

Alzheimer's disease (AD), the predominant form of dementia, is a chronic, incurable neurodegenerative disorder presenting with symptoms including progressive memory loss and disturbed emotional state. It has been estimated that dementia affects over 47 million people worldwide (Prince et al., 2015), and with 60–80% of cases attributable to AD. The primary pathological hallmarks of AD are neuronal death, the deposition of insoluble amyloid-beta (Aβ) plaques, the accumulation of hyperphosphorylated tau neurofibrillary tangles (NFTs), and the widespread dysregulation of neurotransmitter signaling (Francis, 2005; Vinters, 2015). Although the aetiology of the disease is not clearly understood, a range of theories have been proposed over the last few decades. The amyloid hypothesis holds that the anomalous processing of the amyloid precursor protein (APP) to Aβ and its deposition as insoluble plaques is the primary mechanism underlying AD pathogenesis, while the tau hypothesis contends that the hyperphosphorylation of tau and NFT formation is central to the disease process (Mohandas et al., 2009). Others have proposed the contribution of neuroinflammatory pathways, vascular dysfunction, oxidative stress, mitochondrial dysfunction, changes in metal ion regulation, and abnormal insulin signaling (Mohandas et al., 2009). It is widely accepted that the excitatory glutamatergic and cholinergic systems are severely affected in AD, due to the significant loss of cells in these systems and the disruption of their molecular components (Francis, 2005). As a result, the excitatory/inhibitory (E/I) balance is disturbed in the AD brain, and this could well underlie the deficiencies in memory and learning that are characteristic of the condition. At present, all five drugs approved by the US Food and Drug Administration for the symptomatic treatment of AD are targeted towards these systems –including the acetylcholinesterase inhibitors donepezil, rivastigmine and galantamine, and the N-methyl-D-aspartate (NMDA) receptor antagonist memantine (Calvo-Flores Guzmán et al., 2018). However, these therapies do not address the underlying causes of the disease and there is an urgent need for the identification of novel therapeutic targets.
Gamma-aminobutyric acid (GABA) is the primary inhibitory neurotransmitter in the central nervous system. GABA is synthesized by glutamic acid decarboxylase (GAD) and is then recruited into synaptic vesicles. Following membrane depolarization, GABA is released into the synapse and can bind to either ionotropic GABAA receptors (GABAA Rs) or metabotropic GABAB receptors. Released GABA is cleared from the synapse by membrane-bound GABA transporters, localized to neurons and astrocytes. For a long time, it has been believed that GABA signaling is not particularly affected in AD, with some early studies demonstrating a sparing of GABAergic interneurons and others reporting inconsistent and often contradictory changes in GABA levels, GAD enzyme activity and GABA receptor expression in the AD brain. However, study design and case selection have often been inadequate, the results seem to vary depending on the techniques used to detect and quantify these changes, and comparability between studies is problematic (Govindpani et al., 2017). In addition, little attention has been paid to the possibility of highly localized GABAergic disruption, for instance in hippocampal sub-regions and layers, or in the immediate vicinity of amyloid inclusions (Govindpani et al., 2017). The GABAA Rs are pentameric complexes formed by co-assembly from amongst at least 20 different subunit types, and show remarkable complexity within the human and rodent brains. Our recent study is the first to comprehensively demonstrate, using immunohistochemistry and laser-scanning confocal microscopy, brain region- and cell layer-specific alterations in the expressions of the α1–3, α5, β1–3 and γ2 GABAAR subunits in the human AD hippocampus, entorhinal cortex and superior temporal gyrus (STG) (Kwakowsky et al., 2018) - brain regions severely affected in AD. Importantly, this study revealed that even though some GABAAR subunits such as α3 and β1 are well preserved in all the brain regions examined in AD cases, most subunits suffer subregion- and cell layer- specific changes in comparison with control cases. We noted a decrease in GABAAR α1 subunit expression in the stratum (str.) radiatum of the CA1 region, and an increase through all layers of the CA3 region and in the str. granulare and hilus layers of the dentate gyrus (DG), compared with control cases. We also found that GABAAR α2 subunit expression was significantly increased in the str. oriens layers of CA1–3 and in the str. radiatum of CA2–3, but decreased in the str. pyramidale of the CA1 region. We found that the GABAAR α5 subunit, for which expression changes in AD are highly controversial, was decreased in the STG. However, the α5 subunit was upregulated in the str. pyramidale and str. oriens of the CA1 region. A significant increase in GABAAR β2 subunit expression was detected in the str. oriens and str. radiatum of the CA2 region and in the str. radiatum of the CA3 region, and a significant decrease was found in the DG str. moleculare. We also found a significant decrease in GABAAR β3 subunit immunoreactivity in the str. oriens of the CA2, and in the str. granulare and str. moleculare of the DG. We found a significant increase in GABAAR γ2 subunit expression in the str. oriens of the CA1, in the str. pyramidale of the CA3, and in the str. moleculare of the DG. Our study also provided GABAAR expression data on brain areas which have been poorly studied in the past such as the subiculum and entorhinal cortex, where most subunits were found to be preserved in AD. In the entorhinal cortex, only the α1 subunit was downregulated, while the γ2 subunit was upregulated. The γ2 subunit was also upregulated in the subiculum alongside an upregulation in α5 subunit expression (Kwakowsky et al., 2018).
The expression of GABAAR subunits has been examined in previous studies in rodent and healthy human brains, but ours is one of the very few comprehensive studies on GABAAR subunit distribution in the human hippocampus. This work has helped to clarify some of the inconsistencies and controversial results reported previously. We have also provided evidence for the existence of species differences in GABAAR subunit expression that have to be considered when using animal models to study the GABAergic system or to model disease conditions like AD. Furthermore, the examination of subunit expression in specific subregions and cell layers revealed details of their distribution that have not been observed before. The techniques utilized in our study, particularly the use of fluorescent labeling combined with laser-scanning confocal microscopy and semi-high-throughput imaging, allowed us to more accurately identify and analyze in more depth the specific cortical and hippocampal regions and cell layers; this approach is more suitable for receptor density quantification and provides results with more sensitivity and less variability than previous studies that utilized radiolabeled ligands and peroxidase-based immunohistochemistry and which have demonstrated relatively inconsistent results in the hippocampus. We can perhaps conclude that the kind of fine structural resolution utilized in our study is important in understanding AD-associated GABAergic remodeling, as it is possible that a consideration of a hippocampal subfield or cortical region as a whole, as in previous studies, masks more localized or layer-specific changes. The remarkable anatomical and physiological complexity and diversity of hippocampal GABAergic circuits is involved in the regulation of all aspects of cellular and circuit function. The physiological consequences of altered GABAAR α1, α2, α5, β2, β3 and γ2 subunit expression remain to be established, but they are most likely associated with behavioral effects related to cognition, amnesia, anxiety, depression and altered receptor responses – particularly to GABAergic drugs like benzodiazepines and anesthetics (Kwakowsky et al., 2018) (Figure 1). These changes are most likely not just compensatory alterations, but reflect the reorganization of defined neuronal circuits and are critical to produce a stable neuronal network. As an example, we found that in the AD CA3 region the upregulation of the α1 subunit is paralleled by increased β2 and γ2 subunit expression, which suggests the upregulation of the well-established α1β2γ2 subunit-containing benzodiazepine-sensitive receptor in this region.
Figure 1.
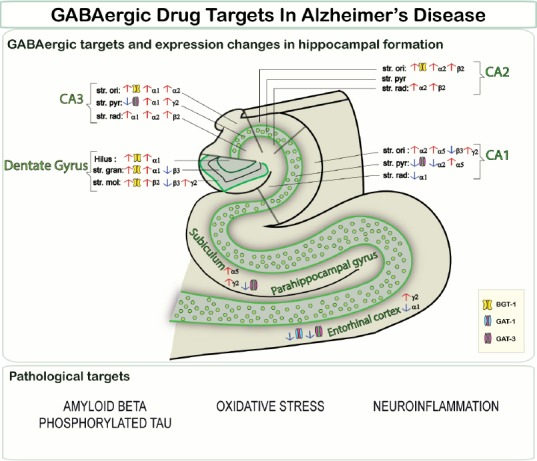
GABAergic drug targets in Alzheimer's disease.
GABA: Gamma-aminobutyric acid; str.: stratum; ori: oriens; pyr: pyramidale; rad: radiatum; hilus: hilus layer; gran: granulare layer; mol: moleculare layer; BGT-1: betaine-GABA transporter; GAT: GABA transporter.
GABA transporter changes in the AD hippocampus also appear to follow an interesting trend, with significant region- and layer-specific alterations (Fuhrer et al., 2017). We have shown that BGT-1, the betaine-GABA transporter, usually expressed at low levels in the healthy human hippocampus, is up-regulated on astrocytes in AD cortical and hippocampal tissue. We also observed a possible compensatory co-regulation of GABA transporters, but a further investigation of this mechanism is warranted (Fuhrer et al., 2017). GABA transporter changes in AD, alongside altered GABAAR subunit expression, might buffer changes in synaptic GABA levels and regulate the disturbances in E/I balance and neuronal excitability associated with AD. Existing and emerging evidence, taken together, seems to indicate that the GABAergic system indeed undergoes remodeling in AD, and that across the hippocampus, cerebral cortex and subcortical structures, there are significant changes in GABA receptor and transporter expression.
Our recent findings, demonstrating subregion- and cell layer-specific changes in most GABAAR subunits in the AD hippocampus, suggest that these changes might underlie some kind of compensatory mechanism, secondary to neurodegeneration, resulting from excitatory dysfunction. Alternatively, GABAergic dysfunction may be a direct consequence of factors such as Aβ deposition. The relationship between GABAergic remodeling and AD pathology appears to be complex, and the mechanisms underlying this remodeling need to be elucidated. Given the compiled evidence for a remodeling of the GABAergic system in AD, potentially affecting many different components of the system (Fuhrer et al., 2017; Govindpani et al., 2017; Kwakowsky et al., 2018), it will be important to consider this system in the development of novel pharmacological and possibly brain region- and cell type-specific treatments for AD. Existing therapies for AD primarily target components of excitatory signaling systems. However, these therapies produce temporary and purely symptomatic effects, are associated with unwanted side-effects, and do not halt disease progression or reverse cognitive decline (Takeda et al., 2006). Experimental therapies have attempted to target features of AD pathology like Aβ deposition (Doody et al., 2014), but these have yet to emerge as viable treatments. GABAA Rs are involved in the regulation of neuronal pathways involved in memory and learning (Chapouthier and Venault, 2002). In the last decade, multiple in vivo studies have demonstrated that the use of GABAergic compounds is linked with enhanced cognition (Calvo-Flores Guzmán et al., 2018). Some of these nootropics have been shown to counteract Aβ neurotoxicity through the activation of GABAergic neurotransmission, and to themselves exert anti-amyloidogenic effects, resulting in reduced Aβ deposition (Calvo-Flores Guzmán et al., 2018). Although the activation of GABAA Rs may mediate these neuroprotective anti-amyloidogenic effects, negative modulators of GABAA Rs, especially those with affinity for α5 subunit-containing receptors, have also been proposed to act as effective cognition-enhancers (Calvo-Flores Guzmán et al., 2018). Given the emerging relevance of GABAergic remodeling in AD and the inconsistency of past findings in this area, more research is needed to elucidate the underlying causes and consequences of this dysfunction. Thus, current and new drugs, as well as novel therapeutic targets for GABA signaling, should be better explored for the enhancement of cognition in AD and other dementias (Calvo-Flores Guzmán et al., 2018). Importantly, since AD is a complex and multifactorial disease with multiple systems involved in its pathology, we strongly suggest further investment and research into multi-drug approaches to treat the complex symptomatology of AD - possibly targeting the GABAergic system together with other systems involved in disease pathogenesis. A recent study combining the GABAB receptor agonist baclofen and the anti-craving agent acamprosate has produced encouraging results, demonstrating the neuroprotective and vasoprotective benefits of this dual-drug therapy against Aβ toxicity and underscoring the potential for combinatorial therapies in the synergistic treatment of the cognitive and pathophysiological symptoms of AD (Chumakov et al., 2015).
Conclusions: Our findings demonstrated that the GABAergic system suffers significant remodeling in the brains of AD patients, yet it is to be determined if this is as a consequence of the disease itself or secondary to neurodegeneration. Most aspects of the GABA signaling system, including GABA levels, GABA currents, and the expression levels and functional characteristics of GABA receptors and transporters, are affected in AD (Fuhrer et al., 2017; Govindpani et al., 2017; Kwakowsky et al., 2018). Our investigation has provided, for the first time, a comprehensive study into subregion- and cell layer-specific GABAAR subunit expression changes in the human AD hippocampus, subiculum, entorhinal cortex and STG. We have demonstrated that the α3 and β1 subunits are well preserved in the AD hippocampus, in contrast with the α1, α2, α5, β2, β3 and γ2 subunits which show subregion- and cell layer-specific alterations. Our finding that the expression of most GABAAR subunits is altered in a layer-specific manner in the AD hippocampus should serve as the basis for future work into understanding the complex mechanisms underlying this remodeling and the contribution of this to cognitive dysfunction, and future work in this area could perhaps lead to the formulation of innovative GABAergic therapies for the restoration of cognition in AD patients (Calvo-Flores Guzmán et al., 2018).
Future directions: Subsequent studies must be directed towards better understanding the mechanisms underlying the regulation of GABAAR subunit expression in the AD cortex and hippocampus. It is yet to be determined not only why these expression changes occur, but why the expression of the same subunit is often differentially regulated in different sub-regions and cell layers within the same brain region, and what the physiological consequences of this might be. The development of AD drugs targeting the GABAergic system in general and GABAARs in particular will need to take into account the complex regulation patterns of GABAAR subunit expression. It is our hope that such findings will open new avenues in the development of disease-modifying therapies and multi-drug approaches for AD, broadening our understanding of the pharmacological basis of nootropic GABAergic drugs in the synergistic alleviation of AD symptoms. The elucidation of GABAergic remodeling patterns and mechanisms will enhance our understanding of the underlying basis of the disease, and may provide new perspectives on the prevention of cognitive decline through the modulation of the disturbed E/I balance and its associated inhibitory deficits.
Additional file: Open peer review report 1 (75KB, pdf) .
Footnotes
Copyright transfer agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Dirk Montag, Leibniz Institute for Neurobiology, Germany.
References
- Calvo-Flores Guzmán B, Vinnakota C, Govindpani K, Waldvogel H, Faull RL, Kwakowsky A. The GABAergic system as a therapeutic target for Alzheimer's disease. J Neurochem. 2018 doi: 10.1111/jnc.14345. doi: 10.1111/jnc.14345. [DOI] [PubMed] [Google Scholar]
- Chapouthier G, Venault P. GABA-A receptor complex and memory processes. Curr Top Med Chem. 2002;2:841–851. doi: 10.2174/1568026023393552. [DOI] [PubMed] [Google Scholar]
- Chumakov I, Nabirotchkin S, Cholet N, Milet A, Boucard A, Toulorge D, Pereira Y, Graudens E, Traore S, Foucquier J, Guedj M, Vial E, Callizot N, Steinschneider R, Maurice T, Bertrand V, Scart-Gres C, Hajj R, Cohen D. Combining two repurposed drugs as a promising approach for Alzheimer's disease therapy. Sci Rep. 2015;5:7608. doi: 10.1038/srep07608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H, Mohs R Alzheimer's Disease Cooperative Study Steering Committee, Solanezumab Study Group. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370:311–321. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- Francis PT. The interplay of neurotransmitters in Alzheimer's disease. CNS Spectr. 2005;10:6–9. doi: 10.1017/s1092852900014164. [DOI] [PubMed] [Google Scholar]
- Fuhrer TE, Palpagama TH, Waldvogel HJ, Synek BJL, Turner C, Faull RL, Kwakowsky A. Impaired expression of GABA transporters in the human Alzheimer's disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. Neuroscience. 2017;351:108–118. doi: 10.1016/j.neuroscience.2017.03.041. [DOI] [PubMed] [Google Scholar]
- Govindpani K, Calvo-Flores Guzman B, Vinnakota C, Waldvogel HJ, Faull RL, Kwakowsky A. Towards a better understanding of GABAergic remodeling in Alzheimer's disease. Int J Mol Sci. 2017;18:E1813. doi: 10.3390/ijms18081813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwakowsky A, Calvo-Flores Guzman B, Pandya M, Turner C, Waldvogel HJ, Faull RL. GABAA receptor subunit expression changes in the human Alzheimer's disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. J Neurochem. 2018 doi: 10.1111/jnc.14325. doi: 10.1111/jnc.14325. [DOI] [PubMed] [Google Scholar]
- Mohandas E, Rajmohan V, Raghunath B. Neurobiology of Alzheimer's disease. Indian J Psychiatry. 2009;51:55–61. doi: 10.4103/0019-5545.44908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince M, Wimo A, Guerchet M, Ali G, Wu Y, Prina M. London, UK: Alzheimer's Disease International; 2015. World Alzheimer Report 2015: The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends. [Google Scholar]
- Takeda A, Loveman E, Clegg A, Kirby J, Picot J, Payne E, Green C. A systematic review of the clinical effectiveness of donepezil, rivastigmine and galantamine on cognition, quality of life and adverse events in Alzheimer's disease. Int J Geriatr Psychiatry. 2006;21:17–28. doi: 10.1002/gps.1402. [DOI] [PubMed] [Google Scholar]
- Vinters HV. Emerging concepts in Alzheimer's disease. Annu Rev Pathol. 2015;10:291–319. doi: 10.1146/annurev-pathol-020712-163927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.