

Abstract
Fluorine-18 labeled phenethylguanidines are currently under development in our laboratory as radiotracers for quantifying regional cardiac sympathetic nerve density using PET imaging techniques. In this study, we report an efficient synthesis of 18F-hydroxyphenethylguanidines consisting of nucleophilic aromatic [18F]fluorination of a protected diaryliodonium salt precursor followed by a single deprotection step to afford the desired radiolabeled compound. This approach has been shown to reliably produce 4-[18F]fluoro-m-hydroxyphenethylguanidine ([18F]4F-MHPG, [18F]1) and its structural isomer 3-[18F]fluoro-p-hydroxyphenethylguanidine ([18F]3F-PHPG, [18F]2) with good radiochemical yields. Preclinical evaluations of [18F]2 in non-human primates were performed to compare its imaging properties, metabolism, and myocardial kinetics with those obtained previously with [18F]1. The results of these studies have demonstrated that [18F]2 exhibits imaging properties comparable to those of [18F]1. Myocardial tracer kinetic analysis of each tracer provides quantitative metrics of cardiac sympathetic nerve density. Based on these findings, first-in-human PET studies with [18F]1 and [18F]2 are currently in progress to assess their ability to accurately measure regional cardiac sympathetic denervation in patients with heart disease, with the ultimate goal of selecting a lead compound for further clinical development.
Keywords: 3-[18F]fluoro-p-hydroxyphenethylguanidine, [18F]3F-PHPG, 4-[18F]fluoro-m-hydroxyphenethylguanidine, [18F]4F-MHPG, norepinephrine transporter, sympathetic nervous system
TABLE OF CONTENTS GRAPHIC
INTRODUCTION
Cardiac autonomic dysfunction contributes to morbidity and mortality in many diseases that damage the heart, including congestive heart failure, myocardial ischemia, myocardial infarction, and diabetic autonomic neuropathy.1–3 Disease-induced alterations in the nervous control of the heart can be caused by changes in the outflow of nervous impulses to the parasympathetic and sympathetic branches of the autonomic nervous system arising from central sites in the brain, and by regional degeneration of postganglionic parasympathetic and/or sympathetic nerve fibers in the heart. The complex interaction between aberrant parasympathetic and sympathetic influences on the heart often evolves with the progression of disease. For example, early heart failure is characterized by hyperactivity of sympathetic nerve pathways, with a concomitant reduction in parasympathetic activation, a process that referred to as ‘parasympathetic withdrawal’.4 The chronically elevated levels of the neurotransmitter norepinephrine that result from this imbalance of parasympathetic and sympathetic influences are cardiotoxic and contribute to the progression of heart failure, including development of left ventricular dilation.5, 6 Throughout the time course of heart failure, autonomic dysfunction can promote malignant arrhythmias, leading to sudden cardiac death.7, 8
Clinical imaging studies of cardiac sympathetic denervation using nuclear scintigraphy techniques have provided important insights into how different diseases affect this nerve population.9, 10 Our laboratory has previously developed several radiotracers for noninvasive imaging studies of cardiac sympathetic nerves, including [123I]metaiodobenzylguanidine ([123I]MIBG) for planar scintigraphy and SPECT imaging, and [11C]-(–)-m-hydroxyephedrine ([11C]HED) for PET imaging (Figure 1).11, 12 These radiolabeled analogs of the endogenous neurotransmitter norepinephrine are transported into presynaptic sympathetic nerve terminals by the norepinephrine transporter (NET). Since NET expression in the heart is only associated with sympathetic nerve varicosities, the cardiac retention levels of these tracers can be used as metrics of regional sympathetic nerve density. Clinical trials with [123I]MIBG and [11C]HED tracers in heart failure patients have demonstrated that cardiac sympathetic denervation is associated with a significantly increased risk of sudden cardiac death. For example, the multi-center ADMIRE-HF study of 961 heart failure patients showed that a low heart-to-mediastinum ratio (H/M) of [123I]MIBG obtained from planar gamma camera images, a global measure of cardiac sympathetic denervation, was a predictor of mortality and fatal arrhythmic events in patients eligible for implantable cardioverter defibrillator (ICD) therapy.13 More recently, the prospective PAREPET trial used [11C]HED to assess regional sympathetic denervation, [13N]ammonia to assess myocardial perfusion, and [18F]FDG to assess myocardial viability in 204 heart failure patients staged for an ICD.14 The PAREPET results showed that the regional extent of sympathetic denervation in the left ventricle (as defined by [11C]HED retention deficits) was the strongest predictor of sudden cardiac arrest among all imaging parameters measured. Patients with regional sympathetic denervation encompassing more than 38% of their left ventricle defined the upper tertile of patients with the highest risk of sudden cardiac arrest (p < 0.001). The results of the ADMIRE-HF and PAREPET trials suggest that noninvasive imaging studies of cardiac sympathetic denervation may find a clinical role in improved risk stratification of heart failure patients for ICD placement.15
Figure 1.

Structures of norepinephrine and some radiotracers for imaging cardiac sympathetic nerve terminals.
We have recently been investigating radiolabeled phenethylguanidines as next generation sympathetic nerve tracers with improved myocardial kinetics for accurate and sensitive quantification of regional sympathetic nerve density using PET and tracer kinetic analysis methods.16 Our initial work focused on the carbon-11 labeled hydroxyphenethylguanidine N-[11C]guanyl-(–)-m-octopamine ([11C]GMO, Figure 1). PET studies with [11C]GMO in non-human primates showed that the myocardial kinetics of this tracer could be successfully analyzed using compartmental modeling methods or Patlak graphical analysis to obtain an estimates of the ‘net uptake rate constant’ Ki (mL/min/g) as a measure of regional sympathetic nerve density.17 In control studies and pharmacological blocking studies with the NET inhibitor desipramine (DMI) in rhesus macaque monkeys, the Ki values from compartmental modeling analysis were found to decrease with increasing DMI doses along a sigmoidal dose-response curve, with an IC50 of 0.087 mg/kg and a Hill slope nH = –0.70. Similar results were obtained using the Patlak slopes Kp (mL/min/g) from Patlak graphical analysis of [11C]GMO kinetics, as an alternative estimate of the Ki values (IC50 = 0.068 mg/kg, nH = –0.54). These results strongly suggest that regional estimates of Ki from tracer kinetic analysis of [11C]GMO kinetics in human hearts would serve as a reproducible metric of regional cardiac sympathetic nerve density.
While the preclinical PET studies with [11C]GMO were highly encouraging, the short half-life of carbon-11 (20.4 min) limits its use to PET centers with an on-site cyclotron. Conversely, fluorine-18 has a sufficiently long half-life (1.83 h) that the production and distribution of 18F-labeled radiopharmaceuticals from a central production facility to stand-alone PET imaging centers is feasible. To develop a fluorine-18 labeled phenethylguanidine, we initially chose to prepare 4-[18F]fluoro-m-hydroxyphenethylguanidine ([18F]4F-MHPG, [18F]1, Figure 1). In previous studies in an isolated rat heart model, the kinetics of [11C]4F-MHPG were comparable to those of [11C]GMO.16
A previous report described several preclinical evaluations of [18F]1, including PET imaging studies in non-human primates.18 The results of the PET studies of [18F]1 closely paralleled those obtained with [11C]GMO,17 supporting further development of this agent for clinical translation. For these pilot studies, our initial approach to the preparation of [18F]1 was a multi-step automated radiosynthesis involving production of a 18F-labeled intermediate using a diaryliodonium salt precursor followed by a deprotection step, an N-guanylation step, a second deprotection step and finally HPLC purification to provide [18F]1.18 Because of the multiple steps required, this initial method relied on the use of two automated radiosynthesis modules in adjacent hot-cells, linked in series, to provide the final product. While this approach was adequate for preclinical evaluations of [18F]1, it was poorly suited to routine production of the radiotracer for clinical studies in human subjects. Thus, one goal of this work was to develop an efficient automated radiosynthesis of [18F]1 capable of providing sufficiently high radiochemical yields for clinical PET studies.
We report here a new two-step approach to the radiosynthesis of [18F]1 that utilizes a novel N,N’,N’’,N’’-tetrakis-Boc protected guanidinyliodoium salt precursor for nucleophilic aromatic [18F]fluorination, followed by a single deprotection step, to provide [18F]1 in good yields and high specific activities for clinical PET studies. This new approach is performed using a single automated radiosynthesis module and has proven to be very reliable.
With the establishment of this new synthetic approach to 18F-hydroxyphenethylguanidines, the second objective of this study was to prepare 3-[18F]fluoro-p-hydroxyphenethylguanidine ([18F]3F-PHPG, [18F]2, Figure 1), the structural isomer of [18F]1. Preclinical evaluations of this compound were performed to compare its characteristics as a cardiac innervation tracer with those of [18F]1. Studies in non-human primates showed that [18F]2 is as good or better than [18F]1 as a cardiac PET imaging agent. Based on the positive preclinical testing results with [18F]1 and [18F]2, these new cardiac sympathetic innervation radiotracers are currently being evaluated in first-in-human PET studies under an exploratory investigational new drug (IND) clearance from the FDA.
RESULTS AND DISCUSSION
Chemistry and Radiochemistry.
Our initial strategy for improving the radiosynthesis of [18F]1 was to utilize a N,N’-bis-Boc-protected diaryliodonium salt precursor for direct no-carrier-added nucleophilic aromatic [18F]fluorination via a two-step automated radiosynthesis (Scheme 1). However, many attempts at 18F-radiofluorination of the N,N′-bis-Boc-guanidinyliodonium salt precursor 3 with different [18F]fluoride sources under a variety of reaction conditions were unsuccessful in providing the desired 18F-labeled intermediate [18F]4. We hypothesized that this disappointing outcome was related to the two unprotected protons of the guanidinyl group being acidic, causing a disturbance in the 18F-radiofluorination reaction. To confirm this hypothesis, we explored the 18F-radiofluorination of a test compound with a fully protected guanidinyl moiety to eliminate the unprotected protons (Scheme 2). The N,N′,N′′,N′′-tetrakis-Boc-guanidinyliodonium salt precursor 5 was synthesized, and radiolabeling tests were carried out under various conditions, including different [18F]fluoride sources, reaction solvents (e.g., DMF and MeCN), reaction times and reaction temperatures to produce the 18F-labeled compound [18F]6 (Table 1).19 The reaction of K[18F]F-K222 or [18F]TBAF with precursor 5 afforded [18F]6 in low yields (0–3%) at 150 °C for 10 min. When precursor 5 was reacted with Cs[18F]F under general anhydrous radiofluorination conditions, the yield of [18F]6 was improved (~ 9% at 150 °C for 10 min). However, when 5 was reacted with Cs[18F]F for a longer reaction time (150 °C for 25 min), the yield of the desired product decreased to ~ 4%. We hypothesized that the reduced radiochemical yields at longer reaction times might be due to the decomposition of the iodonium salt precursor 5 and the radiofluorinated product [18F]6 at the 150 °C reaction temperature. In tests, 5 started slow thermal decomposition at 150 °C in open capillary tubes in a melting point apparatus, supporting this hypothesis. The addition of TEMPO (1.0 mg) as a radical scavenger and a small amount of water (10 μL) in the reaction solvent (DMF, 0.5 mL) to increase the solubility of Cs[18F]F greatly increased the radiochemical yields, up to ∼ 51% for manual syntheses at 150 °C for 5 min. Additional tests using a reduced temperature of 130 °C for 5 min, decreased the yield of [18F]6 to ~ 31%. Tests at temperatures greater than 150 °C did not produce any improvements in the yields.
Scheme 1.

Ineffective Initial Approach to an Improved Radiosynthesis of [18F]1.
Scheme 2.

18F-Fluorination of a Test Compound with a Fully Protected Guanidinyl Moiety.
Table 1.
[18F]Fluorination of diaryliodonium salts (5, 11 and 21) under various conditions.
| Entry | I+ Salta |
18F-fluoride [18F]F−X+ |
Method |
T
(°C) |
t
(min) |
Solventb | TEMPO & H2Ob |
Product | Yield (%)c |
n |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5 | [18F]F−K+ | manual | 150 | 10 | MeCN | – | [18F]6 | NR | 2 |
| 2 | 5 | [18F]F−K+ | manual | 150 | 10 | DMF | – | [18F]6 | 3 ± 2 | 2 |
| 3 | 5 | [18F]F−TBA+ | manual | 150 | 10 | DMF | – | [18F]6 | Trace | 2 |
| 4 | 5 | [18F]F−Cs+ | manual | 150 | 10 | DMF | – | [18F]6 | 9 ± 2 | 2 |
| 5 | 5 | [18F]F−Cs+ | manual | 150 | 5 | DMF | + | [18F]6 | 51 ± 5 | 3 |
| 6 | 5 | [18F]F−Cs+ | manual | 130 | 5 | DMF | + | [18F]6 | 31 ± 5 | 3 |
| 7 | 5 | [18F]F−Cs+ | manual | 120 | 5 | DMF | + | [18F]6 | 28 ± 3 | 3 |
| 8 | 5 | [18F]F−Cs+ | manual | 150 | 15 | DMF | + | [18F]6 | 43 ± 5 | 2 |
| 9 | 5 | [18F]F−Cs+ | manual | 150 | 25 | DMF | + | [18F]6 | 30 ± 5 | 2 |
| 10 | 11 | [18F]F−Cs+ | manual | 150 | 5 | DMF | + | [18F]22 | 16 ± 5 | 5 |
| 11 | 11 | [18F]F−Cs+ | manual | 150 | 15 | DMF | + | [18F]22 | 11 ± 4 | 3 |
| 12 | 11 | [18F]F−Cs+ | automated | 150 | 5 | DMF | + | [18F]1 | 7.0 ± 3.5 d | 13 |
| 13 | 21 | [18F]F−Cs+ | automated | 150 | 5 | DMF | + | [18F]2 | 8.0 ± 3.5 e | 12 |
Amount of diaryliodonium salt precursor used was 4-6 mg.
Reaction solvent (MeCN or DMF; 500 μL) prepared without (−) or with (+) 1 mg TEMPO and 10 μL of H2O.
Progress of the reaction and yields for manual reactions were analyzed by radio-TLC (developing solvent: ethylacetate/hexane = 30:70, vol/vol).
Yield of the isolated pure product [18F]1 by semipreparative column (Phenomenex Synergi 10μ Hydro-RP 80A, 250×10 mm) using HPLC (5% EtOH in 40 mM NH4OAc buffer, λ=254 nm, 4.0 ml/min).
Yield of the isolated pure product [18F]2 by semipreparative column (Phenomenex Synergi 10μ Hydro-RP 80A, 250×10 mm) using HPLC (3.5% EtOH in 40 mM NH4OAc buffer, λ=254 nm, 4.0 ml/min).
This encouraging result led us to prepare the required N,N′,N′′,N′′-tetrakis-Boc-protected guanidinyliodonium salt precursor for a simplified automated production of [18F]1 for clinical studies (Scheme 3). The N′,N′′-bis-Boc-protected iodophenethylguanidine 7 was previously synthesized in our laboratory.18 This compound was converted to the N,N′,N′′,N′′-tetrakis-Boc-protected guanidine 8 by treatment with di-t-butyl dicarbonate in the presence of dimethylaminopyridine and triethylamine in THF. Reaction of 8 with bis(trimethyl)tin in the presence of tetrakis(triphenylphosphine)palladium provided the trimethylstannane compound 9. [2-hydroxy(tosyloxy)iodo]thiophene,20 which was generated in situ by a mixture of 2-(diacetoxyiodo)thiophene21 with p-toluenesulfonic acid under nitrogen atmosphere, was reacted with trimethylstannane 9 to provide (2-thienyl)iodonium tosylate 10 in 87% yield as a yellow solid. Among various counteranions, bromide was found to be especially reactive, increasing the radiochemical yield.22 Therefore, (2-thienyl)iodonium tosylate 10 was converted into the corresponding bromide 11 as a grey powder in 86% yield using KBr in a solution of CH3CN and H2O.
Scheme 3.

Synthetic Route to the Diaryliodonium Salt Precursor 11a
a(a) (Boc)2CO, DMAP, Et3N, THF, rt, 48h, 92%; (b) Sn2Me6, Pd(PPh3)4, toluene, reflux, N2, 30 min, 86%; (c) (i) 2-(Diacetoxyiodo)thiophene, p-TsOH·H2O, MeCN, CH2Cl2, N2, rt, 1 h; (ii) 9, MeCN, CH2Cl2, N2, rt, 20 h, 87%; (d) KBr, MeCN, H2O, 65 °C-rt, 1 h, 86%.
A similar approach was used to prepare the guanidinyl-iodonium salt precursor 21 for production of [18F]2 (Scheme 4). The N-Boc-protected tyramine 13 was iodinated in the presence of sodium iodide and sodium hypochlorite to afford 14 in low yields (43%). The iodophenol 14 was protected by benzylation with benzyl bromide followed by subsequent deprotection of the Boc group to afford the benzylated 3-iodotyramine intermediate 16. Condensation of 16 with 1,3-bis(t-butoxycarbonyl)-2-methyl-2-thiopseudourea afforded the N′,N′′-bis-Boc-protected iodophenethylguanidine 17. This compound was used to convert the N,N′,N′′,N′′-tetrakis-Boc-protected guanidininyliodonium salt 21 using the same sequence of steps used to prepare 11 as shown in Scheme 3. Briefly, reaction of the trimethylstannane 19 with [2-hydroxy(tosyloxy)iodo]-thiophene provided the (2-thienyl)iodonium tosylate 20 in 86% yield as a yellow solid. Conversion of 20 to the corresponding bromide 21 yielded a grey powder in 96% yield. The structures of 11 and 21 were confirmed by 1H NMR, 13C-NMR, high-resolution mass spectrometry (HRMS) spectra and C, H and N elemental analysis.
Scheme 4.

Synthetic Route to the Diaryliodonium Salt Precursor 21a
a(a) (Boc)2CO, Et3N, THF, rt, 24 h, 98%; (b) NaI, NaOCl, KOH, MeOH, 0 °C, 43%; (c) BnBr, K2CO3, Acetone, 4 h, 78%; (d) HCl, 65 °C, 1 h, 97%; (e) 1,3-N,N’-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, Et3N, DMF, 0 °C-rt, 24 h, 58%; (f) (Boc)2CO, DMAP, Et3N, THF, rt, 20 h, 63%; (g) Sn2Me6, Pd(PPh3)4, toluene, reflux, N2, 30 min, 98%; (h) (i) 2-(Diacetoxyiodo)thiophene, p-TsOH·H2O, MeCN, CH2Cl2, N2, rt, 1 h; (ii) 19, MeCN, CH2Cl2, N2, rt, 44 h, 86%; (i) KBr, MeCN, H2O, 65 °C-rt, 1 h, 96%.
A TRACERlab FXFN radiosynthesis module (GE Healthcare) was used to achieve a fully automated radiosynthesis of [18F]1 and [18F]2 using a two-step reaction as shown in Scheme 5. The first step incorporates [18F]F– into the aromatic ring of the protected diaryliodonium salt precursor to yield an 18F-labeled intermediate ([18F]22 or [18F]23) using Cs[18F]F in DMF (150 °C, 5 min, 1 mg TEMPO, 10 μL H2O). The second step is the simultaneous cleavage of the benzyl ether and the N,N′,N′′,N′′-tetrakis-Boc protecting groups by treatment with 3.0 N HBr at 120 °C for 15 min followed by HPLC purification to provide the desired radiotracer, [18F]1 or [18F]2. The structure of the 18F-labeled product was confirmed by comparing its retention time against the corresponding non-radioactive fluorine-19 standard using reverse-phase HPLC. The total synthesis time of the fully automated process is 90 min. For [18F]1, the purified product was obtained in 7.0 ± 3.5% yield at end of synthesis (EOS), (n = 13, decay-corrected based on starting activity) with >98% radiochemical purities. [18F]2 was obtained in 8.0 ± 3.5% yield (n = 12) with >99% radiochemical purities (Table 1). Activity levels in the final product ranged from 0.6–2.0 GBq for [18F]1 and 1.0–2.4 GBq for [18F]2. Specific activities were 56 ± 19 GBq/μmol for [18F]1 and 104 ± 26 GBq/μmol for [18F]2 at EOS. Other than the radiolabeled product, no other non-radioactive compounds were observed in the HPLC/UV trace during quality control of the final product. Mass concentrations averaged 0.48 ± 0.29 μg/mL for [18F]1 and 0.27 ± 0.14 μg/mL for [18F]2. Stability tests of 3 batches of each compound showed that both compounds are completely stable in the ammonium acetate buffer at room temperature for at least 6 hours.
Scheme 5.

Efficient Radiosynthetic Route to [18F]1 and [18F]2a
a(a) Cs[18F]F, TEMPO, H2O, DMF, 150 °C, 5 min; (b) 3.0 N HBr, 120 °C, 15 min.
Isolated Rat Heart Studies.
The kinetics of [18F]2 were measured in the isolated working rat heart model, which has been used previously to measure the neuronal uptake and retention kinetics of [18F]1,18 the carbon-11 analog of MIBG ([11C]MIBG) and [11C]HED.16 Extraneuronal uptake (‘uptake-2’), which transports some norepinephrine analogs into myocytes, was blocked pharmacologically by adding 54 μM corticosterone to all heart perfusates.23 At the beginning of the study, a 10 min constant infusion of tracer in the heart perfusate is performed to measure a neuronal uptake rate, Kup (mL perfusate/min/g wet). After this, the heart is switched to normal heart perfusate to measure clearance rates from neuronal spaces, expressed as a major clearance half-time, T1/2 (h). As shown in Figure 2, [18F]2 had almost identical kinetics as [18F]1 in this model, with neuronal uptake rates Kup = 0.79 mL/min/g wet for [18F]2 and 0.77 mL/min/g wet for [18F]1. Major clearance halftimes were T1/2 > 50 h for [18F]2 and T1/2 > 24 h for [18F]1. The long neuronal retention times of these compounds are due to their efficient uptake and storage in norepinephrine storage vesicles. This shows that [18F]1 and [18F]2 are good substrates of the vesicular monoamine transporter, isoform 2 (VMAT2), which is localized in cell membranes of storage vesicles in peripheral sympathetic nerve terminals.24 In comparison, [11C]MIBG and [11C]HED have much faster neuronal uptake rates, with Kup = 3.65 mL/min/g wet for [11C]MIBG and Kup = 2.35 mL perfusate/min/g wet for [11C]HED. They also have much faster clearance rates in this experimental model. [11C]MIBG has a major clearance half-time T1/2 = 2.1 h and [11C]HED has T1/2 = 1.6 h. These clearance rates are faster than are seen in human imaging studies with [123I]MIBG and [11C]HED because the isolated rat heart has coronary flow rates that are about 10 times higher than resting blood flow in a human heart.25 If tracer molecules diffuse out of sympathetic neurons in the isolated rat heart they are more likely to be cleared from interstitial spaces into the capillary bed rather than re-entering nerve terminals by NET transport. In contrast, several studies have shown that [11C]HED continuously diffuses out of nerve terminals only to be taken back up into the nerves during PET studies in normal human hearts.25–27 The slower neuronal uptake rates and long neuronal retention times of [18F]1 and [18F]2 are favorable for tracer kinetic analysis,17 including Patlak graphical analysis, which requires irreversible trapping of a tracer in a tissue compartment.28
Figure 2.

Kinetics of [18F]2 in isolated rat heart. Data for [18F]1, [11C]MIBG and [11C]HED are shown for comparison. The tracer is infused for 10 min to measure its neuronal uptake rate (Kup, mL/min/g wet), then the heart is switched to normal perfusate to measure tracer clearance rates (T1/2, h). [18F]1 and [18F]2 have very long neuronal retention times due to efficient storage inside norepinephrine storage vesicles. ADV: apparent distribution volume.
PET Studies in Non-human Primates.
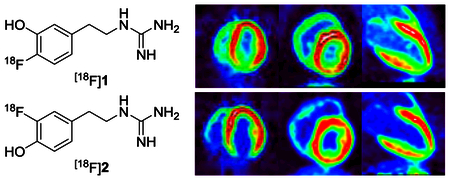
Dynamic PET studies with [18F]2 were performed in rhesus macaque monkeys to characterize the imaging properties, metabolism and myocardial kinetics of the tracer (n = 4). Representative cardiac PET images of [18F]2 show that the tracer provides high quality images of the distribution of sympathetic nerve terminals in the heart (Figure 3). Corresponding PET images for [18F]1 are also provided. There is little retention of either tracer in the lungs, providing very high heart-to-lung contrast. [18F]2 exhibited good heart-to-blood (H/B) contrast in the final PET images (t = 85 min), with H/B = 4.0 ± 0.3. This was better heart-to-blood contrast than the ratio measured for [18F]1, which was H/B = 3.0 ± 0.5.17, 18 Final heart-to-liver (H/L) ratios for [18F]2 were 2.2 ± 0.8. Due to faster clearance of activity from the liver, contrast between heart and liver was a little better for [18F]1, with final H/L = 2.5 ± 0.3. No significant uptake of free fluorine-18 was observed in the vertebral bones of the spine for either compound, indicating that these tracers are not susceptible to defluorination in non-human primates.
Figure 3.

Representative PET images of [18F]1 and [18F]2 in rhesus macaque monkeys.
Metabolism of [18F]2.
To assess the metabolism of [18F]2, six venous blood samples were drawn during the dynamic PET studies. Plasma was separated from blood and processed for analysis on a reverse-phase HPLC system with an in-line radiation detector optimized for detection of 511 keV positron annihilation photons to determine the fraction of plasma activity in the form of radiolabeled metabolites.18 Representative data showing the metabolic breakdown of [18F]2 in plasma are shown in Figure 4. Data on the metabolic breakdown of [18F]1 in rhesus macaques are shown for comparison. For both compounds, the metabolism time course was bi-phasic, with an initially rapid phase of metabolism followed by a slower phase. [18F]2 was metabolized more slowly than [18F]1. The mean time at which 50% of parent radiotracer was still intact was 6.7 ± 2.4 min for [18F]2 vs. 2.3 ± 0.9 min for [18F]1 (n = 4 each).
Figure 4.

Time course of the metabolic breakdown of [18F]1 and [18F]2 in the plasma of rhesus macaque monkeys.
Using in vitro incubations of [18F]1 with a monkey liver cytosol fraction and the cofactor for sulfur conjugation, 3’-phospho-adenosine-5’-phosphosulfate (PAPS), we previously showed that the main metabolic pathway for this compound in rhesus macaques was sulfate conjugation at its m-hydroxyl group.18 Incubations of [18F]2 under identical conditions were unable to produce any radiometabolite, suggesting that this compound is not a substrate for sulfate conjugation in rhesus monkeys. Additional in vitro tests with [18F]2 using a monkey liver microsomal fraction and the glucuronidation cofactor uridine 5’-diphospho-glucuronic acid (UDPGA) were also negative. Further tests are needed to identify the unknown polar metabolite of [18F]2 produced in rhesus macaque monkeys.
Blood Activity Partitioning.
Using aliquots of plasma and whole blood, the ratio of the activity concentration in plasma (Cp) over that in whole blood (Cwb) was determined for each venous blood sample. For [18F]2, the mean ratio Cp/Cwb measured was 1.23 ± 0.05 (n = 4). The Cp/Cwb ratio tended to be constant throughout the PET study. This is similar to previous results for [18F]1 which found Cp/Cwb = 1.25 ± 0.06 (n = 4).18
Tracer Kinetic Analysis.
The myocardial tissue kinetics Ct(t) of [18F]2 and its estimated kinetics in plasma Cp(t) were analyzed using compartmental modeling and Patlak graphical analysis as previously described.17, 18 For compartmental modeling analysis, the model used has two tissue compartments, one for extraction into and clearance from extracellular spaces, with rate constants K1 (mL/min/g) and k2 (min–1), respectively, and the second for irreversible uptake into sympathetic neurons, with rate constant k3 (min–1). A blood volume fraction term BV (dimensionless) was also included to account for activity in myocardial blood. The estimates of K1, k2, and k3 from compartmental modeling were used to calculate a ‘net uptake rate constant’ Ki (mL/min/g) = (K1k3)/(k2 + k3), which reflects the net rate of tracer influx into tissue compartments. K1 estimates were fairly consistent for [18F]2, while k2 and k3 were more variable, similar to previous findings with [18F]1. The mean values of K1 for [18F]2 were K1 = 0.46 ± 0.05 mL/min/g, compared with K1 = 0.56 ± 0.07 mL/min/g for [18F]1. Estimates of k3 (the rate constant related to neuronal uptake of the tracer) were too variable to be useful as a quantitative measure of nerve density, but the values of the net uptake rate constant Ki calculated from the rate constant estimates were very consistent. For [18F]2, the mean net uptake rate constant was Ki = 0.221 ± 0.029 mL/min/g, which can be compared with Ki = 0.341 ± 0.041 mL/min/g for [18F]1. Patlak graphical analysis also gave consistent results. This analysis approach uses a mathematical transformation of Cp(t) and Ct(t) to construct a ‘Patlak plot’ which has a distinct linear phase. The slope of the linear phase, the Patlak slope Kp (mL/min/g), is theoretically equal to the calculated net uptake rate constant Ki derived from the compartmental modeling rate constants. For [18F]2, the measured Patlak slopes averaged Kp = 0.169 ± 0.021 mL/min/g, comparied with Kp = 0.302 ± 0.031 mL/min/g for [18F]1. For any given study, relative to their corresponding Ki values, the Patlak slopes are biased a little lower in magnitude because Patlak analysis does not account for myocardial blood activity in the tissue kinetics Ct(t), while the blood volume term BV used in compartmental modeling does account for the blood activity. Representative examples of the results of compartmental modeling and Patlak analysis of [18F]2 are shown in Figure 5. In previous PET studies in non-human primates with [11C]GMO and [18F]1, using the NET inhibitor desipramine (DMI) to pharmacologically induce different degrees of NET transporter occlusion showed that net uptake rate constants Ki and Patlak slopes Kp each tracked sensitively and reproducibly with DMI-induced declines in available NET transporters.17, 18 Taken together, the results of these studies suggest that values of Ki or Kp obtained from tracer kinetic analysis of the kinetics of [18F]1 or [18F]2 can be used as quantitative metrics of regional sympathetic nerve density in the heart.
Figure 5.

Myocardial tracer kinetic analyses for [18F]2 kinetics in the same monkey. Parameter estimates from compartment modeling (A) were used to calculate a net uptake rate constant Ki (mL/min/g). Patlak graphical analysis (B) provided a Patlak slopes Kp (mL/min/g).
Advantages of 18F-Hydroxyphenethylguanidines.
[18F]1 and [18F]2 offer some advantages over existing cardiac sympathetic innervation radiotracers. The longer half-life of a fluorine-18 (1.83 h) compared with the 20.4 min half-life of carbon-11 in compounds such as [11C]HED and [11C]GMO was one advantage discussed previously. In terms of structure-activity relationships, there are four major factors that govern the neuronal uptake and retention kinetics of sympathetic nerve radiotracers: (a) the rate of neuronal uptake by NET (equal to Vmax/Km for NET transport); (b) the rate of vesicular uptake (equal to Vmax/Km for VMAT2 transport); (c) vulnerability to intraneuronal metabolism by enzymes such as monoamine oxidase (MAO); and (d) membrane diffusion rates, which influence vesicular storage and neuronal retention, and are partly determined by a tracer’s lipophilicity (log P). [18F]1 and [18F]2 were developed in an effort to design a tracer with optimal properties in each of these four categories. We hypothesized that such a tracer would possess myocardial kinetics that could be successfully analyzed using established tracer kinetic analysis techniques to provide quantitative measures of regional sympathetic nerve density. Specifically, the targeted properties were: (a) slower NET transport rates than [11C]HED and [123I]MIBG; (b) rapid vesicular uptake due to efficient VMAT2 transport; (c) resistance to intraneuronal metabolism; and (d) low lipophilicity to promote long neuronal retention times. The slower neuronal uptake rate makes the rate constant associated with this process more identifiable from the cardiac PET kinetics, and the rapid uptake and long retention in storage vesicles eliminates one rate constant from the kinetic model, leading to more reliable and consistent parameter estimates.17
To achieve this goal, radiolabeled phenethylguanidines were investigated because several phenethylguanidines are potent neuron blocking agents due to their prolonged retention inside norepinephrine storage vesicles.29–31 The isolated rat heart studies with [18F]1 and [18F]2 presented above (Figure 2) demonstrate that these compounds satisfy the targeted properties (a), (b) and (d). PET studies showing very long neuronal retention times for [18F]1 and [18F]2 in non-human primate myocardium are consistent with (b) rapid vesicular uptake and (d) very little diffusion of the tracers from storage vesicles. For (c), the guanidine group of the side chain of [18F]1 and [18F]2 confers stability against neuronal enzymes such as tyrosine hydroxylase, MAO, dopamine-β-hydroxylase, or DOPA decarboxylase.32 Previous studies with the carbon-11 analog of [18F]1 in rats showed that 100% of the activity in the myocardium at t = 30 min after tracer administration was in the form of the parent tracer, consistent with no intraneuronal metabolism of the tracer.33 Thus, [18F]1 and [18F]2 have been found to satisfy all of our targeted neuronal tracer properties.
Structure-activity studies of a series of 11C-labeled phenethylguanidines in the isolated rat heart showed that several hydroxyphenethylguanidines had extremely long retention times in this model. This was particularly true for ring hydroxylation at the m- and p- positions, the beta- position of the side chain, or combinations of these hydroxyl substitutions.16 In contrast, none of the 11C-labeled benzylguanidine compounds tested, including [11C]-p-hydroxybenzylguanidine, were found to have long neuronal retention times in the isolated rat heart. These results suggest that hydroxyphenethylguanidines are better VMAT2 substrates than benzylguanidines, or are retained in storage vesicles much more efficiently, leading to high uptake and prolonged retention in storage vesicles. The highly acidic interior of vesicles has been described as an ‘amine trap’, since the low pH inside vesicles further encourages high pKa molecules to exist in their ionized form.34 The very high pKa of the guanidine group of hydroxyphenethylguanidines (pKa = 10 to 13) likely causes them to be highly protonated inside vesicles, further enhancing its vesicular retention.
Two 18F-labeled analogs of [123I]MIBG, m-[18F]fluorobenzylguanidine ([18F]MFBG) and p-[18F]fluorobenzylguanidine ([18F]PFBG) were synthesized previously for PET studies of cardiac sympathetic innervation and neuroendocrine tumors.35, 36 Studies of [18F]PFBG in the isolated rat heart model showed it had a neuronal uptake rate Kup = 2.80 mL/min/g wet, ~ 77% of the rate of 3.65 mL/min/g wet measured for [123I]MIBG.16, 37 The major neuronal clearance halftime for [18F]PFBG was T1/2 = 0.59 h, compared with T1/2 = 2.1 h for [123I]MIBG. Thus, [18F]PFBG has rapid NET transport rate into sympathetic neurons and clears from the neurons faster than [123I]MIBG. We are not aware of any human cardiac imaging studies with [18F]PFBG or [18F]MFBG, although one report used [18F]PFBG to assess cardiac nerve damage in a canine model of infarction.38 While these tracers are certainly capable of imaging cardiac sympathetic nerves, their kinetics would not satisfy the four tracer properties we had targeted in the development of [18F]1 and [18F]2. Thus, another advantage of the 18F-hydroxyphenethylguanidines [18F]1 and [18F]2 over radiolabeled benzylguanidines is the ability to analyze their irreversible myocardial kinetics using standard tracer kinetic methods, such as Patlak analysis, to obtain quantitative regional estimates of sympathetic nerve density.
CONCLUSION
Our previous multi-step radiosynthesis of [18F]1 was not a practical approach for routine production of [18F]fluoro-hydroxyphenethylguanidines in a clinical PET radiochemistry facility. A more efficient method involving 18F-labeling of a N,N’,N’’,N’’-tetrakis-Boc protected guanidinyliodoium salt precursor followed by a single deprotection step and HPLC purification has been developed. This new radiosynthetic approach has proven to be very reliable and provides sufficient radiochemical yields and specific activities for clinical studies in human subjects. This new method was used to synthesize [18F]2, a structural isomer of [18F]1. Preclinical tests of [18F]2 demonstrated that it has imaging properties and kinetics that are very similar to those observed with [18F]1, and that quantitative metrics of regional cardiac sympathetic nerve density can be obtained from tracer kinetic analyses of its myocardial kinetics.
Due to the different metabolic pathways of [18F]1 and [18F]2, both agents are currently being evaluated in first-in-human studies under an exploratory IND clearance from the FDA (ClinicalTrials.gov NCT02385877). The results of these studies will be used to select a lead radiotracer for further clinical development. If PET studies with one of these radiotracers can provide accurate and sensitive regional measures of cardiac sympathetic nerve density in human hearts, it could be used to investigate the contribution of cardiac sympathetic denervation to mechanisms that lead to sudden cardiac death from heart diseases. Based on previous clinical studies with [123I]MIBG and [11C]HED, a potential clinical role for [18F]1 or [18F]2 would be in improved risk stratification of heart failure patients being staged for implantable cardioverter defibrillator (ICD) therapy.
METHODS
Chemicals and General Instrumentation.
NMR spectra were obtained on a Varian Inova 500 (499.90 MHz for 1H; 125.70 MHz for 13C) spectrometer. 1H and 13C NMR chemical shifts (δ) are reported in parts per million (ppm) relative to internal standard TMS and coupling constants (J) are in Hz. High-resolution mass spectra were obtained on a VG (Micromass) 70–250S spectrometer using electrospray ionization (ESI) in positive ion mode, direct chemical ionization (DCI) or electron impact (EI) at 70 eV. Melting points were determined on a Mel-Temp capillary melting point apparatus in open capillary tubes. Flash column chromatography was performed with E. Merck 230–400 mesh silica gel. Analytical TLC was performed with Analtech 0.25 mm glass-backed plates with fluorescent background. Visualization of TLC plates was achieved by UV illumination or treatment with phosphomolybdic acid (PMA). High pressure liquid chromatography (HPLC) was performed on a Hitachi pump L-7100 instrument equipped with Hitachi D-7500 integrator and Hitachi L-4000 UV detector. Radioactivity detection was done with Bioscan coincidence (model B-FC-4000) detector. Reverse-phase HPLC analysis of the formation of radiometabolites in plasma samples was performed on a Perkin-Elmer Series 410 LC instrument equipped with an Ortec Model 905–4 NaI(Tl) radiodetector (Oak Ridge, TN).
Reagents and solvents were purchased from commercial sources and used without further purification unless otherwise noted. [19F]4F-MHPG and [19F]3F-PHPG (standards for HPLC analysis) and N,N′-bis(tert-butoxycarbonyl)-N-3-benzyloxy-4-iodophenethylguanidine (7) were prepared in our laboratory using previously reported methods.18, 39
N,N′,N′′,N′′-Tetrakis(tert-butoxycarbonyl)-N-3-benzyloxy-4-iodophenethylguanidine (8).
A solution of di-tert-butyl dicarbonate (6.1 mmol, 6.1 mL of 1.0 M solution in THF) was added to a solution of N′,N′′-bis(tert-butoxycarbonyl)-N-3-benzyloxy-4-iodophenethylguanidine 7 (600 mg, 1.0 mmol), dimethylaminopyridine (74 mg, 0.61 mmol) and triethylamine (0.85 mL, 6.1 mmol) in anhydrous THF (12 mL) at room temperature. The mixture was stirred for 48 h and then poured over water (50 mL). The mixture was diluted with ethyl acetate (50 mL) and extracted with ethyl acetate (2 × 50 mL). The combined extracts were washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, 15% ethyl acetate in hexane) to afford the product 8 (730 mg, 92%) as a white oil; 1H NMR (500 MHz, CDCl3) δ 7.69 (d, J = 7.9 Hz, 1H), 7.53 (d, J = 7.4 Hz, 2H), 7.40 (t, J = 7.4 Hz, 2H), 7.32 (t, J = 7.4 Hz, 1H), 6.83 (d, J = 1.7 Hz, 1H), 6.66 (dd, J = 7.9, 1.7 Hz, 1H), 5.15 (s, 2H), 3.94 (td, J = 8.0, 4.9 Hz, 2H), 2.90 (td, J = 8.0, 4.9 Hz, 2H), 1.52–1.46 (m, 36H); 13C NMR (125 MHz, CDCl3) δ 157.95, 157.48, 151.39, 147.63, 143.88, 141.05,139.51,136.74, 131.09, 128.72, 128.05, 127.40, 123.71, 113.89, 84.30, 83.92, 83.90, 82.29, 71.05, 48.78, 33.36, 28.25, 28.17, 28.13; MS (ESI) m/z 796 (M+H)+, HRMS (ESI) calcd for C36H50IN3O9 818.2484 (M+Na)+, found 818.2491; Anal. Calcd. For C36H50IN3O9: C, 54.34; H, 6.34; N, 5.28. Found: C, 54.54; H, 6.41; N, 5.14.
N,N′,N′′,N′′-Tetrakis(tert-butoxycarbonyl)-N-3-benzyloxy-4-trimethylstannylphenethyl guanidine (9).
Hexamethylditin (0.36 mL, 1.73 mmol) was added to a solution of compound 8 (688 mg, 0.86 mmol) and tetrakis(triphenylphosphine)palladium (50 mg, 0.04 mmol) in anhydrous toluene (8.0 mL) at room temperature under nitrogen atmosphere. The resulting mixture was heated to 130 oC for 30 min, cooled down to room temperature and filtered through a Celite pad. Celite pad was washed with ethyl acetate and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography (silica gel, 100% hexane to 10% ethyl acetate in hexane) to afford the product 9 (621 mg, 86%) as a yellow oil; 1H NMR (500 MHz, CDCl3) δ 7.40–7.34 (m, 4H), 7.31–7.29 (m, 2H), 6.88 (d, J = 7.2 Hz, 1H), 6.82 (s, 1H), 5.02 (s, 2H), 3.96 (t, J = 8.1 Hz, 2H), 2.92 (t, J = 8.1 Hz, 2H), 1.53–1.31 (m, 36H), 0.17 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 158.0, 157.5, 151.4, 147.6, 143.9, 141.1 139.5, 136.7, 128.7, 128.5, 127.4, 123.7, 113.9, 84.3, 83.9, 83.9, 82.3, 71.1, 48.8, 33.4, 28.3, 28.2, 28.1, 0.2; MS (ESI) m/z 834 (M+H)+, HRMS (ESI) calcd for C39H59N3O9Sn 856.3166 (M+Na)+, found 856.3183; Anal. Calcd. For C39H59N3O9Sn: C, 56.26; H, 7.14; N, 5.05. Found: C, 56.00; H, 7.14; N, 4.88.
2-Benzyloxy-4-{2′-(N,N′,N′′,N′′-tetrakis(tert-butoxycarbonyl)guanidinyl)ethyl}phenyl (2-thienyl)iodonium tosylate (10).
A solution of 2-(diacetoxy)iodothiophene (92 mg, 0.278mmol) in CH2Cl2 (1.0 mL) was added to a solution of p-toluenesulfonic acid hydrate (53 mg, 0.278 mmol) in MeCN (1.0 mL) at room temperature under nitrogen atmosphere. The white precipitate was immediately generated and the mixture was stirred for 1 h. A solution of compound 9 (232 mg, 0.278 mmol) in CH2Cl2 (1.0 mL) and MeCN (1.0 mL) was added slowly to the reaction mixture. After the white precipitate was disappeared, the mixture was stirred at room temperature for 20 h under nitrogen atmosphere. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (silica gel, 100% CH2Cl2 to 20:1=CH2Cl2:MeOH) to afford the tosilate salt 10 (255 mg, 87%) as a yellow solid. mp 79–84 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.26 (d, J = 8.1 Hz, 1H), 7.89 (dd, J = 5.3, 1.3 Hz, 1H), 7.76 (dd, J = 3.8, 1.3 Hz, 1H), 7.52–7.37 (m, 7H), 7.24 (d, J = 1.6, 1H), 7.11–7.08 (m, 3H), 6.95 (dd, J = 8.2, 1.6 Hz, 1H), 5.34 (s, 2H), 3.91 (t, J = 7.4 Hz, 2H), 2.91 (t, J = 7.4 Hz, 2H), 2.28 (s, 3H), 1.43–1.39 (m, 27H), 1.29 (s, 9H); 13C NMR (126 MHz, DMSO-d6) δ 157.05, 155.10, 150.54, 146.676, 146.23, 145.82, 143.64, 139.44, 137.51, 136.87, 136.33, 135.67, 129.23, 128.63, 128.40, 128.01, 127.96, 127.28, 125.48, 123.77, 114.20, 108.08, 101.56, 83.43, 83.21, 81.51, 70.90, 48.86, 47.40, 46.17, 40.01, 32.64, 27.49, 27.41, 27.19, 20.77; HRMS (EI) calcd for C40H53IN3O9S 878.2542 (M-OTs)+, found 878.2546.
2-Benzyloxy-4-{2′-(N,N′,N′′,N′′-tetrakis(tert-butoxycarbonyl)guanidinyl)ethyl}phenyl (2-thienyl)iodonium bromide (11).
A solution of KBr (98 mg, 0.82 mmol) in H2O (1.0 mL) was added to a solution of compound 10 (200 mg, 0.19 mmol) in MeCN (1.0 mL) at 60 oC for 5 min. The reaction mixture was stirred at room temperature for 1 h. The precipitate was washed with ice H2O (10 mL), filtered, washed further with hexane several times and dried in vacuo to afford the bromide salt 11 (156 mg, 86%) as a light yellow solid. mp 116–119°C; 1H NMR (500 MHz, DMSO-d6) δ 8.25 (d, J = 8.1 Hz, 1H), 7.86 (d, J = 5.3 Hz, 1H), 7.73 (d, J = 3.7 Hz, 1H), 7.52–7.38 (m, 5H), 7.22 (s, 1H), 7.08 (dd, J = 5.3, 3.7 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 5.33 (s, 2H), 3.91 (t, J = 7.4 Hz, 2H), 2.90 (t, J = 7.4 Hz, 2H), 1.43–1.39 (m, 27H), 1.30 (s, 9H); 13C NMR (126 MHz, DMSO-d6) δ 157.05, 155.06, 150.53, 146.73, 146.00, 143.63, 139.03, 136.90, 135.96, 135.70, 129.10, 128.61, 128.35, 127.92, 123.72, 114.17, 109.53, 108.87, 102.82, 83.43, 83.19, 81.49, 70.87, 47.38, 32.64, 27.48, 27.40, 27.20; HRMS (ESI) calcd for C40H53IN3O9S 878.2542 (M-Br)+, found 878.2555; Anal. Calcd. For C40H53BrIN3O9S: C, 50.11; H, 5.57; N, 4.38. Found: C, 50.04; H, 5.69; N, 4.37.
2-(4-hydroxyphenyl)ethylamine tert-butylcarbamate (13).
To a solution of 2-(4-hydroxy phenyl)ethylamine 12 (4.00 g, 29.16 mmol) in THF (48 mL) was added triethylamine (4.3 mL, 30.82 mmol). Di-tert-butyl-dicarbonate (6.70 g, 30.70 mmol) was added to the resulting solution and the reaction mixture was stirred at room temperature overnight. The solvent was evaporated under reduced pressure and the residue was purified by flash column chromatography (silica gel, 20% ethyl acetate in hexane) to afford the desired compound 13 (6.78 g, 98%) as a colourless oil; 1H NMR (500 MHz, CDCl3) δ 7.03 (d, J = 8.1 Hz, 2H), 6.77 (d, J = 8.1 Hz, 2H), 4.57 (br. s, NH), 3.33 (t, J = 6.5 Hz, 2H), 2.71 (t, J = 6.5 Hz, 2H), 1.47 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 154.43, 129.88, 115.43, 98.46, 67.47, 42.01, 33.25, 28.46. CAS Registry Number: 64318–28-1.
2-(3-iodo-4-hydoxyphenyl)ethylamine tert-butylcarbamate (14).
Sodium iodide (7.41 g, 49.45 mmol) and NaOH (1.54 g, 38.50 mmol) were added to a solution of compound 13 (7.82 g, 32.98 mmol) in MeOH (60 mL). The resulting solution was cooled to 0 °C. Sodium hypochlorite (4.0–4.9% in water, 81.8 mL, 49.45 mmol) was added slowly to the solution by dropping funnel. The reaction temperature was kept at 0–3 °C. After adding NaOCl, the reaction mixture was stirred for one more hour at 0–5 °C. A solution of sodium thiosulfate (10% in H2O, 70 mL) was added, and the pH was adjusted to 6.5 by addition of HCl (2.0 N solution). The product was extracted by ethyl acetate (3 × 100 mL), and the combined extracts were washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, 20% ethyl acetate in hexane) to afford the compound 14 (5.15 g, 43%) as a white powder; mp 112–115 °C; 1H NMR (500 MHz, CDCl3) δ 7.49 (s, 1H), 7.06 (d, J = 8.0 Hz, 1H), 6.92 (d, J = 8.0 Hz, 1H), 4.53 (br. s, NH), 3.31 (t, J = 6.5 Hz, 2H), 2.70 (t, J = 6.5 Hz, 2H), 1.47 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 155.86, 153.51, 138.23, 133.11, 130.64, 115.02, 85.67, 79.40, 44.56, 34.78, 28.54. CAS Registry Number: 788824–50-0.
2-(4-benzyloxy-3-iodophenyl)ethylamine tert-butylcarbamate (15).
Benzyl bromide (0.52 g, 3.03 mmol) and K2CO3 (0.63 g, 4.55 mmol) were added to a solution of compound 14 (1.10 g, 3.03 mmol) in acetone (17 mL). The resulting solution was stirred at 70 °C for 4 hours. The reaction mixture was filtered and the solvent was evaporated under reduced pressure. Water was added to the residue and the product was extracted by ethyl acetate (3 × 100 mL), and the combined extracts were washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, 10% ethyl acetate in hexane) to afford the compound 15 (1.07 g, 78%) as a white powder; mp 70–72 °C; 1H NMR (500 MHz, CDCl3) δ 7.63 (s, 1H), 7.49 (d, J = 7.4 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.32 (t, J = 7.4 Hz, 1H), 7.09 (d, J = 8.3 Hz, 1H), 6.79 (d, J = 8.3 Hz, 1H), 5.13 (s, 2H), 4.52 (br. s, NH), 3.32 (t, J = 6.5 Hz, 2H), 2.70 (t, J = 6.5 Hz, 2H), 1.44 (S, 9H); 13C NMR (126 MHz, CDCl3) δ 155.90, 155.80, 139.67, 136.56, 133.56, 130.87, 128.79, 128.54, 127.86, 126.98, 112.72, 86.92, 79.33, 70.97, 41.78, 34.80, 28.45, 28.41. CAS Registry Number: 788824–73-7.
2-(4-Benzyloxy-3-iodophenyl)ethylamine hydrochloride (16).
Hydrochloric acid (45 mL of 4.0M solution in 1,4-dioxane, 180 mmol) was added to a solution of compound 15 (0.80 g, 17.6 mmol) in ethyl acetate (5 mL). The resulting solution was stirred at 65 °C for 1 hour. The solvent was evaporated under reduced pressure. The crude product was dissolved again in ethyl acetate and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, 5% methanol and 0.3% NH4OH in methylene chloride) to afford the compound 16 (0.61 g, 97%) as a yellow powder; mp 162–167 °C; 1H NMR (500 MHz, CDCl3) δ 7.70 (s, 1H), 7.46 (d, J = 7.4 Hz, 2H), 7.37 (t, J = 7.4 Hz, 2H), 7.30 (t, J = 7.4 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 8.4 Hz, 1H), 5.07 (s, 2H), 3.20 (t, J = 7.8 Hz, 2H), 3.00 (t, J = 7.8 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 156.50, 139.75, 136.35, 130.52, 129.83, 128.55, 127.89, 126.97, 112.91, 110.01, 87.48, 87.22, 70.92, 45.82, 41.12. CAS Registry Number: 794507–50-9.
N′, N′′-Bis(tert-butoxycarbonyl)-N-4-benzyloxy-3-iodophenethylguanidine (17).
To a cooled (0 °C) solution of compound 16 (0.37 g, 1.06 mmol) and triethylamine (0.75 mL, 5.38 mmol) in anhydrous DMF (3.5 mL) was added in portion 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (0.34 g, 1.16 mmol). The resulting mixture was stirred at 0 °C for 1 h, warmed to room temperature and stirred overnight. The mixture was diluted with ethyl acetate (50 mL), washed with saturated NH4Cl solution (200 mL), and extracted with ethyl acetate (2 × 150 mL). The combined extracts were washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, 10% ethyl acetate in hexane) to afford the product 17 (0.37 g, 58%) as a white solid; mp 115–117 °C. 1H NMR (500 MHz, CDCl3) δ 11.46 (br. s, 1NH), 8.38 (br. s, 1NH), 7.67 (s, 1H), 7.49 (d, J = 7.6 Hz, 2H), 7.39 (t, J = 7.6 Hz, 2H), 7.32 (t, J = 7.6 Hz, 1H), 7.11 (d, J = 9.0 Hz, 1H), 6.78 (d, J = 9.0 Hz, 1H), 5.13 (s, 2H), 3.62 (q, J = 6.7 Hz, 2H), 2.77 (t, J = 6.7 Hz, 2H), 1.50 (s, 18H); 13C NMR (126 MHz, CDCl3) δ 156.11, 155.99, 139.76, 136.57, 133.15, 129.69, 128.53, 127.84, 126.98, 112.74, 86.91, 83.12, 79.26, 70.96, 42.15, 33.93, 28.31, 28.09; HRMS calcd for C26H34IN3O5 596.1616, found 596.1615.
N, N′,N′′,N′′-Tetrakis(tert-butoxycarbonyl)-N-4-benzyloxy-3-iodophenethylguanidine (18).
A solution of di-tert-butyl dicarbonate (40.7 mmol, 40.7 mL of 1.0 M solution in THF) was added to a solution of compound 17 (4.04 g, 6.78 mmol), N,N-dimethylaminopyridine (497 mg, 4.07 mmol) and triethylamine (5.67mL, 40.1 mmol) in anhydrous THF (82 mL) at room temperature. The mixture was stirred for 48 h and then poured over water (200 mL). The mixture was diluted with ethyl acetate (200 mL). After decantation, the aqueous layer was extracted with ethyl acetate (2 × 200 mL). The combined extracts were washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, 10% ethyl acetate in hexane) to afford the product 18 (3.41 g, 63%) as a white oil; 1H NMR (500 MHz, CDCl3) δ 7.71 (s, 1H), 7.49 (d, J = 7.6 Hz, 2H), 7.39 (t, J = 7.6 Hz, 2H), 7.32 (t, J = 7.6 Hz, 1H), 7.17 (d, J = 8.4 Hz, 1H), 6.78 (d, J = 8.4 Hz, 1H), 5.13 (s, 2H), 3.93 (t, J = 8.0 Hz, 2H), 2.85 (t, J = 8.0 Hz, 2H), 1.50 (s, 36H); 13C NMR (126 MHz, CDCl3) δ 157.68, 155.85, 147.34, 139.77, 136.58, 133.51, 129.92, 128.54, 127.85, 126.98, 112.71, 86.84, 83.66, 82.03, 70.96, 48.73, 31.87, 28.04, 27.98, 27.91; HRMS calcd for C36H50IN3O9 818.2484, found 818.2479.
N,N′,N′′,N′′-Tetrakis(tert-butoxycarbonyl)-N-4-benzyloxy-3-trimethylstannylphenethylguanidine (19).
Hexamethylditin (2.0 mL, 9.60 mmol) was added to a solution of compound 18 (2.79 g, 3.50 mmol) and tetrakis(triphenylphosphine)palladium (200 mg, 0.16 mmol) in anhydrous toluene (30 mL) at room temperature under nitrogen atmosphere. The resulting mixture was heated to 130 °C for 30 min, cooled down to room temperature and filtered through a Celite pad. Celite pad was washed with ethyl acetate and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography (silica gel, 100% hexane to 10% ethyl acetate in hexane) to afford the product 19 (2.87, 98%) as a yellow oil; 1H NMR (500 MHz, CDCl3) δ 7.42–7.37 (m, 4H), 7.32 (t, J = 6.5 Hz, 2H), 6.91 (d, J = 7.1 Hz, 1H), 6.84 (s, 1H), 5.04 (s, 2H), 3.98 (t, J = 8.1 Hz, 2H), 2.94 (t, J = 8.1 Hz, 2H), 1.51– 1.49 (m, 36H), 0.19 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 163.2, 157.8, 151.2, 147.4, 143.6, 141.2, 137.1, 136.5, 128.4, 128.1, 127.8, 127.7, 121.8, 110.9, 83.6, 83.4, 82.0, 70.0, 48.9, 33.6, 28.1, 28.0, 27.9, 14.1; HRMS (ESI) calcd for C39H59N3O9Sn 856.3166 (M+Na)+, found 856.3172; Anal. Calcd. For C39H59N3O9Sn: C, 56.26; H, 7.14; N, 5.05. Found: C, 56.00; H, 7.23; N, 4.90.
2-Benzyloxy-5-{2′-(N,N′,N′′,N′′-tetrakis(tert-butoxycarbonyl)guanidinyl)ethyl}phenyl(2-thienyl)iodonium tosylate (20).
A solution of 2-(diacetoxy)iodothiophene (209 mg, 0.637 mmol) in CH2Cl2 (5.0 mL) was added to a solution of p-toluenesulfonic acid hydrate (121 mg, 0.637 mmol) in MeCN (5.0 mL) at room temperature under nitrogen atmosphere. The white precipitate was immediately generated and the mixture was stirred for 1 h. A solution of compound 19 (530 mg, 0.637 mmol) in CH2Cl2 (3.0 mL) and MeCN (3.0 mL) was added slowly to the reaction mixture. After the white precipitate disappeared, the mixture was stirred at room temperature for 20 h under nitrogen atmosphere. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (silica gel, 100% CH2Cl2 to 20:1=CH2Cl2:MeOH) to afford the tosylate salt 20 (482 mg, 86%) as a yellow solid; mp 140–141°C; 1H NMR (500 MHz, DMSO-d6) δ 8.21 (s, 1H), 7.90 (t, J = 5.3 Hz, 1H), 7.77 (t, J = 5.3 Hz, 1H), 7.49–7.37 (m, 7H), 7.32 (d, J = 8.5 Hz, 2H), 7.12–7.08 (m,3H), 5.33 (s, 2H), 3.88 (t, J = 7.8 Hz, 2H), 2.85 (t, J = 7.8 Hz, 2H), 2.28 (s, 3H), 1.43–1.36 (m, 36H); 13C NMR (126 MHz, DMSO-d6) δ 157.1, 153.7, 150.6, 146.7, 145.8, 143.6, 139.5, 137.5, 136.5, 136.3, 135.8, 134.7, 133.7, 129.2, 128.6, 128.3, 128.0, 127.7, 125.5, 113.9, 110.3, 101.3, 83.6, 83.2, 81.6, 70.9, 47.5, 31.1, 27.9, 27.52, 27.4, 27.3, 27.2, 20.9.
2-Benzyloxy-5-{2′-(N,N′,N′′,N′′-tetrakis(tert-butoxycarbonyl)guanidinyl)ethyl}phenyl(2- thienyl)iodonium bromide (21).
A solution of KBr (583 mg, 4.90 mmol) in H2O (5.0 mL) was added to a solution of compound 20 (430 mg, 0.49 mmol) in MeCN (7.0 mL) at 60 °C for 5 min. The reaction mixture was stirred at room temperature for 1 h. The precipitate was washed with ice H2O (10 mL), filtered, washed further with hexane several times and dried in vacuo to afford the bromide salt 21 (411 mg, 96%) as a light yellow solid; mp 140–141 °C; 1H NMR (500 MHz, CDCl3) δ 7.97 (s, 1H), 7.53 (d, J = 5.2 Hz, 1H), 7.46 (d, J = 5.2 Hz, 1H), 7.41–7.35 (m, 6H), 6.96–6.91 (m, 2H), 5.21 (s, 2H), 3.92 (t, J = 8.1 Hz, 2H), 2.90 (t, J = 8.1 Hz, 2H), 1.52–1.48 (m, 36H); 13C NMR (126 MHz, CDCl3) δ 157.6, 153.6, 151.1, 147.3, 143.5, 137.9, 136.5, 135.2, 134.7, 134.6, 133.8, 128.8, 128.7, 128.5, 127.5, 114.4, 113.8, 83.89, 83.8, 82.2, 71.9, 48.4, 33.8, 32.0, 29.7, 28.1, 28.0, 27.9; HRMS (ESI) calcd for C40H53IN3O9S 878.2542 (M-Br)+, found 878.2540; Anal. Calcd. For C40H53BrIN3O9S: C, 50.11; H, 5.57; N, 4.38. Found: C, 51.86; H, 5.95; N, 4.24.
Radiosynthesis of [18F]1.
A TRACERlab FXFN computer-controlled radiosynthesis module (GE Healthcare) was used to achieve fully automated radiosyntheses of [18F]1 and [18F]2. [18F]F– was prepared by the 18O(p,n)18F reaction using H218O as the target material in a GE PETrace cyclotron. [18F]F– was isolated from the enriched water by trapping on a Waters Sep-Pak® Light QMA cartridge (pre-activated with 10 mL of ethanol and 10 mL of H2O) and eluted from the cartridge into the glassy-carbon reactor vial of the TRACERlab FXFN system with a solution of 0.5 mL Cs2CO3 (0.05 M in H2O). MeCN (1.0 mL) was added to the reactor vessel and then water/acetonitrile is evaporated at 80 °C under vacuum with a nitrogen stream to yield dried Cs[18F]F. After cooling to 60 °C, a mixed solution of 0.5 mL of DMF and 20 μL of MilliQ H2O, containing 5.5–6.0 mg of the diaryliodonium salt precursor 11 and 1.0 mg of TEMPO (2,2,6,6-tetramethylpiperidine-N-oxyl) were added to the reactor vessel containing Cs[18F]F. The sealed reaction mixture was heated at 150 °C for 5 min to produce 3-benzyloxy-4-[18F]fluorophenethyl-N,N′,N′′,N′′-tetrakis-BOC-guanidine [18F]22 as intermediate. After cooling to 70 °C, a solution of 48% HBr (0.5 mL) and MeCN (0.5 mL) was added to the reaction mixture. The reaction solution was heated at 120 °C for 15 min. and then cooled to 50 °C. Next, a mixture solution of NaOH solution (1.0 mL, 4.0 M in H2O) and buffer solution (1.8 mL, 5% EtOH in 40 mM NH4OAc) was added into the reactor vessel. This mixture was injected onto a reverse-phase HPLC column (Phenomenex Synergi 10μ Hydro-RP 80A, 250×10 mm, 5% EtOH in 40 mM NH4OAc buffer, flow rate 4.0 mL/min, λ = 254 nm) and [18F]1 was collected at Rt = 30–32 min. The collected [18F]1 fraction was passed through a 0.22 μm sterilizing filter directly into a 10 mL septum-sealed sterile pyrogen-free glass vial.
Specific activity (SA) was determined by injecting a sample of [18F]1 with known activity (kBq) onto an HPLC system used for quality control. The area under the UV absorbance peak associated with the [18F]1 radioactivity peak was compared against a predetermined standard curve to estimate the total mass ([18F]4F-MHPG + [19F]4F-MHPG) in μg. The ratio of 18F-activity to total mass (converted from μg to μmol using the molecular weight of [19F]4F-MHPG) gave the specific activity.
Radiosynthesis of [18xsF]2.
The same methods described for the synthesis of [18F]1 were used to prepare [18F]2, except that the appropriate precursor 21 was used instead of precursor 11. HPLC purification conditions were slightly different: Phenomenex Synergi 10μ Hydro-RP 80A, 250×10 mm, 3.5% EtOH in 40 mM NH4OAc buffer, flow rate 4.0 mL/min, λ = 254 nm. In this system, [18F]2 was collected at Rt = 31–33 min.
Isolated Rat Heart Studies.
Hearts from male Sprague-Dawley rats (225 – 500 g) were perfused under moderate workload conditions (7.3 mmHg preload, 73 mmHg afterload) using a working heart preparation.40 Two parallel perfusion circuits were connected to the left atrial cannula with a 3-way connector to allow for rapid switching from one circuit to the other. The heart perfusate was Krebs-Henseleit (KH) bicarbonate buffer (118 nM NaCl, 4.7 mM KCl, 2.55 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, and 25 mM NaHCO3) containing 5 mM glucose, oxygenated with a 95% O2/5% CO2 gas mixture and held at 37 °C. Corticosterone (54 μM) was added to the perfusate to block extraneuronal uptake (uptake-2) of the radiotracer into the rat myocardium.23
Fluorine-18 activity in the heart was measured externally using a pair of cesium fluoride (CsF) scintillation detectors with crystal size 51 cm diameter and 51 cm thick (Crismatec 51Y51; Saint-Gobain, Nemours, France). The front faces of the two CsF detectors were positioned directly opposite each other, ~ 4 cm apart, with the heart centered between them. Each detector was enclosed in a large cylindrical lead collimator (2 cm wall thickness, 25 cm long) to shield against background counts from radioactive sources outside the heart. Two coincidence detection circuits were established between the detectors using standard Nuclear Instrumentation Module (NIM) electronic modules. One circuit measured total coincident events between the two detectors (true + random coincident events), and the second measured only random coincident events. A computerized data acquisition system interfaced to the NIM-module coincidence circuits was used to acquire and record the whole-heart radioactivity data throughout the study.41
Hearts were initially perfused for a 30 min stabilization period using KH buffer in the first perfusion circuit. During this time, [18F]2 was added to 1.0 L of KH buffer circulating in the second perfusion circuit and allowed to equilibrate over several minutes. Three 1.0 mL aliquots were drawn from the second perfusion circuit for counting in a gamma counter (Cobra II Auto-Gamma, Perkin-Elmer, Waltham, MA) to determine the radioactivity concentration in the perfusate (Cp), which was ~ 74 kBq/mL perfusate. After initiating data acquisition from the CsF detectors, the heart was rapidly switched to the second perfusion circuit to begin a constant infusion of [18F]2 for 10 min. Then the heart was switched back to the first perfusion circuit for 120 min to measure clearance rates of the tracer from the heart.
The acquired whole-heart radioactivity data (counts per second; cps) at each time point were converted to an ‘apparent distribution volume’ (ADV; mL perfusate/g wet), by dividing by the perfusate radioactivity concentration Cp (kBq/mL perfusate), the detector system calibration factor Zcalib (cps/kBq), and the measured wet mass of the heart Mw (g wet). Neuronal uptake rates of the radiotracers (Kup; mL perfusate/min/g wet) were calculated as the slope of a linear regression of the ADV data between t = 1 min and t = 4 min of the 10 min infusion study. Clearance rates were estimated by fitting the ADV data during the clearance phase of the study to multiple exponential decay processes. The exponential clearance rate constants (λi) were used to calculate corresponding clearance half-times: T1/2 = ln(2)/λi. The slowest rate, associated with clearance from sympathetic neurons, is reported for each compound.
PET imaging Studies with [18F]2.
Cardiac PET studies (n = 4) were performed in rhesus macaque monkeys using a microPET P4 primate scanner (Siemens/CTI Concorde Microsystems, Knoxville, TN). After anesthetizing the animal, a percutaneous angiocather was placed in the saphenous vein of each leg, one for tracer injection, the other for blood sampling. Vital signs, including heart rate (bpm), blood oxygen saturation levels (SpO2) and body temperature were monitored continuously (model V3404P, SurgiVet, Norwell, MA). Dynamic PET data were acquired in list-mode for 90 min after intravenous injection of 155 – 230 MBq of [18F]2. List-mode emission data were rebinned into a 27-frame dynamic sequence (12×10 s, 2×30 s, 2×60 s, 2×150 s, 2×300 s, 7×600 s). Rebinned emission data were corrected for attenuation and scatter, and transaxial images reconstructed using maximum a posteriori (MAP) reconstruction42, an iterative method that accounts for the detector point spread function in the model of the system.
Blood Partitioning and Radiometabolite Analysis.
Venous blood samples (1.5–2.0 mL) were centrifuged for 1 min at 12000 × g to separate plasma and red blood cells. Plasma was deproteinized by adding perchloric acid (HClO4; final concentration 0.4N) and centrifuging for 5 min at 12000 × g. The supernatant was neutralized with KOH (pH 7.0–7.5) and filtered twice (Millex GS 0.22 μm, Millipore, Billerica, MA). Aliquots (0.1 mL) of whole blood, plasma and the final supernatant were counted in a gamma counter. Count data for plasma and whole blood aliquots were decay corrected and used to calculate the relative concentrations of [18F]2 in plasma and whole blood (Cp/Cwb). The final supernatant was analyzed by HPLC (Synergi 10 μm Hydro-RP column, 4.6 × 250 mm, 60 mM sodium phosphate buffer, pH 5.4 with 8% ethanol, flow rate 1.0 mL/min) with an in-line radiation detector. Under the HPLC conditions used [18F]2 had a retention time Rt = 12.7 min, while the main polar radiometabolite formed had Rt = 9.3 min. Peak area analysis of the radiation detection curve was used to estimate the percentage intact parent tracer fraction (fintact) for each plasma sample.
In Vitro Metabolism Tests.
To test for sulfate conjugation of [18F]2, a 20 μL aliquot of monkey liver cytosol (#452461, BD Biosciences, San Jose, CA) was added to a 10 μL aliquot of 10 mM sulfotransferase cofactor PAPS (adenosine-3′-phosphate-5′-phosphosulfate lithium salt hydrate; #A1651, Sigma-Aldrich, Milwaukee, WI) dissolved in 50 μL of 1.0 mM Tris-HCl buffer (pH 7.4) and 170 μL of ultrapure water (18 MΩ·cm MilliQ, Millipore, Billerica, MA) and incubated at 37 °C for 5 min.43 Next, 740 kBq of [18F]2 in 250 μL of ultrapure water was added to the reaction mixture (final volume 500 μL) and incubated at 37 °C for 20 min. The reaction was terminated by centrifugation at 16,000 × g for 5 min at 4 °C. The supernatant was filtered (Millex GS 0.22 μm, Millipore, Billerica, MA) and analyzed using HPLC with radiation detection as described above for the rhesus macaque plasma samples. To test for glucuronidation of [18F]2, a 25 μL aliquot containing 0.5 mg of monkey liver microsomes (#452413, BD Biosciences, San Jose, CA) was added to a reaction mixture of 50 μL of 20 mM glucuronidation cofactor UDPGA (uridine 5′-diphosphoglucuronic acid; #U5625 Sigma-Adrich, Milwaukee, WI) and 50 μL of 30 mM DTT (dithiothreitol, #D0632, Sigma-Aldrich, Milwaukee, WI) dissolved in 1.0 M glycine/NaOH buffer containing 50 mM MgCl2 (pH 9.2). An additional 125 μL of the glycine/NaOH buffer was added and the mixture incubated at 37 °C for 5 min. Next, 740 kBq of [18F]2 in 250 μL of ultrapure water was added to the reaction mixture (final volume 500 μL) and incubated at 37 °C for 20 min. Using the same procedures for the sulfate conjugation test, the reaction mixture was centrifuged and the supernatant filtered for analysis using radio-HPLC.
Plasma Time-Activity Curve.
Region-of-interest analysis of the dynamic PET images was used to determine a time-activity curve of the activity concentration in whole blood, Cwb(t). The whole blood time-activity curve Cwb(t) was then multiplied by the measured ratio of activity in plasma over whole blood (Cp/Cwb) and by the percentage of intact parent tracer fraction in plasma data, fintact(t), to estimate the kinetics of the plasma concentration of intact tracer, Cp(t). The estimated plasma curve Cp(t) was used as the input function for tracer kinetic analyses.
Tracer Kinetic Analysis.
For each PET study, the final four dynamic image frames were summed and used to draw a region-of-interest on the left ventricular wall, encompassing 3–4 transaxial slices, to extract a time-activity curve for myocardial tissue Ct(t). For compartmental modeling analysis, Ct(t) and the plasma kinetics Cp(t) for [18F]2 were analyzed using a two-tissue compartment model with irreversible trapping to estimate the rate constants K1 (mL/min/g), k2 (min–1), k3 (min–1) and a blood volume fraction BV (dimensionless). The Ct(t) and Cp(t) data were also analyzed using Patlak analysis to estimate a Patlak slope, Kp (mL/min/g).
Animal Care.
The care of all animals used in this study was done in accordance with the Animal Welfare Act and the National Institute of Health’s Guide for the Care and use of Laboratory Animals.44 Animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan.
ACKNOWLEDGMENTS
The authors thank the staff of the University of Michigan Cyclotron Facility for their many contributions to this study.
Funding
We gratefully acknowledge the support of this work through PHS grant R01-HL079540 from the National Heart Lung and Blood Institute, National Institutes of Health, Bethesda, MD USA.
ABBREVIATIONS USED
- DMI
desipramine
- [11C]HED
[11C]-(–)-m-hydroxyephedrine
- [11C]GMO
N-[11C]guanyl-(–)-m-octopamine
- [123I]MIBG
[123I]m-iodobenzylguanidine
- NET
norepinephrine transporter
- TEMPO
2,2,6,6-tetramethylpiperidine-N-oxyl
- VMAT2
vesicular monoamine transporter, isoform 2.
REFERENCES
- 1.Fukuda K, Kanazawa H, Aizawa Y, Ardell JL, and Shivkumar K (2015) Cardiac innervation and sudden cardiac death. Circ. Res 116, 2005–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen MJ, and Zipes DP (2014) Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ. Res 114, 1004–1021. [DOI] [PubMed] [Google Scholar]
- 3.Kuehl M, and Stevens MJ (2012) Cardiovascular autonomic neuropathies as complications of diabetes mellitus. Nat. Rev. Endocrinol 8, 405–416. [DOI] [PubMed] [Google Scholar]
- 4.Floras JS (1993) Clinical aspects of sympathetic activation and parasympathetic withdrawal in heart failure. J Am Coll Cardiol 22 (4 Suppl. A), 72A–82A. [DOI] [PubMed] [Google Scholar]
- 5.Baker AJ (2014) Adrenergic signaling in heart failure: a balance of toxic and protective effects. Pflugers Arch. - Eur. J. Physiol 466, 1139–1150. [DOI] [PubMed] [Google Scholar]
- 6.Gardner RT, Ripplinger CM, Myles RC, and Habecker BA (2016) Molecular mechanisms of sympathetic remodeling and arrhythmias. Circ. Arrhythm. Electrophysiol 9, e001359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaseghi M, and Shivkumar K (2008) The role of the autonomic nervous system in sudden cardiac death. Prog. Cardiovasc. Dis 50, 404–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zipes DP, and Rubart M (2006) Neural modulation of cardiac arrhythmias and sudden cardiac death. Heart Rhythm 3, 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henneman MM, Bengel FM, van der Wall EE, Knuuti J, and Bax JJ (2008) Cardiac neuronal imaging: application in the evaluation of cardiac disease. J. Nucl. Cardiol 15, 442–455. [DOI] [PubMed] [Google Scholar]
- 10.Wollenweber T, and Bengel FM (2014) Molecular imaging to predict ventricular arrhythmia in heart failure. J. Nucl. Cardiol 21, 1096–1109. [DOI] [PubMed] [Google Scholar]
- 11.Wieland DM, Brown LE, Rogers WL, Worthington KC, Wu J-L, Clinthorne NH, Otto CA, Swanson DP, and Beierwaltes WH (1981) Myocardial imaging with a radioiodinated norepinephrine storage analog. J. Nucl. Med 22, 22–31. [PubMed] [Google Scholar]
- 12.Rosenspire KC, Haka MS, Van Dort ME, Jewett DM, Gildersleeve DL, Schwaiger M, and Wieland DM (1990) Synthesis and preliminary evaluation of carbon-11-meta-hydroxyephedrine: a false transmitter agent for heart neuronal imaging. J. Nucl. Med 31, 1328–1334. [PubMed] [Google Scholar]
- 13.Jacobson AF, Senior R, Cerqueira MD, Wong ND, Thomas GS, Lopez VA, Agostini D, Weiland F, Chandna H, and Narula J (2010) Myocardial iodine-123 meta-iodobenzylguanidine imaging and cardiac events in heart failure: results of the prospective ADMIRE-HF (AdreView Myocardial Imaging for Risk Evaluation in Heart Failure) study. J. Am. Coll. Cardiol 55, 2212–2221. [DOI] [PubMed] [Google Scholar]
- 14.Fallavollita JA, Heavey BM, Luisi AJ, Michalek SM, Baldwa S, Mashtare TL, Hutson AD, DeKemp RA, Haka MS, Sajjad M, Cimato TR, Curtis AB, Cain ME, and Canty JM (2014) Regional myocardial sympathetic denervation predicts the risk of sudden cardiac arrest in ischemic cardiomyopathy. J. Am. Coll. Cardiol 63, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deyell MW, Krahn AD, and Goldberger JJ (2015) Sudden cardiac death risk stratification. Circ. Res 116, 1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raffel DM, Jung YW, Gildersleeve DL, Sherman PS, Moskwa JJ, Tluczek LJ, and Chen W (2007) Radiolabeled phenethylguanidines: novel imaging agents for cardiac sympathetic neurons and adrenergic tumors. J. Med. Chem 50, 2078–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raffel DM, Koeppe RA, Jung YW, Gu G, Jang KS, Sherman PS, and Quesada CA (2013) Quantification of cardiac sympathetic nerve density of N-11C-guanyl-meta-octopamine and tracer kinetic analysis. J. Nucl. Med 54, 1645–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jang KS, Jung YW, Gu G, Koeppe RA, Sherman PS, Quesada CA, and Raffel DM (2013) 4-[18F]fluoro-m-hydroxyphenethylguanidine: a radiopharmaceutical for quantifying regional cardiac sympathetic nerve density with positron emission tomography. J. Med. Chem 56, 7312–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jang KS, Jung YW, Song HC, and Raffel DM (2015) Strategies for radiolabeling of 4-[18F]fluoro-m-hydroxyphenethylguandine ([18F]4F-MHPG): a novel imaging agent for cardiac sympathetic innervation. Mol. Imaging Biol 17 (Suppl 1), S997. [Google Scholar]
- 20.Kazmierczak P, Skulski L, and Kraszkiewicz L (2001) Synthesis of (diacetoxyiodo)arenes or iodylarenes from iodoarenes, with sodium periodate as the oxidant. Molecules 6, 881–891. [Google Scholar]
- 21.Chun J-H, and Pike VW (2012) Regiospecific syntheses of functionalized diaryliodonium tosylates via [hydroxy(tosyloxy)iodo]arenes generated in site from (diacetoxyiodo)arenes. J. Org. Chem 77, 1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ross TL, Ermert J, Hocke C, and Coenen HH (2007) Nucleophilic 18F-fluorination of heteroaromatic iodonium salts with no-carrier-added [18F]fluoride. J. Am. Chem. Soc 129, 8018–8025. [DOI] [PubMed] [Google Scholar]
- 23.Salt PJ (1972) Inhibition of noradrenaline uptake2 in the isolated rat heart by steroids, clonidine and methoxylated phenylethylamines. Eur. J. Pharmacol 20, 329–340. [DOI] [PubMed] [Google Scholar]
- 24.Schäfer MKH, Weihe E, and Eiden LE (2013) Localization and expression of VMAT2 across mammalian species: a translational guide for its visualization and targeting in health and disease. Adv. Pharmacol 63, 319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeGrado TR, Hutchins GD, Toorongian SA, Wieland DM, and Schwaiger M (1993) Myocardial kinetics of carbon-11-meta-hydroxyephedrine: retention mechanisms and effects of norepinephrine J. Nucl. Med 34, 1287–1293. [PubMed] [Google Scholar]
- 26.Raffel DM, and Wieland DM (2001) Assessment of cardiac sympathetic nerve integrity with positron emission tomography. Nucl. Med. Biol 28, 541–559. [DOI] [PubMed] [Google Scholar]
- 27.Raffel DM (2012) Targeting norepinephrine transporters in cardiac sympathetic nerve terminals In Targeted Molecular Imaging (Welch MJ, and Eckelman WC, Eds.), pp 305–320, CRC Press, Boca Raton. [Google Scholar]
- 28.Patlak CS, and Blasberg RG (1985) Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J. Cereb. Blood Flow 5, 584–590. [DOI] [PubMed] [Google Scholar]
- 29.Costa E, Kunstman R, Gessa GL, and Brodie BB (1962) Structural requirements for bretylium and guanethidine-like activity in a series of guanidine derivatives. Life Sci. 3, 75–80. [DOI] [PubMed] [Google Scholar]
- 30.Fielden R, and Green AL (1965) The effects of some aralkylguanidines in mice. Brit. J. Pharmacol 24, 408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green AL, Fielden R, Bartlett DC, Cozens MJ, Eden RJ, and Hills DW (1967) New norepinephrine-depleting agents. β-hydroxyphenethylguanidine and related compounds. J. Med. Chem 10, 1006–1008. [DOI] [PubMed] [Google Scholar]
- 32.Maxwell RA, and Wastila WB (1977) Adrenergic Neuron Blocking Drugs In Antihypertensive Agents. Handbook of Pharmacology XXXIX. (Gross F, Ed.), pp 161–211, Springer-Verlag, New York. [Google Scholar]
- 33.Raffel DM (2015) Preclinical evaluations of cardiac sympathetic innervation radiotracers In Autonomic Innervation of the Heart: Role of Molecular Imaging. (Slart RHJA, Tio RA, Elsinga PH, and Schwaiger M, Eds.), pp 201–234, Springer, Berlin. [Google Scholar]
- 34.Graefe K-H, and Bönisch H (1988) The transport of amines across axonal membranes of noradrenergic and dopaminergic neurones In Catecholamines I, Handbook of Experimental Pharmacology (Trendelenburg U, and Werner N, Eds.), pp 192–245, Springer-Verlag, Berlin. [Google Scholar]
- 35.Garg PK, Garg S, and Zalutsky MR (1994) Synthesis and preliminary evaluation of para- and meta-[18F]fluorobenzylguanidine. Nucl. Med. Biol 21, 97–103. [DOI] [PubMed] [Google Scholar]
- 36.Zhang H, Huang R, Pillarsetty N, Thorek DLJ, Vaidyanathan G, Serganova I, Blasberg RG, and Lewis JS (2014) Synthesis and evaluation of 18F-labeled benzylguanidine analogs for targeting the human norepinephrine transporter. Eur. J. Nucl. Med. Mol. Imaging 41, 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berry CR, Garg PK, Zalutsky MR, Coleman RE, and DeGrado TR (1996) Uptake and retention kinetics of para-fluorine-18-fluorobenzylguanidine in isolated rat heart J. Nucl. Med 37, 2011–2016. [PubMed] [Google Scholar]
- 38.Berry CR, Garg PK, DeGrado TR, Hellyer P, Weber W, Garg S, Hansen B, Zalutsky MR, and Coleman RE (1996) Para-[18F]fluorobenzylguanidine kinetics in a canine coronary artery occlusion model. J. Nucl. Cardiol 3, 119–129. [DOI] [PubMed] [Google Scholar]
- 39.Jang KS, Jung YW, Sherman PS, Quesada CA, Gu G, and Raffel DM (2013) Synthesis and bioevaluation of [18F]4-fluoro-m-hydroxyphenethylguanidine ([18F]4F-MHPG): a novel radiotracer for quantitative PET studies of cardiac sympathetic innervation. Bioorg. Med. Chem. Lett 23, 1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taegtmeyer H, Hems R, and Krebs HA (1980) Utilization of energy providing substrates in the isolated working rat heart Biochemistry Journal 186, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raffel D, Loc’h C, Mardon K, Mazière B, and Syrota A (1998) Kinetics of the norepinephrine analog [Br-76]-meta-bromobenzylguanidine in isolated working rat heart. Nucl. Med. Biol 25, 1–16. [DOI] [PubMed] [Google Scholar]
- 42.Qi J, and Leahy RM (2000) Resolution and noise properties of MAP reconstruction for fully 3D-PET. IEEE Trans. Med. Imaging 19, 493–506. [DOI] [PubMed] [Google Scholar]
- 43.Narimatsu S, Kobayashi N, Asaoka K, Masubuchi Y, Horie T, Hosokawa M, Ishikawa T, Ohmori S, Kitada M, Miyano J, Kataoka H, and Yamamoto S (2001) High-performance liquid chromatographic analysis of the sulfation of 4-hydroxypropranolol enantiomers by monkey liver cytosol. Chirality 13, 140–147. [DOI] [PubMed] [Google Scholar]
- 44.National Research Council (1985) Guide for the Care and Use of Laboratory Animals., U.S. Department of Health and Human Services, National Institutes of Health, Bethesda, MD. [Google Scholar]