

Abstract
We herein report a gram-scale, enantioselective synthesis of Tamiflu, in which the key trans-diamino moiety has been efficiently installed via an iron-catalyzed stereo-selective olefin diazidation. This significantly improved, iron- catalyzed method is uniquely effective for highly functionalized yet electronically deactivated substrates that have been previously problematic. Preliminary catalyst structure—reactivity—stereoselectivity relationship studies revealed that both the iron catalyst and the complex substrate cooperatively modulate the stereoselectivity for diazidation. Safety assessment using both differential scanning calorimetry (DSC) and the drop weight test (DWT) has also demonstrated the feasibility of carrying out this iron-catalyzed olefin diazidation for large-scale Tamiflu synthesis.
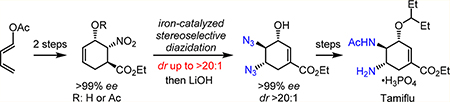
INTRODUCTION
Oseltamivir phosphate 1 (Tamiflu), the pro-drug of a potent viral neuraminidase inhibitor, has been used as an effective medicine to treat and prevent influenza A and influenza B.1a Designed and developed by scientists at Gilead and Hoffman— La Roche, it effectively mimics the transition state of enzymatic hydrolysis of terminal sialic acids 2 from cell-surface glycoconjugates, a step postulated to be necessary for elution of newly formed viruses from infected cells (Scheme 1a). From a synthetic chemistry perspective, the structure of Tamiflu can be simplified to a functionalized trans,trans-diamino cyclic allylic alcohol 3, in which the stereochemical alignment of three contiguous stereogenic centers is critical for Tamiflu’s antiviral activity;1b however, the stereoselective synthesis of 3 from a readily available starting material is not straightforward. As a result, numerous efforts have been devoted to search for an expedient strategy to produce Tamiflu and a range of efficient Tamiflu syntheses have been reported (Scheme 2).2,3
Scheme 1.

Enantioselective Synthesis of Tamiflu via the Iron-Catalyzed Stereoselective Olefin Diazidation
Scheme 2.

Selected Examples of the Previous Tamiflu Syntheses
In Roche’s Tamiflu production route, the starting material, shikimic acid 4, is converted to a key homoallylic epoxide intermediate 5 (Scheme 2a).2 The trans-diamino moiety in Tamiflu is installed through the stereoselective ring opening of both epoxide 5 and a homoallylic N-H aziridine 6 using NaN3 (Scheme 2a).2 Azide-free procedures were later developed from 5 as well; however, additional epimerization steps are necessary.2c,d To search for an efficient synthetic strategy from readily available achiral starting materials, chemists have also extensively investigated the de novo enantioselective synthesis of Tamiflu.3
Among these efforts, Corey developed a chiral oxazabor- olidinium-catalyzed asymmetric Diels—Alder method for the synthesis of a chiral cyclohexene 7, which then underwent several transformations, including iodo-lactamization and N- acyl aziridine ring opening, to furnish 1 (Scheme 2b).3a Shibasaki reported an enantioselective Tamiflu synthesis that involves an asymmetric ring opening of a meso N-acyl aziridine 8 to provide the key trans-amino azide intermediate 9 (Scheme 2c).3 During development of several generations of synthetic strategies, he also capitalized on a barium-catalyzed asymmetric Diels—Alder reaction and a stereospecific Curtius rearrangement of a trans-diacyl azide intermediate 10 to afford the key oxazolidinone intermediate 11, which was later converted to 1 (Scheme 2c).3d
In Fukuyama’s Tamiflu synthesis,3e an organo-catalytic acrolein Diels—Alder reaction was applied to assemble a chiral 2-azabicyclo[2.2.2]octene building block 12 (Scheme 2d). Furthermore, a palladium-catalyzed asymmetric allylic alkylation and a rhodium-catalyzed dienoate aziridination were used to prepare a key intermediate 13 in Trost’s synthesis of Tamiflu (Scheme 2e).3f Moreover, Hayashi developed an organo-catalytic conjugate addition of an α-alkyoxyaldehyde 14 with a nitro-olefin 15 in an efficient synthesis of 1 (Scheme 2f).3g,h Notably, Ma independently reported the asymmetric conjugate addition of 14 with (Z)-2-nitroethenamide 16 in another expedient synthesis of Tamiflu (Scheme 2g).3i
These existing Tamiflu syntheses have showcased the power of catalytic reactions in assembling stereochemically complex synthetic targets; however, a strategically unique synthetic approach has not been reported that can directly install the trans-diamino moiety within Tamiflu via stereoselective diamination or diazidation of a highly functionalized synthetic intermediate, such as 17 or 18 (Scheme 3). Implementation of this diazidation-diamination strategy will provide a mechanistically distinct approach for an efficient synthesis of Tamiflu in complement to the previous syntheses.
Scheme 3.

Retrosynthetic Analysis Based upon the Iron- Catalyzed Stereoselective Olefin Diazidation
In 2015, we reported an iron-catalyzed direct diazidation method for a broad range of olefins, in which an iron catalyst activates TMSN3 in the presence of bench-stable benziodoxole 19a4 to achieve direct olefin diazidation (Scheme 4a).5 This reaction occurs at room temperature, and it is effective for a wide variety of olefins, including those that are incompatible with the previously reported diazidation methods.6 Coupled with facile reduction, it readily provides an array of valuable vicinal primary diamines. Recently, we disclosed a second iron- catalyzed olefin diazidation method via ligand-promoted activation of bench-stable peroxyesters.7 In this method, nearly a stoichiometric amount of commercially available tert-butyl perbenzoate 20 and TMSN3 are sufficient for high-yielding diazidation of a wide variety of olefins and N-heterocycles (Scheme 4b).7 These and other concurrent olefin diazidation methods8 add useful tools in the repertoire of synthetic chemists.
Scheme 4.

Iron-Catalyzed Direct Olefin Diazidation Methods
Our initial attempts to directly apply these two methods under the standard conditions to a range of designed late-stage intermediates for Tamiflu synthesis were not successful. Most of these complex substrates, including 18, suffer from lack of reactivity, while functionalized 1,4-cyclohexadiene 17 readily undergoes aromatization.9 Guided by mechanistic analysis, we have improved these iron-catalyzed methods such that they become effective with these highly functionalized substrates. Herein, we disclose these new discoveries that have enabled the iron-catalyzed stereoselective diazidation of these previously challenging substrates and thereby facilitated a gram- scale, enantioselective synthesis of Tamiflu in short steps (Scheme 1b).
RESULTS AND DISCUSSION
To explore the feasibility of achieving an expedient Tamiflu synthesis via the iron-catalyzed stereoselective olefin diazida- tion, we initially chose a chiral 1,4-cyclohexadiene 17 as a possible substrate (Scheme 5). Since 17 readily underwent aromatization under the diazidation conditions,9 we further targeted a functionalized chiral cyclic allylic alcohol 18 as an alternative substrate and expected that facile elimination under mild conditions would unveil the key enoate moiety within Tamiflu (Scheme 3).
Scheme 5.

Enantioselective Synthesis of Highly Functionalized Cyclic Allylic Alcohols for the Iron- Catalyzed Olefin Diazidation
The enantioselective synthesis of both 1,4-cyclohexadiene 17 and cyclic allylic alcohol 18 has not been reported; however, Danishefsky’s pioneering studies of a Diels—Alder reaction between a siloxy diene 21a and a nitroacrylate 22 have laid the foundation for the synthesis of 18 as a racemate (Scheme 5).10a Since nitroacrylate 22 tends to decompose under the reaction conditions, we speculated that a more efficient cycloaddition could be achieved when 22 was gradually generated in situ from its precursor, bromo-3- nitropropanoate 23 (Scheme 5). As a promising lead, we discovered that solid NaOAc-3H2O effectively promotes the reaction between siloxy diene 21a and 23 at room temperature, which readily affords the Diels—Alder products 24a and 24b in a high combined yield, albeit with a low dr. Inspired by this observation, we further discovered that acetoxy diene 21b undergoes a highly endo-selective Diels—Alder reaction, delivering 25 as a single diastereomer. Notably, this reaction has been consistently scaled up to 30 g scale and 25 can be easily purified through recrystallization (Scheme 5).9
To search for an enantioselective variant of this transformation, we have explored a range of catalytic enantiose-lective strategies which prove less effective, presumably due to the high reactivity associated with nitroacrylate 22. Therefore, as an alternative strategy, we envision that an efficient kinetic resolution of 25 may provide both of its enantiomers with high enantiopurity.
Although the chemo- and enzymatic kinetic resolution of cyclic allylic alcohols have been precedented,11 the kinetic resolution of highly functionalized substrates with three contiguous stereogenic centers has not been reported. We thereby investigated an array of chemo- and enzymatic catalysts for the proposed kinetic resolution and discovered that Amano Lipase from Pseudomonas fluorescens is uniquely effective (Scheme 5): the highly enantioenriched product (—)-26 (>99% ee) was isolated in 48% yield, and the starting material (+)-25 was recovered in a good yield and excellent ee (44% yield, 98% ee). A single recrystallization affords (+)-25 essentially in its enantiopure form (40% yield, >99% ee).9
To our surprise, neither of the aforementioned iron- catalyzed diazidation methods was effective for the highly functionalized (+)-25 when it was directly applied under the standard reaction conditions (Scheme 6). In the benziodoxole-mediated diazidation,5 (+)-25 was fully recovered while benziodoxole 19a completely decomposed to o-iodobenzoic acid.9 Notably, both (+)-25 and tert-butyl perbenzoate 20 were largely recovered using the iron-catalyzed, peroxyester-based8 diazidation method.9
Scheme 6.

Initial Attempts for the Direct Diazidation of (+)-25 Using the Standard Iron-Catalyzed Diazidation Procedures
In order to significantly improve these iron-catalyzed methods such that they can become effective with (+)-25, we carried out detailed mechanistic analysis of both iron- catalyzed olefin diazidation reactions.5,7 The mechanistic studies5 have uncovered that TMSN3 may reversibly convert otherwise insoluble benziodoxole 19a to azidoiodinane 19b, and then to a transient iodine(III)-diazide species 19c, with which an iron catalyst may be oxidized to a high-valent iron- azide species that promotes the stepwise olefin diazidation (Scheme 7). A variety of experiments suggest that the iron- ligand complexes are involved in the second C-N3 bond forming step which is rate-determining.5
Scheme 7.

Mechanistic Analysis of the Iron-Catalyzed Olefin Diazidation Using Benziodoxole 19a
Furthermore, we observed that, in the absence of an olefin, an iron catalyst completely decomposes benziodoxole 19a together with TMSN3 (Scheme 7), while 19a is stable toward TMSN3 without an iron catalyst (Scheme 7).8 These results suggest that competing reaction pathways do exist and they presumably proceed through the iron-promoted nonproductive decomposition of iodine(III)-diazide species 19c (Scheme 7).8 These competing pathways may become particularly detrimental for electronically deactivated substrates that are less reactive.
Based upon this analysis, we envision that the rate of olefin diazidation may have an order-dependence on both [iron catalyst] and [olefin] since the first C—N3 bond forming step is reversible and the second C—N3 bond forming step is rate- limiting (Scheme 7). Furthermore, the rate of iron-mediated nonproductive decomposition of 19c is likely dependent on both [19c] and [iron catalyst] (Scheme 7). Therefore, we suspect that an effective diazidation of (+)-25 may be achieved if a high concentration of (+)-25 and a low concentration of 19c can be maintained at the same time through the reaction such that nonproductive decomposition of 19c can be largely suppressed. Built upon this mechanistic proposal, we have drastically modulated the previously reported method to increase the concentration of (+)-25 up to 0.8 M and to decrease the concentration of TMSN3 through slow addition (up to 8 h, Scheme 8).
Scheme 8.

Iron-Catalyzed Stereoselective Diazidation of (+)-25 for the Expedient Synthesis of 3 2TsOH
We discovered that the Fe(OAc)2-L1 catalyst (5 mol %) effectively promotes the stereoselective diazidation of (+)-25 with a good combined yield and excellent dr (dr 7.4:1). Notably, an array of control experiments revealed that deviation from the newly discovered condition leads to an incomplete reaction. The desired trans,trans-diazide 27a can be readily separated from the cis,trans-diazide 27b through either recrystallization or flash-column chromatography. Their structures were initially assigned with 2D-NMR experiments and later corroborated by X-ray crystallographic analysis of 27a (Figure 1).9 A straightforward hydrolysis—elimination procedure converts 27a to trans,trans-diazido alcohol 28, which can be easily converted to trans,trans-hydroxyl diaminium tosylates 3 2TsOH via a standard reduction—protonation procedure (Scheme 8).
Figure 1.

X-ray crystallographic analysis of diazide 27a.
Organic azides, especially those with lower molecular weights, may present potential safety concerns for their handling, with regard to their thermo- and mechanical impact stabilities.12 In order to explore the feasibility of the iron-catalyzed olefin diazidation for Tamiflu production on a larger scale, it is imperative to carry out chemical hazard assessment of this olefin diazidation process. A reactive chemical hazard assessment refers the identification and possibly quantification of dangerous energy release scenarios for a chemical process of interest. Differential Scanning Calorimetry (DSC) is one of the most commonly applied thermal stability testing methods for organic compounds, while the Drop Weight Test (DWT) has been routinely applied to detect the sensitivity of a chemical toward mechanical impact. In 2017, we reported a process safety assessment of the iron-catalyzed olefin diazidation using benziodoxole 19a.13 DSC analysis of the corresponding reagents, intermediates presented in sufficient concentrations, and a list of representative diazide products revealed that all of them are thermal stable at the reaction temperature.13 Based upon these results, we carried out DSC analysis of 27a and observed that it does not decompose until 189 °C,9 which allows for a convenient operating margin in carrying out the diazidation at room temperature. Most notably, the diazide 27a is insensitive to mechanical impact during DWT studies. Encouraged by these results, this iron-catalyzed diazidation has been consistently scaled up to 5 g scale without compromising the yield and dr of the product (Scheme 8).9
The observed promising stereoselectivity at both C3 (dr 7.4:1) and C4 (dr >20:1) positions is mechanistically interesting. During the diazidation (Scheme 9), we envision that, based upon electronic effect, β-azido-C3 carboradical 29a, a putative intermediate reversibly generated from (+)-25, is likely more reactive than β-azido-C4 carboradical 29b toward the rate-determining oxidative C—N3 bond formation; there-fore, we suspect that the dr at C3 may be further improved through structural modulation of both iron catalysts and substrates.
Scheme 9.

Proposed Reversible Azido-Radical Addition Step during the Diazidation of (+)-25
Extensive explorations of a range of iron catalysts and ligands for the diazidation of (+)-25 provided modest improvement over the dr: notably, the Fe(NTf2)2—ligand complexes induced an even lower dr at the C3 position (dr 4.8:1).9 We thereby investigated the possibility of achieving enhanced stereo-selectivity via substrate control.
The diazidation of a 3-pentyl-substituted allylic ether 30 was first evaluated since it could lead to a more streamlined Tamiflu synthesis (Scheme 10). We observed that the Fe(OAc)2-L1 catalyst promotes an efficient diazidation of 30, albeit with a low dr at C3 (dr: 2.2:1). Considering the β-branch effect that might be associated with 30, we further explored the reaction with a TMS-protected allyl silyl ether 32; however, a modest level of dr was again observed (dr: 1.7:1). To our surprise, the iron catalyst promotes a highly stereoselective diazidation with an unprotected allylic alcohol (+)-26, affording a trans,trans-diazido alcohol 34 and a small amount of its TMS-protected ether 33a with excellent dr (dr > 20:1 at both C3 and C4 positions in 33a and 34). It is worth noting that (+)-26 is evidently more reactive than (+)-25 and acid workup readily converts 33a back to 34. Since TMSN3 is gradually released under the diazidation condition, 33a is likely derived from 34 by the residue TMSN3 and it is unlikely the diazidation product from 32.
Scheme 10.

Substrate Structure-Stereoselectivity Relationship Studies for the Iron-Catalyzed Olefin Diazidation
The excellent stereoselectivity achieved with (+)-26 urged us to further evaluate 24b and 35, two exo-Diels-Alder products, for the diazidation (Scheme 10). Interestingly, both 24b and 35 present excellent reactivity and stereoselectivity, affording either a trans,cis-diazido silyl ether 36 or an alcohol 37 as a single diastereomer (dr >20:1 at both the C3 and C4 positions in 36 and 37). Again, a small amount of TMS- protected allyl silyl ether 38 was also obtained (dr >20:1).
These observations evidently suggest that both iron catalysts and substrates cooperatively influence the stereoselectivity of the diazidation. In order to propose a plausible stereochemical model, the possible structure of a catalytically active iron complex needs to be considered. Our mechanistic studies have revealed that the Fe(OAc)2-L1 complex readily reacts with TMSN3, furnishing an iron-azide complex 39 (Scheme 11).14 IR analysis of 39 uncovered strong azido group absorptions (2047 and 2060 cm−1) shifted to lower energy in comparison to free azide, characteristic of iron—azide complexes.15 Subsequent X-ray crystallographic analysis of 39 revealed a unique iron coordination polymer with all iron centers equivalent and generated by symmetry—(Fe(L1)(N3)2)n (Scheme 11).14 In addition to the rigid tridentate ligand L1, three remaining coordination sites of the iron center are occupied by three azides with one being terminal and the other two azides cis to each other bridging adjacent iron centers to form the coordination polymer.14 Importantly, this polymeric iron catalyst 39 is catalytic active and it catalyzes the diazidation of (+)-26 with essentially the same yield and dr (Scheme 11). Surprisingly, 39 can also promote diazidation of (+)-26 with 19b in the absence of TMSN3, albeit with a low yield (Scheme 11, 17% yield, dr >20:1). Therefore, it is likely that iron—azide complex 39 becomes oxidized to a high-valent iron-azide species that facilitates the olefin diazidation and that the terminal iron-azide bond in 39 may be involved in the rate- limiting azido-group transfer.
Scheme 11.

Catalyst Structure—Reactivity Relationship Studies of the Polymeric Iron—Azide Complex (Fe(L1)(N3)2)n
Based upon these catalyst structure-reactivity relationship studies, a plausible stereochemical model that fits the observations is presented in Scheme 12. We envision that the endo-Diels—Alder product 40 can adopt either of the two conformations (40a and 40b) that may be in rapid equilibrium. Since the azido-radical addition to 40 is reversible and it likely occurs at the C4 position from the axial trajectory in order to avoid a twist-boat conformation (Scheme 9),16 β- azido carboradical species 42a and 42b may be two reactive intermediates in equilibrium. In the subsequent rate-determining C—N3 bond forming step, the bulky iron(III) azide species derived from 39 may oxidize 42a or 42b through direct azido-ligand transfer from the iron center.5,7 Considering the significant unfavorable 1,3-diaxial interactions that may build up in 42b (between the iron-azide complex and the CO2Et or NO2 group) along the reaction trajectory, it is less likely that this oxidation occurs with 42b from either the α or β face.
Scheme 12.

Proposed Stereochemical Model for the Iron- Catalyzed Olefin Diazidation of Cyclic Allylic Alcohols
However, the azido-group transfer may occur with 42a in which these unfavorable interactions no longer exist. If R is a less sterically demanding group, such as hydrogen or an acetyl group, the iron complex may readily deliver the azido-group to 42a from the α face.16 The axial hydroxyl group may also direct the iron catalyst to achieve enhanced α-selectivity. Alternatively, when R becomes more sterically demanding, the iron complex may thereby be forced to deliver the azido-group from the β face. Therefore, excellent stereoselectivity can be achieved with an unprotected allylic alcohol (+)-26 using a bulky polymeric iron-azide catalyst (Fe(L1)(N3)2)n 39.
This proposed model can also rationalize the excellent stereoselectivity observed with exo-Diels-Alder product 44 (Scheme 12). Locked in a conformation where the OR, CO2Et, and NO2 groups all reside in equatorial positions, the polymeric iron(III)-azide intermediate should be able to approach the β-azido carboradical species 45 from its α face regardless of the R substituent.
With the success of this improved diazidation method using benziodoxole 19a, we further explored the possibility of developing a new peroxyester-based diazidation approach for the synthesis of trans,trans-diazido alcohol 28. Under the standard peroxyester-based diazidation conditions, both (+)-25 and tert-butyl perbenzoate 20 were largely recovered (Scheme 6), which suggests that the energy barrier of the rate-determining step may be too high for this electronically deactivated substrate.
Based upon our mechanistic studies,7 we envision that the Fe(NTf2)2-ligand complex may reductively cleave the O—O bond in a peroxyester 20 to generate a tert-butoxyl radical that is associated with a high-valent iron complex 47 (Scheme 13a). ‘iPrOH presumably facilitates gradual release of HN3 from TMSN3, and the tert-butoxyl radical may thereby be rapidly sequestered by HN3 to liberate the azido radical. The azido radical may reversibly add to an olefin to afford carbo-radical species 48 and TMSN3 may also convert the iron(III) species 47 to a high-valent iron—azide species 49, which presumably mediates the rate-determining azido-group transfer to the carbo-radical species 48 and afford the diazidation product.7
Scheme 13.

(a) Mechanistic Working Hypothesis of the Fe(NTf2)2-Catalyzed Olefin Diazidation Using Peroxyesters and (b) Iron-Catalyzed Peroxyester Activation for Diazidation of (+)-25
Since the O-O bond cleavage is unlikely rate-determining, we suspect that a more electron-deficient iron(III) species 49 may accelerate the rate-determining C—N3 bond forming step. Therefore, we evaluated peroxyester 50 with a more-electron- withdrawing acyl group and observed that the Fe(NTf2)2— racemic L2 catalyst promotes an efficient diazidation with (+)-25, affording 27 in an excellent yield albeit moderate dr (Scheme 13b). It is worth noting that no significant match/ mismatch effect was observed using the enantiopure L2 ligand9 and that a high concentration of TMSN3 is necessary for the reactivity. Further evaluation of the unprotected allylic alcohol (+)-26 under the newly discovered condition furnished decreased stereoselectivity, presumably due to the readily conversion of (+)-26 to its TMS-protected ether 32 in situ.
The gram-scale preparation of trans,trans-diazido alcohol 28 in short steps and high enantiopurity have allowed us to explore the selective incorporation of both 3-pentyl and acetyl groups for a short Tamiflu synthesis (Scheme 14). Although a list of standard 3-pentyl-derived electrophiles are unreactive toward this diazido cyclic allylic alcohol 28, alkylation with trichloroacetimidate 51 proves uniquely effective under acidic conditions.17
Scheme 14.

Expedient Tamiflu Synthesis from 3 2TsOH
We observed that MsOH promotes the difficult alkylation of 28 to afford 52, albeit in a low yield (Scheme 14); however, 3- pentylmesylate 53, generated in situ, is an ineffective electrophile toward 28. We subsequently discovered that a catalytic amount of TfOH promotes the alkylation of diazido allylic alcohol 28; however, a small amount of regioisomeric 2- pentyl-alkylation product 54 was formed as an inseparable mixture with 52.9 We suspected that 54 may be generated from 52 via TfOH-catalyzed rearrangement, which was confirmed by a subsequent experiment (Scheme 14).
Given the pitfalls of alkylation with diazido alcohol 28, we further evaluated bis-carbamates that are more nucleophilic. Straightforward reduction of 28 and N-Boc protection affords 55, which demonstrates excellent reactivity in the acid-catalyzed alkylation with trichloroacetimidate 51; however, N- Boc groups surprisingly participate in the alkylation as well (Scheme 14). Fortunately, bis-methyl carbamate 57 can be engaged in this alkylation and it was converted to 58 in an excellent yield (Scheme 14). White crystalline solid 58 can be further converted to 59, the penultimate synthetic target, via a gram-scale procedure that involves TMSCl-NaI-mediated carbamate deprotection18 and selective N-acylation19 of both Boc and Ac groups. Subsequently, N-Boc deprotection of 59 using H3PO4 in hot EtOH readily affords Tamiflu 1 (Scheme 14).
CONCLUSIONS
In conclusion, we have reported a gram-scale, enantioselective Tamiflu synthesis, in which the key trans-diamino moiety within Tamiflu has been efficiently installed via an iron- catalyzed stereoselective olefin diazidation (Scheme 15). This improved, iron-catalyzed method is effective for highly functionalized yet electronically deactivated substrates that have been otherwise problematic. Preliminary catalyst structure-reactivity-stereoselectivity relationship studies revealed that both the iron catalyst and the complex substrate cooperatively modulate the stereoselectivity for diazidation. Most notably, an oligomeric iron-azide catalyst proves uniquely effective for the stereoselective diazidation. Process safety assessment using both differential scanning calorimetry. (DSC) and the drop weight test (DWT) has also demonstrated the feasibility of carrying out this iron-catalyzed olefin diazidation for large-scale Tamiflu synthesis.
Scheme 15.

Summary of the Enantioselective Synthesis of Tamiflu via the Iron-Catalyzed Stereoselective Olefin Diazidation
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Institutes of Health (GM110382). We thank Luca Arosio, Dr. Roberto Villa, and Professor Marino Nebuloni for the DSC and DWT safety assessment of compound 27a. We thank Peijing Jia and Naixin Qian for their assistance in lipase-catalyzed kinetic resolution of 25. P.J. and N.Q. were supported by a Li-Yun Summer Undergraduate Research Scholarship.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b06900.
Experimental procedure, characterization data for all new compounds, selected NMR spectra and HPLC traces (PDF)
Crystallographic data for (±)-27a (CIF)
The authors declare the following competing financial interest(s): The subject matter described in this article is included in patent applications filed by Georgia State University.
REFERENCES
- (1).(a) Kim CU; Lew W; Williams MA; Liu H; Zhang L; Swaminathan S; Bischofberger N; Chen MS; Mendel DB; Tai CY; Laver WG; Stevens RC J. Am. Chem. Soc 1997, 119, 681. [DOI] [PubMed] [Google Scholar]; (b) Hajzer V; Fisera R; Latika A; Durmis J; Kollar J; Frecer V; Tucekova Z; Miertus S; Kostolansky F; Vareckova E; Sebesta R Org. Biomol. Chem 2017, 15, 1828. [DOI] [PubMed] [Google Scholar]
- (2).(a) Rohloff JC; Kent KM; Postich MJ; Becker MW; Chapman HH; Kelly DE; Lew W; Louie MS; McGee LR; Prisbe EJ; Schultze LM; Yu RH; Zhang L J. Org. Chem 1998, 63, 4545. [Google Scholar]; (b) Federspiel M; Fischer R; Hennig M; Mair H-J; Oberhauser T; Rimmler G; Albiez T; Bruhin J; Estermann H; Gandert C; Göckel V; Götzö S; Hoffmann U; Huber G; Janatsch G; Lauper S; Röckel-Stöbler O; Trussardi R; Zwahlen AG Org. Process Res. Dev 1999, 3, 266. [Google Scholar]; (c) Karpf M; Trussardi R J. Org. Chem 2001, 66, 2044. [DOI] [PubMed] [Google Scholar]; (d) Harrington PJ; Brown JD; Foderaro T; Hughes RC Org. Process Res. Dev 2004, 8, 86. [Google Scholar]; (e) Karpf M; Trussardi R Angew. Chem. Int. Ed. 2009, 48, 5760. [DOI] [PubMed] [Google Scholar]
- (3).(a) Yeung YY; Hong S; Corey EJ J. Am. Chem. Soc 2006, 128, 6310. [DOI] [PubMed] [Google Scholar]; (b) Fukuta Y; Mita T; Fukuda N; Kanai M; Shibasaki MJ Am. Chem. Soc 2006, 128, 6312. [DOI] [PubMed] [Google Scholar]; (c) Mita T; Fukuda N; Roca FX; Kanai M; Shibasaki M Org. Lett 2007, 9, 259. [DOI] [PubMed] [Google Scholar]; (d) Yamatsugu K; Yin L; Kamijo S; Kimura Y; Kanai M; Shibasaki M Angew. Chem., Int. Ed 2009, 48, 1070. [DOI] [PubMed] [Google Scholar]; (e) Satoh N; Akiba T; Yokoshima S; Fukuyama T Angew. Chem. Int. Ed 2007, 46, 5734. [DOI] [PubMed] [Google Scholar]; (f) Trost BM; Zhang T Angew. Chem. Int. Ed 2008, 47, 3759. [DOI] [PubMed] [Google Scholar]; (g) Ishikawa H; Suzuki T; Hayashi Y Angew. Chem., Int. Ed 2009, 48, 1304. [DOI] [PubMed] [Google Scholar]; (h) Hayashi Y; Ogasawara S Org. Lett 2016, 18, 3426. [DOI] [PubMed] [Google Scholar]; (i) Zhu S; Yu S; Wang Y; Ma D Angew. Chem., Int. Ed 2010,49, 4656. [DOI] [PubMed] [Google Scholar]; (j) Shie J-J; Fang J-M; Wang S-Y; Tsai K-C; Cheng Y-SE; Yang A-S; Hsiao S-C; Su C-Y; Wong C-H J. Am. Chem. Soc 2007, 129, 11892. [DOI] [PubMed] [Google Scholar]; (k) Sullivan B; Carrera I; Drouin M; Hudlicky T Angew. Chem., Int. Ed 2009, 48, 4229. [DOI] [PubMed] [Google Scholar]; (l) Bromfield KM; Graden H; Hagberg DP; Olsson T; Kann N Chem. Commun 2007, 3183. [DOI] [PubMed] [Google Scholar]; (m) Matveenko M; Willis AC; Banwell MG Tetrahedron Lett. 2008, 49, 7018. [Google Scholar]; (n) Mandai T; Oshitari T Synlett 2009, 2009, 783. [Google Scholar]; (o) Osato H; Jones IL; Chen A; Chai CL L. Org. Lett 2010, 12, 60. [DOI] [PubMed] [Google Scholar]; (p) Zutter U; Iding H; Spurr P; Wirz B J. Org. Chem 2008, 73, 4895 See also: [DOI] [PubMed] [Google Scholar]; (q) Cong X; Yao Z-JJ Org. Chem 2006, 71, 5365. [DOI] [PubMed] [Google Scholar]; (r) Nie L-D; Shi X-X; Ko KH; Lu W-D J. Org. Chem 2009, 74, 3970. [DOI] [PubMed] [Google Scholar]; (s) Weng J; Li Y-B; Wang RB; Li F-Q; Liu C; Chan ASC; Lu GJ Org. Chem 2010, 75, 3125. [DOI] [PubMed] [Google Scholar]; (t) Rawat V; Dey S; Sudalai A Org. Biomol Chem 2012, 10, 3988. [DOI] [PubMed] [Google Scholar]; (u) Chen CA; Fang JM Org. Biomol. Chem 2013, 11, 7687. [DOI] [PubMed] [Google Scholar]
- (4).Zhdankin VV; Krasutsky AP; Kuehl CJ; Simonsen AJ; Woodward JK; Mismash B; Bolz JT J.Am. Chem. Soc 1996, 118, 5192. [Google Scholar]
- (5).Yuan Y-A; Lu D-F; Chen Y-R; Xu H Angew. Chem., Int. Ed 2016, 55, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Minisci F; Galli R Tetrahedron Lett. 1962, 3, 533. [Google Scholar]; (b) Fristad WE; Brandvold TA; Peterson JR; Thompson SR J. Org. Chem 1985, 50, 3647. [Google Scholar]; (c) Moriarty RM; Khosrowshahi JS Tetrahedron Lett. 1986, 27, 2809. [Google Scholar]; (d) Arimoto M; Yamaguchi H; Fujita E; Nagao Y; Ochiai M Chem. Pharm. Bull 1989, 37, 3221. [Google Scholar]; (e) Magnus P; Lacour J J. Am. Chem. Soc 1992, 114, 767. [Google Scholar]; (f) Chung R; Yu E; Incarvito CD; Austin DJ Org. Lett 2004, 6, 3881. [DOI] [PubMed] [Google Scholar]
- (7).Shen S-J; Zhu C-L; Lu D-F; Xu H ACS Catal 2018, 8, 4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Fu N; Sauer GS; Saha A; Loo A; Lin S Science 2017, 357, 575. [DOI] [PubMed] [Google Scholar]; (b) Peng H; Yuan Z; Chen P; Liu G Chin. J. Chem 2017, 35, 876. [Google Scholar]; (c) Zhou H; Jian W; Qian B; Ye C; Li D; Zhou J; Bao H Org. Lett 2017, 19, 6120 For recent reviews for olefin azidation, see: [DOI] [PubMed] [Google Scholar]; (d) Wu K; Liang Y; Jiao N Molecules 2016, 21, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sauer GS; Lin S ACS Catal. 2018, 8, 5175. [Google Scholar]
- (9).For experimental details, see Supporting Information.
- (10).(a) Danishefsky S; Prisbylla MP; Hiner S J. Am. Chem. Soc 1978, 100, 2918 see also:. [Google Scholar]; (b) Stoodley RJ; Yuen W-H Chem. Commun 1997, 1371. [Google Scholar]
- (11).(a) Martin VS; Woodard SS; Katsuki T; Yamada Y; Ikeda M; Sharpless KB J. Am. Chem. Soc 1981, 103, 6237. [Google Scholar]; (b) Kitamura M; Kasahara I; Manabe K; Noyori R; Takaya H J. Org. Chem 1988, 53, 708. [Google Scholar]; (c) Lüssem BJ; Gais H-JJ Am. Chem. Soc 2003, 125, 6066. [DOI] [PubMed] [Google Scholar]; (d) Lihammar R; Millet R; Bäckvall J-E J.Org. Chem. 2013, 78, 12114 For an enzymatic kinetic resolution to obtain enantio- enriched acyclic allylic alcohols, see: [DOI] [PubMed] [Google Scholar]; (e) Burgess K; Jennings LD J. Am. Chem. Soc. 1991, 113, 6129. [Google Scholar]
- (12).Bräse S; Gil C; Knepper K; Zimmermann V Angew. Chem. Int. Ed 2005, 44, 5188. [DOI] [PubMed] [Google Scholar]
- (13).Zhu H-T; Arosio L; Villa R; Nebuloni M; Xu H Org. Process Res. Dev 2017, 21, 2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhu C-L; Wang C; Qin Q-X; Yruegas S; Martin CD; Xu H ACS Catal 2018, 8, 5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).For a selected reference of characterized monomeric iron-azide complexes and their IR measurements, see: Grove LE; Hallman JK; Emerson JP; Halfen JA; Brunold TC Inorg. Chem 2008, 47, 5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Rondan NG; Paddon-Row MN; Caramella P; Houk KN J. Am. Chem. Soc 1981, 103, 2436. [Google Scholar]; (b) Paddon-Row MN; Rondan NG; Houk KN J. Am. Chem. Soc 1982, 104, 7162. [Google Scholar]
- (17).(a) Overman LE J. Am. Chem. Soc 1976, 98, 2901. [Google Scholar]; (b) Schmidt RR; Michel J Angew. Chem., Int. Ed 1980, 19, 731. [Google Scholar]; (c) Zhang Q; Stockdale DP; Mixdorf JC; Topczewski JJ; Nguyen HM J. Am. Chem. Soc 2015, 137, 11912. [DOI] [PubMed] [Google Scholar]
- (18).Jung ME; Lyster MA J. Chem. Soc., Chem. Commun 1978, 315. [Google Scholar]
- (19).Konno F; Arai T; Zhang M-R; Hatori A; Yanamoto K; Ogawa M; Ito G; Odawara C; Yamasaki T; Kato K; Suzuki K Bioorg. Med. Chem. Lett 2008, 18, 1260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.