

Abstract
Background: Phenylketonuria as the most common genetic metabolic disorder is the result of disruption of the phenylalanine hydroxylase gene. This study was carried out to explore the phenylalanine hydroxylase gene mutation status of Iranian phenylketonuria patients.
Methods: Blood samples were collected from 30 patients, and hot spot areas of the phenylalanine hydroxylase gene, including exons 6, 7, 8, 11, and 12 were studied through polymerase chain reaction and sequencing techniques.
Results: Eight different mutations, including 5 missense mutations, 1 splice mutation, 1 nonsense mutation, and 1 Silent/Splice mutation were detected. These mutations were R243X, R261Q, R261X, P281L, R241C, V399V, E280K, and IVS11+1G>C. V399V and R241C were reported for the first time in Iranian population. Three polymorphisms including Q232Q, V245V and L385L and 3 novel intronic variants including IVS10-15A>C, IVS6+44T>G, and IVS6+36 T>G were also detected in this study.
Conclusion: The results of this study prove the heterogeneous status of phenylalanine hydroxylase gene mutations in the Iranian population, which can be useful in carrier testing and genetic counseling.
Keywords: Phenylketonuria, Phenylalanine hydroxylase gene, Sequencing, Mutation, Iran
↑ What is “already known” in this topic:
Phenylketonuria is one of the most prevalent metabolic genetic disorders. Mutations in phenylalanine hydroxylase gene cause this disease. More than 800 different mutations are registered on the PAH mutation database.
→ What this article adds:
Mutation analysis of PAH gene study (exons 6, 7, 8, 11 and 12) in Iranian patients with PKU was screened. These mutations showed regional variations useful in genetic counselling and prenatal testing in different ethnic groups. We found three novel intronic variants and two previously unreported mutations. Our data proved the heterogenic status of PAH gene mutations.
Introduction
Phenylketonuria (PKU, OMIM 261600), a common metabolic genetic disorder, is identified by the lack of phenylalanine hydroxylase (PAH) enzyme (EC 1.14.16.1) activity (1). Phenylalanine (Phe) conversion to tyrosine by this enzyme is the main pathway for Phe metabolism, and this activity is dependent on tetrahydrobiopterin (BH4) presence as a cofactor (2). If this disease cannot be diagnosed in the first days after birth, it will contribute to the accumulation of phenylalanine in the patient’s body fluids with side effects such as failure in growth and brain damages (3). Newborn screening programs for PKU diagnosis are important postnatal requirement tests in many countries. The Guthrie test is the most applicable method for this purpose and has been used for more than 40 years (4).Based on blood Phe concentrations, PKU can be classified to classic PKU, mild PKU and mild hyperphenylalaninemia (HPA) (5).
The human PAH gene (GenBank: AF404777) with 13 exons is located on chromosome 12q22-q24.1, spanning about 90 kb, which encodes a polypeptide with 452 amino acids (6). The broad spectrum of mutations in PAH gene is the main etiology of this disease, and more than 800 different mutations are registered on the PAH mutation database; most of them are missense.
These mutations can make the enzyme structure and activity dysregulated which leads to variable phenotypes based on their severity to be observed (7). The presence of different disease-causing mutations in the biallelic PAH gene contributes to the compound heterozygosity status in most patients (8). The regional prevalence of PKU varies worldwide, and the highest frequency of this disease is observed among Caucasians with a rate of 1/10,000 per live births (5).
Iran is a country with a high number of consanguine marriages showing a rate of 1.6 in 3,627 (9). Surveying PAH gene mutations is applicable in genetic counseling for prenatal diagnosis and presentation of an efficient dietary program for PKU patients (10). This study identifies the genetic mutation status of a group of Iranian PKU patients to expand the mutation database of different ethnic groups within this population proving beneficial for mutation panel designs of laboratories and prevention of the birth of children affected with PKU.
Methods
Subjects
A total of 30 unrelated PKU patients were chosen from the Institute of Endocrinology and Metabolism of Ali Asghar Hospital between August 2015 and July 2016.
They were accepted into this study after their parents filled out the official consent form provided by the Ethical Committee of Iran University of Medical Sciences. The classification of patients into classic and mild PKU was based on their blood phenylalanine concentrations. Most of them were identified during neonatal screening programs by a Guthrie test. These patients, between 8 months to 18 years old, were from different ethnic groups in Iran.
DNA Extraction:
A 5 ml blood sample was collected from each patient. The genomic DNA was extracted from leukocytes isolated from EDTA anticoagulated blood, using a DNA purification Genet Bio kit (South Korea). DNA Purity and quality were assessed by NanoDrop Spectrophotometer (Thermo Scientific™, USA) and 1% agarose gel respectively.
PCR:
The choice of PAH gene exons including 6, 7, 8, 11, and 12 was based on their high mutation rates in previous literature (6, 11). These 5 exons and their adjacent intronic regions were separately amplified by PCR on the T100™ Thermal Cycler system (Bio-Rad Laboratories, Hercules, CA, USA) (Fig. 1).
Fig. 1.
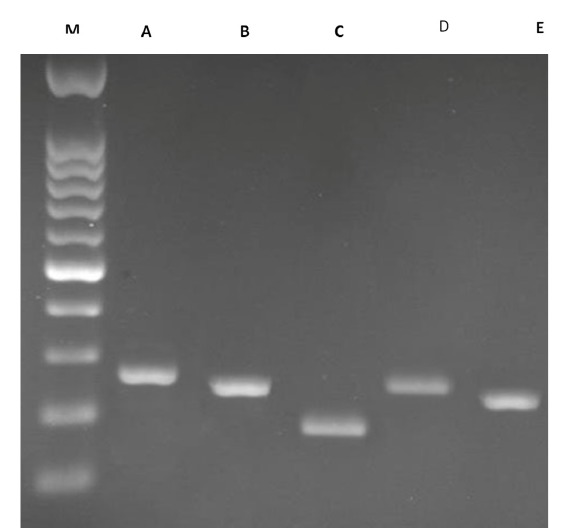
The analysis of PCR amplification of exons 6, 7, 8, 11 and 12 on 1% agarose gel; M- Marker; A- exon 6 (283 bp); B- exon 7 (156); C- exon 8 (194); D- exon 11 (260); E- exon 12 (232).
The primers were chosen from previous studies (Table 1) (12). PCR reactions were carried out in a total volume of 25 μl containing 2.4 µL of 2× reaction buffer, 2 µL of genomic DNA templates, 0.1 µL of Taq DNA polymerase (5 U/µL), 0.7 µL of each primer, 1.5 µL MgCl2 (50 mM) and 0.5 µM of dNTP (10 mM). The cycling program of PCR was 94 ˚C denaturation for 3 min, followed by 35 cycles of denaturation at 94 ºC for 45 s, annealing at 59 ˚C for 30 s, extension at 72 ºC for 45 s, and a final extension step at 72 ºC for 3 min. The 1% agarose gel was used for electrophoresis of PCR amplicons.
Table 1. The list of primer sequences and expected PCR product sizes .
| Exons | Primers | Annealing temperature (°C) | PCR product length |
| 6 |
F: TGCCCTGCTTGAGACACCTA R: TCCTCCCCCAACTTTCTGC |
60 | 283 |
| 7 |
F: CCTCTGACTCAGTGGTGA R: CTTGCAGCAGGAAAAG |
59 | 256 |
| 8 |
F: CTGAGTCTGGCTTGGCTTAA R: CTCATTTGAGAAATTCAGGT |
58 | 194 |
| 11 |
F: TGCAGCAGGGAATACTGA R: AGATGAGTGGCACCAGT |
60 | 260 |
| 12 |
F: TCCAAATGGTGCCCT R: GGCGATGGTAGGGAA |
57 | 232 |
DNA Sequencing:
The PCR products were purified and sequenced using PCR forward and reverse primers by the Applied Biosystems 3730 DNA Analyzer (Macrogen, Seoul, Korea) using the classic Sanger’s method.
Data Analysis
Mutations and polymorphisms were detected by software and programs including ChromasPro (Technelysium, South Brisbane Australia), Sequence Scanner (Applied Biosystems, US), Mutation Surveyor (SoftGenetics, Pennsylvania, USA), and nucleotide blast program of NCBI. These ones were then identified by comparison with the reference sequence in GenBank (www.ncbi.nlm.nih. gov/genbank), available genetic data in PAH database (http://www.PAHdb.mcgill.ca), and SNP database of NCBI (www.ncbi.nlm.nih.gov/snp).
The clinical significance of mutations was characterized by using ClinVar (www.ncbi.nlm.nih.gov/clinvar/) database. The analysis of new variants was carried out by Mutation Taster (http://www.mutationtaster.org/) and CADD (http://cadd.gs.washington.edu/) online bioinformatics tools.
Results
The patients (15 females and 15 males), aged between 8 months to 18 years old, were examined. They were from different parts of Iran, and their ethnicity was also diverse such as Kurdish, Azeri, and Persian. The rate of consanguine marriage was 53%, and most of the parents were third cousins. A total of 8 different mutations consisting of 5 missense mutations, 1 splice mutation, 1 nonsense mutation, and 1 Silent/Splice mutation were characterized in 14 alleles. The rate of mutation detection was about 23.3%. Three polymorphisms and four intronic novel variations were characterized as well.
The mutations, including c.782G>A, c.838 G>A, c.842C>T, c.721C>T, c.727C>T, c.1197A>T, c.781C>T and IVS11+1G>C, the polymorphisms including c.696A>G, c.735G>A and c.1155G>C and the novel intronic variants including IVS6+44T>G, IVS6+36T>G, and IVS10-15A>C were identified (Table 2). The survey of pathogenic variations in ClinVar database also proved their pathogenic effect (Table 3). The genotype and phenotype information of patients were demonstrated in Table 4. Some associations were also observed in this study and one of them, including R243X mutation and V245V polymorphism was shown in Fig. 2. The mean quality value of surrounding nucleotides of these variants was higher than 50 which proved their accuracy. The PHRED score of these novel variants showed their non-pathogenic effect (Table 5).
Table 2. Relative frequency of variations and prediction of their effec .
| Location |
Systematic name (DNA level) |
Trivial name (protein effect) |
No. of alleles |
Relative frequency (%) |
phyloP | phastCons | Prediction |
| Exon7 | c.782G>A | p .R261Q | 4 | 6.66 | 6.198 | 1 | Disease causing |
| Exon7 | c.781C>T | p .R261X | 2 | 3.33 | 2.582 | 1 | Disease causing |
| Exon7 | c.838G>A | p .E280K | 1 | 1.66 | 6.203 | 1 | Disease causing |
| Exon7 | c.842C>T | p .P281L | 2 | 3.33 | 6.203 | 1 | Disease causing |
| Exon7 | c.721C>T | p .R241C | 1 | 1.66 | 4.439 | 1 | Disease causing |
| Exon7 | c.727C>T | p .R243X | 2 | 3.33 | 2.751 | 1 | Disease causing |
| Exon 11 | c.1197A>T | p .V399V | 1 | 1.66 | 0.423 | 1 | Disease causing |
| Intron11 | c.1199+1G>C | IVS11+1G>C | 1 | 1.66 | 6.012 | 1 | Disease causing |
| Exon 6 | c.696A>G | p .Q232Q | 7 | 11.6 | -3.252 | 0.005 | polymorphism |
| Exon7 | c.735G>A | p .V245V | 7 | 11.6 | 0.873 | 1 | Polymorphism |
| Exon11 | c.1155G>C | p .L385L | 2 | 3.3 | -0.213 | 0.749 | Polymorphism |
| Intron6 | c.706+36T>G | IVS6+36 T>G | 1 | 1.6 | -0.172 | 0 | Polymorphism |
| Intron 6 | c.706+44T>G | IVS6+44T>G | 1 | 1.6 | -0.986 | 0.004 | Polymorphism |
| Intron 10 | c.1066-15A>C | IVS10-15A>C | 3 | 5 | 0.479 | 0 | Polymorphism |
Table 3. Clinical significance of mutations .
| Mutation | rs Number | Clinical significance |
| R261Q | rs62508698 | Pathogenic |
| R261X | rs5030850 | Pathogenic |
| E280K | rs62508698 | Pathogenic |
| P281L | rs5030851 | Pathogenic |
| R241C | rs76687508 | Pathogenic |
| R243X | rs5030846 | Pathogenic |
| V399V | rs199475584 | Pathogenic |
| IVS11+1G>C | rs62509015 | Likely pathogenic |
Table 4. Genotypes and characteristics of 12 patients .
|
Patients ID |
Location | Genotypes | Classification | HPA Type | Ethnicity | Pre-treatment Phe levels (mg/dl) |
| 5 | Exon 11 | L385L/U | Heterozygote | Classic | Azeri | 21.5 |
| 6 | Exon 7 |
V245V/U E280K/U |
Heterozygote | Mild HPA | Persian | 18.4 |
| 7 | Exon 7 | R261Q/ U | Heterozygote | Mild HPA |
Persian |
16.7 |
| 7 | Exon 6 | Q232Q/ U | Heterozygote | |||
| 8 | Exon 7 | V245V/U | Heterozygote | Mild HPA | Kurdish | 14 |
| 9 | Exon 6 | Q232Q/ Q232Q | Homozygote | Mild HPA | Persian | 15 |
| 11 | Exon 7 | P281L/ P281L | Homozygote | Classic | Persian | 25 |
| 12 | Exon 7 |
R241C/U V245V/U |
Heterozygote | Classic | Azeri | 21.1 |
| 14 | Exon 7 | R261Q/U | Heterozygote | Classic | Persian | 24 |
| 15 | Exon 6 | Q232Q/ Q232Q | Homozygote | Classic | Persian | 26 |
| R261X/R261X | Homozygote | |||||
| 16 | Exon 6 | Q232Q/ Q232Q | Homozygote | Classic | Azeri | 25 |
| 18 | Exon 11 | V399V/U | Heterozygote | Classic | Persian | 28.8 |
| Exon 7 | V245V | Homozygote | ||||
| 19 | Exon 7 |
R243X/ R243X V245V/ V245V |
Homozygote | Classic | Azeri | 22.9 |
| 22 |
Exon 11 Intron 11 |
L385L/U IVS11+1G>C/U |
Heterozygote | Classic | Persian | 23.4 |
| 25 | Exon 7 | R261Q | Homozygote | Classic | Azeri | 25.5 |
Fig. 2.
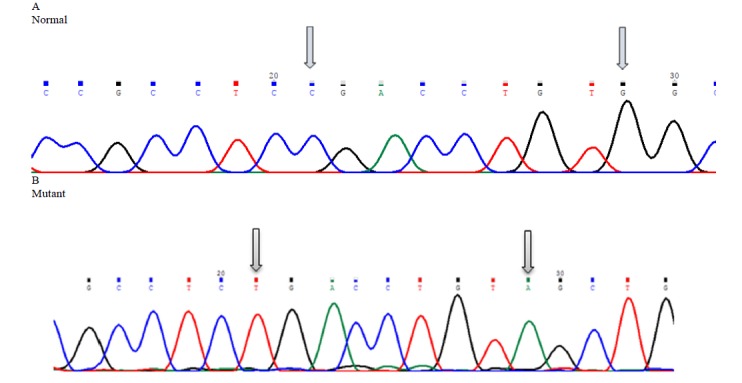
Mutation analysis of R243X (c.727C>T) nonsense mutation and V245V (c.735G>A) polymorphism in exon 7 of PAH gene
Table 5. The PHRED score of new variants .
| Variant | PHRED Score |
| IVS6+36 T>G | 2.980 |
| IVS6+44T>G | 2.380 |
| IVS10-15A>C | 2.560 |
Discussion
Phenylketonuria, a metabolic autosomal recessive genetic disorder, is the consequence of PAH gene deficiency. The hallmark of this disease is high blood serum Phe levels with a low-Phe diet as the cornerstone of its treatment (13). Mutations in the BH4 gene also resulted in hyperphenylalaninemia, and identification of related mutations to PAH enzyme can be applicable in the prescription of BH4 based therapies (14). Patients were diagnosed based on their high blood Phe and in order to confirm mild HPA cases for having PAH mutations, analysis of DHPR and biopterin-neopterin was carried out.
In this study, molecular characterization of exons 6, 7, 8, 11, and 12 in 30 Iranian patients showed a 23.3% mutation detection rate. Ten percent of patients were homozygote for mutations. Three polymorphisms including Q232Q (11.6%) in exon 6, V245V (11.6%) in exon 7 (11.6%) and L385L (3.33%) in exon 11 were also identified. The R261Q mutation (6.66%) in exon 7 was observed in two patients in heterozygote form and one patient in homozygote form which affects a CpG mutation hot spot in nucleotide 782 of PAH gene. The explanation of this mutation mechanism is by the 5-methylcytosine deamination at a CpG dinucleotide site which contributes to the change of amino acid Arg to Glutamine. This one is the second most common mutation in Turkey, which is also common in European nations like France (17%), and Italy (16%) (15, 16). The presence of Mediterranean mutations in our patients may prove the historical connection between Iranian and Mediterranean populations.
The R261X mutation which is the result of C to T conversion at codon 781 of PAH gene is placed in its catalytic domain (residues 118-427) (17). P281L (3.33%), a missense mutation in homozygote form, is the result of C to T substitution at codon 842 of PAH gene. This nucleotide position is in the region of cofactor binding domain. P281L has been identified in classic PKU patients frequently. Previous studies in Iran have also reported this mutation (11).
R243X (3.3%) as a nonsense mutation is located in exon 7 of PAH gene, which was observed along with V245V as a polymorphism in one classic patient. These ones were in homozygote form and had located in 21th and 29th base of exon 7 respectively. Two associations in heterozygote patients, including R241C (1.66%) with V245V and E280K (1.66%) with V245V were also reported, which have not been reported previously in PAHdb. R241C is classified as the BH4-responsiveness mutation reported for the first time in the Iranian population.
The splice mutation IVS11+1G>C/U (1.66%) and L385L polymorphism were observed in intron 11 and exon 11 of one classic PKU patient. The V399V (1.66%) mutation had been identified in the Chinese population for the first time (17). The c.1197 nucleotide is located at the 3'-end of exon 11 at position -3′ of the exon-intron boundary, and transition of A to T in this position cause exon skipping as a post-transcriptional event which can result in deficient PAH mRNA (18). This heterozygote one was observed in a classic patient and was the first report of this mutation in Iran.
Conclusion
The results of our study proved the existence of heterogeneous mutations in different ethnic groups in Iran. The highest rate of mutations was observed in exon 7, and no mutation was observed in exon 8. It should be considered that our results showed two mutations which had not been reported previously in Iran.
So by monitoring diverse ethnic groups in Iran, other novel mutations will be easier to find and identify. This data can be useful for genetic counseling and carrier detection tests and as a base to start further research from.
Acknowledgment
This study was supported by research committee foundation grant no 26080-30-02-94 of Iran University of Medical Sciences. We wish to thank the patients and their families for participating in this study, and the staff at the department of genetics at Iran University of Medical Sciences.
Conflict of Interests
The authors declare that they have no competing interests.
Cite this article as: Rastegar Moghadam M, Shojaei A, Babaei V, Rohani F, Ghazi F. Mutation analysis of Phenylalanine hydroxylase gene in Iranian patients with Phenylketonuria. Med J Islam Repub Iran. 2018(11 Mar);32:21. https://doi.org/10.14196/mjiri.32.21
References
- 1.Scriver CR. The PAH gene, phenylketonuria, and a paradigm shift . Hum Mutat. 2007;28(9):831–845. doi: 10.1002/humu.20526. [DOI] [PubMed] [Google Scholar]
- 2.Setoodeh A, Yarali B, Rabbani A, Khatami S, Shams S. Tetrahydrobiopterin responsiveness in a series of 53 cases of phenylketonuria and hyperphenylalaninemia in Iran. Mol Genet Metab Rep. 2015 Mar 31;2:77–9. doi: 10.1016/j.ymgmr.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. The Lancet. 2010 Oct 29;376(9750):1417–27. doi: 10.1016/S0140-6736(10)60961-0. [DOI] [PubMed] [Google Scholar]
- 4.Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011 Dec 31;104:S2–9. doi: 10.1016/j.ymgme.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 5.Biglari A, Saffari F, Rashvand Z, Alizadeh S, Najafipour R, Sahmani M. Mutations of the phenylalanine hydroxylase gene in Iranian patients with phenylketonuria. SpringerPlus. 2015 Dec 1;4(1):542. doi: 10.1186/s40064-015-1309-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ajami N, Kazeminezhad SR, Foroughmand AM, Hasanpour M, Aminzadeh M. A preliminary mutation analysis of phenylketonuria in southwest Iran. Genet Mol Res. 2013 Jan 1;12(4):4958–66. doi: 10.4238/2013.October.24.7. [DOI] [PubMed] [Google Scholar]
- 7.Cabalska B, Cyrytowski L, Nowacki P, Walczak M, Koziarska D. Different presentations of late‐detected phenylketonuria in two brothers with the same R408W/R111X genotype in the PAH gene . J Intellect Disabil Res. 2003 Feb 1;47(2):146–52. doi: 10.1046/j.1365-2788.2003.00449.x. [DOI] [PubMed] [Google Scholar]
- 8.Bercovich D, Elimelech A, Zlotogora J, Korem S, Yardeni T, Gal N. et al. Genotype–phenotype correlations analysis of mutations in the phenylalanine hydroxylase (PAH) gene . J Hum Genet. 2008 May 1;53(5):407–18. doi: 10.1007/s10038-008-0264-4. [DOI] [PubMed] [Google Scholar]
- 9.Koochmeshgi J, Bagheri A, Hosseini-Mazinani SM. Incidence of phenylketonuria in Iran estimated from consanguineous marriages. J Inherit Metab Dis. 2002 Feb 1;25(1):80–1. doi: 10.1023/a:1015154321142. [DOI] [PubMed] [Google Scholar]
- 10.Li N, Jia H, Liu Z, Tao J, Chen S, Li X. et al. Molecular characterisation of phenylketonuria in a Chinese mainland population using next-generation sequencing. Sci Rep. 2015;5 doi: 10.1038/srep15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamzehloei T, Hosseini SA, Vakili R, Mojarad M. Mutation spectrum of the PAH gene in the PKU patients from Khorasan Razavi province of Iran . Gene 2012. Sep 10;506(1):230–2. doi: 10.1016/j.gene.2012.06.043. [DOI] [PubMed] [Google Scholar]
- 12.Dobrowolski SF, Ellingson C, Coyne T, Grey J, Martin R, Naylor EW. et al. Mutations in the phenylalanine hydroxylase gene identified in 95 patients with phenylketonuria using novel systems of mutation scanning and specific genotyping based upon thermal melt profiles. Mol Genet Metab. 2007 Jul 31;91(3):218–27. doi: 10.1016/j.ymgme.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 13.MacDonald A, Rocha JC, Van Rijn M, Feillet F. Nutrition in phenylketonuria. Mol Genet Metab. 2011;104:S10–S18. doi: 10.1016/j.ymgme.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 14. Staudigl M, Gersting SW, Danecka MK, Messing DD, Woidy M, Pinkas D, et al. The interplay between genotype, metabolic state and cofactor treatment governs phenylalanine hydroxylase function and drug response. Hum Mol Genet, 2011. 20(13): p. 2628-2641. [DOI] [PubMed]
- 15.Staudigl M, Gersting SW, Danecka MK, Messing DD, Woidy M, Pinkas D. et al. The interplay between genotype, metabolic state and cofactor treatment governs phenylalanine hydroxylase function and drug response. Hum Mol Genet. 2011 Apr 28;20(13):2628–41. doi: 10.1093/hmg/ddr165. [DOI] [PubMed] [Google Scholar]
- 16.Rivera I, Mendes D, Afonso Â, Barroso M, Ramos R, Janeiro P. et al. Phenylalanine hydroxylase deficiency: molecular epidemiology and predictable BH 4-responsiveness in South Portugal PKU patients. Mol Genet Metab. 2011 Dec 31;104:S86–92. doi: 10.1016/j.ymgme.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 17.Yu WZ, Qiu DH, Song F, Liu L, Liu SM, He XJ. et al. Characteristics of the PAH gene mutation in Chinese patients with phenylketonuria in Xinjiang . Chinese J Med Genetic. 2009 Feb;26(1):26–30. doi: 10.3760/cma.j.issn.1003-9406.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 18.Zhu T, Qin S, Ye J, Qiu W, Han L, Zhang Y. et al. Mutational spectrum of phenylketonuria in the Chinese Han population: a novel insight into the geographic distribution of the common mutations. Pediatr Res. 2010 Mar 1;67(3):280–5. doi: 10.1203/PDR.0b013e3181c9fb85. [DOI] [PubMed] [Google Scholar]