

Abstract
The mitochondrion serves many functions in the central nervous system (CNS) and other organs beyond the well-recognized role of adenosine triphosphate (ATP) production. This includes calcium-dependent cell signaling, regulation of gene expression, synthesis and release of cytotoxic reactive oxygen species, and the release of cytochrome c and other apoptotic cell death factors. Traumatic injury to the CNS results in a rapid and, in some cases, sustained loss of mitochondrial function. One consequence of compromised mitochondrial function is induction of the mitochondrial permeability transition (mPT) state due to formation of the cyclosporine A sensitive permeability transition pore (mPTP). In this mini-review, we summarize evidence supporting the involvement of the mPTP as a mediator of mitochondrial and cellular demise following CNS traumatic injury and discuss the beneficial effects and limitations of the current ex-perimental strategies targeting the mPTP.
Keywords: mitochondrial permeability transition, cyclophilin-D; cyclosporine A, NIM811, spinal cord injury, traumatic brain injury, secondary injury, functional recovery
Introduction
The ability of cells in the central nervous system (CNS) to survive and maintain a functional level of homeostasis following a traumatic injury depends on a number of critical factors. At a relatively gross level, the type of insult (vascular, structural, etc.) as well as the severity and proximity of cells to the injury site are obvious factors. However, many pathophysiological events occur at the subcellular and molecular levels that ultimately determine cellular demise or recovery. These events have been well studied and include, but are not limited to, glutamate excitotoxicity, Ca2+ overload, inflammation, free radical mediated oxidative damage, and a loss of mitochondrial bioenergetics. Some of these secondary injury events are intimately interconnected, which is one rationale for developing neuroprotective therapeutic strategies targeting cell death signaling pathways at the molecular and cellular level.
The Mitochondrial Permeability Transition
Our research has focused on characterizing changes in mitochondrial function after spinal cord (SCI) and traumatic brain (TBI) injuries (Sullivan et al., 2005; McEwen et al., 2011). This is based on well-documented observations that mitochondria play a critical role in determining cellular fate, and compromised mitochondrial function is a prominent feature in both SCI and TBI. Under physiological conditions, mitochondria exhibit a high transmembrane potential generated by the proton pumping components of the respiratory electron transport system. This transmembrane potential is a driving force in the phosphorylation of adenosine diphosphate (ADP) and sequestering Ca2+ from the cytosol. However, following SCI or TBI, mitochondria rapidly become dysfunctional resulting in a loss of cytosolic Ca2+ buffering capacity due to influx of massive pathophysiological levels of Ca2+ through glutamate receptor subtypes, free radical mediated oxidative damage to mitochondrial complex proteins, and a subsequent compromise in bioenergetic capacity. This process becomes insidious as the loss of energy production further reduces the ability of adenosine triphosphate (ATP)-dependent Ca2+ channels to regulate Ca2+ cytosolic levels. Prolonged Ca2+ overload can push mitochondria to the next pathophysiological stage- induction of the mitochondrial permeability transition (mPT) state due to mPT pore (mPTP) formation (Halestrap and Brenner, 2003). The mPT state uncouples respiration from ATP production and by this point recovery of mitochondrial function is limited at best. Cell survival is then compromised when sufficient numbers of mitochondria undergo mPT. Given the importance of mitochondria in cellular function, inhibiting formation of the mPTP is a strategy for limiting cell death in both SCI and TBI. In this perspective article, we summarize what is known about the mPTP and discuss the possible pros and cons of targeting the mPTP as a therapeutic strategy in CNS injury.
The mPTP is a multiprotein mega-channel complex spanning both the inner and outer mitochondrial membranes, essentially allowing for communication of small molecules between the matrix and cytosol. The structural components of the pore are not well understood and the subject of continuing debate. Initial studies suggested that the pore was made up of the voltage-dependent anion channel (VDAC) located on the outer mitochondrial membrane, the adenine nucleotide translocator (ANT) on the inner mitochondrial membrane, and cyclophilin-D (Cyp-D) in the matrix (Halestrap and Brenner, 2003). However, follow-up genetic deletion studies revealed that ANT and VDAC are not required for mPTP formation, which may be explained, in part, by the presence of different isoforms of these putative pore components (Bernardi et al., 2015). Despite these observations, there is a general consensus that Cyp-D functions as a major contributor to mPTP formation.
Inhibiting mPTP Formation in TBI and SCI
Cyclosporine A (CsA) is an immunosuppressant that inhibits mPT by binding to Cyp-D. It has been shown repeatedly to be neuroprotective in TBI, although similar effects in SCI remain controversial based on conflicting reports. This differential efficacy of CsA in TBI versus SCI may be related to properties inherent to mitochondria in the respective CNS regions. Moreover, it is difficult to examine a wide dose range of CsA due to its potent side effects and potential toxicity. In fact, doses of CsA above 20 mg/kg were, for the most part, ineffective in providing neuroprotection following TBI. Given these caveats, our group started investigating the use of NIM811, a Cyp-D binding CsA derivate lacking any immunosuppressive properties and having relatively minimal toxicity (Waldmeier et al., 2002). Our early studies provided evidence that NIM811 reduces oxidative damage while improving mitochondrial function and tissue sparing following TBI or SCI (McEwen et al., 2007; Mbye et al., 2008). The outcome of these studies provided strong evidence that NIM811 and CsA exhibited neuroprotective effects by inhibiting mPTP formation. A recent report by our group revealed a dose dependent effect of post-injury NIM811 treatment in experimental SCI (Springer et al., 2018). Interestingly, we observed that a low dose of NIM811 significantly improved locomotor recovery, while higher doses significantly increased tissue sparing and reflexive bladder control but not recovery of locomotor function. The reasons for these dose-related differences are not clear at this time, although it can be suggested that conducting a follow-up study examining additional doses in the range used in this study might prove revealing.
As stated above, there is strong evidence that targeting the mPTP has therapeutic potential in the treatment of TBI and SCI and numerous studies have examined this hypothesis with mixed results. We conducted a PubMed search to identify published studies investigating CsA or NIM811 in either TBI or SCI and the results of this search are summarized in Tables 1 and 2, respectively. What is clear from this analysis is that the many labs found CsA or NIM811 to be effective in the treatment of various TBI-related outcomes across a range of doses, routes of administration and injury models. NIM811 was reported to be effective in one TBI study while all but two studies (see Fukui et al. (2003) and Dixon et al. (2016) in Table 1) reported structural, physiological, or functional efficacy with CsA treatment. It is interesting to point out that these two negative outcome studies used an intravenous route of delivery, while the positive outcome studies employed intrathecal, subcutaneous or intraperitoneal routes of administration. Given the pharmacokinetics, clearance rates, etc., associated with different routes of administration, it is possible to speculate that an intravenous route may not be optimal. One of these studies (Fukui et al., 2003) recommended that another route of administration be considered as there was no effect of intravenous CsA administration at least in terms of reducing brain edema. In addition, we have avoided an intravenous route of administration in our TBI and SCI studies due to the relative toxicity of the vehicle. However, the results of a prospective randomized clinical trial indicate a good safety profile when using intravenous CsA administration in severe TBI patients (Mazzeo et al., 2009).
Table 1.
Experimental studies testing the efficacy of CsA or NIM811 in the treatment of TBI
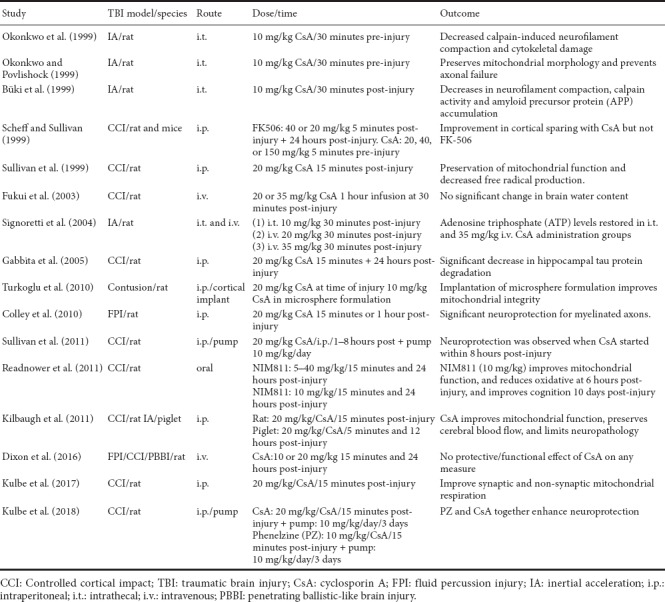
Table 2.
Experimental studies testing the efficacy of CsA or NIM811 in the treatment of traumatic SCI
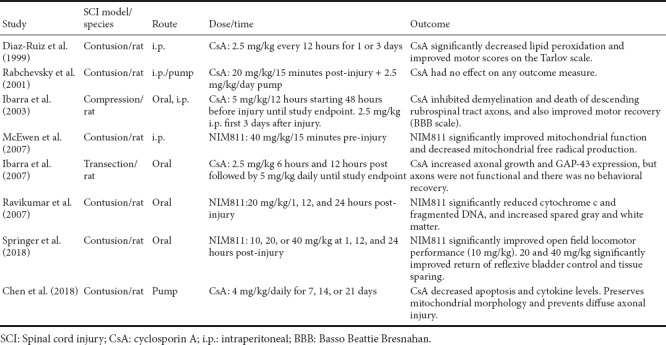
CsA treatment in compression, contusion, or transection models of SCI has been reported to be either effective or ineffective in promoting functional recovery and tissue sparing (see Table 2). For example, work from Ibarra and colleagues report that CsA reduces lipid peroxidation, increases survival of rubrospinal tract axons, and improves functional recovery, while Rabchevsky et al. (2001) report no effect of CsA treatment on any outcome measure including functional recovery, tissue sparing. The reason for the discrepancies is not entirely clear, but may be related to differences in the injury models, injury severities, dosing, and routes of administration. We also demonstrated that the Cyp-D and oxidative stress levels are higher in uninjured spinal cord relative to cortex, and that it takes a higher dose of CsA to inhibit mPTP in mitochondria isolated from spinal cord compared to cortex (Sullivan et al., 2004). This is an important observation as our recent and previous studies have focused exclusively on NIM811, which can be used at higher doses than CsA. These studies demonstrate that NIM811 treatment increases mitochondrial function, spinal cord tissue sparing, and functional recovery in a contusion injury model (Table 2).
Future Considerations
One caveat of comparing CsA efficacy is the fact that many negative reports go unpublished making it difficult to ascertain the true impact of CsA treatment in TBI and SCI. Regardless, given the detrimental side effects of CsA and its vehicle, as well as its immunosuppressive properties, we support the idea of targeting mPTP in both TBI and SCI using reformulated CsA and non-immunosuppressive CsA derivatives such as NIM811. However, some effort should go into understanding the impact of targeting mPTP formation in cells that are significantly compromised and may not survive without intervention. There is evidence that mitochondrial dysfunction persists well beyond the acute stages following TBI and SCI, and it is not clear whether rescuing compromised mitochondria in minimally functioning cells is beneficial. For example, the benefit of CsA administration seems to follow an inverted U-shape highlighting the need for robust therapeutic window data in preclinical studies. It also is not clear what cellular energy demands are placed on surrounding cells in order to maintain minimally functioning cells that might die off in the absence of mPTP targeted drugs. Finally, the current mPTP drugs do not exhibit any cell specificity, which might prove detrimental if treatment promotes survival and life span of pro-inflammatory and destructive microglia, neutrophils, macrophages, and astrocytes.
In conclusion, there is compelling evidence that targeting the mPTP with CsA or NIM811 limits secondary injury and promotes functional recovery after TBI and SCI. Future studies examining the therapeutic potential of this strategy will rely, in part, on 1) identification of other CsA analogues or other compounds that selectively target the mPTP, 2) replication of existing studies using identical methodologies, and 3) examination of mPTP targeted drugs in other species and other injury models in which mPTP is known to occur.
Additional file: Open peer review report 1 (115KB, pdf) .
Footnotes
Conflicts of interest: None declared.
Financial support: This work was supported by a grant from the Kentucky Spinal Cord and Head Injury Research Trust.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Petra Henrich-Noack, Otto von Guericke Universitat Magdeburg, Germany.
Funding: This work was supported by a grant from the Kentucky Spinal Cord and Head Injury Research Trust.
References
- Büki A, Okonkwo DO, Povlishock JT. Postinjury cyclosporin A administration limits axonal damage and disconnection in traumatic brain injury. J Neurotrauma. 1999;16:511–521. doi: 10.1089/neu.1999.16.511. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Rasola A, Forte M, Lippe G. The mitochondrial permeability transition pore: channel formation by f-atp synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev. 2015;95:1111–1155. doi: 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZR, Ma Y, Guo HH, Lu ZD, Jin QH. Therapeutic efficacy of cyclosporin A against spinal cord injury in rats with hyperglycemia. Mol Med Rep. 2018;17:4369–4375. doi: 10.3892/mmr.2018.8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley BS, Phillips LL, Reeves TM. The effects of cyclosporin-A on axonal conduction deficits following traumatic brain injury in adult rats. Exp Neurol. 2010;224:241–251. doi: 10.1016/j.expneurol.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Ruiz A, Rios C, Duarte I, Correa D, Guizar-Sahagun G, Grijalva I, Ibarra A. Cyclosporin-A inhibits lipid peroxidation after spinal cord injury in rats. Neurosci Lett. 1999;266:61–64. doi: 10.1016/s0304-3940(99)00255-4. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Bramlett HM, Dietrich WD, Shear DA, Yan HQ, Deng-Bryant Y, Mondello S, Wang KK, Hayes RL, Empey PE, Povlishock JT, Tortella FC, Kochanek PM. Cyclosporine treatment in traumatic brain injury: operation brain trauma therapy. J Neurotrauma. 2016;33:553–566. doi: 10.1089/neu.2015.4122. [DOI] [PubMed] [Google Scholar]
- Fukui S, Signoretti S, Dunbar JG, Marmarou A. The effect of cyclosporin A on brain edema formation following experimental cortical contusion. Acta Neurochir Suppl. 2003;86:301–303. doi: 10.1007/978-3-7091-0651-8_65. [DOI] [PubMed] [Google Scholar]
- Gabbita SP, Scheff SW, Menard RM, Roberts K, Fugaccia I, Zemlan FP. Cleaved-tau: a biomarker of neuronal damage after traumatic brain injury. J Neurotrauma. 2005;22:83–94. doi: 10.1089/neu.2005.22.83. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem. 2003;10:1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- Ibarra A, Correa D, Willms K, Merchant MT, Guizar-Sahagún G, Grijalva I, Madrazo I. Effects of cyclosporin-A on immune response, tissue protection and motor function of rats subjected to spinal cord injury. Brain Res. 2003;979:165–178. doi: 10.1016/s0006-8993(03)02898-1. [DOI] [PubMed] [Google Scholar]
- Ibarra A, Hernandez E, Lomeli J, Pineda D, Buenrostro M, Martinon S, Garcia E, Flores N, Guizar-Sahagun G, Correa D, Madrazo I. Cyclosporin-A enhances non-functional axonal growing after complete spinal cord transection. Brain Res. 2007;1149:200–209. doi: 10.1016/j.brainres.2007.02.056. [DOI] [PubMed] [Google Scholar]
- Kilbaugh TJ, Bhandare S, Lorom DH, Saraswati M, Robertson CL, Margulies SS. Cyclosporin A preserves mitochondrial function after traumatic brain injury in the immature rat and piglet. J Neurotrauma. 2011;28:763–774. doi: 10.1089/neu.2010.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbe JR, Hill RL, Singh IN, Wang JA, Hall ED. Synaptic mitochondria sustain more damage than non-synaptic mitochondria after traumatic brain injury and are protected by cyclosporine A. J Neurotrauma. 2017;34:1291–1301. doi: 10.1089/neu.2016.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbe JR, Singh IN, Wang JA, Cebak JE, Hall ED. Continuous infusion of phenelzine, cyclosporine A, or their combination: evaluation of mitochondrial bioenergetics, oxidative damage, and cytoskeletal degradation following severe controlled cortical impact traumatic brain injury in rats. J Neurotrauma. 2018;35:1280–1293. doi: 10.1089/neu.2017.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzeo AT, Brophy GM, Gilman CB, Alves OL, Robles JR, Hayes RL, Povlishock JT, Bullock MR. Safety and tolerability of cyclosporin a in severe traumatic brain injury patients: results from a prospective randomized trial. J Neurotrauma. 2009;26:2195–2206. doi: 10.1089/neu.2009.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Sullivan PG, Springer JE, Hall ED. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Exp Neurol. 2008;209:243–253. doi: 10.1016/j.expneurol.2007.09.025. [DOI] [PubMed] [Google Scholar]
- McEwen ML, Sullivan PG, Springer JE. Pretreatment with the cyclosporin derivative, NIM811, improves the function of synaptic mitochondria following spinal cord contusion in rats. J Neurotrauma. 2007;24:613–624. doi: 10.1089/neu.2006.9969. [DOI] [PubMed] [Google Scholar]
- McEwen ML, Sullivan PG, Rabchevsky AG, Springer JE. Targeting mitochondrial function for the treatment of acute spinal cord injury. Neurotherapeutics. 2011;8:168–179. doi: 10.1007/s13311-011-0031-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab. 1999;19:443–451. doi: 10.1097/00004647-199904000-00010. [DOI] [PubMed] [Google Scholar]
- Okonkwo DO, Büki A, Siman R, Povlishock JT. Cyclosporin A limits calcium-induced axonal damage following traumatic brain injury. Neuroreport. 1999;10:353–358. doi: 10.1097/00001756-199902050-00026. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Fugaccia I, Sullivan PG, Scheff SW. Cyclosporin A treatment following spinal cord injury to the rat: behavioral effects and stereological assessment of tissue sparing. J Neurotrauma. 2001;18:513–522. doi: 10.1089/089771501300227314. [DOI] [PubMed] [Google Scholar]
- Ravikumar R, McEwen ML, Springer JE. Post-treatment with the cyclosporin derivative, NIM811, reduced indices of cell death and increased the volume of spared tissue in the acute period following spinal cord contusion. J Neurotrauma. 2007;24:1618–1630. doi: 10.1089/neu.2007.0329. [DOI] [PubMed] [Google Scholar]
- Readnower RD, Pandya JD, McEwen ML, Pauly JR, Springer JE, Sullivan PG. Post-injury administration of the mitochondrial permeability transition pore inhibitor, NIM811, is neuroprotective and improves cognition after traumatic brain injury in rats. J Neurotrauma. 2011;28:1845–1853. doi: 10.1089/neu.2011.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma. 1999;16:783–792. doi: 10.1089/neu.1999.16.783. [DOI] [PubMed] [Google Scholar]
- Signoretti S, Marmarou A, Tavazzi B, Dunbar J, Amorini AM, Lazzarino G, Vagnozzi R. The protective effect of cyclosporin A upon N-acetylaspartate and mitochondrial dysfunction following experimental diffuse traumatic brain injury. J Neurotrauma. 2004;21:1154–1167. doi: 10.1089/neu.2004.21.1154. [DOI] [PubMed] [Google Scholar]
- Springer JE, Visavadiya NP, Sullivan PG, Hall ED. Post-injury treatment with NIM811 promotes recovery of function in adult female rats after spinal cord contusion: a dose-response study. J Neurotrauma. 2018;35:492–499. doi: 10.1089/neu.2017.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Sebastian AH, Hall ED. Therapeutic window analysis of the neuroprotective effects of cyclosporine A after traumatic brain injury. J Neurotrauma. 2011;28:311–318. doi: 10.1089/neu.2010.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death. J Neurosci Res. 2005;79:231–239. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Keller JN, Lovell M, Sodhi A, Hart RP, Scheff SW. Intrinsic differences in brain and spinal cord mitochondria: Implication for therapeutic interventions. J Comp Neurol. 2004;474:524–534. doi: 10.1002/cne.20130. [DOI] [PubMed] [Google Scholar]
- Turkoglu OF, Eroglu H, Gurcan O, Bodur E, Sargon MF, Oner L, Beskonakli E. Local administration of chitosan microspheres after traumatic brain injury in rats: a new challenge for cyclosporine--a delivery. Br J Neurosurg. 2010;24:578–583. doi: 10.3109/02688697.2010.487126. [DOI] [PubMed] [Google Scholar]
- Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–29. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.