (m/z 371 > 283), M3 (m/z 397 > 281), M5 (m/z 702 > 586), and sunitinib-d4 (internal standard). Metabolites were generated from incubation of sunitinib (10 μM) with recombinant P450 1A2.
Figure 3.
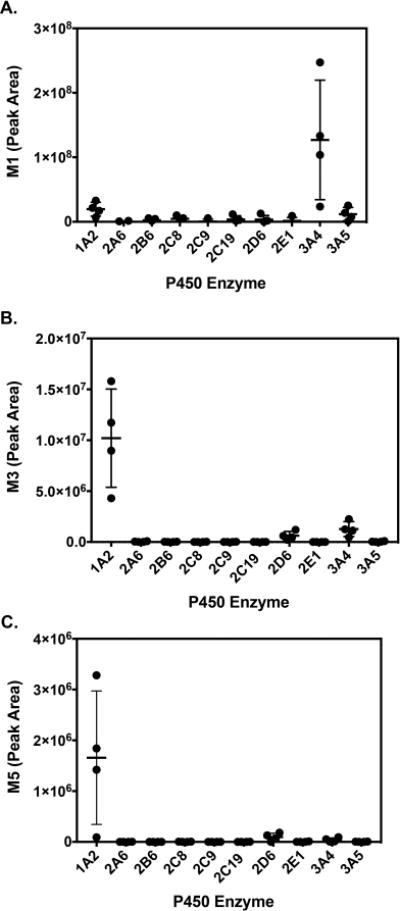
Relative P450 enzyme contribution to sunitinib metabolism. (A) M1, (B) M3, and (C) M5. Sunitinib (10 μM) was incubated with a panel of recombinant human P450 Supersomes (20 nM) in the presence of an NADPH regenerating system and supplemented with GSH (5 mM) for 10 min. Sunitinib metabolites were analyzed by LC–MS/MS analysis utilizing MRM. The MRM transitions for the metabolites indicated were as follows: M1(m/z 371 > 283), M3 (m/z 397 > 281), M5 (m/z 702 > 586). The MRM peak areas are shown for each metabolite. The results are shown as the means ± SD from four independent experiments (n = 4) performed in triplicate each.
Among selected extrahepatic P450 enzymes, P450 1A1 showed activity toward formation of M3 and M5 (Figure S5). P450 2J2 and 1B1 were also capable of forming M3 and M5, although to a markedly less extent compared to P450 1A1.
Effect of P450 Chemical Inhibitors on Metabolite Formation
Inhibition of P450 enzymes using selective chemical inhibitors in pooled human liver microsomal incubations confirmed the results from experiments with recombinant P450 enzymes. Co-incubation with ketoconazole (P450 3A inhibitor) and CYP3cide (P450 3A4-selective inactivator) decreased M1 levels by 88 ± 5% (mean ± SD, n = 3, p < 0.0001) and 88% ± 5% (n = 3, p < 0.0001), respectively, compared to those of the vehicle control (Figure 4A).
Figure 4.
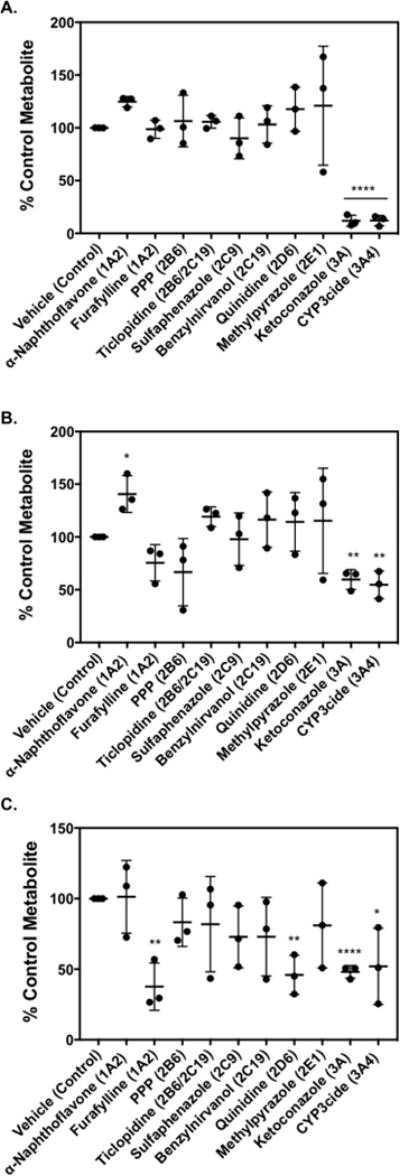
Effect of P450 inhibitors on sunitinib metabolite formation: (A) M1, (B) M3, and (C) M5. Sunitinib (10 μM) was incubated with pooled human liver microsomes (0.1 mg/mL) supplemented with 5 mM GSH in the presence or absence of P450-selective chemical inhibitors. The following chemical inhibitors were used to block the respective P450: α-naphthoflavone (1 μM, P540 1A2); furafylline (25 μM, P450 1A2); PPP, phenyl-piperidinyl propane (15 μM, P450 2B6); ticlopidine (5 μM, P450 2B6/2C19); sulfaphenazole (5 μM, P450 2C9); (+)-N-3-benzylnirvanol (5 μM, P450 2C19); quinidine (2 μM, P450 2D6); 4-methylpyrazole (100 μM, P450 2E1); ketoconazole (1 μM, P450 3A); and CYP3cide (2 μM, P450 3A4). Control incubations were carried out with the appropriate vehicle solvent control in the absence of inhibitor. Relative levels of metabolite formed were measured by LC–MS/MS. Metabolite levels (expressed as peak area ratio) under control conditions were: (A) 6.6 ± 1.4 for M1, (B) 0.04 ± 0.02 for M3, and (C) 0.01 ± 0.008 for M5. Percent (%) metabolite formation was based on comparison to vehicle control. The results are shown as the means ± SD from three independent experiments (n = 3) performed in triplicate each. Comparisons of inhibitor vs vehicle control were performed by unpaired two-tailed t test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In initial experiments, coincubation with the P450 1A inhibitor and P450 3A4 activator α-naphthoflavone25 significantly increased formation of M3 and M5 (data not shown). In subsequent experiments, α-naphthoflavone increased levels of M3 by 41 ± 17% (mean ± SD, n = 3, p = 0.0156) (Figure 4B). This increase in metabolite formation was likely due to the ability of α-naphthoflavone to activate P450 3A4 rather than from inhibition of P450 1A2.25 Ketoconazole (P450 3A inhibitor) reduced M3 levels by 40 ± 9% (n = 3, p = 0.0018), and CYP3cide (P450 3A4 inactivator) decreased M3 by 45 ± 13% (n = 3, p = 0.0038) compared to control.
In analysis of quinoneimine–GSH formation, the P450 1A2 inhibitor furafylline26,27 reduced M5 by 62 ± 17% (mean ± SD, n = 3, p = 0.0030), compared to vehicle (Figure 4C). The P450 2D6 inhibitor quinidine decreased M5 formation by 54 ± 14% (n = 3, p = 0.0026). Ketoconazole reduced M5 levels by 52 ± 4% (n = 3, p < 0.0001), and CYP3cide decreased M5 by 48 ± 27% (n = 3, p = 0.0371), compared to the control values.
Kinetic Analysis of Sunitinib Metabolite Formation with Recombinant P450 Enzymes
In kinetic analysis of sunitinib N-deethylation (M1 formation), the reaction did not reach saturation at substrate concentrations up to 50 μM (Table 1, Figure S5A). However, the estimated kcat/Km, calculated by the slope of the rate vs. substrate concentration graph, for P450 3A4 was 2.1 ± 0.58 min−1 μM−1 (n = 3). The kcat/Km for P450 1A2 was 0.45 ± 0.02 min−1 μM−1 (n = 3). This indicates that P450 3A4 was ∼5-fold more efficient in sunitinib N-deethylation compared to P450 1A2. The results from kinetic assays with recombinant P450 1A2 and 3A4 are summarized in Table 1.
Table 1.
Kinetic Parameters of Sunitinib Metabolite Formation by Recombinant P450 1A2 and P450 3A4a
| kinetic parameter | M1b | M3 | M5 |
|---|---|---|---|
| P450 1A2 | |||
| Km, μM | – | 22 ± 5 | 9.4 ± 5.3 |
| kcatc | – | 3.6 ± 0.4 | 0.09 ± 0.02 |
| kcat/Kmd | 0.45 ± 0.02 | 0.17 ± 0.05 | 0.01 ± 0.001 |
| P450 3A4 | |||
| Km, μM | – | 42 ± 9.7 | 26 ± 17 |
| kcatc | – | 1.2 ± 0.2 | 0.01 ± 0.003 |
| kcat/Kmd | 2.1 ± 0.58 | 0.03 ± 0.01 | 0.0004 ± 0.0002 |
Kinetic parameters were estimated using GraphPad Prism software. The results are shown as the means ± SD from three independent experiments (n = 3) performed in triplicate each.
Formation of M1 did not saturate at substrate concentrations up to 50 μM; therefore, the Km and kcat values for M1 were not determined; the linear slope of the rate vs substrate concentration graph was used to estimate catalytic efficiency of M1 formation (kcat/Km).
kcat for M3 and M5 is shown as the maximum peak area ratio for metabolite to internal standard.
kcat/Km for M1 is in units of min−1 μM−1 based on calculation of M1 formation using an authentic standard. kcat/Km for M3 and M5 is in units of peak area ratio/μM.
For sunitinib defluorination (M3 formation), the mean apparent Km and kcat values for P450 1A2 were 22 ± 5 μM and 3.6 ± 0.4 (peak area ratio, n = 3), and the mean apparent Km and kcat values for P450 3A4 were 42 ± 9.7 μM and 1.2 ± 0.2 (peak area ratio, n = 3) (Table 1, Figure S5B). These data indicate that P450 1A2 was ∼6-fold more efficient in sunitinib defluorination compared to P450 3A4, as determined by relative kcat/Km (P450 1A2 kcat/Km = 0.17 ± 0.05 vs P450 3A4 kcat/Km = 0.03 ± 0.01). Generation of M5 was also more efficient by recombinant P450 1A2 compared to P450 3A4 (Table 1, Figure S5C). The relative maximal M5 formation (kcat) was measured as the peak area ratio of M5 to sunitinib-d4 internal standard. The apparent Km and kcat values for P450 1A2 were 9.4 ± 5.3 μM and 0.09 ± 0.02 (peak area ratio, n = 3), respectively. The apparent Km and kcat for P450 3A4 were 26 ± 17 μM and 0.01 ± 0.003 (peak area ratio, n = 3), respectively (Table 1, Figure S5C). The overall catalytic efficiency (kcat/Km) for forming the quinoneimine trapped as a GSH conjugate (M5) was ∼25-fold higher for P450 1A2 (relative kcat/Km = 0.01 ± 0.001) compared to P450 3A4 (relative kcat/Km = 0.0004 ± 0.0002). Kinetic assays were also performed with P450 2D6 to examine M3 and M5 formation; however, the efficiency for metabolite formation by P450 2D6 was much lower compared to P450s 1A2 and 3A4 (data not shown). In a separate experiment, kinetic assays were performed to examine M3 and M5 formation by P450 1A1 compared to P450 1A2. P450 1A1 formed M3 with higher affinity (apparent Km = 3.1 ± 1.3) and lower capacity (kcat = 0.27 ± 0.03) than P450 1A2 (Figure S6A). The overall catalytic efficiency for M3 formation by P450 1A1 was kcat/Km = 0.09. Formation of M5 by P450 1A1 demonstrated biphasic kinetics, in which M5 generation increased with increasing concentrations of sunitinib up to 5 μM; however, lower levels of M5 were formed from 10–50 μM sunitinib (Figure S6B).
Kinetic Analysis of Sunitinib Metabolite Formation with Human Liver Microsomes
To further investigate the roles of P450 1A2 and 3A4 in sunitinib bioactivation, kinetic experiments were performed using human liver microsomes from single donors characterized for having “high” vs “low” P450 1A2 activity (characterization provided by supplier). Human liver microsomes from Lot HH581 was reported to have high P450 1A2 activity, as measured by phenacetin O-deethylase activity (2100 pmol/min/mg protein), and Lot HH741 was characterized as having low P450 1A2 activity (phenacetin O-deethylase activity 240 pmol/min/mg protein). The P450 3A4 activity was similar between the two donors, with slightly higher activity for Lot HH741 compared to HH581, as measured by testosterone 6β-hydroxylase activity (7400 pmol/min/mg protein for HH741 vs 5400 pmol/min/mg protein for HH581).
Again, in kinetic analysis of sunitinib N-deethylation (M1 formation), the reaction did not reach saturation at substrate concentrations up to 50 μM (Table 2, Figure S7A). Nonetheless, the estimated Vmax/Km, measured by the slope of the rate vs. substrate concentration graph, for HH741 was 32 ± 4.2 μL min−1 mg−1 protein, compared to an estimated Vmax/Km of 23 ± 1.7 μL min1 mg−1 protein for HH581 (n = 3). This indicates that the catalytic efficiency for sunitinib N-deethylation was 1.4-fold higher by HH741 compared to HH581 human liver microsomes.
Table 2.
Kinetic Parameters of Sunitinib Metabolite Formation in Single Donor Human Liver Microsomes (HH581 and HH741)a
| kinetic parameter | M1b | M3e | M5 |
|---|---|---|---|
| HH581 | |||
| Km, μM | – | 13 ± 3.0 | 7.5 ± 3.4 |
| Vmaxc | – | 0.4 ± 0.04 | 0.006 ± 0.0008 |
| Vmax/Kmd | 23 ± 1.7 | 0.022 ± 0.008 | 0.0008 ± 0.0005 |
| HH741 | |||
| Km, μM | – | – | 12 ± 8.8 |
| Vmaxc | – | – | 0.003 ± 0.0009 |
| Vmax/Kmd | 32 ± 4.2 | 0.004 ± 0.001 | 0.0003 ± 0.0003 |
Kinetic parameters were estimated using GraphPad Prism software. The results are shown as the means ± SD from three independent experiments (n = 3) performed in triplicate each.
Formation of M1 did not saturate at substrate concentrations up to 50 μM; therefore, the Km and Vmax values for M1 were not determined, and the linear slope of the rate vs substrate concentration graph was used to estimate catalytic efficiency of M1 formation (Vmax/Km).
Vmax for M3 and M5 is shown as the maximum peak area ratio for metabolite to internal standard.
Vmax/Km for M1 is in units of μL min−1 μM−1 based on calculation of M1 formation using an authentic standard. Vmax/Km for M3 and M5 is in units of peak area ratio/μM.
Formation of M3 did not saturate at substrate concentrations up to 50 μM for HH741; therefore, the linear slopes of the M3 peak area ratio vs substrate concentration graphs were calculated to estimate Vmax/Km for both HH741 and HH581 for comparison.
The relative kinetic parameters were also determined for M3 and M5 formation from microsomal incubations (Table 2, Figures S7B and S7C). The mean apparent Km for sunitinib defluorination was 13 ± 3 μM (n = 3) for HH581, although the reaction did not reach saturation for HH741. The estimated maximal M3 formation (Vmax), as measured by peak area ratios between M3 and internal standard, was 0.4 ± 0.04 for HH581. The overall catalytic efficiency (Vmax/Km) for sunitinib defluorination, estimated from the linear slope of the M3 peak area ratio vs substrate concentration graph, was ∼6-fold higher by HH581 compared to HH741 (HH581 Vmax/Km = 0.022 ± 0.008 vs HH741 Vmax/Km = 0.004 ± 0.001). The apparent Km for M5 formation was 7.5 ± 3.4 μM (n = 3) for HH581 and 12 ± 8.8 μM (n = 3) for HH741. The relative maximal M5 formation (Vmax), as measured by peak area ratios between M5 and internal standard, was 0.006 ± 0.0008 (n = 3) for HH581 and 0.003 ± 0.0009 (n = 3) for HH741. Thus, the overall catalytic efficiency (Vmax/Km) for formation of M5 was 2.7-fold higher by HH581 (Vmax/Km = 0.0008 ± 0.0005) compared to HH741 (Vmax/Km = 0.0003 ± 0.0003).
Identification of Putative Quinoneimine–dGSH Conjugates
In addition to trapping studies with GSH, dansylated GSH was used as a trapping agent to assess formation of reactive metabolites from sunitinib. Dansyl GSH was synthesized according to procedures described previously,19 and product formation was confirmed by LC-UV-MS analysis and fluorescence detection. LC–MS/MS analysis using ESI in positive ion mode verified the presence of the precursor ion m/z 541 for dGSH, as in the literature.19
Because recombinant P450 1A2 was found to generate the highest levels of the reactive metabolite–GSH conjugate (M5), sunitinib was incubated with dGSH and recombinant P450 1A2 to test whether reactive metabolite–dGSH conjugates are also generated. Putative quinoneimine–dGSH conjugates of sunitinib were detected by LC–MS/MS analysis following incubation of sunitinib (25 μM) with recombinant P450 1A2 in the presence of dGSH (Figure 5). Moreover, quinoneimine–dGSH conjugates were detected from incubation of sunitinib (50 μM) with pooled human liver microsomal fractions. In both systems, dGSH conjugates were formed in an NADPH-dependent manner.
Figure 5.
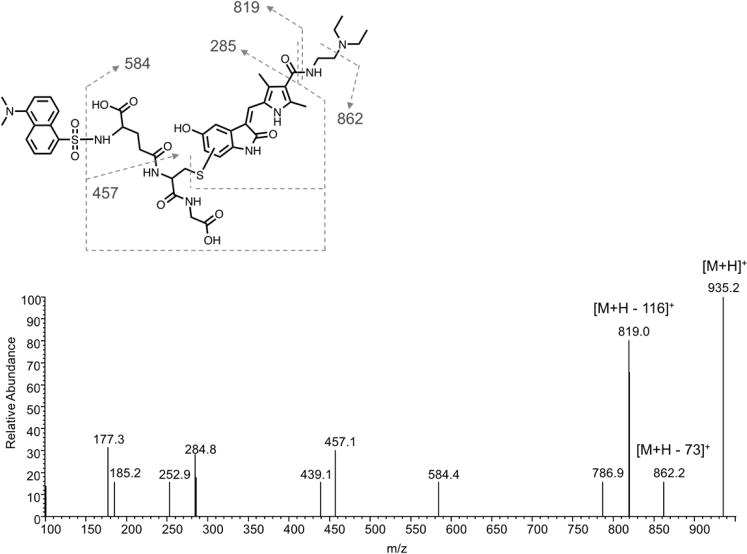
LC–MS/MS analysis of the putative sunitinib quinoneimine dansyl–GSH conjugate. Sunitinib (25 μM) was incubated with recombinant P450 1A2 Supersomes (20 nM) in the presence of an NADPH-regenerating system and supplemented with dansyl–GSH (1 mM) for 30 min. Shown is a representative product ion spectrum from LC–MS/MS analysis of the precursor ion m/z 935, corresponding to the quinoneimine dansyl–GSH conjugate. The predicted structure of the sunitinib quinoneimine dansyl–GSH conjugate is shown above.
When analyzed by ESI in positive ion mode, the predicted precursor ion for a sunitinib quinoneimine–dGSH conjugate is m/z 935, which is 540 mass units higher than the putative quinoneimine metabolite, consistent with addition of the dansyl–GSH moiety (Figure 5). MS/MS fragmentation of m/z 935 in positive mode generated major product ions at m/z 862 (loss of the diethylamine moiety) and m/z 819 (cleavage of the aliphatic amide bond on the drug portion of the molecule). Additionally, secondary fragment ions were observed at m/z 457 (cleavage of the amide bond of the drug portion and loss of the pyroglutamic acid-dansyl moiety), m/z 285 (fragmentation about the amide bond and cleavage of the thioether moiety of dGSH), and m/z 177 (fragmentation of the cysteinyl–glycine portion of the adduct).
Danysl–GSH was originally utilized to quantify formation of dansyl–GSH conjugates using the fluorescence properties of the dansyl group based on the methods described by Gan et al.19,20 Microsomal incubations of sunitinib (25 μM) with dGSH were analyzed by UPLC–UV–fluorescence. The following controls were used for these experiments: (1) without NADPH, (2) without dGSH, and (3) without substrate. Unfortunately, dansyl–GSH conjugates of the sunitinib quinoneimine metabolite were not readily detected by UPLC–fluorescence analysis. This may be due to a lack of sensitivity of our method to detect quinoneimine–dGSH conjugates above the background fluorescence interference or insufficient chromatographic separation. However, dGSH conjugates were readily detectable by LC–MS/MS analysis utilizing MRM for the precursor-to-product transition m/z 935 > 862, corresponding to the quinoneimine–dGSH conjugate of sunitinib. As shown, the quinoneimine–dGSH conjugate was formed in an NADPH-dependent manner (Figure 6A).
Figure 6.
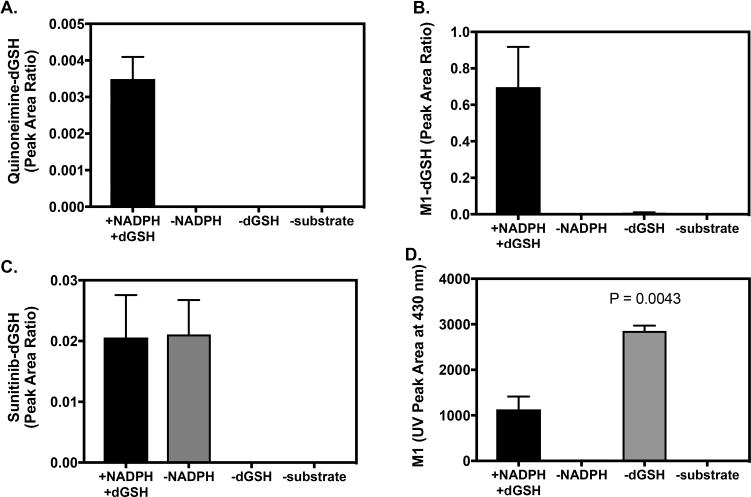
Analysis of dansyl–GSH Conjugates and M1 by LC–MS/MS and UPLC–UV Detection. Sunitinib (25 μM) was incubated with pooled human liver microsomes (1 mg/mL) in the presence of dGSH (1 mM) and an NADPH-regenerating system for 30 min. Control incubations were without NADPH, without dGSH, or without substrate (blank sample). Each reaction condition was performed in triplicate. Samples were analyzed by LC–MS/MS utilizing MRM for the (A) quinoneimine–dGSH conjugate of sunitinib: m/z 935 > 862, (B) M1–dGSH conjugate: m/z 911 > 371, and sunitinib–dGSH conjugate(s): m/z 939 > 399. (D) Relative levels of M1 measured by UPLC–UV absorbance at 430 nm. Levels of M1 were compared from incubations with and without dGSH by unpaired two-tailed t test (n = 2-3).
Evidence for Direct dGSH Conjugation of Sunitinib and M1
Dansyl–GSH conjugates of sunitinib and M1 were also detected via LC–MS/MS analysis of microsomal incubations with sunitinib and dGSH. The predicted direct sunitinib–dGSH conjugate had a m/z of 939, and the M1–dGSH conjugate had a m/z of 911 (data not shown). HRMS analysis of microsomal incubations with sunitinib containing dGSH and NADPH regenerating system confirmed the formation of the M1–dGSH conjugate. The accurate mass detected was m/z 911.3222 (C42H52FN8O10S +2, Δ0.4 ppm). Sunitinib–dGSH was detected by LC–MRM analysis of the precursor-to-product ion m/z 939 > 399, likely corresponding to cleavage of the dGSH moiety (M + H − 540). LC–MS/MS product ion analysis of m/z 911 for the M1–dGSH conjugate yielded a fragment ion at m/z 371, which also corresponds to cleavage of the dGSH moiety (M + H − 540). Sunitinib–dGSH conjugates were detected in microsomal incubations with and without NADPH-regenerating system (Figure 6B), while M1–dGSH conjugates were solely detected in incubations with NADPH, as expected because formation of M1 requires P450-mediated metabolism (Figure 6C). After M1 is formed, this metabolite may undergo conjugation with GSH due to the presence of an electrophilic α,β-unsaturated carbonyl moiety in the metabolite’s chemical structure, leading to formation of an M1–dGSH conjugate. To investigate this possibility, we analyzed the levels of M1 in incubations with dGSH compared to without dGSH. As shown in Figure 6D, levels of M1 were 2.5-fold higher in the absence of dGSH compared to M1 levels in the presence of dGSH (n = 2-3, p = 0.0043) based on the UV absorbance peak area at 430 nm. Further, analysis of UV data permitted estimation of the % sunitinib turnover via the N-deethylation pathway in microsomal incubations.20 In the presence of dGSH, the ratio of M1 to sunitinib (as measured by UV peak area at 420–440 nm) was 0.41, indicating that M1 accounted for 41% of the metabolic turnover of sunitinib. In the absence of dGSH, the ratio of M1 to sunitinib UV peak area increased to 0.70 (70%). Collectively, these data suggest that conjugation with glutathione may contribute to clearance of M1 under the experimental conditions tested. Other oxidative metabolites of sunitinib accounted for an estimated 5–10% of substrate turnover based on UV peak area.
DISCUSSION
In the present investigation, we identified the P450 enzymes involved in sunitinib metabolic activation. The results of this study demonstrated that sunitinib undergoes P450-mediated bioactivation to form a putative reactive quinoneimine, which was trapped as a GSH conjugate (M5). Reaction phenotyping studies using recombinant P450 enzymes, selective chemical inhibitors, and kinetic assays indicate that P450 1A2 is a major contributor to the bioactivation of sunitinib; P450 3A4 may also play an important role (Scheme 1). The mechanism of sunitinib metabolic activation is proposed to involve epoxidation and/or quinoneimine formation and oxidative defluorination, as described for the tyrosine kinase inhibitors gefitinib14 and famitinib.13
Scheme 1. Proposed Metabolism and Bioactivation Pathway of Sunitiniba.
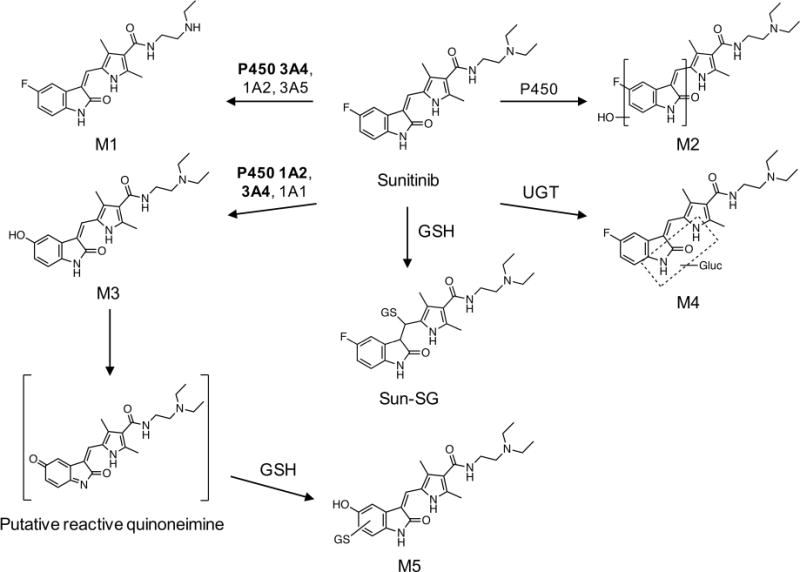
aThe metabolic scheme is based on pathways proposed by Speed et al.9 and Xie et al.13 The enzymes indicated for each route of metabolism are based on evidence from the literature as well as results from the studies described herein. Bold indicates the major enzymes involved.
The primary products of sunitinib identified from human liver microsomal incubations are consistent with the metabolites reported in vivo by Speed et al.9 Findings in this study and previous studies indicate that sunitinib undergoes extensive biotransformation.9 The major routes of oxidative metabolism include N-deethylation, N-oxidation, hydroxylation, oxidative defluorination, and conjugative metabolism by direct glucuronidation; sulfation of hydroxylated metabolites has also been described.9 Products of sunitinib biotransformation identified herein were N-desethylsunitinib (M1), monooxygenated metabolites (M2), defluorinated sunitinib (M3), and glucuronide conjugates (M4). The defluorinated metabolite contains a para-hydroxyaniline moiety, which can be further oxidized to an electrophilic quinoneimine.14,16,28,29
A GSH conjugate of the putative quinoneimine product of sunitinib (M5) was detected by LC–MS/MS from incubations with recombinant P450 enzymes and human liver microsomal fractions. M5 was generated in an NADPH-dependent manner, indicative of cytochrome P450-mediated bioactivation. The detection of dansyl–GSH conjugates provided further confirmation for the formation of electrophilic metabolites of sunitinib. An attempt was made to quantify dansyl–GSH conjugates of the putative quinoneimine metabolite based on the fluorescence-based method described by Gan et al.19,20 However, dansyl–GSH conjugates of the sunitinib reactive metabolite were not readily quantifiable by fluorescence above the background. Previous studies have suggested the generation of reactive metabolites from sunitinib in vitro based on detection of GSH conjugates by LC–MS analysis. Kenny et al. reported identification of two GSH conjugates of sunitinib.30 Xie et al. demonstrated formation of reactive metabolite–GSH conjugates from sunitinib and its structural analogue famitinib in human liver microsomal incubations.13 P450 1A1/2 was shown to catalyze famitinib bioactivation in vitro.13 Cysteine conjugates of famitinib were also identified in primary human hepatocytes incubated with famitinib.13
P450s 1A2 and 3A4 were the main enzymes involved in sunitinib oxidative defluorination and formation of quinoneimine–GSH conjugates. Kinetic assays with recombinant enzymes demonstrated that P450 1A2 had greater efficiency for M3 and M5 formation compared to P450 3A4. Moreover, higher levels of M3 and M5 were generated in human liver microsomal incubations from an individual donor with high P450 1A2 activity (HH581) compared to the donor with low P450 1A2 activity (HH741). Taken together, these data support the contention that P450 1A2 may play an important role in the bioactivation of sunitinib in vivo. It is worth noting that the product information for human liver microsomes HH581 (high P450 1A2 activity) indicated that this donor was a smoker (1 pack per day for 40 years; Corning Gentest inventory datasheet), while the donor for HH741 (low P450 1A2 activity) was listed as a nonsmoker. This is relevant because cigarette smoking is known to induce P450 1A.31 P450 2D6 was capable of generating M5 in vitro, as demonstrated from incubations with recombinant enzyme and human liver microsomal fractions; however, P450 2D6 is likely not a significant contributor to sunitinib reactive metabolite generation based on its low catalytic efficiency observed in kinetic assays. M3 and M5 were also formed by the extrahepatic P450s 1A1 and 2J2. Studies are currently underway to further investigate the impact of smoking on the generation of sunitinib reactive metabolites in hepatic and extrahepatic tissues (i.e., lung).
Our results confirm that P450 3A4 is the primary enzyme responsible for formation of the major active metabolite of sunitinib, M1. Other P450 enzymes, including P450s 3A5 and 1A2, were shown to have less activity toward sunitinib N-deethylation. This finding is consistent with a previous report showing that recombinant P450 3A4 was approximately 10-fold more efficient at sunitinib N-deethylation compared to P450 3A5.32 Narjoz et al. previously showed that sunitinib can also be metabolized by P450s 1A1, 1B1, and 2J2, leading to formation of M1.12
Identification of the enzymes involved in sunitinib metabolism and disposition provides insight into the potential risk factors for drug toxicity. The current study supports ongoing work to define factors that contribute to the large interindividual variability in sunitinib response and toxicity.33–35 A number of reports suggest that genetic variation in genes related to sunitinib metabolism and transport significantly impact drug exposure and the risk for sunitinib-related toxicities.10,11,34–37 Single nucleotide polymorphisms in CYP3A4, CYP3A5, and CYP1A1 have been associated with sunitinib pharmacokinetics, response, and toxicities.10,11,38 In addition, polymorphisms in genes encoding nuclear receptors, NR1I2 (PXR) and NR1I3 (CAR), which regulate P450 expression, and variations in ATP binding cassette (ABC) transporters, have been implicated to affect sunitinib drug exposure and toxicity.10,11,37,39–44 Other factors such as cigarette smoking, gender, body weight, and age have also been associated with sunitinib toxicity and treatment outcomes.33,42,43,45–47 Individualization of sunitinib treatment based on genetic and nongenetic factors has been proposed to help improve sunitinib efficacy and reduce toxicity.10,48,49
The mechanisms and risk factors of sunitinib-induced liver injury remain largely undefined, although one study has begun to investigate the pharmacogenetics related to liver toxicity.37 Hepatotoxicity associated with sunitinib is characterized by marked elevations in alanine aminotransferase, aspartate aminotransferase, and hyperbilirubinemia.3,50,51 The onset of liver injury is delayed and predominately resembles hepatocellular necrosis.50,51 In one case, fatal hepatotoxicity from sunitinib was reported with concomitant use of acetaminophen at therapeutic doses.51 Clinically significant hepatotoxicity has been reported with several other tyrosine kinase inhibitors, including lapatinib, pazopanib, regorafenib, imatinib, gefitinib, and erlotinib.52,53 Metabolic activation by cytochrome P450 enzymes is proposed to play a role in initiating tyrosine kinase inhibitor-mediated liver injury for some agents; however, the link between metabolism and drug toxicity is unclear and is the subject of ongoing investigations.14–16,54,55 Paech et al. recently reported an investigation on the mechanisms of hepatocellular toxicity of sunitinib and other tyrosine kinase inhibitors (imatinib, erlotinib, and lapatinib) in HepG2 and HepaRG cells.56 Treatment of HepG2 cells with sunitinib was shown to increase the production of reactive oxygen species, disrupt the mitochondrial membrane potential, deplete glutathione content, and induce apoptosis in a concentration-dependent manner.56 In the metabolically competent cell line Hep-aRG,57,58 the cytotoxic effects of sunitinib increased following induction by rifampin, compared with sunitinib treatment alone.56 This finding suggests that formation of toxic metabolite(s) may be involved in sunitinib-induced hepatotoxicity, but the parent drug itself may also be involved in mediating some of the hepatotoxic effects associated with sunitinib.
We report for the first time the identification of GSH conjugates formed directly from sunitinib and M1 in vitro. Sunitinib contains an α,β-unsaturated carbonyl moiety which can undergo Michael addition with cellular thiols. The identification of GSH and dGSH conjugates of sunitinib provides evidence of direct thiol conjugation to the electrophilic moiety of the parent drug, independent of metabolic activation. Sunitinib–GSH and dGSH conjugates were detected with the precursor ions m/z 706 and 939, respectively, consistent with addition of GSH (+307) and dGSH (+540) to sunitinib. Formation of Sun–SG (m/z 706) was observed in microsomal incubations in the absence of NADPH. Moreover, metabolites of sunitinib, which retain the electrophilic α,β-unsaturated carbonyl moiety, are also expected to be susceptible to nucleophilic addition with GSH and dGSH. As anticipated, dGSH conjugates of M1 were detected in microsomal incubations supplemented with dGSH and NADPH. This suggests that M1, a major metabolite of sunitinib formed in vivo, may be susceptible to covalent binding to cellular nucleophiles and/or may contribute to GSH depletion. Stable GSH adducts of sunitinib or M1 have not yet been reported in vivo; however, urinary excretion of a cysteine conjugate of sunitinib in monkey was reported in the NDA document of sunitinib, suggesting in vivo metabolism of sunitinib via the mercapturic acid pathway (sunitinib NDA document). The toxicological consequences of the α,β-unsaturated carbonyl moiety within the structure of sunitinib warrant further investigation. An additional GSH conjugate was observed by HRMS analysis at m/z 704. The exact identity of this compound is unknown; however, the observed accurate mass m/z 704.2874 and MS/MS fragmentation pattern are consistent with a GSH conjugate of sunitinib in which the sunitinib portion of the molecule has undergone desaturation (C32H43FN7O8S+, Δ0.3 ppm). An alternative possibility is that the compound at m/z 704 is derived from GSH conjugation of an oxidative defluorination product of sunitinib or a reduced derivative of M5 (704.3072, C32H46N7O9S+, Δ28 ppm). However, the observation that m/z 704 was also present in incubations without NADPH suggests this potential GSH conjugate is not derived from P450-mediated metabolism of sunitinib.
The in vivo exposure to sunitinib reactive metabolites remains unknown. An important remaining question is what fraction of sunitinib undergoes bioactivation via the oxidative defluorination pathway vs what fraction of sunitinib is metabolized via the major N-deethylation pathway. Based on mass balance studies in humans reported by Speed et al., sunitinib metabolites accounted for 41% out of 61% of the total recoverable radioactivity (∼67%), and the N-desethyl metabolite (M1) accounted for 32–52% of the total dose.9 Our in vitro finding that M1 accounted for 41% or greater of sunitinib metabolic turnover in human liver microsomes is consistent with the results reported in vivo. In monkeys administered [14C] sunitinib L-malate, the defluorinated metabolite of sunitinib accounted for 6–8% of the total radioactivity recovered in feces, while N-desethylsunitinib (M1) accounted for 20–40% of the total radioactivity in feces in monkey (sunitinib NDA document).9 Additional studies are needed to determine what fraction of sunitinib metabolism is accounted for by the defluorination pathway in vivo in humans.
In summary, our findings support the conclusion that P450s 1A2 and 3A4 play a predominate role in the formation of a reactive, potentially toxic metabolite of sunitinib. Further, the chemical reactivity of sunitinib itself may account for some of the drug’s biological and toxicological properties. Future studies are needed to better understand the mechanisms of sunitinib-induced liver injury and examine the impact of genetic variations and environmental factors on sunitinib metabolism and toxicity.
Supplementary Material
Acknowledgments
The authors thank Dr. F. Peter Guengerich and his group (Vanderbilt University) for valuable scientific discussions, editing during the manuscript preparation, and for generously allowing us to use analytical instrumentation. We thank Matthew Albertolle of the Guengerich laboratory for assistance with high resolution mass spectrometric analysis and Michael Reddish for assistance with UPLC–UV–fluorescence instrumentation for dansyl–glutathione analysis (Vanderbilt University). We also thank Matthew Vergne for analytical instrumentation assistance in the Lipscomb University College of Pharmacy Bioanalytical Core Laboratory.
Funding
This research was supported by a New Investigator Award from the American Association of Colleges of Pharmacy, Lipscomb University College of Pharmacy and Health Sciences, and the National Cancer Institute of the National Institutes of Health (K01CA190711). Research reported in this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- ABC
ATP binding cassette
- dGSH
dansyl–glutathione
- ESI
electrospray ionization
- HRMS
high resolution mass spectrometry
- ITMS
ion trap mass spectrometry
- MRM
multiple reaction monitoring
- PXR
pregnane X receptor
- TCEP
tris(2-carboxylethyl)phosphine hydrochloride
- UGT
UDP-glucuro-nosyltransferase
- UHPLC
ultrahigh performance liquid chromatography
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrestox.8b00005.
LC–MS/MS and HRMS analyses, relative contributions of extrahepatic P450s to sunitinib metabolism, kinetic analyses, UPLC–UV–fluorescence analyses, and additional references (PDF)
ORCID
R. Nathan Daniels: 0000-0001-8482-8208
Klarissa D. Jackson: 0000-0002-9388-9800
The authors declare no competing financial interest.
References
- 1.Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JV, Booth BP, Verbois SL, Morse DE, Liang CY, Chidambaram N, Jiang JX, Tang S, Mahjoob K, Justice R, Pazdur R. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13:1367–1373. doi: 10.1158/1078-0432.CCR-06-2328. [DOI] [PubMed] [Google Scholar]
- 2.Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884–896. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- 3.FDA. Sutent (Sunitinib malate) Prescribing Information 2015 [Google Scholar]
- 4.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- 5.Sun L, Liang C, Shirazian S, Zhou Y, Miller T, Cui J, Fukuda JY, Chu JY, Nematalla A, Wang X, Chen H, Sistla A, Luu TC, Tang F, Wei J, Tang C. Discovery of 5-[5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem. 2003;46:1116–1119. doi: 10.1021/jm0204183. [DOI] [PubMed] [Google Scholar]
- 6.Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, Bello C, Deprimo S, Brega N, Massimini G, Armand JP, Scigalla P, Raymond E. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol. 2006;24:25–35. doi: 10.1200/JCO.2005.02.2194. [DOI] [PubMed] [Google Scholar]
- 7.Kollmannsberger C. Sunitinib side effects as surrogate biomarkers of efficacy. Can Urol Assoc J. 2016;10:S245–S247. doi: 10.5489/cuaj.4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speed B, Bello C, Peng GW, Patyna S, Wu EY. In vitro and in vivo metabolism of sunitinib in nonclinical species and humans. Cancer Res. 2008;68:1285. [Google Scholar]
- 9.Speed B, Bu HZ, Pool WF, Peng GW, Wu EY, Patyna S, Bello C, Kang P. Pharmacokinetics, distribution, and metabolism of [14C]sunitinib in rats, monkeys, and humans. Drug Metab Dispos. 2012;40:539–555. doi: 10.1124/dmd.111.042853. [DOI] [PubMed] [Google Scholar]
- 10.van Erp NP, Eechoute K, van der Veldt AA, Haanen JB, Reyners AK, Mathijssen RH, Boven E, van der Straaten T, Baak-Pablo RF, Wessels JA, Guchelaar HJ, Gelderblom H. Pharmacogenetic pathway analysis for determination of sunitinib-induced toxicity. J Clin Oncol. 2009;27:4406–4412. doi: 10.1200/JCO.2008.21.7679. [DOI] [PubMed] [Google Scholar]
- 11.Diekstra MHM, Klümpen HJ, Lolkema MPJK, Yu H, Kloth JSL, Gelderblom H, van Schaik RHN, Gurney H, Swen JJ, Huitema ADR, Steeghs N, Mathijssen RHJ. Association analysis of genetic polymorphisms in genes related to sunitinib pharmacokinetics, specifically clearance of sunitinib and SU12662. Clin Pharmacol Ther. 2014;96:81–89. doi: 10.1038/clpt.2014.47. [DOI] [PubMed] [Google Scholar]
- 12.Narjoz C, Favre A, McMullen J, Kiehl P, Montemurro M, Figg WD, Beaune P, de Waziers I, Rochat B. Important role of CYP2J2 in protein kinase inhibitor degradation: a possible role in intratumor drug disposition and resistance. PLoS One. 2014;9:e95532. doi: 10.1371/journal.pone.0095532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie C, Zhou J, Guo Z, Diao X, Gao Z, Zhong D, Jiang H, Zhang L, Chen X. Metabolism and bioactivation of famitinib, a novel inhibitor of receptor tyrosine kinase, in cancer patients. Br J Pharmacol. 2013;168:1687–1706. doi: 10.1111/bph.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Kamenecka TM, Cameron MD. Bioactivation of the epidermal growth factor receptor inhibitor gefitinib: implications for pulmonary and hepatic toxicities. Chem Res Toxicol. 2009;22:1736–1742. doi: 10.1021/tx900256y. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Kamenecka TM, Cameron MD. Cytochrome P450-mediated bioactivation of the epidermal growth factor receptor inhibitor erlotinib to a reactive electrophile. Drug Metab Dispos. 2010;38:1238–1245. doi: 10.1124/dmd.109.030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teng WC, Oh JW, New LS, Wahlin MD, Nelson SD, Ho HK, Chan ECY. Mechanism-Based Inactivation of Cytochrome P450 3A4 by Lapatinib. Mol Pharmacol. 2010;78:693–703. doi: 10.1124/mol.110.065839. [DOI] [PubMed] [Google Scholar]
- 17.Li X, He Y, Ruiz CH, Koenig M, Cameron MD, Vojkovsky T. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab Dispos. 2009;37:1242–1250. doi: 10.1124/dmd.108.025932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Towles JK, Clark RN, Wahlin MD, Uttamsingh V, Rettie AE, Jackson KD. Cytochrome P450 3A4 and CYP3A5-Catalyzed Bioactivation of Lapatinib. Drug Metab Dispos. 2016;44:1584–1597. doi: 10.1124/dmd.116.070839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gan J, Harper TW, Hsueh MM, Qu Q, Humphreys WG. Dansyl glutathione as a trapping agent for the quantitative estimation and identification of reactive metabolites. Chem Res Toxicol. 2005;18:896–903. doi: 10.1021/tx0496791. [DOI] [PubMed] [Google Scholar]
- 20.Gan J, Ruan Q, He B, Zhu M, Shyu WC, Humphreys WG. In vitro screening of 50 highly prescribed drugs for thiol adduct formations - Comparison of potential for drug-induced toxicity and extent of adduct formation. Chem Res Toxicol. 2009;22:690–698. doi: 10.1021/tx800368n. [DOI] [PubMed] [Google Scholar]
- 21.Patras A, Julakanti S, Yannam S, Bansode RR, Burns M, Vergne MJ. Effect of UV irradiation on aflatoxin reduction: a cytotoxicity evaluation study using human hepatoma cell line. Mycotoxin Res. 2017;33:343–350. doi: 10.1007/s12550-017-0291-0. [DOI] [PubMed] [Google Scholar]
- 22.Baratte S, Sarati S, Frigerio E, James CA, Ye C, Zhang Q. Quantitation of SU11248, an oral multi-target tyrosine kinase inhibitor, and its metabolite in monkey tissues by liquid chromatograph with tandem mass spectrometry following semi-automated liquid–liquid extraction. J Chromatogr A. 2004;1024:87–94. doi: 10.1016/j.chroma.2003.10.085. [DOI] [PubMed] [Google Scholar]
- 23.Sun L, Tran N, Tang F, App H, Hirth P, McMahon G, Tang C. Synthesis and biological evaluations of 3-substituted indolin-2-ones: a novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases. J Med Chem. 1998;41:2588–2603. doi: 10.1021/jm980123i. [DOI] [PubMed] [Google Scholar]
- 24.Baillie TA, Davis MR. Mass spectrometry in the analysis of glutathione conjugates. Biol Mass Spectrom. 1993;22:319–325. doi: 10.1002/bms.1200220602. [DOI] [PubMed] [Google Scholar]
- 25.Shou M, Grogan J, Mancewicz JA, Krausz KW, Gonzalez FJ, Gelboin HV, Korzekwa KR. Activation of CYP3A4: evidence for the simultaneous binding of two substrates in a cytochrome P450 active site. Biochemistry. 1994;33:6450–6455. doi: 10.1021/bi00187a009. [DOI] [PubMed] [Google Scholar]
- 26.Kunze KL, Trager WF. Isoform-selective mechanism-based inhibition of human cytochrome P450 1A2 by furafylline. Chem Res Toxicol. 1993;6:649–656. doi: 10.1021/tx00035a009. [DOI] [PubMed] [Google Scholar]
- 27.Clarke SE, Ayrton AD, Chenery RJ. Characterization of the inhibition of P4501A2 by furafylline. Xenobiotica. 1994;24:517–526. doi: 10.3109/00498259409043254. [DOI] [PubMed] [Google Scholar]
- 28.Baillie TA, Rettie AE. Role of biotransformation in drug-induced toxicity-influence of intra- and inter-species differences in drug metabolism. Drug Metab Pharmacokinet. 2011;26:15–29. doi: 10.2133/dmpk.dmpk-10-rv-089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem Res Toxicol. 2011;24:1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 30.Kenny JR, Mukadam S, Zhang C, Tay S, Collins C, Galetin A, Khojasteh SC. Drug-drug interaction potential of marketed oncology drugs: in vitro assessment of time-dependent cytochrome P450 inhibition, reactive metabolite formation and drug-drug interaction prediction. Pharm Res. 2012;29:1960–1976. doi: 10.1007/s11095-012-0724-6. [DOI] [PubMed] [Google Scholar]
- 31.Conney AH, Reidenberg MM. Cigarette smoking, coffee drinking, and ingestion of charcoal-broiled beef as potential modifiers of drug therapy and confounders of clinical trials. J Pharmacol Exp Ther. 2012;342:9–14. doi: 10.1124/jpet.112.193193. [DOI] [PubMed] [Google Scholar]
- 32.Sugiyama M, Fujita K, Murayama N, Akiyama Y, Yamazaki H, Sasaki Y. Sorafenib and sunitinib, two anticancer drugs, inhibit CYP3A4-mediated and activate CY3A5-mediated midazolam 1′-hydroxylation. Drug Metab Dispos. 2011;39:757–762. doi: 10.1124/dmd.110.037853. [DOI] [PubMed] [Google Scholar]
- 33.Houk BE, Bello CL, Kang D, Amantea M. A Population Pharmacokinetic Meta-analysis of Sunitinib Malate (SU11248) and Its Primary Metabolite (SU12662) in Healthy Volunteers and Oncology Patients. Clin Cancer Res. 2009;15:2497–2506. doi: 10.1158/1078-0432.CCR-08-1893. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Donas J, Esteban E, Leandro-García LJ, Castellano DE, del Alba AG, Climent MA, Arranz JA, Gallardo E, Puente J, Bellmunt J, Mellado B, Martínez E, Moreno F, Font A, Robledo M, Rodríguez-Antona C. Single nucleotide polymorphism associations with response and toxic effects in patients with advanced renal-cell carcinoma treated with first-line sunitinib: a multicentre, observational, prospective study. Lancet Oncol. 2011;12:1143–1150. doi: 10.1016/S1470-2045(11)70266-2. [DOI] [PubMed] [Google Scholar]
- 35.van der Veldt AA, Eechoute K, Gelderblom H, Gietema J, Guchelaar HJ, van Erp NP, van den Eertwegh AJ, Haanen JB, Mathijssen RH, Wessels JA. Genetic polymorphisms associated with a prolonged progression-free survival in patients with metastatic renal cell cancer treated with sunitinib. Clin Cancer Res. 2011;17:620–629. doi: 10.1158/1078-0432.CCR-10-1828. [DOI] [PubMed] [Google Scholar]
- 36.Diekstra MH, Swen JJ, Gelderblom H, Guchelaar HJ. A decade of pharmacogenomics research on tyrosine kinase inhibitors in metastatic renal cell cancer: a systematic review. Expert Rev Mol Diagn. 2016;16:605–618. doi: 10.1586/14737159.2016.1148601. [DOI] [PubMed] [Google Scholar]
- 37.Teo YL, Wee HL, Chue XP, Chau NM, Tan MH, Kanesvaran R, Wee HL, Ho HK, Chan A. Effect of the CYP3A5 and ABCB1 genotype on exposure, clinical response and manifestation of toxicities from sunitinib in Asian patients. Pharmacogenomics J. 2016;16:47–53. doi: 10.1038/tpj.2015.13. [DOI] [PubMed] [Google Scholar]
- 38.Diekstra MH, Belaustegui A, Swen JJ, Boven E, Castellano D, Gelderblom H, Mathijssen RH, Garcia-Donas J, Rodriguez-Antona C, Rini BI, Guchelaar HJ. Sunitinib-induced hypertension in CYP3A4 rs4646437 A-allele carriers with metastatic renal cell carcinoma. Pharmacogenomics J. 2017;17:42–46. doi: 10.1038/tpj.2015.100. [DOI] [PubMed] [Google Scholar]
- 39.Beuselinck B, Karadimou A, Lambrechts D, Claes B, Wolter P, Couchy G, Berkers J, Paridaens R, Schoffski P, Mejean A, Verkarre V, Lerut E, de la Taille A, Tourani JM, Bigot P, Linassier C, Negrier S, Berger J, Patard JJ, Zucman-Rossi J, Oudard S. Single-nucleotide polymorphisms associated with outcome in metastatic renal cell carcinoma treated with sunitinib. Br J Cancer. 2013;108:887–900. doi: 10.1038/bjc.2012.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beuselinck B, Lambrechts D, Van Brussel T, Wolter P, Cardinaels N, Joniau S, Lerut E, Karadimou A, Couchy G, Sebe P, Ravaud A, Zerbib M, Caty A, Paridaens R, Schoffski P, Verkarre V, Berger J, Patard JJ, Zucman-Rossi J, Oudard S. Efflux pump ABCB1 single nucleotide polymorphisms and dose reductions in patients with metastatic renal cell carcinoma treated with sunitinib. Acta Oncol. 2014;53:1413–1422. doi: 10.3109/0284186X.2014.918276. [DOI] [PubMed] [Google Scholar]
- 41.Diekstra MHM, Swen JJ, Boven E, Castellano D, Gelderblom H, Mathijssen RHJ, Rodríguez-Antona C, García-Donas J, Rini BI, Guchelaar HJ. CYP3A5 and ABCB1 Polymorphisms as Predictors for Sunitinib Outcome in Metastatic Renal Cell Carcinoma. Eur Urol. 2015;68:621–629. doi: 10.1016/j.eururo.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 42.Chae JW, Teo YL, Ho HK, Lee J, Back HM, Yun HY, Karlsson MO, Kwon KI, Chan A. BSA and ABCB1 polymorphism affect the pharmacokinetics of sunitinib and its active metabolite in Asian mRCC patients receiving an attenuated sunitinib dosing regimen. Cancer Chemother Pharmacol. 2016;78:623–632. doi: 10.1007/s00280-016-3104-9. [DOI] [PubMed] [Google Scholar]
- 43.Narjoz C, Cessot A, Thomas-Schoemann A, Golmard JL, Huillard O, Boudou-Rouquette P, Behouche A, Taieb F, Durand JP, Dauphin A, Coriat R, Vidal M, Tod M, Alexandre J, Loriot MA, Goldwasser F, Blanchet B. Role of the lean body mass and of pharmacogenetic variants on the pharmacokinetics and pharmacodynamics of sunitinib in cancer patients. Invest New Drugs. 2015;33:257–268. doi: 10.1007/s10637-014-0178-2. [DOI] [PubMed] [Google Scholar]
- 44.Numakura K, Tsuchiya N, Kagaya H, Takahashi M, Tsuruta H, Inoue T, Narita S, Huang M, Satoh S, Niioka T, Miura M, Habuchi T. Clinical effects of single nucleotide polymorphisms on drug-related genes in Japanese metastatic renal cell carcinoma patients treated with sunitinib. Anti-Cancer Drugs. 2017;28:97–103. doi: 10.1097/CAD.0000000000000425. [DOI] [PubMed] [Google Scholar]
- 45.Keizman D, Gottfried M, Ish-Shalom M, Maimon N, Peer A, Neumann A, Hammers H, Eisenberger MA, Sinibaldi V, Pili R, Hayat H, Kovel S, Sella A, Boursi B, Weitzen R, Mermershtain W, Rouvinov K, Berger R, Carducci MA. Active smoking may negatively affect response rate, progression-free survival, and overall survival of patients with metastatic renal cell carcinoma treated with sunitinib. Oncologist. 2014;19:51–60. doi: 10.1634/theoncologist.2012-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Segarra I, Modamio P, Fernandez C, Marino EL. Sunitinib possible sex-divergent therapeutic outcomes. Clin Drug Invest. 2016;36:791–799. doi: 10.1007/s40261-016-0428-5. [DOI] [PubMed] [Google Scholar]
- 47.van der Veldt AAM, Boven E, Helgason HH, van Wouwe M, Berkhof J, de Gast G, Mallo H, Tillier CN, van den Eertwegh AJM, Haanen JBAG. Predictive factors for severe toxicity of sunitinib in unselected patients with advanced renal cell cancer. Br J Cancer. 2008;99:259–265. doi: 10.1038/sj.bjc.6604456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ganapathi RN, Bukowski RM. Predicting responses to sunitinib using single nucleotide polymorphisms: Progress and recommendations for future trials. Genome Med. 2011;3:79. doi: 10.1186/gm295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Velasco G, Gray KP, Hamieh L, Urun Y, Carol HA, Fay AP, Signoretti S, Kwiatkowski DJ, McDermott DF, Freedman M, Pomerantz MM, Choueiri TK. Pharmacogenomic Markers of Targeted Therapy Toxicity in Patients with Metastatic Renal Cell Carcinoma. Eur Urol Focus. 2016;2:633–639. doi: 10.1016/j.euf.2016.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mueller EW, Rockey ML, Rashkin MC. Sunitinib-related fulminant hepatic failure: case report and review of the literature. Pharmacotherapy. 2008;28:1066–1070. doi: 10.1592/phco.28.8.1066. [DOI] [PubMed] [Google Scholar]
- 51.Weise AM, Liu CY, Shields AF. Fatal liver failure in a patient on acetaminophen treated with sunitinib malate and levothyroxine. Ann Pharmacother. 2009;43:761–766. doi: 10.1345/aph.1L528. [DOI] [PubMed] [Google Scholar]
- 52.Teo YL, Ho HK, Chan A. Risk of tyrosine kinase inhibitors-induced hepatotoxicity in cancer patients: A meta-analysis. Cancer Treat Rev. 2013;39:199–206. doi: 10.1016/j.ctrv.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 53.Shah R, Morganroth J, Shah D. Hepatotoxicity of Tyrosine Kinase Inhibitors: Clinical and Regulatory Perspectives. Drug Saf. 2013;36:491–503. doi: 10.1007/s40264-013-0048-4. [DOI] [PubMed] [Google Scholar]
- 54.Teo YL, Saetaew M, Chanthawong S, Yap YS, Chan EC, Ho HK, Chan A. Effect of CYP3A4 inducer dexamethasone on hepatotoxicity of lapatinib: clinical and in vitro evidence. Breast Cancer Res Treat. 2012;133:703–711. doi: 10.1007/s10549-012-1995-7. [DOI] [PubMed] [Google Scholar]
- 55.Hardy KD, Wahlin MD, Papageorgiou I, Unadkat JD, Rettie AE, Nelson SD. Studies on the role of metabolic activation in tyrosine kinase inhibitor–dependent hepatotoxicity: Induction of CYP3A4 enhances the cytotoxicity of lapatinib in HepaRG cells. Drug Metab Dispos. 2014;42:162–171. doi: 10.1124/dmd.113.054817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paech F, Bouitbir J, Krahenbuhl S. Hepatocellular toxicity associated with tyrosine kinase inhibitors: Mitochondrial damage and inhibition of glycolysis. Front Pharmacol. 2017;8:367. doi: 10.3389/fphar.2017.00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aninat C, Piton A, Glaise D, Le Charpentier T, Langouet S, Morel F, Guguen-Guillouzo C, Guillouzo A. Expression of cytochromes P450, conjugating enzymes and nuclear receptors in human hepatoma HepaRG cells. Drug Metab Dispos. 2006;34:75–83. doi: 10.1124/dmd.105.006759. [DOI] [PubMed] [Google Scholar]
- 58.Guillouzo A, Corlu A, Aninat C, Glaise D, Morel F, Guguen-Guillouzo C. The human hepatoma HepaRG cells: a highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem-Biol Interact. 2007;168:66–73. doi: 10.1016/j.cbi.2006.12.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.