

Abstract
Arsenate is an abundant oxyanion that, because of its ability to mimic the phosphate group, is toxic to cells. Arsenate reductase (EC 1.97.1.5; encoded by the arsC gene in bacteria) participates to achieve arsenate resistance in both prokaryotes and yeast by reducing arsenate to arsenite; the arsenite is then exported by a specific transporter. The crystal structure of Bacillus subtilis arsenate reductase in the reduced form with a bound sulfate ion in its active site is solved at 1.6-Å resolution. Significant structural similarity is seen between arsenate reductase and bovine low molecular weight protein tyrosine phosphatase, despite very low sequence identity. The similarity is especially high between their active sites. It is further confirmed that this structural homology is relevant functionally by showing the phosphatase activity of the arsenate reductase in vitro. Thus, we can understand the arsenate reduction in the light of low molecular weight protein tyrosine phosphatase mechanism and also explain the catalytic roles of essential residues such as Cys-10, Cys-82, Cys-89, Arg-16, and Asp-105. A “triple cysteine redox relay” is proposed for the arsenate reduction mechanism.
The chemical similarity of arsenate and phosphate enables arsenate ions to enter the cell through the phosphate transport systems (1). However, high concentration of arsenate is toxic to cells. In bacteria, arsenate resistance is achieved by first reducing the arsenate ion (H2AsO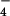 ) to arsenite ion (AsO
) to arsenite ion (AsO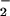 ), then the arsenite is exported through an efficient and specific transport system (2). In general, the arsenate reductase from Gram-negative (represented by a plasmid R773-encoded enzyme in Escherichia coli), and Gram-positive (represented by a plasmid pI258-encoded enzyme in Staphylococcus aureus) bacteria share very low sequence homology (about 10% sequence identity) and distinct catalytic features (3, 4). Nevertheless, they can catalyze the same chemical reaction and utilize a similar cysteine residue (Cys-10 in the S. aureus arsenate reductase) at the active site (5, 6). Whereas E. coli arsenate reductase is coupled to the glutathione and glutaredoxin system for its enzyme activity, S. aureus arsenate reductase requires thioredoxin and thioredoxin reductase to work. This difference in redox partner requirement explains the sequence differences. It has experimentally been shown that in S. aureus arsenate reductase a cysteine pair, Cys-82 and Cys-89, is essential for the enzyme activity, and these two cysteines form a disulfide bridge upon oxidation (6). However, such a cysteine pair is missing in E. coli arsenate reductase. The arsC gene of the Gram-positive bacterium Bacillus subtilis (strain 168) is located in the skin element in the chromosome, and this gene is required to confer arsenate resistance in vivo (7, 8). B. subtilis arsenate reductase shares about 65% identity with the S. aureus arsenate reductase, and the three important catalytic cysteines are conserved. The Gram-positive family of arsenate reductases is distantly related to the mammalian low molecular weight protein tyrosine phosphatase (LMW PTPase) family with about 18% sequence identity (Fig. 1A). Recently, an arsenate reductase encoded by the ACR2 gene has been identified in the yeast Saccharomyces cerevisiae (9). The yeast enzyme is similar to the catalytic domain of a protein tyrosine phosphatase, cdc25, and it has been shown that the key residues identified from the cdc25 active site are also required in the arsenate reductase for its function (10).
), then the arsenite is exported through an efficient and specific transport system (2). In general, the arsenate reductase from Gram-negative (represented by a plasmid R773-encoded enzyme in Escherichia coli), and Gram-positive (represented by a plasmid pI258-encoded enzyme in Staphylococcus aureus) bacteria share very low sequence homology (about 10% sequence identity) and distinct catalytic features (3, 4). Nevertheless, they can catalyze the same chemical reaction and utilize a similar cysteine residue (Cys-10 in the S. aureus arsenate reductase) at the active site (5, 6). Whereas E. coli arsenate reductase is coupled to the glutathione and glutaredoxin system for its enzyme activity, S. aureus arsenate reductase requires thioredoxin and thioredoxin reductase to work. This difference in redox partner requirement explains the sequence differences. It has experimentally been shown that in S. aureus arsenate reductase a cysteine pair, Cys-82 and Cys-89, is essential for the enzyme activity, and these two cysteines form a disulfide bridge upon oxidation (6). However, such a cysteine pair is missing in E. coli arsenate reductase. The arsC gene of the Gram-positive bacterium Bacillus subtilis (strain 168) is located in the skin element in the chromosome, and this gene is required to confer arsenate resistance in vivo (7, 8). B. subtilis arsenate reductase shares about 65% identity with the S. aureus arsenate reductase, and the three important catalytic cysteines are conserved. The Gram-positive family of arsenate reductases is distantly related to the mammalian low molecular weight protein tyrosine phosphatase (LMW PTPase) family with about 18% sequence identity (Fig. 1A). Recently, an arsenate reductase encoded by the ACR2 gene has been identified in the yeast Saccharomyces cerevisiae (9). The yeast enzyme is similar to the catalytic domain of a protein tyrosine phosphatase, cdc25, and it has been shown that the key residues identified from the cdc25 active site are also required in the arsenate reductase for its function (10).
Figure 1.

(A) Structure-based multiple sequence alignment by CLUSTAL X (36) with Gram-positive bacterial arsenate reductases, bacterial PTPase homologues, and mammalian LMW PTPases. The protein sequences are obtained from the SWISS-PROT database, and the alignment is drawn by the program ALSCRIPT (37). AB loop refers to the CX5R motif with Cys-10 and Arg-16 as filled and open circles; flexible region is indicated. Filled arrowheads refer to the Cys-82, Cys-89 pair and the open arrowhead to Asp-105. The PTPase homologues are widespread in most bacteria with known genome sequences. However, the functions of these bacterial PTPase homologues are largely unknown. (B) The overall structure of B. subtilis arsenate reductase in the reduced form with the secondary structure elements and N and C termini labeled. Some key residues are also shown. (C) The Cα trace of B. subtilis arsenate reductase (yellow) superimposed with bovine LMW PTPase (BPTP) (blue) with N and C termini labeled. Note the different positions of the sulfate ions (yellow sulfate for B. subtilis arsenate reductase and blue sulfate for BPTP) in these two structures.
The arsenate reductase (EC 1.97.1.5) is a newly characterized enzyme (11); structural data are accumulating for this diverse family of proteins (12–14). The B. subtilis arsenate reductase is chosen for crystallographic studies to understand the arsenate resistance mechanism.
Materials and Methods
Protein Purification and PTPase Activity Assay.
The preparation of wild-type and selenomethionine (Se-Met)-substituted B. subtilis arsenate reductase is as described in ref. 14.
B. subtilis arsenate reductase PTPase activity assays using p-nitrophenyl phosphate (PNPP; Sigma) as substrate were performed in a total volume of 200 μl containing 0.2 M Tris⋅Cl (pH 7.6), 5 mM DTT, 70 mM substrate, and 40 μg of B. subtilis arsenate reductase at 40°C. The reactions were stopped by adding 400 μl of 0.5 M NaOH at reaction time 2, 3, 5, 10, 15, 20, and 30 min, respectively. The concentration of the product, p-nitrophenolate ion, is measured by the absorbance at 410 nm and calculated by using a millimolar extinction coefficient of 17.8. The control experiments with the cysteine-containing proteins BSA and hen egg lysozyme (both from Sigma) were performed under the same conditions, only the arsenate reductase was replaced by the same amount of control proteins. The Se-Met protein showed identical activity profiles as the wild-type B. subtilis arsenate reductase.
Protein Crystallization and Data Collection.
The crystallization of B. subtilis arsenate reductase is as described in ref. 14 for both wild-type and Se-Met-substituted proteins. Briefly, the hanging-drop vapor diffusion method was used to crystallize the arsenate reductase with the best conditions around pH 4.4–4.6 in 0.1 M sodium acetate and 0.2 M ammonium sulfate with the presence of 30–35% polyethyleneglycol methyl ether and 5 mM DTT. Thus, the crystal structure is in the reduced form. Although the crystallization condition and space group (P212121) of the wild-type and Se-Met proteins are identical, the cell parameters differ considerably, with a = 51.22 Å, b = 91.62 Å, and c = 101.93 Å for wild type and a = 50.94 Å, b = 94.31 Å, and c = 118.70 Å for Se-Met arsenate reductase.
All data collection experiments were performed on the beamline BL711 at the synchrotron lab MAX-II in Lund (Sweden) (15). BL711 is not a multiple-wavelength anomalous dispersion beamline; there is no scanning facility to locate the selenium absorption edge, thus all of the wavelength values used in this work are nominal, either calibrated from a powder sample or calculated from the angle setting of the monochromator. Diffraction data were processed by using the DENZO and SCALEPACK (16) packages or XDS package (17). The data collection statistics are listed in Table 1.
Table 1.
Statistics of crystallographic data, phasing, and refinement
| Se0* | Se1 | Se2 | Se3 | SeHg1 | SeHg2 | |
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Wavelength,† Å | 1.09 | 0.97 | 0.95 | 1.00 | 1.00 | 1.06 |
| Resolution, Å | 26.8–1.60 | 15.0–1.8 | 15.0–2.0 | 15.0–1.8 | 15.0–2.4 | 15.0–2.4 |
| (1.70–1.60) | (1.84–1.80) | (2.05–2.00) | (1.84–1.80) | (2.46–2.40) | (2.46–2.40) | |
| Completeness, % | 97.3 (97.4) | 99.8 (99.2) | 99.5 (98.5) | 99.3 (97.8) | 98.8 (99.8) | 100.0 (100.0) |
| Redundancy | 4.0 | 4.6 | 3.9 | 4.2 | 3.8 | 3.9 |
| Mean I/σ | 19.0 (4.7) | 22.1 (3.6) | 21.1 (4.9) | 21.5 (5.2) | 18.3 (4.4) | 18.4 (6.3) |
| Rsym,‡ % | 0.044 (0.30) | 0.045 (0.27) | 0.047 (0.21) | 0.043 (0.23) | 0.067 (0.27) | 0.061 (0.18) |
| All Se acentric/centric | All Se and SeHg acentric/centric | |||||
| Phasing§ | ||||||
| Phasing power¶ | 1.44/0.86 | 1.35/0.74 | ||||
| Rcullis‖ | 0.78/0.86 | 0.84/0.88 | ||||
| Mean figure of merit | 0.45/0.16 | 0.38/0.14 |
| Refinement | |||
| Resolution, Å | 26.8–1.60 | Rcryst** | 0.226 |
| No. of atoms | Rfree‡‡ | 0.241 | |
| Protein | 4,090 | rms bond lengths, Å | 0.007 |
| Ions | 20 | rms bond angles, ° | 1.20 |
| Solvent | 260 | Ramachandran angles most favored, % | 95.8 |
| No. of reflections | 74,177 | Ramachandran angles additional allowed, % | 4.0 |
Numbers in parentheses are the resolution limits of the outmost shells.
Data set used for the final refinement.
The wavelength values are nominal as described.
Rsym = (Σ |I − 〈I〉|/Σ I), where I is the observed intensity and 〈I〉 is the average intensity of symmetry-related reflections.
The phasing statistics are calculated at 3.0 Å from solve. Although the statistics of selenium multiple-wavelength anomalous dispersion phasing alone seems better than that of all data phasing, the structure was solved from all data-phasing phases as described.
Rcullis = (Σ∥FPH− FP − FH(calc)/Σ|FPH − FP|), where FP and FH are protein and heavy-atom structure factors, respectively, and FH(calc) is the calculated heavy-atom structure factor.
Phasing power = (FH(calc)/E), where E is the estimated lack-of-closure error (isomorphous/anomalous).
Rcryst = Σh∥Fobs| − |Fcalc∥/Σh|Fobs|, where Fcalc and Fobs are calculated and observed structure factors.
Rfree = Rcryst for a 5% subset of the reflections that were not included in the refinement.
Structure Solution and Refinement.
The crystal structure of B. subtilis arsenate reductase was solved by a combination of multiple-wavelength anomalous dispersion and isomorphous replacement methods. Three-wavelength selenium and two-wavelength mercury (sodium methylmercurithiosalicylate-soaked Se-Met crystal) data sets were used for the structure determination. Four arsenate reductase molecules per asymmetric unit were expected from the self-rotation function. Although the multiple-wavelength anomalous dispersion data collected were not optimal, the anomalous differences were sufficient to find eight selenium sites both in SHELXS (18) and SOLVE (19). Four of these selenium sites were put into SOLVE again and used to identify the remaining selenium and mercury sites. Altogether, the phases were calculated from the three-wavelength selenium anomalous differences, the two-wavelength mercury anomalous differences (the selenium contribution was ignored), and the isomorphous differences between the selenium and mercury versus the selenium alone. From these phases, four noncrystallographic symmetry operations were determined by using GETAX (CCP4) (20). Noncrystallographic symmetry averaging and solvent-flattening were then performed by using the program DM (21). The backbone was automatically traced by using ARP/WARP5.1 (22); a total of 440 residues in 12 chains were picked. The backbone chains were grouped together into four molecules; their side chains were then automatically fitted.
This structure was initially refined in SHELX97 (18); further refinements were done by using cns (23) with a 1.6-Å resolution data set (see Table 1). No noncrystallographic symmetry restrain was applied because of the high resolution. The final model consists of 519 residues for all four molecules, 4 sulfate ions, and 261 water molecules with good stereochemistry (Table 1). The missing residues are not visible in the density map. The structural evaluation by using PROCHECK (24) indicated that 95.8% of the residues are in the most favorable regions of the Ramachandran plot. The R factor and Rfree are 0.226 and 0.241, respectively. The graphics programs XFIT/XtalView (25) and O (26) were used to examine the maps and build the structures.
Results and Discussion
Overall Structure of B. subtilis Arsenate Reductase.
The B. subtilis arsenate reductase contains 139 amino acid residues and is a monomer in solution. In the crystal, arsenate reductase packs into four molecules (chains A, B, C, and D) per asymmetric unit. Despite only 18% identity between B. subtilis arsenate reductase and bovine LMW PTPase (BPTP) sequences and unsuccessful trials of using the molecular replacement methods, the refined arsenate reductase structure is surprisingly similar to that of BPTP (27) (Protein Data Bank ID code 1PHR). The root-mean-square deviation is 2.2 Å between these two structures for 114 Cα atoms compared. An overlay of B. subtilis arsenate reductase (yellow) with BPTP (blue) shows their striking similarity (Fig. 1C). The structure can be described as a single α/β domain containing a central four-stranded, parallel open-twisted β-sheet flanked by α-helices on both sides (Fig. 1B).
Most parts of the different molecules are well ordered; chains A and B contain all residues from 3 to 139. However, the helix α4 plus the connecting loop to strand β4, i.e., residues from Cys-82 to Val-96, are mostly invisible in chains C and D, implying a flexible structure in this region (see Fig. 1A). The well-ordered chains A and B have good protein–protein contacts in the regions from Cys-82 to Val-96, whereas in chains C and D such contacts are missing. Preliminary analysis of other crystal forms also indicated flexibility in this region (data not shown). This flexibility is of profound importance in the understanding of enzyme mechanism, as this region harbors the catalytically important cysteines 82 and 89.
Our Se-Met-substituted crystals diffracted to much higher resolution (14); the Met residues are far away from the active site, and the activity on a PTPase substrate (see below) showed no difference between the wild-type and Se-Met enzymes. Thus, the high-resolution structure of the Se-Met enzyme is reported here. Chain B is chosen for detailed discussions.
Comparison of the Active Sites of Arsenate Reductase and PTPases.
As was expected from the sequence alignment, the residues CTGNSCR (residues 10–16) in B. subtilis form an oxyanion binding site named arsenate binding loop (AB loop). This loop resembles the so called PTP loop that is the catalytic site for all classes of PTPases and has a signature motif CX5R (28). The Cα root-mean-square deviation of the AB loop and the PTP loop in BPTP is 0.6 Å (for residues between 9 and 17). There is an oxyanion, identified as a sulfate ion because of the crystallization conditions, tightly bound in the loop. All of the backbone amide groups, except for Asn-13, are turned to face to and within hydrogen bond distance to this oxyanion as seen for other PTP loops. The amide group of Asn-13 is pointed away from the oxyanion because of a flip of the peptide bond of Gly-12 (Fig. 2A). This configuration change of Gly-12 makes the AB loop slightly larger than the PTP loop. A similar kind of peptide-bond flip has been observed before only in an empty PTP loop of cdc25A (29). The AB loop is expected to better accommodate the slightly larger arsenate ion; the sulfate ion bound to the AB loop sits more than 1.4 Å deeper than the sulfate ion in the PTP loop of BPTP (see Fig. 1C). The sulfate binding geometry depicted here can be used only as the first-order approximation to the arsenate binding, as the charge and other chemical properties differ significantly between sulfate and arsenate ions. Further investigations are needed to elucidate the substrate binding and product release.
Figure 2.

(A) Stereoview of the active site with the residues labeled. (B) The AB loop, sulfate ion, and surroundings are superimposed with 3Fobs − 2Fcalc density map calculated from the refined model contoured at 1.0 σ. (C) The space-filling model to show the half-buried active site and the triple cysteine residues, Cys-10, Cys-82, and Cys-89. The structure figures in Figs. 1 and 2 are made by the programs RIBBONS (38) and MOLSCRIPT (39).
Another major difference in the arsenate reductase active site compared with that of BPTP is the Sγ atom of the key residue Cys-10. This atom points away from the center of the AB loop because of a rotation of about 150° of the χ angle relative to the Cys-12 in BPTP (Fig. 2 A and B). Furthermore, the Cys-10 Sγ is about 3.9 Å away from Cys-82 Sγ. Whereas Cys-82 Sγ is hydrogen bonded to Thr-11 Oγ and a water molecule, the Cys-10 Sγ is within hydrogen-bond distance (about 3.3 Å averaged from all four molecules) with an oxygen atom of the bound sulfate ion. The conserved basic residue Arg-16 is found nearby the cysteine pair 10 and 82 with the distances Cys-10 Sγ to Arg-16 Nη2 3.5 Å, and Cys-82 Sγ to Arg-16 Nη1 3.7 Å (see Fig. 2A). The placement of Arg-16 is ideal for a role to lower the pKa values of the cysteines and stabilize the thiolate ion(s) needed during catalysis. This intriguing positioning of the three residues Cys-10, Arg-16, and Cys-82 together with the sulfate ion (presumably an arsenate mimic) makes the previously proposed mechanism plausible (3). The position of Cys-10 makes it possible to place the Sγ atom back and forth from the oxyanion to the Cys-82. In this reduced form of arsenate reductase structure, the other essential residue, Cys-89, is situated at the other end of the helix α4 (see Fig. 1A), about 11 Å away from Cys-82. Because the α4 and nearby region is flexible as observed in chains C and D, the Cys-89 can be expected to come close to the active site to form a disulfide bridge with Cys-82 during a reaction cycle. It is believed that this reduced structure with closely placed Cys-10 and Cys-82 reported here represents the most active form of the enzyme during a catalytic cycle.
An aspartate, Asp-105, is found to be quite close to the active site, about 4.1 Å away from the sulfate ion; a well-ordered water molecule is found to form hydrogen bonds to both the sulfate and the Asp-105 (Fig. 2 A and B). This aspartate is absolutely conserved in all Gram-positive arsenate reductases and all kinds of LMW PTPases (see Fig. 1A). Such an aspartate positioned close to the active site can also be found in all classes of PTPases (28), playing a key role as general acid/general base catalyst. The corresponding aspartate in other PTPases such as Yop51 (30) and PTP1B (31, 32) is located on a mobile loop that can swing toward the active site upon substrate binding. Thus, we have reasons to believe that the Asp-105 can come closer to the active site during the enzymatic reaction.
In BPTP, the active site is surrounded by hydrophobic residues such as Leu-13, Ile-16, Trp-49, Tyr-131, and Tyr-132; these residues are believed to participate in protein–substrate interactions (27, 33). In B. subtilis arsenate reductase, those residues are replaced by more polar residues Thr-11, Ser-14, His-42, Ala-107, and Arg-108 to facilitate the entry of arsenate ions to the active site.
The Arsenate Reduction Mechanism.
B. subtilis arsenate reductase can hydrolyze the PTPase substrate PNPP.
B. subtilis arsenate reductase can hydrolyze PNPP, which is commonly used as the substrate in PTPase assays (Fig. 3A) in vitro. The estimated kcat for this reaction is about 0.5 per min. Although this rate is manyfold slower than that of the PNPP hydrolysis by true PTPases, it suggests that the PTP catalytic mechanism is functioning in B. subtilis arsenate reductase. By analogy to the PTPases, the PNPP hydrolysis activity of arsenate reductase involves a nucleophilic attack on the phosphorus atom of the substrate by Cys-10 in an in-line associative mechanism (27). By comparison with the BPTP structure, the essential general acid/general base catalyst was identified as Asp-105; this identification could not be made unambiguously from multiple sequence alignment alone. We thus propose that the first step in the arsenate reduction of Gram-positive bacteria is similar to the first step in the PTP mechanism. Namely, arsenate reductase Cys-10 acts as an attacking nucleophile to form an arsenylated enzyme–substrate (ES) intermediate as suggested for E. coli arsenate reductase earlier (3). However, because the E. coli arsenate reductase is not a BPTP homolog, the detailed mechanism to form an ES intermediate in E. coli enzyme is expected to be different.
Figure 3.

(A) The PNPP hydrolysis by B. subtilis arsenate reductase. See Materials and Methods for detailed description of the experiments; cysteine-containing proteins lysozyme and BSA are used as controls. pNP, p-nitrophenolate. (B) The proposed catalytic mechanism for Gram-positive bacterial arsenate reductase. The notation H+S− refers to the sulfhydryl group of a cysteine residue that is prone to be deprotonated when placed close to a NH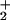 group. The first half of arsenate reduction is analogous to the first step in the PTPase mechanism. Namely, the arsenic atom in H2AsO
group. The first half of arsenate reduction is analogous to the first step in the PTPase mechanism. Namely, the arsenic atom in H2AsO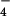 is subjected to a nucleophilic attack by Cys-10 thiolate formed because of the lowered pKa and helped by Asp-105 in an in-line associative mechanism to form an arsenylated enzyme–substrate (ES) intermediate; a water molecule is the leaving group. Then, this ES intermediate is attacked by the adjacent Cys-82 thiolate ion stabilized by Arg-16. The bound arsenate ion gets reduced to arsenite (first H2AsO
is subjected to a nucleophilic attack by Cys-10 thiolate formed because of the lowered pKa and helped by Asp-105 in an in-line associative mechanism to form an arsenylated enzyme–substrate (ES) intermediate; a water molecule is the leaving group. Then, this ES intermediate is attacked by the adjacent Cys-82 thiolate ion stabilized by Arg-16. The bound arsenate ion gets reduced to arsenite (first H2AsO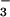 , then most likely loses one water molecule quickly to become AsO
, then most likely loses one water molecule quickly to become AsO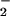 ) by obtaining two electrons from cysteines 10 and 82. Cys-10 and Cys-82 form a transient mixed disulfide bond similar to the mechanism of disulfide reduction involving Cys-32 in E. coli thioredoxin (40). On the other hand, the reduced Cys-89 can come close to the active site because of the flexible region. By the same token, the positive charge of Arg-16 nearby can lower the pKa value of the Cys-89 as well and make it prone to become an activated thiolate. Thus, upon the activation of Cys-89, Cys-82 and Cys-89 can be oxidized to form a disulfide bridge and leave the Cys-10 reduced for the next cycle. The disulfide bond Cys-82–Cys-89 will be reduced by the thioredoxin and thioredoxin reductase system to regenerate the whole system. In E. coli and yeast, arsenate reductases lack the essential cysteine pair; the role of the cysteine pair is proposed to be carried out by glutathione molecules.
) by obtaining two electrons from cysteines 10 and 82. Cys-10 and Cys-82 form a transient mixed disulfide bond similar to the mechanism of disulfide reduction involving Cys-32 in E. coli thioredoxin (40). On the other hand, the reduced Cys-89 can come close to the active site because of the flexible region. By the same token, the positive charge of Arg-16 nearby can lower the pKa value of the Cys-89 as well and make it prone to become an activated thiolate. Thus, upon the activation of Cys-89, Cys-82 and Cys-89 can be oxidized to form a disulfide bridge and leave the Cys-10 reduced for the next cycle. The disulfide bond Cys-82–Cys-89 will be reduced by the thioredoxin and thioredoxin reductase system to regenerate the whole system. In E. coli and yeast, arsenate reductases lack the essential cysteine pair; the role of the cysteine pair is proposed to be carried out by glutathione molecules.
A triple cysteine redox relay system.
The cysteines Cys-10, Cys-82, and Cys-89 have been identified as key residues in the arsenate reductase redox reaction in S. aureus, and Cys-82 and Cys-89 would form a disulfide bond upon oxidation (6). In the B. subtilis arsenate reductase structure, these cysteines are all located close to the protein surface, but mostly buried, with Cys-10 in the AB loop and Cys-82 Sγ more exposed to the solvent (Fig. 2C).
The simplest reaction scheme for arsenate reduction would involve a reduced cysteine pair, Cys-10 and Cys-82, to provide the electrons to the bound arsenate ion, and the cysteines get oxidized to form a disulfide bond as directly suggested in the structure. However, this scheme would prevent the Cys-10 from binding to another arsenate to form a new ES intermediate until this disulfide gets reduced by the thioredoxin system, which may be the rate-limiting step for this enzyme. The participation of Cys-89 from a flexible loop to form a disulfide bond away from the active site would solve this problem. This triple cysteine redox relay would facilitate the enzyme turnover by separating the arsenate binding from the disulfide reduction; see Fig. 3B for details.
The proposed catalytic mechanism.
Summarizing the above two steps, a complete arsenate reduction mechanism is proposed and shown in full detail in Fig. 3B. Briefly, the first step (the ES complex formation) involves the activated Cys-10 thiolate attacking the substrate, arsenate, to form an ES complex with the help of Asp-105 as a general acid in an in-line associative manner. A water molecule will leave to facilitate the complex formation. The second step (the arsenate reduction) involves the three catalytic essential cysteines identified earlier in the S. aureus arsenate reductase (6), Cys-10, Cys-82, and Cys-89, in a so-called triple cysteine redox relay system to produce the Cys-82–Cys-89 disulfide bond and an arsenite ion. The most important residue identified in this mechanism is the positive-charged Arg-16, which has the roles of stabilizing the AB loop and binding the arsenate ion; more important, its positive charge can lower the pKa values of all three cysteines to activate them for the reactions. The other important residue, Asp-105, works as a general acid to facilitate the leaving of a water molecule, thus stabilizing the ES complex.
The product, the arsenite ion, is most likely coupled to the membrane arsenite-specific transporter and exported immediately rather than being released into the cell, as the arsenite itself is even more toxic than the arsenate ion. More studies are needed to identify the binding and release modes of the substrate and product in this interesting system. It is believed that this mechanism deduced from the B. subtilis arsenate reductase structure is testable; thus, more experiments that either confirm or disprove this mechanism are expected. Studying the effects of the alterations of the identified residues important to catalysis will provide us with further details of this enzyme mechanism.
In this study, the B. subtilis arsenate reductase and BPTP are shown to share a common evolutionary origin functionally and structurally, although the evolution history between E. coli and B. subtilis arsenate reductases is most likely convergent. It is interesting to note that perhaps not by coincidence the yeast arsenate reductase is a homologue of the catalytic domain of cdc25A, which is also a CX5R motif-containing PTPase. We suggest that all of the arsenate reductases containing the CX5R motif would have a two-step catalytic mechanism similar to the one proposed here.
The PTP loop or the CX5R motif has been shown to catalyze diversified biological reactions. It not only participates in dephosphorylating phosphotyrosine or phosphoserine/phosphothreonine on protein substrates but also works as an RNA triphosphatase (34) in mRNA capping events and as a dephosphorylase acting on phosphorylated carbohydrate substrates (35). The common theme of all of the reactions catalyzed in this active site is dephosphorylation. The arsenate reduction mechanism proposed here has added another function to this well-conserved active site. Further structural and functional investigations on the arsenate reductase should contribute not only to deciphering the arsenate detoxification mechanism but also to a more detailed understanding of the PTPase mechanism from another angle.
Acknowledgments
We thank Drs. Lars Hederstedt, Per Nordlund, and Anders Liljas for invaluable discussions, and Mr. Michel Fodje for kind help with computer graphics. M.S.B. is supported by the Medical Faculty, Lund University; X.D.S. is supported by the Swedish Foundation for Strategic Research and the Structural Biology Network.
Abbreviations
- PTPase
protein tyrosine phosphatase
- LMW
low molecular weight
- BPTP
bovine low molecular weight protein tyrosine phosphatase
- Se-Met
selenomethionine
- PNPP
p-nitrophenyl phosphate
- ES
enzyme–substrate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1JL3).
References
- 1.Silver S, Keach D. Proc Natl Acad Sci USA. 1982;79:6114–6118. doi: 10.1073/pnas.79.20.6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rensing C, Ghosh M, Rosen B P. J Bacteriol. 1999;181:5891–5897. doi: 10.1128/jb.181.19.5891-5897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gladysheva T B, Oden K L, Rosen B P. Biochemistry. 1994;33:7288–7293. doi: 10.1021/bi00189a033. [DOI] [PubMed] [Google Scholar]
- 4.Ji G, Garber E A, Armes L G, Chen C M, Fuchs J A, Silver S. Biochemistry. 1994;33:7294–7299. doi: 10.1021/bi00189a034. [DOI] [PubMed] [Google Scholar]
- 5.Liu J, Gladysheva T B, Lee L, Rosen B P. Biochemistry. 1995;34:13472–13476. doi: 10.1021/bi00041a026. [DOI] [PubMed] [Google Scholar]
- 6.Messens J, Hayburn G, Desmyter A, Laus G, Wyns L. Biochemistry. 1999;38:16857–16865. doi: 10.1021/bi9911841. [DOI] [PubMed] [Google Scholar]
- 7.Takemaru K, Mizuno M, Sato T, Takeuchi M, Kobayashi Y. Microbiology. 1995;141:323–327. doi: 10.1099/13500872-141-2-323. [DOI] [PubMed] [Google Scholar]
- 8.Sato T, Kobayashi Y. J Bacteriol. 1998;180:1655–1661. doi: 10.1128/jb.180.7.1655-1661.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bobrowicz P, Wysocki R, Owsianik G, Goffeau A, Ulaszewski S. Yeast. 1997;13:819–828. doi: 10.1002/(SICI)1097-0061(199707)13:9<819::AID-YEA142>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 10.Mukhopadhyay R, Rosen B P. J Biol Chem. 2001;276:34738–34742. doi: 10.1074/jbc.M103354200. [DOI] [PubMed] [Google Scholar]
- 11.Ji G, Silver S. Proc Natl Acad Sci USA. 1992;89:9474–9478. doi: 10.1073/pnas.89.20.9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Mel V S, Doyle M A, Gladysheva T B, Oden K L, Martin P D, Rosen B P, Edwards B F. J Mol Biol. 1994;242:701–702. doi: 10.1006/jmbi.1994.1618. [DOI] [PubMed] [Google Scholar]
- 13.Jacobs D M, Messens J, Wechselberger R W, Brosens E, Willem R, Wyns L, Martin J C. J Biomol NMR. 2001;20:95–96. doi: 10.1023/a:1011207007790. [DOI] [PubMed] [Google Scholar]
- 14.Guan, Z., Hederstedt, L., Li, J. P. & Su, X. D. (2001) Acta Crystallogr. D, in press. [DOI] [PubMed]
- 15.Cerenius Y, Ståhl K, Svensson L A, Ursby T, Oskarsson Å, Albertsson J, Liljas A. J Synchrotron Radiat. 2000;7:203–208. doi: 10.1107/S0909049500005331. [DOI] [PubMed] [Google Scholar]
- 16.Otwinowski Z, Minor W. Methods Enzymol. 1996;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 17.Kabsch W. J Appl Crystallogr. 1988;21:916–924. [Google Scholar]
- 18.Sheldrick G M. Acta Crystallogr A. 1990;46:467–473. [Google Scholar]
- 19.Terwilliger T C, Berendzen J. Acta Crystallogr D. 1996;52:749–757. doi: 10.1107/S0907444996000832. [DOI] [PubMed] [Google Scholar]
- 20.CCP4 (Collaborative Computational Project 4) (1994) Acta Crystallogr. D50, 760–763. [DOI] [PubMed]
- 21.Cowtan K D. Joint CCP4 and ESF-EACBM Newsletter Protein Crystallography. 1994;31:34–38. [Google Scholar]
- 22.Lamzin V S, Wilson K S. Acta Crystallogr D. 1993;49:129–149. doi: 10.1107/S0907444992008886. [DOI] [PubMed] [Google Scholar]
- 23.Brunger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 24.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 25.McRee D E. J Mol Graphics. 1992;10:44–46. [Google Scholar]
- 26.Jones T A, Zou J-Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 27.Su X D, Taddei N, Stefani M, Ramponi G, Nordlund P. Nature (London) 1994;370:575–578. doi: 10.1038/370575a0. [DOI] [PubMed] [Google Scholar]
- 28.Fauman E B, Saper M A. Trends Biochem Sci. 1996;21:413–417. doi: 10.1016/s0968-0004(96)10059-1. [DOI] [PubMed] [Google Scholar]
- 29.Fauman E B, Cogswell J P, Lovejoy B, Rocque W J, Holmes W, Montana V G, Piwnica-Worms H, Rink M J, Saper M A. Cell. 1998;93:617–625. doi: 10.1016/s0092-8674(00)81190-3. [DOI] [PubMed] [Google Scholar]
- 30.Stuckey J A, Schubert H L, Fauman E B, Zhang Z Y, Dixon J E, Saper M A. Nature (London) 1994;370:571–575. doi: 10.1038/370571a0. [DOI] [PubMed] [Google Scholar]
- 31.Barford D, Flint A J, Tonks N K. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- 32.Pannifer A D, Flint A J, Tonks N K, Barford D. J Biol Chem. 1998;273:10454–10462. doi: 10.1074/jbc.273.17.10454. [DOI] [PubMed] [Google Scholar]
- 33.Zhang M, Van Etten R L, Stauffacher C V. Biochemistry. 1994;33:11097–11105. doi: 10.1021/bi00203a006. [DOI] [PubMed] [Google Scholar]
- 34.Changela A, Ho C K, Martins A, Shuman S, Mondragon A. EMBO J. 2001;20:2575–2586. doi: 10.1093/emboj/20.10.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee J O, Yang H, Georgescu M M, Di Cristofano A, Maehama T, Shi Y, Dixon J E, Pandolfi P, Pavletich N P. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 36.Thompson J D, Gibson T J, Plewniak F, Jeanmougin F, Higgins D G. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barton G J. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]
- 38.Carson M. J Appl Crystallogr. 1991;24:958–961. [Google Scholar]
- 39.Kraulis P. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 40.Holmgren A. Structure. 1995;3:239–243. doi: 10.1016/s0969-2126(01)00153-8. [DOI] [PubMed] [Google Scholar]