

Huntington's disease (HD) is an autosomal dominant, monogenic, progressive, neurodegenerative and rare disease with a frequency of 10 per 100,000 in the Caucasian population and occurring more rarely in other races (Squitieri et al., 1994). HD is, nevertheless, one of the most frequently and extensively studied diseases of those caused by a dynamic mutation. The HD mutation is located on the short arm of the 4th chromosome within the HTT gene. This mutation consists of cytidine, adenosine and guanosine (…CAG CAG CAG…), namely trinucleotide repetition exceeding 35 repeats, with a higher CAG expansion leading to disease onset at a younger age (HSG, 1993). It also results in a stronger manifestation of HD symptoms and faster clinical progression. Although the mutation is present in each cell from conception, HD symptoms usually appear between 30 and 50 years of age (HSG, 1993). HD symptoms were first precisely described by George Huntington in a paper titled “On Chorea”, published in the Medical and Surgical Reporter in 1872. Huntington was the descendant of 2 generations of physicians who had been treating HD patients living in the vicinity of East Hampton on Long Island, USA. Earlier, HD had been known as St. Vitus’ Dance; in Huntington's times it was called chorea. The term “Huntington's chorea” is still used today but was recalled as a “Huntington's disease” when other symptoms were identified, including other movement disturbances such as dystonia, stiffness (most prevalent in later stages), oculomotor disturbances, slowness, psychiatric symptoms such as irritability, aggression, apathy and depression, obsessive-compulsive disorders or impulsive suicidal attempts, cognitive disturbances (subcortical dementia), e.g., thinking stiffness and slowness, and attention and executive function disturbances. Both the sequence and intensity of symptoms constitute unique features of every patient, although diagnosis can be established when typical motor symptoms appear. Psychiatric symptoms or isolated cognitive disturbances might preclude HD diagnosis for many years (Martinez-Horta et al., 2016) There are also specific HD presentations such as juvenile HD with onset age before 20 years of age, faster disease progression with frequent symptoms such as stiffness and severe oculomotor disturbances (Westphal variant), or senile HD that usually appears after 60 years of age with a display of predominantly choreic movements. Although the CAG repeats number might indicate the severity of symptoms as well their progression with age, additional modifying factors play a crucial role and often might result in a shift of disease onset for a few or even over a dozen years (Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium, 2015).
There is no known cure or modifying therapy for HD, therefore currently only symptomatic therapy is available (Zielonka et al., 2015). Thus there is an important need to conduct new clinical trials of novel medicines for symptomatic HD therapy. One crucial element to fulfill this purpose is the correct tool for clinical measurements. Called the Unified Huntington's Disease Rating Scale (UHDRS), this tool was formed in 1996 (HSG, 1996). Another element of clinical care is to investigate the natural HD course along with course modifiers and their mechanisms of action. A complex characterization of factors contributing to clinical phenomenology, HD progression, health status and HD patient behavior that might be implemented in symptomatic therapies constitutes an important aim for research from this perspective.
In 2002 it was found that there is a significant correlation (P = 0.013) between the number of CAG repeats and the progression rate of motor and cognitive symptoms in HD (Zielonka et al., 2002). This observation was further confirmed in a large cohort study published by another team in 2008 (Ravina et al., 2008). Subsequent, and important, findings from 2008 showed that gender contributes to the clinical HD picture, namely that HD progression differs in women and men (Zielonka et al., 2008). The plausibility of these findings seemed to be low at the beginning, as gender contribution to this autosomal disorder was unexpected, yet the results of the study indicated that there is a statistically significant correlation between the number of CAG repeats in exon 1 of the HTT gene and scores in UHDRS motor (P = 0.0016), cognitive (P = 0.03), functional (P = 0.001), independence (P = 0.004) and total functional capacity (TFC) (P = 0.002) in 23 women out of 41 subjects. This correlation was not present in men; moreover, these scores correlated with disease duration in women. Because the data were obtained based on a small group of subjects, it was important to confirm these observations in a larger cohort. It was also important to answer the question as to what kind of modifying factors might contribute to the above phenomena. Such an extended analysis was published in 2013 (Zielonka et al., 2013).
The 2013 study was conducted based on a much larger cohort, and it was possible due to collaboration with colleagues from other countries (HD centers) across European Huntington's Disease Network (EHDN). In this study, data from 1267 eligible subjects were collected to analyze the modifying factors that might contribute to HD progression. The enrolled HD patients displayed a (classic) presentation of HD, i.e., motor manifestation. The explanatory variables for gender differences in the clinical picture and HD progression were the following: years of education, presence of depression, depressive episodes in the past, history of psychotic disturbances, history of obsessive-compulsive disorders, history of suicidal ideation, nicotine poisoning, alcohol abuse and drug usage. The following were also considered: symptoms at onset, onset age, disease duration and “disease burden” (DB), calculated as follows: DB = (number of CAG repeats in the larger allele – 35.5) × age in years. DB reflects the stage of brain pathology in HD (Penney et al., 1997). Disease progression was assessed using UHDRS.
Statistically significant differences were found in: years of education (men studied longer) (P = 0.01), presence of depression (more prevalent in women) (P = 0.03), history of depression (more prevalent in women) (P < 0.001), alcohol abuse (more prevalent in men) (P = 0.001), and nicotine poisoning (more prevalent in men) (P = 0.004). All of the above variables were included as confounders in the gender differences analysis conducted in a cross-sectional and longitudinal manner, yet they did not explain the differences observed in the cross-sectional analysis of the UHDRS motor, functional and TFC scores and in the longitudinal analysis of UHDRS motor, functional, independence and TFC scores. It was therefore established that HD displays faster progression in women than in men when measured in the same period from onset and after one year of observation. The lack of the above confounders’ contribution (after preliminary screening for a large number of potential modifiers) suggested that gender factors should be considered as crucial for this difference and emphasized the importance of these factors, thus indicating that gender differences should be considered in new clinical trials.
Although the above study emphasized the differences between genders in HD progression that were taken into account for new clinical trials, it was still important to estimate the contribution of particular HD symptoms in the independence and functional abilities of HD patents for both genders separately and comparatively, for which another study was needed.
Studies conducted throughout the world partially addressed the contribution of certain HD symptoms to the independence and function of patients, but this contribution was never analyzed comprehensively and comparatively. The lack of such an analysis made it impossible to target an efficient therapy for function and quality of life (QoL) improvement. The study (Zielonka et al., 2018) was conducted on 2191 HD affected subjects (1080 women). Subjects enrolled in the study displayed motor symptoms (mostly in middle-advanced HD stages) to ensure that individuals could benefit the most from potential symptomatic therapy. The aim of the study was to assess and compare the contribution of 1) motor symptoms measured in the UHDRS motor, 2) cognitive symptoms measured in UHDRS cognitive, 3) behavioral disturbances measured in UHDRS behavioral to independence and functional abilities measured in 1) UHDRS functional, 2) independence and 3) TFC. This contribution was compared between genders. Moreover, in 1166 subjects of the investigated cohort, function was measured in TFC and was correlated with QoL as measured in 36-Item Short Form Survey Instrument (SF-36). The authors intended to answer the question as to how QoL (physical and mental components of the SF-36) changed within the TFC stage.
The results of the above study confirmed that all of the analyzed symptoms, or rather groups of symptoms, correlated significantly (P < 0.0001) with independence and functional abilities in both women and men. Based on the whole cohort analysis, motor symptoms contributed (correlated) the strongest, followed by cognitive and then behavioral. In women, however, UHDRS motor the correlations with the function and independence were significantly stronger than in men (UHDRS functional scale: P < 0.001; Independence scale: P = 0.003; TFC: P = 0.002). Moreover, linear regression showed that motor symptoms were responsible for more variability in functional abilities in women than in men, while cognitive symptoms had an opposite contribution (more in men than in women). This means that motor symptoms were the strongest contributors to functional abilities in both genders, particularly in women, although cognitive symptoms are more important in men for functional abilities than in women. Psychiatric symptoms are less important for functional abilities and independence in both women and men. The next step of analysis tested how functional abilities measured in the commonly used TFC scale contributed to QoL measured in the SF-36. A statistically significant correlation (P < 0.01) was found between functional deterioration and lower QoL. Based on the TFC scale, a statistically significant relation (P < 0.001) was found between stage worsening and the SF-36 total score (Figure 1), in the physical component but not in the mental component. In general, the study allowed to target future clinical trials at precisely defined factors that worsen both function and QoL in HD patients.
Figure 1.
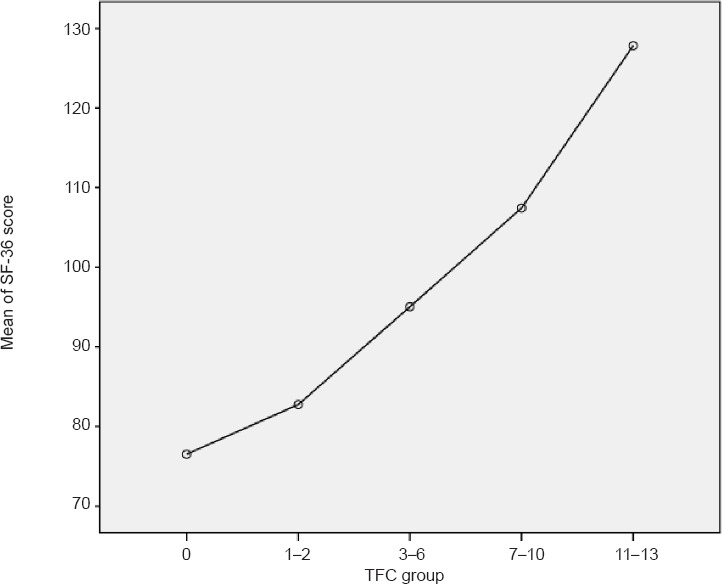
Relation between total functional capacity (TFC) based stage of Huntington's disease (HD) and 36-Item Short Form Survey Instrument (SF-36) global score in all study's participants.
TFC group reflect stage of HD where 11–13 TFC scores reflect stage I, 7–10 – stage II, 3–6 – stage 3, 1–2 stage IV and 0 stage V.
In summary, diseases conditioned by an autosomal mutation are usually considered gender-independent, therefore the role of gender in these disorders has not been emphasized. HD is an unusual disease with regard to gender role in its clinical phenomenology and disease progression, and gender role was observed in humans in 2008 and emphasized in follow-up studies. This review presents the current state of knowledge in this field. Long-term HD studies as described above resulted in findings of several important clinical features that have allowed for the implementation of new symptomatic and course-modifying therapies in HD. The above-mentioned studies cover almost all factors that are potentially able to modify HD clinical phenomenology and progression besides the potential contribution of HD concomitant disorders. The most important finding is gender contribution in HD assessment and symptoms contribution to functional abilities, patients’ independence and QoL. Future exploratory studies in this field should concern the role of concomitant disorders shaping the HD clinical picture and inter-gender differences. The role of medications as used by patients, instead of reducing symptoms only, should be analyzed as clinical status confounders in the moment of examination, moreover some of them could modify HD course, but this has not been investigated so far. Besides the above-mentioned external factors, recent studies have highlighted the meaning of HD course modifiers on chromosomes 15, 8 and probably 3 as well as the role of tissue mosaicism. A summary of all these potential contributors could either explain or underline the meaning of gender in the observed differences.
Footnotes
Copyright transfer agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
References
- Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington's disease. Cell. 2015;162:516–526. doi: 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Horta S, Perez-Perez J, van Duijn E, Fernandez-Bobadilla R, Carceller M, Pagonabarraga J, Pascual-Sedano B, Campolongo A, Ruiz-Idiago J, Sampedro F, Landwehrmeyer GB. Spanish REGISTRY investigators of the European Huntington's Disease Network, Kulisevsky J. Neuropsychiatric symptoms are very common in premanifest and early stage Huntington's disease. Parkinsonism Relat Disord. 2016;25:58–64. doi: 10.1016/j.parkreldis.2016.02.008. [DOI] [PubMed] [Google Scholar]
- HSG. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- HSG Unified Huntington's Disease Rating Scale: reliability and consistency. Huntington Study Group. Mov Disord. 1996;11:136–142. doi: 10.1002/mds.870110204. [DOI] [PubMed] [Google Scholar]
- Penney JB, Jr, Vonsattel JP, MacDonald ME, Gusella JF, Myers RH. CAG repeat number governs the development rate of pathology in Huntington's disease. Ann Neurol. 1997;41:689–692. doi: 10.1002/ana.410410521. [DOI] [PubMed] [Google Scholar]
- Ravina B, Romer M, Constantinescu R, Biglan K, Brocht A, Kieburtz K, Shoulson I, McDermott MP. The relationship between CAG repeat length and clinical progression in Huntington's disease. Mov Disord. 2008;23:1223–1227. doi: 10.1002/mds.21988. [DOI] [PubMed] [Google Scholar]
- Squitieri F, Andrew SE, Goldberg YP, Kremer B, Spence N, Zeisler J, Nichol K, Theilmann J, Greenberg J, Goto J, et al. DNA haplotype analysis of Huntington disease reveals clues to the origins and mechanisms of CAG expansion and reasons for geographic variations of prevalence. Hum Mol Genet. 1994;3:2103–2114. doi: 10.1093/hmg/3.12.2103. [DOI] [PubMed] [Google Scholar]
- Zielonka D, Mielcarek M, Landwehrmeyer GB. Update on Huntington's disease: advances in care and emerging therapeutic options. Parkinsonism Relat Disord. 2015;21:169–178. doi: 10.1016/j.parkreldis.2014.12.013. [DOI] [PubMed] [Google Scholar]
- Zielonka D, de Mezer M, Niezgoda A, Reperowicz K, Krzyzosiak W, Kozubski W. Clinical picture of patients with Huntington's disease in relation to the number of trinucleotide CAG repeats in IT-15 gene. Neurol Neurochir Pol. 2002;36:903–909. [PubMed] [Google Scholar]
- Zielonka D, Niezgoda A, Olejniczak M, Krzyzosiak W, Marcinkowski J, Kozubski W. Gender differences in the CAG repeats and clinical picture correlations in Huntington's disease. Cesk Neurol Neurochir. 2008;71:688–694. [Google Scholar]
- Zielonka D, Ren M, De Michele G, Roos RAC, Squitieri F, Bentivoglio AR, Marcinkowski JT, Landwehrmeyer GB. The contribution of gender differences in motor, behavioral and cognitive features to functional capacity, independence and quality of life in patients with Huntington's disease. Parkinsonism Relat Disord. 2018;49:42–47. doi: 10.1016/j.parkreldis.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Zielonka D, Marinus J, Roos RA, De Michele G, Di Donato S, Putter H, Marcinkowski J, Squitieri F, Bentivoglio AR, Landwehrmeyer GB. The influence of gender on phenotype and disease progression in patients with Huntington's disease. Parkinsonism Relat Disord. 2013;19:192–197. doi: 10.1016/j.parkreldis.2012.09.012. [DOI] [PubMed] [Google Scholar]